Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases


Abstract
:1. Introduction
2. HDACs in the Brain
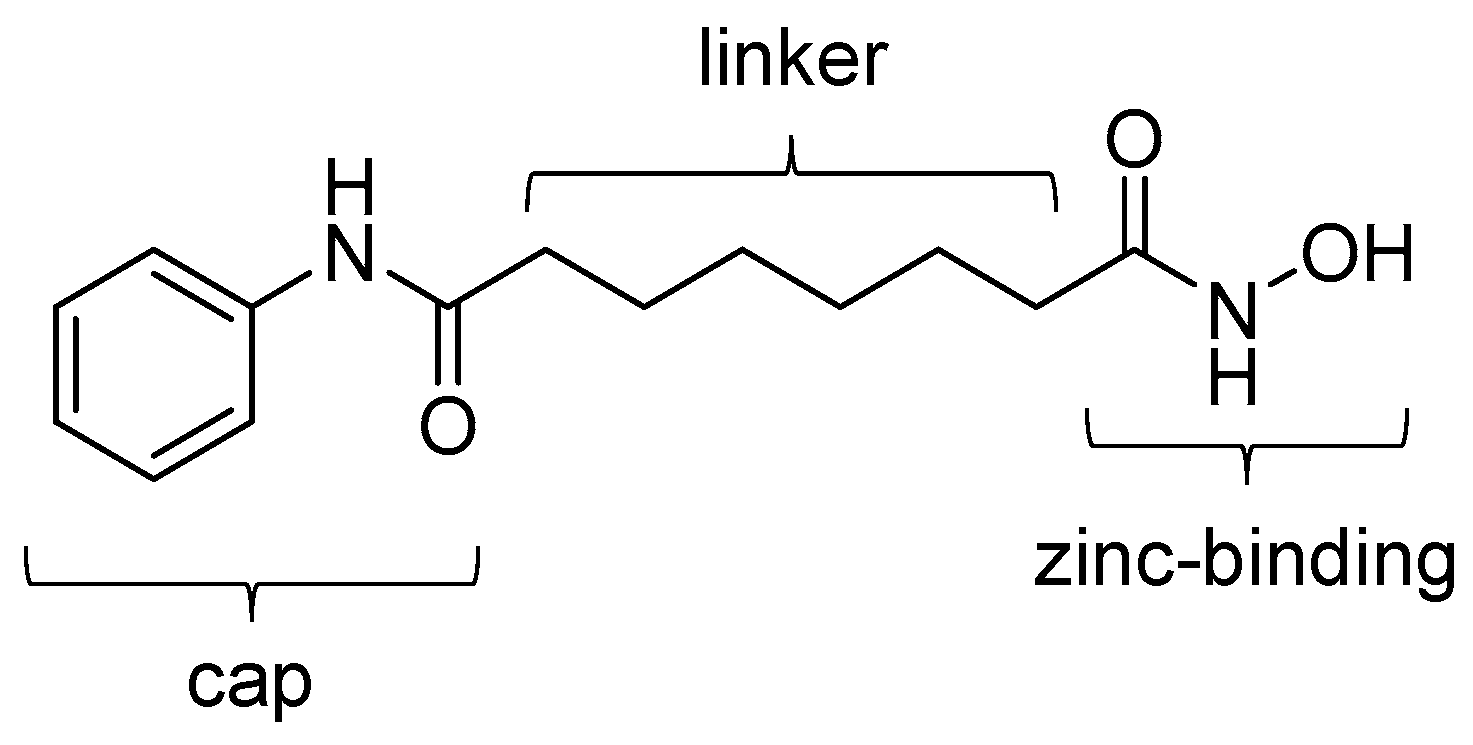
3. HDAC Inhibitors
4. Radioligands for HDACs
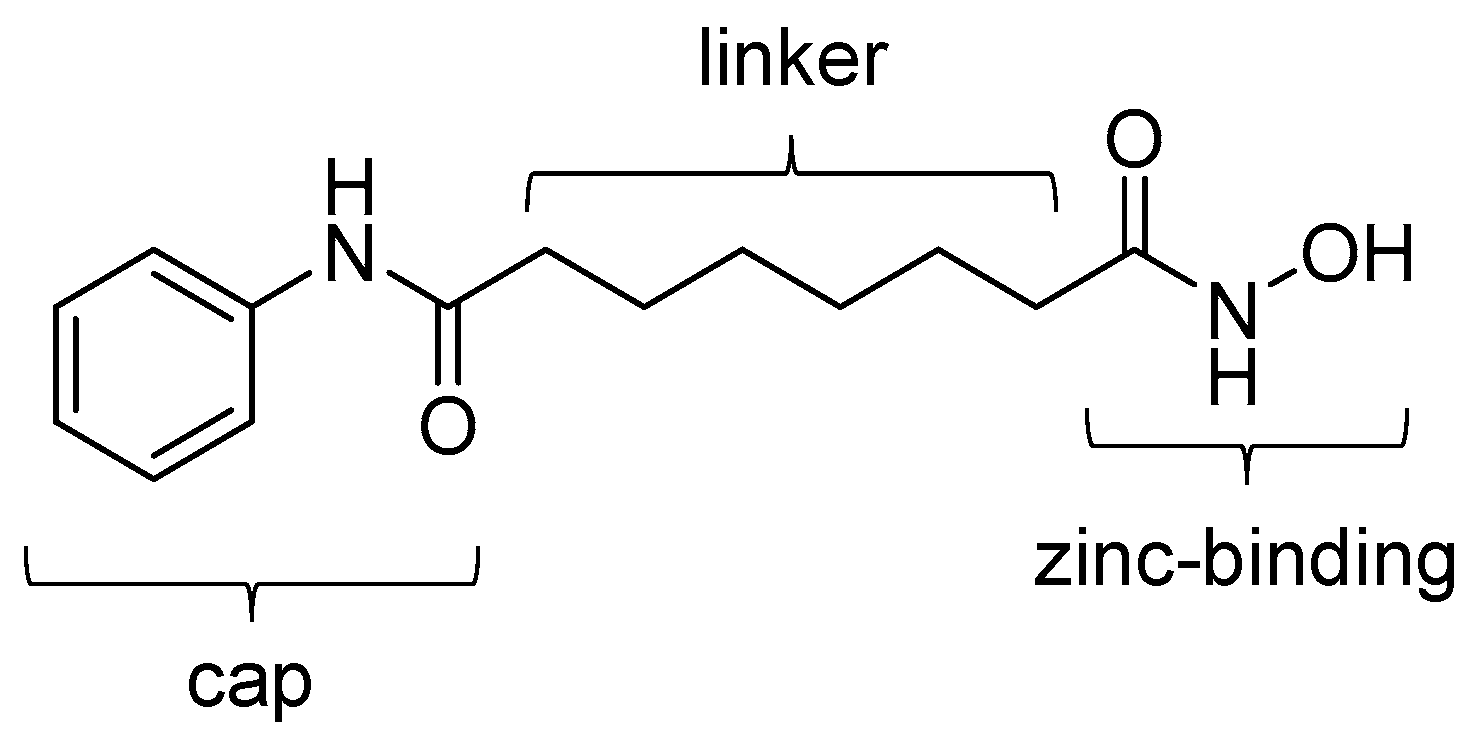
4.1. SAHA-Based Ligands
4.2. Adamantane-Conjugated Ligands
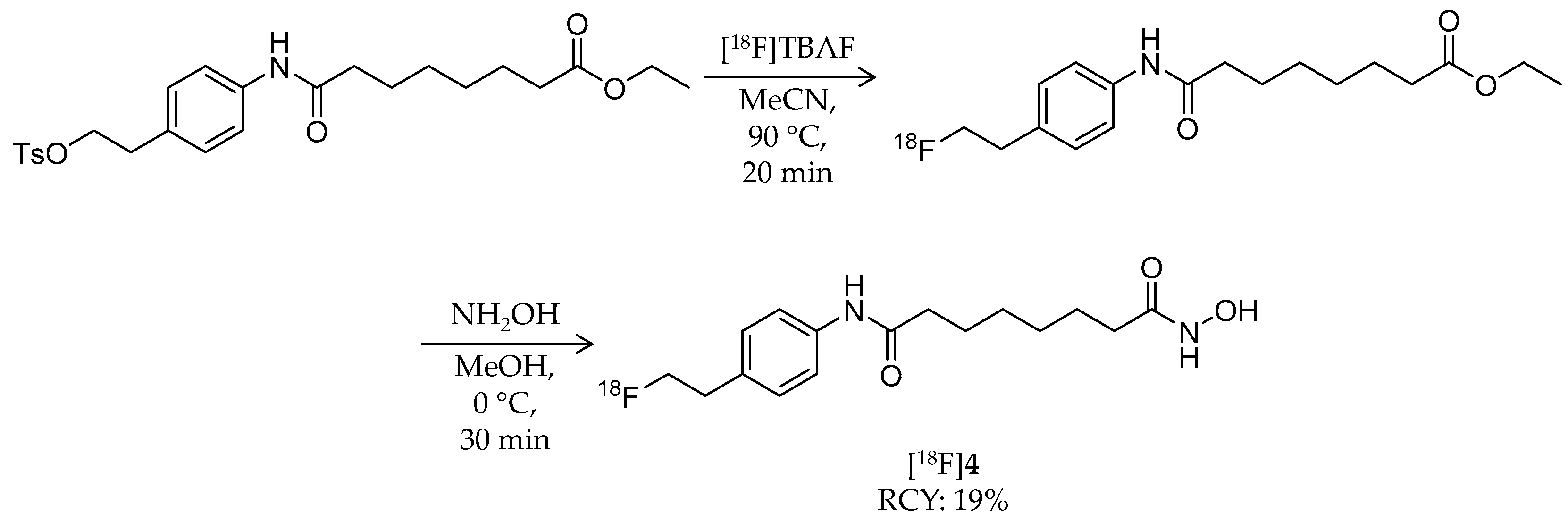
4.3. Other Carboxylic Acid- and Hydroxamic Acid-Based Ligands
4.4. Ortho-Aminoanilide-Based Ligands
4.5. First-in-Human PET Study
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Landgrave-Gomez, J.; Mercado-Gomez, O.; Guevara-Guzman, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Lovrečić, L.; Maver, A.; Zadel, M.; Peterlin, B. The Role of Epigenetics in Neurodegenerative Diseases. In Neurodegenerative Diseases; InTech: London, UK, 2013; pp. 345–365. ISBN 978-953-51-1088-0. [Google Scholar]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A. On the Enzymatic Properties of Dnmt1: Specificity, Processivity, Mechanism of Linear Diffusion and Allosteric Regulation of the Enzyme. Epigenetics 2014, 1, 63–66. [Google Scholar] [CrossRef]
- Turek-Plewa, J.; Jagodzinski, P.P. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell. Mol. Biol. Lett. 2005, 10, 631–647. [Google Scholar] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Wood, M.A. Beyond transcription factors: The role of chromatin modifying enzymes in regulating transcription required for memory. Learn. Mem. 2008, 15, 460–467. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Joseph, N.F.; Horn, M.E.; Samiei, A.; Meng, J.; Seo, J.; Rei, D.; Bero, A.W.; Phan, T.X.; Wagner, F.; et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 2014, 156, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Nestler, E.J. Epigenetic mechanisms in drug addiction. Trends Mol. Med. 2008, 14, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Langley, B.; Lubin, F.D.; Renthal, W.; Wood, M.A.; Yasui, D.H.; Kumar, A.; Nestler, E.J.; Akbarian, S.; Beckel-Mitchener, A.C. Epigenetics in the nervous system. J. Neurosci. 2008, 28, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 2009, 4, e6617. [Google Scholar] [CrossRef] [PubMed]
- Politis, M. Neuroimaging in Parkinson disease: From research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.P.; Liang, S.H.; Rotstein, B.H.; Collier, T.L.; Stephenson, N.A.; Greguric, I.; Vasdev, N. Alternative approaches for PET radiotracer development in Alzheimer’s disease: Imaging beyond plaque. J. Label. Comp. Radiopharm. 2014, 57, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Wesmall yi, U.M.; Lewis, M.C.; Holson, E.B. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 2013, 10, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.L.; Yang, W.M. Beyond histone and deacetylase: An overview of cytoplasmic histone deacetylases and their nonhistone substrates. J. Biomed. Biotechnol. 2011, 2011, 146493. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Anna, G.D.S.S.; Elsner, V.R.; Moyses, F.; Cechinel, R.L.; Lovatel, G.A.; Siqueira, I.R. Histone deacetylase activity is altered in brain areas from aged rats. Neurosci. Lett. 2013, 556, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.W.; Chen, J.; Wang, M.; Mast, N.; Pikuleva, I.A.; Turko, I.V. Quantification of histone deacetylase isoforms in human frontal cortex, human retina, and mouse brain. PLoS ONE 2015, 10, e0126592. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Tian, M.; Hinz, R.; Young, D.; Shavrin, A.; Mukhapadhyay, U.; Flores, L.G.; Balatoni, J.; Soghomonyan, S.; Jeong, H.J.; et al. Imaging epigenetic regulation by histone deacetylases in the brain using PET/MRI with 18F-FAHA. Neuroimage 2013, 64, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, A.; Doherty, K.; Yeh, H.H.; Robinson, A.C.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.; Thompson, J.C.; Davidson, Y.S.; Mann, D.M. Histone deacetylases (HDACs) in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 2015, 41, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.C.; Casaccia, P. HDAC inhibitors and neurodegeneration: At the edge between protection and damage. Pharmacol. Res. 2010, 62, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Webb, Y.; Merger, R.; Sheppard, T.; Jursic, B.; Ngo, L.; Civoli, F.; Breslow, R.; Rifkind, R.A.; Marks, P.A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Opal, P. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015, 2, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Frechilla, D.; Del Rio, J.; Garcia-Osta, A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 2009, 34, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Yang, L.; Cleren, C.; Calingasan, N.Y.; Klivenyi, P.; Beal, M.F. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromol. Med. 2004, 5, 235–241. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.; Tong, W.P.; Gelovani, J.G.; Alauddin, M.M. Radiosynthesis of 6-([18F]fluoroacetamido)-1-hexanoicanilide ([18F]FAHA) for PET imaging of histone deacetylase (HDAC). J. Label. Compd. Radiopharm. 2006, 49, 997–1006. [Google Scholar] [CrossRef]
- Nishii, R.; Mukhapadhyay, U.; Yeh, H.; Soghomonyan, S.; Volgin, A.; Alauddin, M.; Tong, W.; Gelovani, J. PET imaging of histone deacetylase activity in a rat brain using 6-([18F]-fluoroacetamide)-1-hexanoicanilide ([18F]-FAHA). J. Nucl. Med. 2007, 48 (Suppl. 2), 336. [Google Scholar]
- Nishii, R.; Mukhapadhyay, U.; Yeh, H.; Soghomonyan, S.; Volgin, A.; Alauddin, M.; Tong, W.; Gelovani, J. Non-invasive imaging of histone deacetylase activity in human breast carcinoma xenografts in rats using positron emission tomography (PET) with [18F]-FAHA. J. Nucl. Med. 2007, 48 (Suppl. 2), 34. [Google Scholar]
- Reid, A.E.; Hooker, J.; Shumay, E.; Logan, J.; Shea, C.; Kim, S.W.; Collins, S.; Xu, Y.; Volkow, N.; Fowler, J.S. Evaluation of 6-([18F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain. Nucl. Med. Biol. 2009, 36, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.L.; Ackermann, R.F. Evaluation of radiolabeled acetate and fluoroacetate as potential tracers of cerebral oxidative metabolism. Metab. Brain Dis. 1990, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Pourghiasian, M.; Hundal, N.; Lau, J.; Benard, F.; Dedhar, S.; Lin, K.S. 2-[18F]fluoroethanol and 3-[18F]fluoropropanol: Facile preparation, biodistribution in mice, and their application as nucleophiles in the synthesis of [18F]fluoroalkyl aryl ester and ether PET tracers. Nucl. Med. Biol. 2013, 40, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Luurtsema, G.; Schuit, R.C.; Takkenkamp, K.; Lubberink, M.; Hendrikse, N.H.; Windhorst, A.D.; Molthoff, C.F.; Tolboom, N.; van Berckel, B.N.; Lammertsma, A.A. Peripheral metabolism of [18F]FDDNP and cerebral uptake of its labelled metabolites. Nucl. Med. Biol. 2008, 35, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET imaging of the dopamine transporter with 18F-FECNT: A polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Ponde, D.E.; Dence, C.S.; Oyama, N.; Kim, J.; Tai, Y.C.; Laforest, R.; Siegel, B.A.; Welch, M.J. 18F-fluoroacetate: A potential acetate analog for prostate tumor imaging—In vivo evaluation of 18F-fluoroacetate versus 11C-acetate. J. Nucl. Med. 2007, 48, 420–428. [Google Scholar] [PubMed]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfi, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Kuruvilla, S.A.; Galitovskiy, V.; Pan, M.L.; Grando, S.A.; Mukherjee, J. Targeting histone deacetylase in lung cancer for early diagnosis: 18F-FAHA PET/CT imaging of NNK-treated A/J mice model. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 324–332. [Google Scholar] [PubMed]
- Neal, J.W.; Sequist, L.V. Exciting new targets in lung cancer therapy: ALK, IGF-1R, HDAC, and Hh. Curr. Treat. Opt. Oncol. 2010, 11, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.; Galitovskiy, V.; Edwards, R.; Andersen, B.; Grando, S.A. The tobacco carcinogen nitrosamine induces a differential gene expression response in tumour susceptible A/J and resistant C3H mouse lungs. Eur. J. Cancer 2013, 49, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Galitovskiy, V.; Kuruvilla, S.A.; Sevriokov, E.; Corches, A.; Pan, M.L.; Kalantari-Dehaghi, M.; Chernyavsky, A.I.; Mukherjee, J.; Grando, S.A. Development of novel approach to diagnostic imaging of lung cancer with 18F-Nifene PET/CT using A/J mice treated with NNK. J. Cancer Res. Ther. 2013, 1, 128–137. [Google Scholar]
- Bonomi, R.; Mukhopadhyay, U.; Shavrin, A.; Yeh, H.H.; Majhi, A.; Dewage, S.W.; Najjar, A.; Lu, X.; Cisneros, G.A.; Tong, W.P.; et al. Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Pillarsetty, N.; Divilov, V.; Blasberg, R.A.; Lewis, J.S. The synthesis and evaluation of N1-(4-(2-[18F]-fluoroethyl)phenyl)-N8-hydroxyoctanediamide ([18F]-FESAHA), a PET radiotracer designed for the delineation of histone deacetylase expression in cancer. Nucl. Med. Biol. 2011, 38, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Muench, L.; Reid, A.; Chen, J.; Kang, Y.; Hooker, J.M.; Volkow, N.D.; Fowler, J.S.; Kim, S.W. Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain. Bioorg. Med. Chem. Lett. 2013, 23, 6700–6705. [Google Scholar] [CrossRef] [PubMed]
- Majdzadeh, N.; Morrison, B.E.; D’Mello, S.R. Class IIA HDACs in the regulation of neurodegeneration. Front. Biosci. 2008, 13, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Riester, D.; Hildmann, C.; Grunewald, S.; Beckers, T.; Schwienhorst, A. Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun. 2007, 357, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zou, X.; Berger, A.D.; Twiss, C.; Peng, Y.; Li, Y.; Chiu, J.; Guo, H.; Satagopan, J.; Wilton, A.; et al. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am. J. Transl. Res. 2009, 1, 62–71. [Google Scholar] [PubMed]
- Gediya, L.K.; Chopra, P.; Purushottamachar, P.; Maheshwari, N.; Njar, V.C. A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells. J. Med. Chem. 2005, 48, 5047–5051. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Strobel, E.; Ralbovsky, J.; Galemmo, R.A., Jr. Improved solution- and solid-phase preparation of hydroxamic acids from esters. J. Org. Chem. 2005, 70, 4873–4875. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Xu, Y.; Schiffer, W.; Shea, C.; Carter, P.; Fowler, J.S. Pharmacokinetics of the potent hallucinogen, salvinorin A in primates parallels the rapid onset and short duration of effects in humans. Neuroimage 2008, 41, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schroeder, F.A.; Wey, H.Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Banister, S.D.; Wilkinson, S.M.; Longworth, M.; Stuart, J.; Apetz, N.; English, K.; Brooker, L.; Goebel, C.; Hibbs, D.E.; Glass, M.; et al. The synthesis and pharmacological evaluation of adamantane-derived indoles: Cannabimimetic drugs of abuse. ACS Chem. Neurosci. 2013, 4, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, N.; Hama, T.; Kawada, M.; Hasui, A.; Konishi, R.; Shiwa, S.; Ochi, Y.; Futaki, S.; Kitagawa, K. Adamantane as a brain-directed drug carrier for poorly absorbed drug. 2. AZT derivatives conjugated with the 1-adamantane moiety. J. Pharm. Sci. 1994, 83, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, B.; Ponpandian, T.; Kachhadia, V.; Bharathimohan, K.; Vignesh, R.; Sivasudar, V.; Narayanan, S.; Mandar, B.; Praveen, R.; Saranya, N.; et al. Discovery of adamantane based highly potent HDAC inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2532–2537. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Oda, K.; Ishiwata, K. Age-related changes of the [11C]CFT binding to the striatal dopamine transporters in the Fischer 344 rats: A PET study. Ann. Nucl. Med. 2003, 17, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.A.; Wang, C.; Van de Bittner, G.C.; Neelamegam, R.; Takakura, W.R.; Karunakaran, A.; Wey, H.Y.; Reis, S.A.; Gale, J.; Zhang, Y.L.; et al. PET imaging demonstrates histone deacetylase target engagement and clarifies brain penetrance of known and novel small molecule inhibitors in rat. ACS Chem. Neurosci. 2014, 5, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- Binaschi, M.; Boldetti, A.; Gianni, M.; Maggi, C.A.; Gensini, M.; Bigioni, M.; Parlani, M.; Giolitti, A.; Fratelli, M.; Valli, C.; et al. Antiproliferative and differentiating activities of a novel series of histone deacetylase inhibitors. ACS Med. Chem. Lett. 2010, 1, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.A.; Lewis, M.C.; Fass, D.M.; Wagner, F.F.; Zhang, Y.L.; Hennig, K.M.; Gale, J.; Zhao, W.N.; Reis, S.; Barker, D.D.; et al. A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS ONE 2013, 8, e71323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wey, H.Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [11C]Martinostat for In Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Strebl, M.G.; Wang, C.; Schroeder, F.A.; Placzek, M.S.; Wey, H.Y.; Van de Bittner, G.C.; Neelamegam, R.; Hooker, J.M. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem. Neurosci. 2016, 7, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [18F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Beyzavi, M.H.; Mandal, D.; Strebl, M.G.; Neumann, C.N.; D’Amato, E.M.; Chen, J.; Hooker, J.M.; Ritter, T. 18F-Deoxyfluorination of Phenols via Ru pi-Complexes. ACS Cent. Sci. 2017, 3, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Ritter, T. PhenoFluorMix: Practical chemoselective deoxyfluorination of phenols. Org. Lett. 2015, 17, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.M.; Shah, R.; Ghosh, B.; Hennig, K.; Norton, S.; Zhao, W.N.; Reis, S.A.; Klein, P.S.; Mazitschek, R.; Maglathlin, R.L.; et al. Short-Chain HDAC Inhibitors Differentially Affect Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2010, 2, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, P.; Poddar, B.; Parmar, V. Fatal cardiac malformation in fetal valproate syndrome. Indian J. Pediatr. 2001, 68, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Eessalu, T.E.; Barth, V.N.; Mitch, C.H.; Wagner, F.F.; Hong, Y.; Neelamegam, R.; Schroeder, F.A.; Holson, E.B.; Haggarty, S.J.; et al. Design, synthesis, and evaluation of hydroxamic acid-based molecular probes for in vivo imaging of histone deacetylase (HDAC) in brain. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 29–38. [Google Scholar] [PubMed]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar] [PubMed]
- Giles, F.; Fischer, T.; Cortes, J.; Garcia-Manero, G.; Beck, J.; Ravandi, F.; Masson, E.; Rae, P.; Laird, G.; Sharma, S.; et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res. 2006, 12, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, L.; Kalin, J.; Liow, J.S.; Gladding, R.L.; Innis, R.B.; Kozikowski, A.P.; Pike, V.W. Synthesis and evaluation of [methyl-11C]KB631—A candidate radioligand for histone deacetylase isozyme 6 (HDAC6). J. Label. Comp. Radiopharm. 2013, 56, S319. [Google Scholar]
- Kalin, J.H.; Butler, K.V.; Akimova, T.; Hancock, W.W.; Kozikowski, A.P. Second-generation histone deacetylase 6 inhibitors enhance the immunosuppressive effects of Foxp3+ T-regulatory cells. J. Med. Chem. 2012, 55, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, Y.; Kalin, J.H.; Cai, L.; Kozikowski, A.P.; Pike, V.W. Exploration of the labeling of [11C]tubastatin A at the hydroxamic acid site with [11C]carbon monoxide. J. Label. Comp. Radiopharm. 2016, 59, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Li, F.; Jiang, S.; Li, Z. Novel 64Cu-Labeled CUDC-101 for In Vivo PET Imaging of Histone Deacetylases. ACS Med. Chem. Lett. 2013, 4, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew. Chem. Int. Ed. Engl. 2008, 47, 8998–9033. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Lu, Z.; Luo, R.Z.; Zhang, X.; Seto, E.; Liao, W.S.; Yu, Y. Multiple histone deacetylases repress tumor suppressor gene ARHI in breast cancer. Int. J. Cancer 2007, 120, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [11C]MS-275 using Positron Emission Tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Dul, E.; Sung, C.M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Simonini, M.V.; Camargo, L.M.; Dong, E.; Maloku, E.; Veldic, M.; Costa, E.; Guidotti, A. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc. Natl. Acad. Sci. USA 2006, 103, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Reibel, A.T.; Hill, S.M.; Schueller, M.J.; Fowler, J.S. One-pot, direct incorporation of [11C]CO2 into carbamates. Angew. Chem. Int. Ed. Engl. 2009, 48, 3482–3485. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Kang, Y.; Muench, L.; Reid, A.; Caesar, S.; Jean, L.; Wagner, F.; Holson, E.; Haggarty, S.J.; Weiss, P.; et al. Image-guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem. Neurosci. 2014, 5, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.L.; Hennig, K.; Gale, J.P.; Hong, Y.; Cha, A.; Riley, M.; Wagner, F.; Haggarty, S.J.; Holson, E.; et al. Class I HDAC imaging using [3H]CI-994 autoradiography. Epigenetics 2013, 8, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
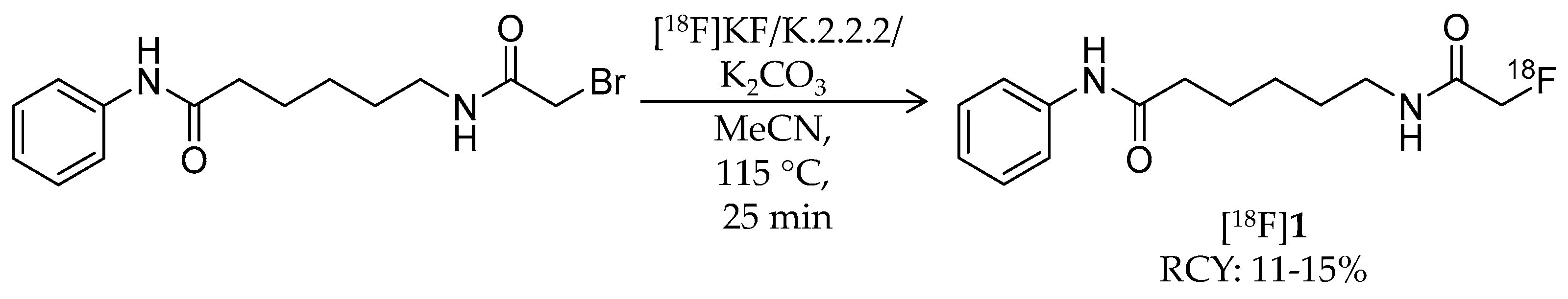



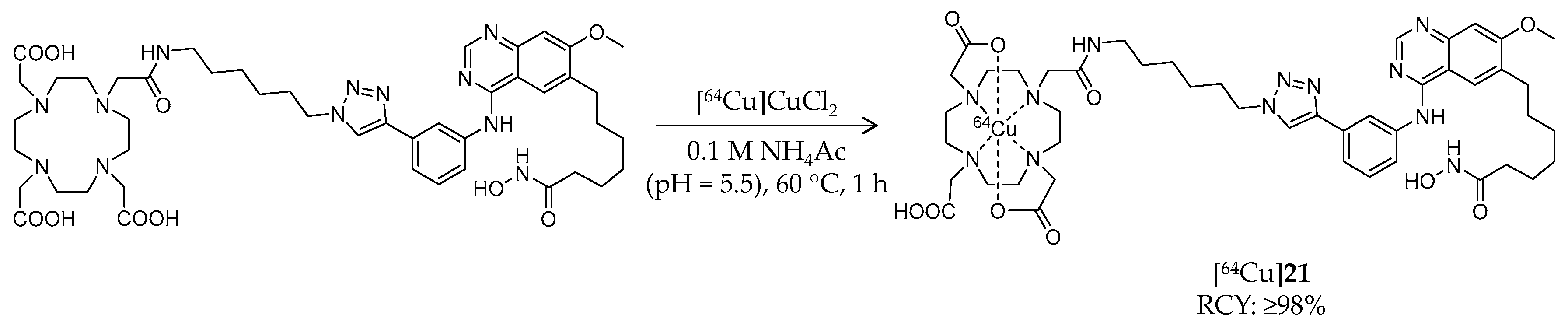

{kind=link}
{kind=link}
{kind=link}
{kind=link}
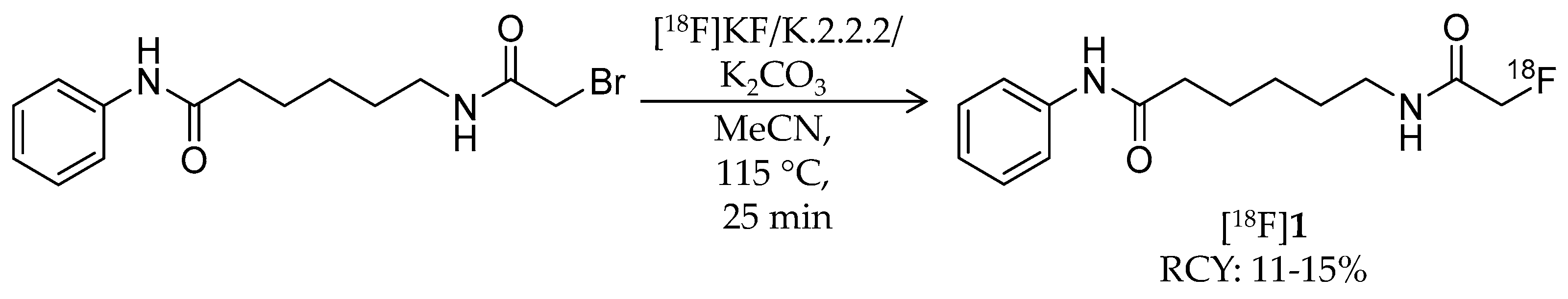{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
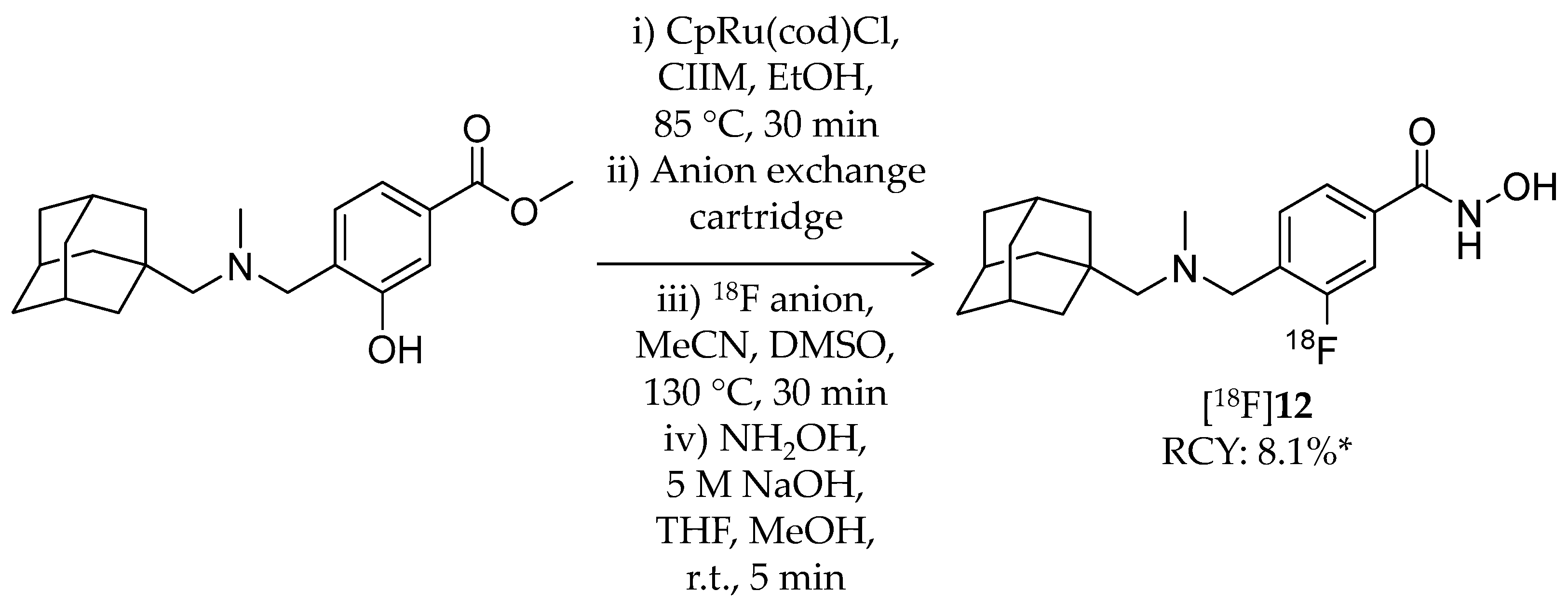{kind=link}
{kind=link}
{kind=link}
{kind=link}
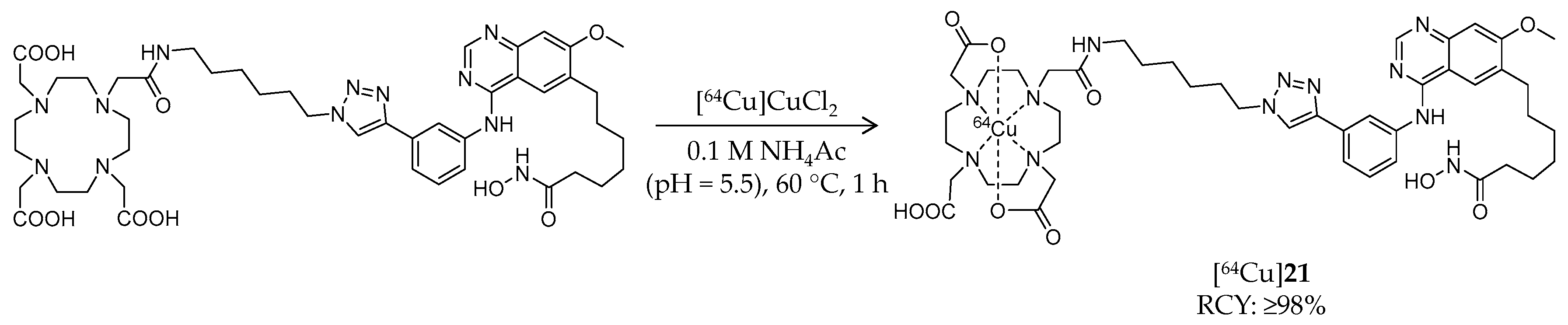{kind=link}
{kind=link}
{kind=link}
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
| [18F]1 | 265.32 | 1.39 | 11–15% | Class IIa | – | Brain uptake: 0.44 and 0.40% ID/g at 5 and 60 min, respectively (rat) | [26,43,44,46] |
| [18F]2 | 283.31 | – | 25% | Class IIa | – | Brain uptake: SUVs were around 1 in various brain regions during the 60-min scan (rat) | [57] |
| [18F]3 | 301.30 | – | 22% | Class IIa | – | Brain uptake: SUVs were around 1 in various brain regions during the 60-min scan (rat) | [57] |
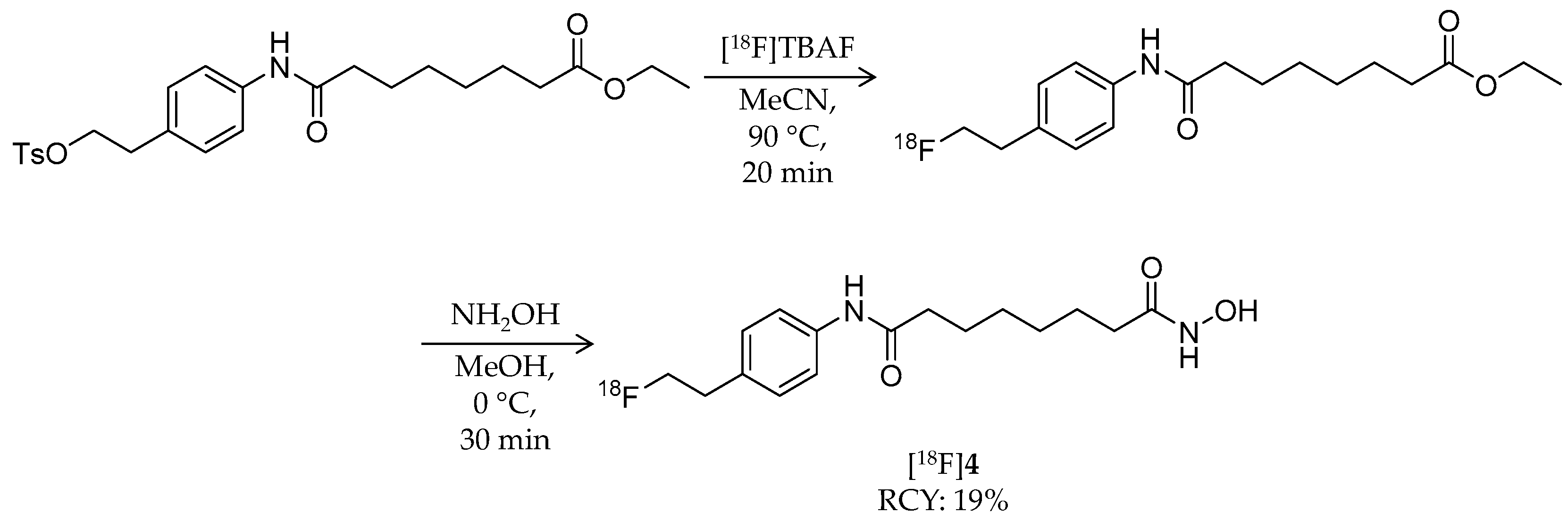
| [18F]4 | 309.37 | 1.01 | 19% | Non-selective | HDAC1: 56 nM HDAC5: 67 nM HDAC6: 3 nM HDAC7: 85 nM | Brain uptake: 1.0, 0.7, and 0.4% ID/g at 30, 60, and 120 min, respectively Defluorination: 9.6, 11.1, and 13.4% ID/g bone uptake at 30, 60, and 120 min, respectively (tumor-bearing mouse) | [58] |
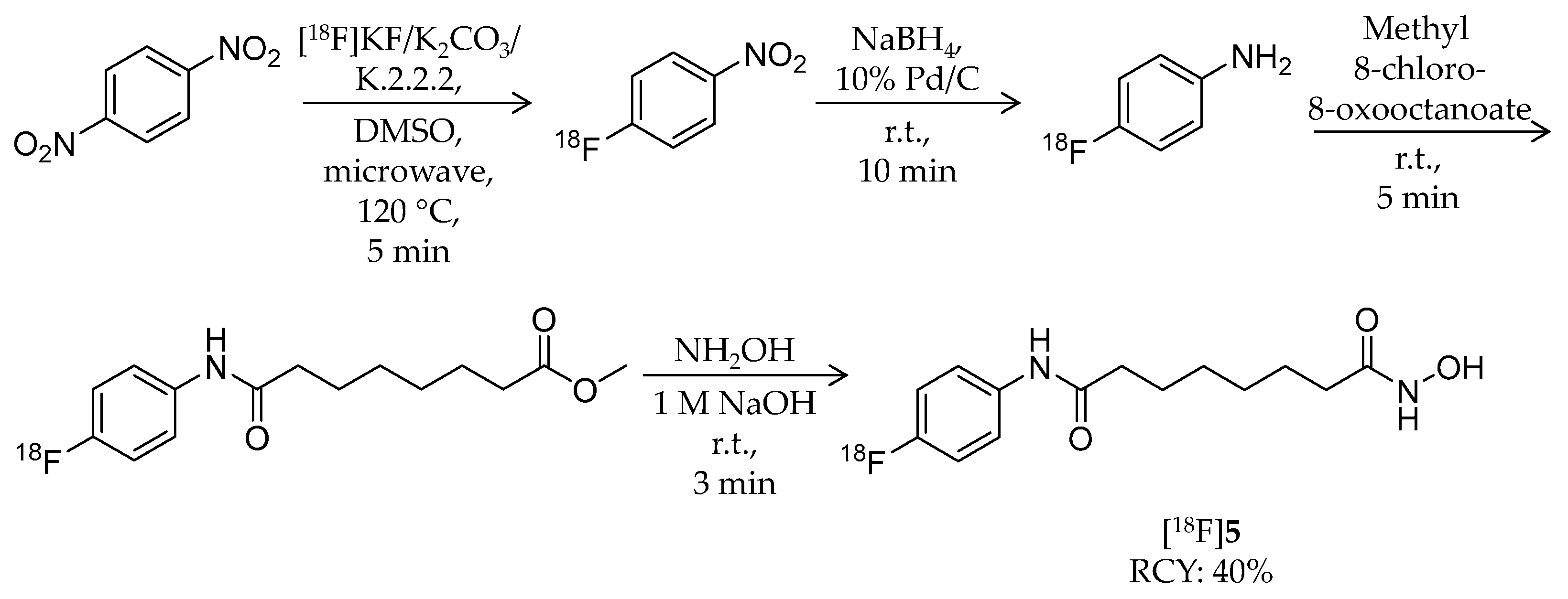
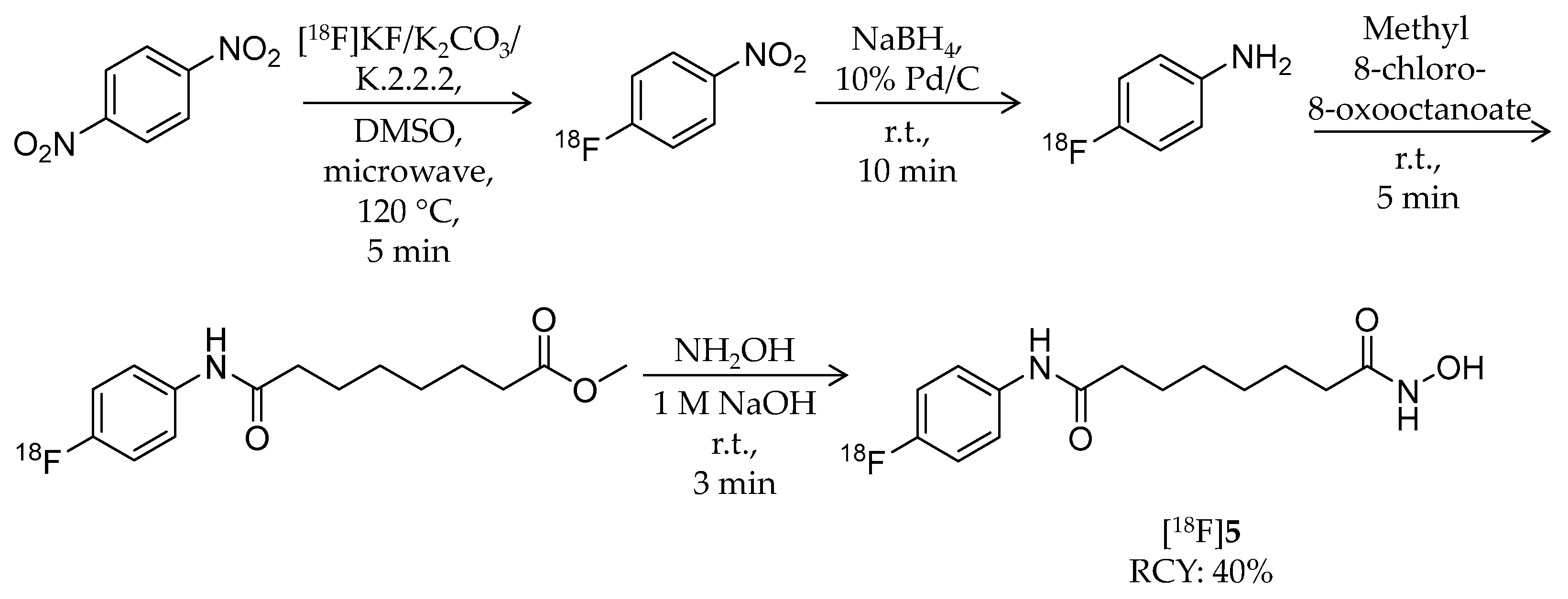
| [18F]5 | 281.32 | – | 40% | HDAC1–3/6 | HDAC1: 9.0 nM HDAC2: 13 nM HDAC3: 24 nM HDAC6: 50 nM | Brain uptake: Very limited in biodistribution study (mouse/tumor-bearing mouse) | [59] |
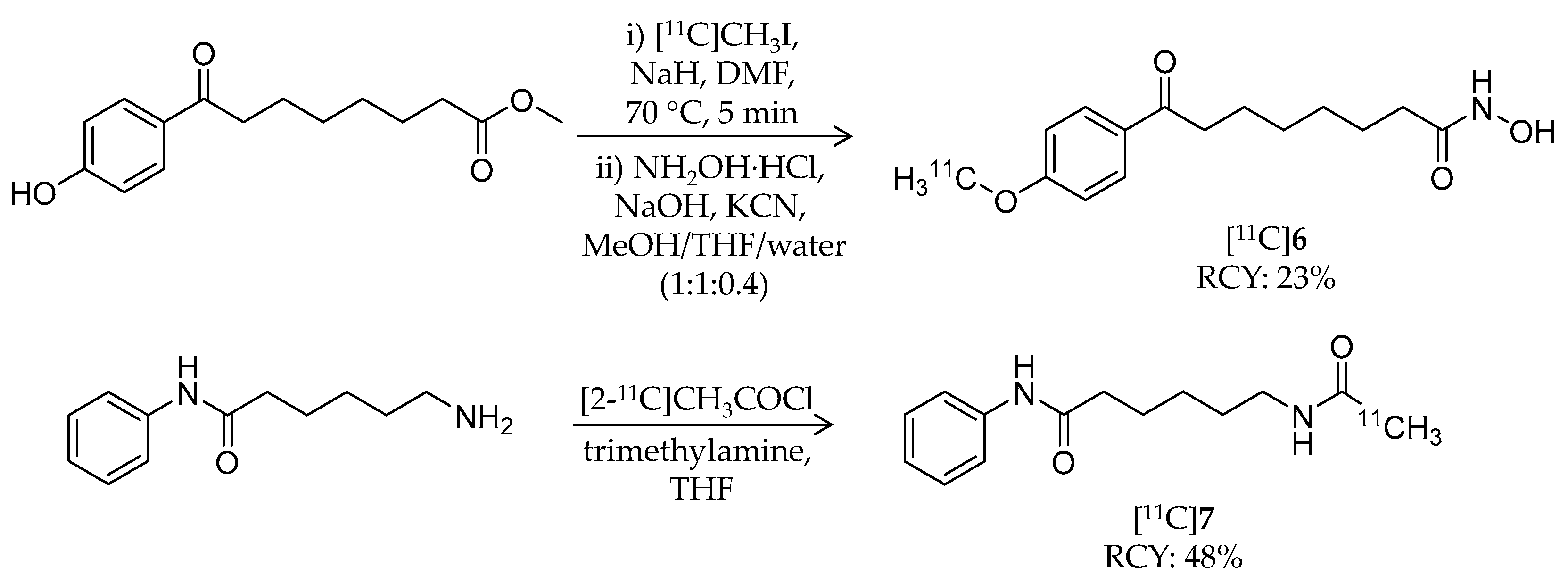
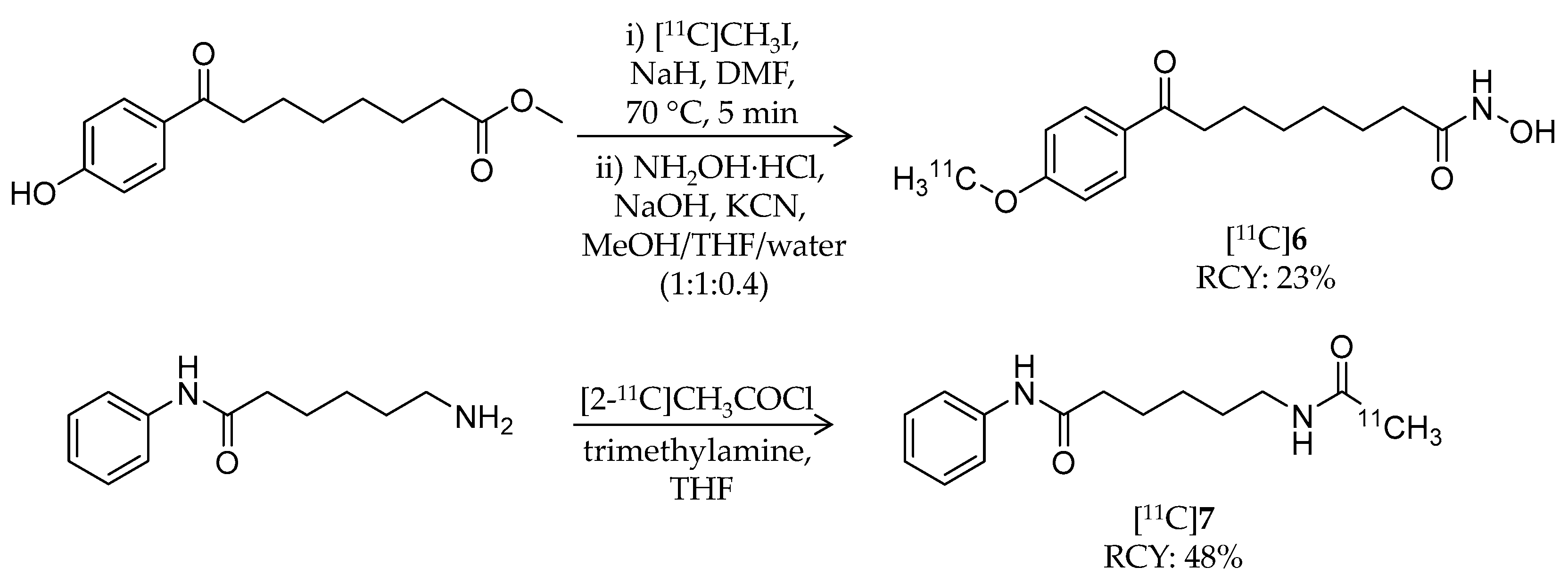
| [11C]6 | 278.34 | 0.5 | 23% | – | – | – | [60] |
| [11C]7 | 247.33 | 1.7 1 | 48% | – | – | – | [60] |
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
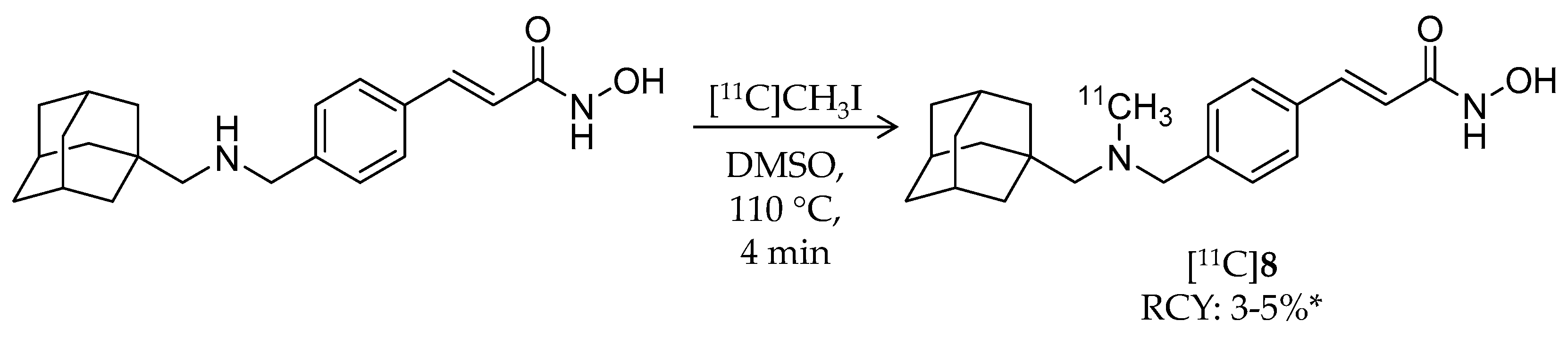
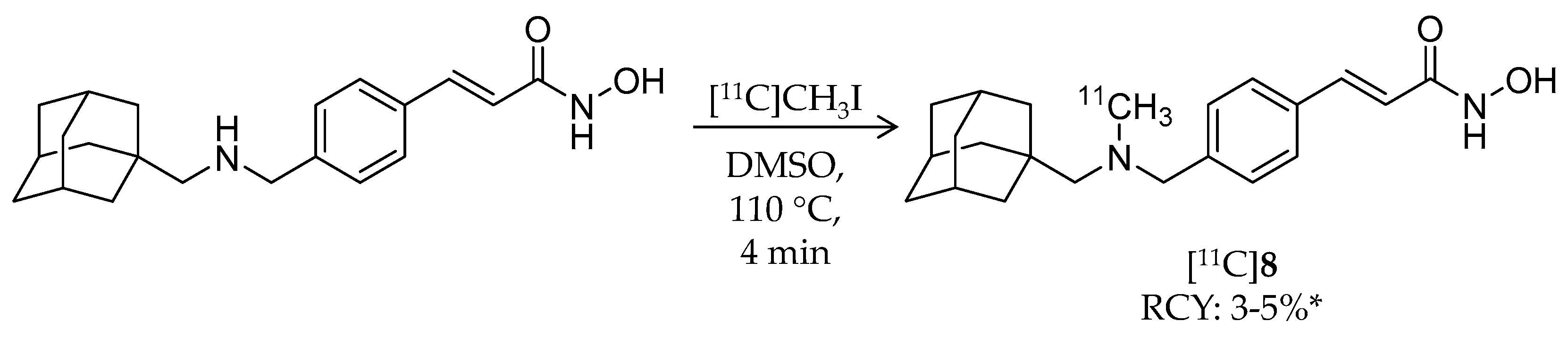
| [11C]8 | 353.49 | 2.03 | 3–5% 2 | Class I/IIb | HDAC1: 0.3 nM HDAC2: 2.0 nM HDAC3: 0.6 nM HDAC6: 4.1 nM | Brain uptake: Around 0.5% ID/cc during the 60-min scan Metabolism: Radiometabolites in the brain were limited (rat) | [28,68,74,78] |
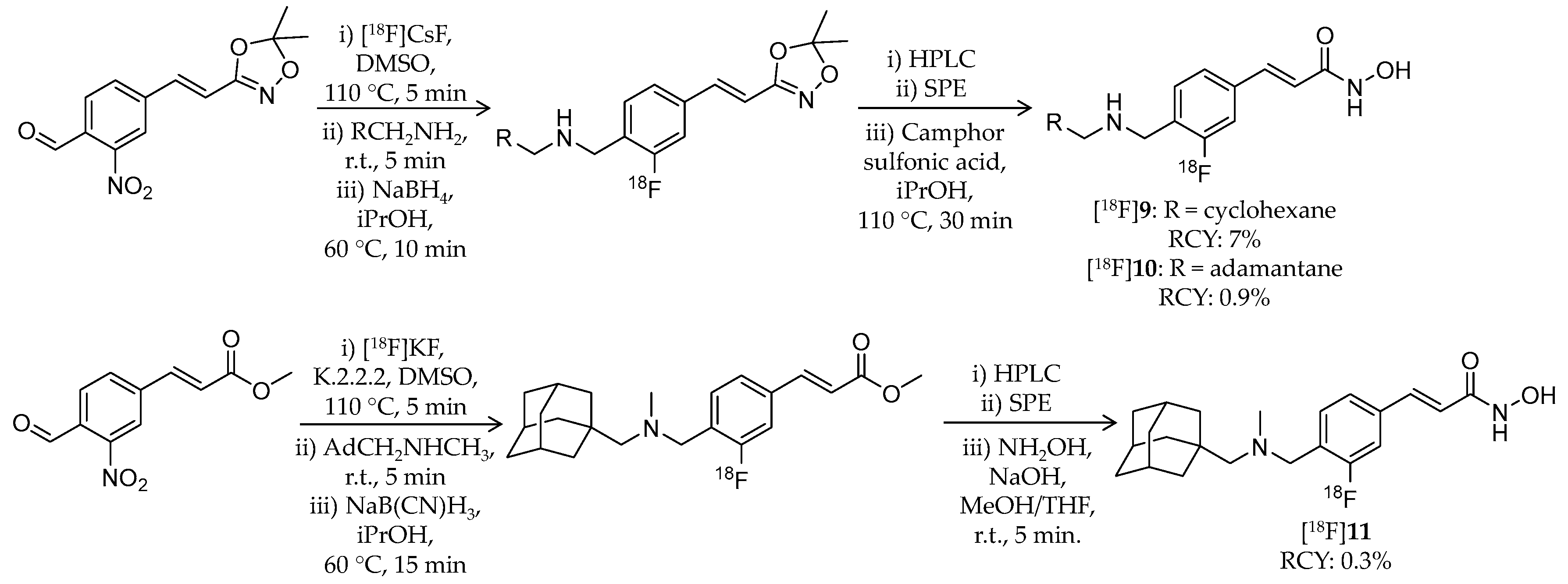
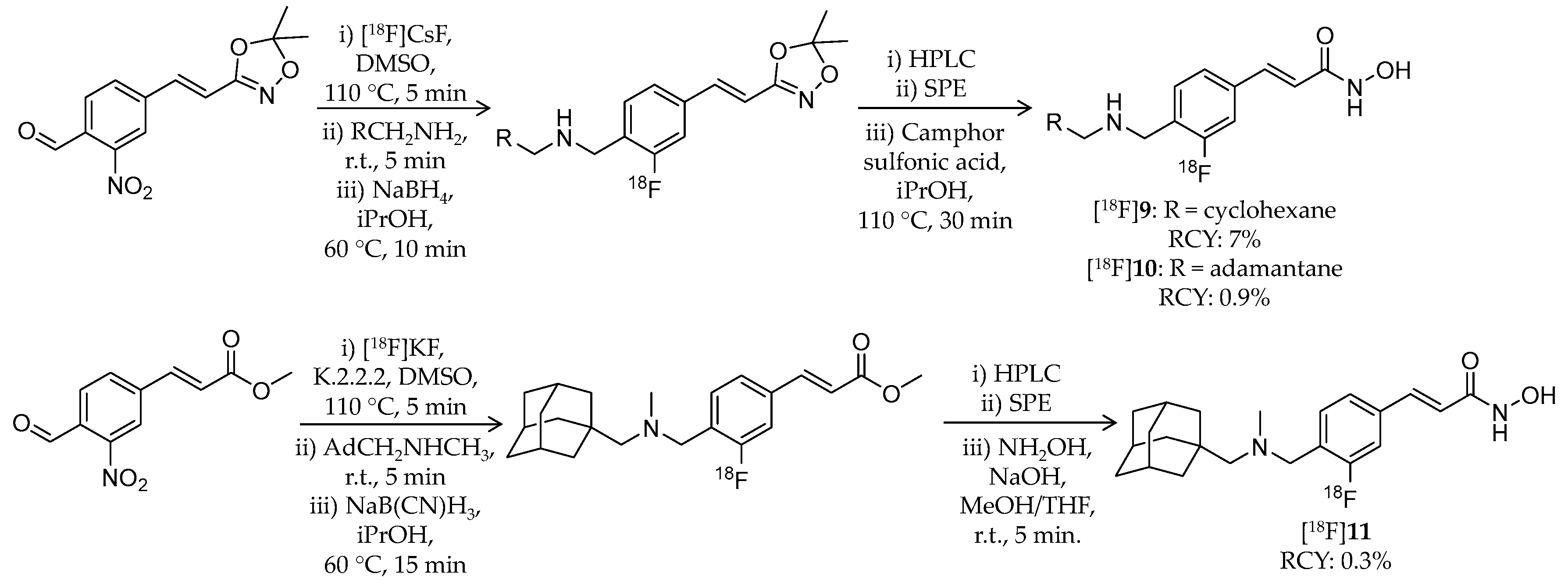
| [18F]9 | 305.38 | 3.07 1 | 7% | Class I/IIb | HDAC1: 1.6 nM HDAC2: 14 nM HDAC3: 0.5 nM HDAC6: 12 nM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [79] |
| [18F]10 | 357.46 | 3.77 1 | 0.9% | Class I/IIb | HDAC1: 0.8 nM HDAC2: 6.4 nM HDAC3: 0.5 nM HDAC6: 9.5 nM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [79] |
| [18F]11 | 371.49 | 4.17 1 | 0.3% | Class I/IIb | HDAC1: 0.8 nM HDAC2: 7.0 nM HDAC3: 0.8 nM HDAC6: 12 nM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [79] |
| [18F]12 | 345.45 | – | 8.1% 2 | HDAC6 | HDAC6: 60 nM Others: ≥1 μM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [80] |
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
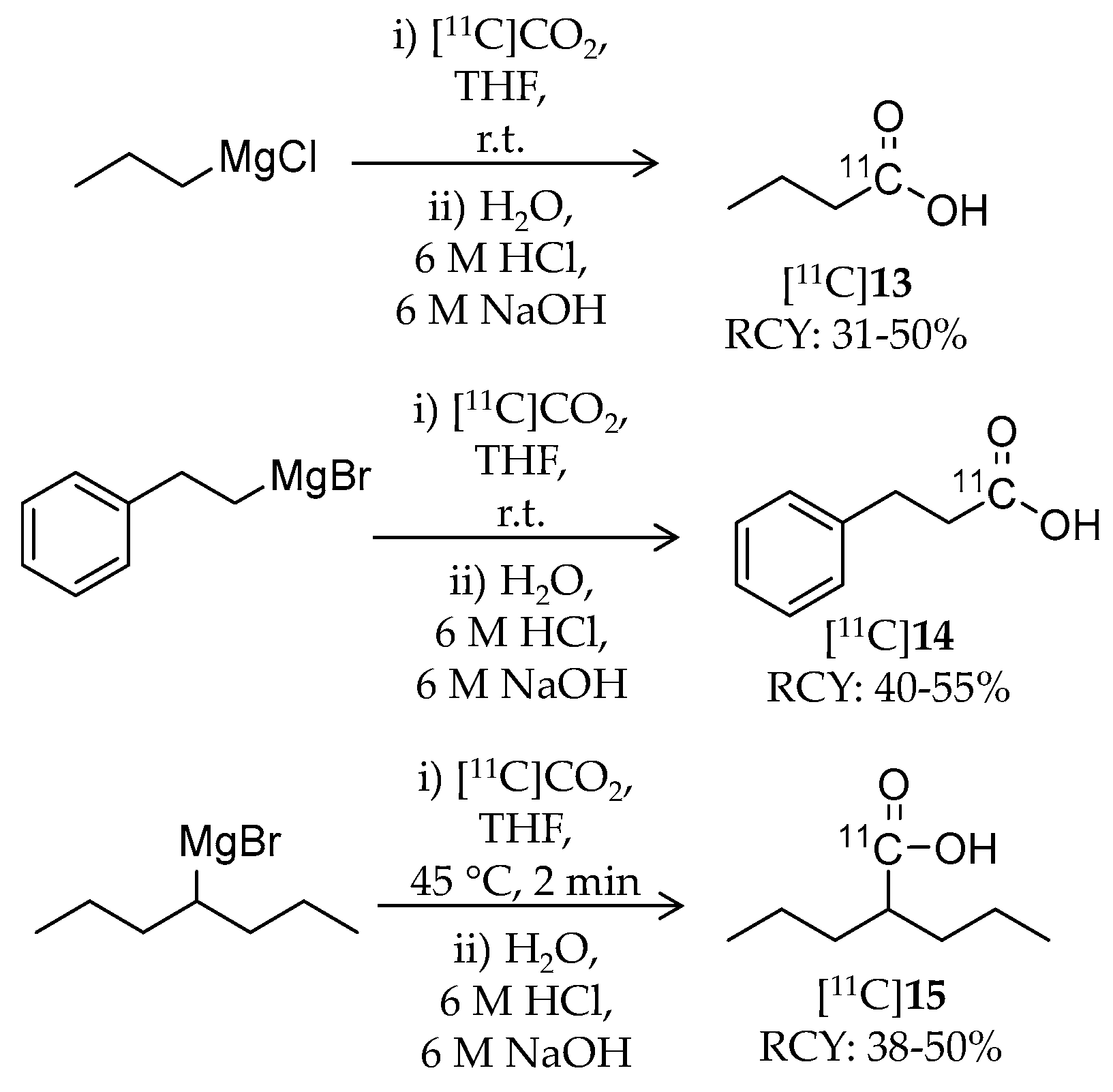
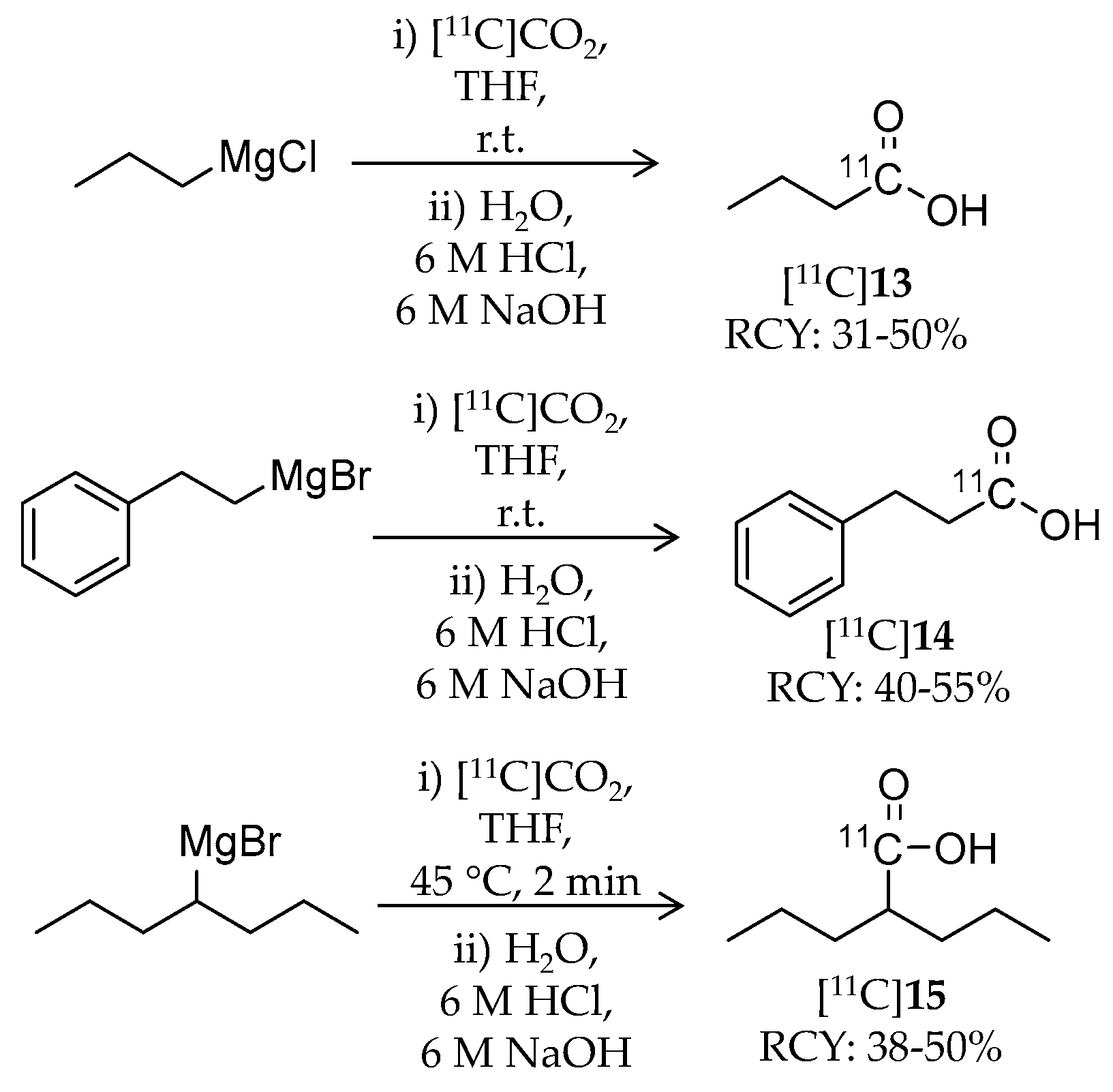
| [11C]13 | 87.11 | 1.02 | 31–50% | Class I | HDAC1: 16 μM HDAC2: 12 μM HDAC3: 9 μM HDAC8: 15 μM | – | [86,87] |
| [11C]14 | 149.18 | −0.20 | 40–55% | Class I | HDAC1: 64 μM HDAC2: 65 μM HDAC3: 260 μM HDAC8: 93 μM | – | [86,87] |
| [11C]15 | 143.21 | 0.26 | 38–50% | Class I | HDAC1: 39 μM HDAC2: 62 μM HDAC3: 161 μM HDAC8: 1103 μM | – | [86,87] |
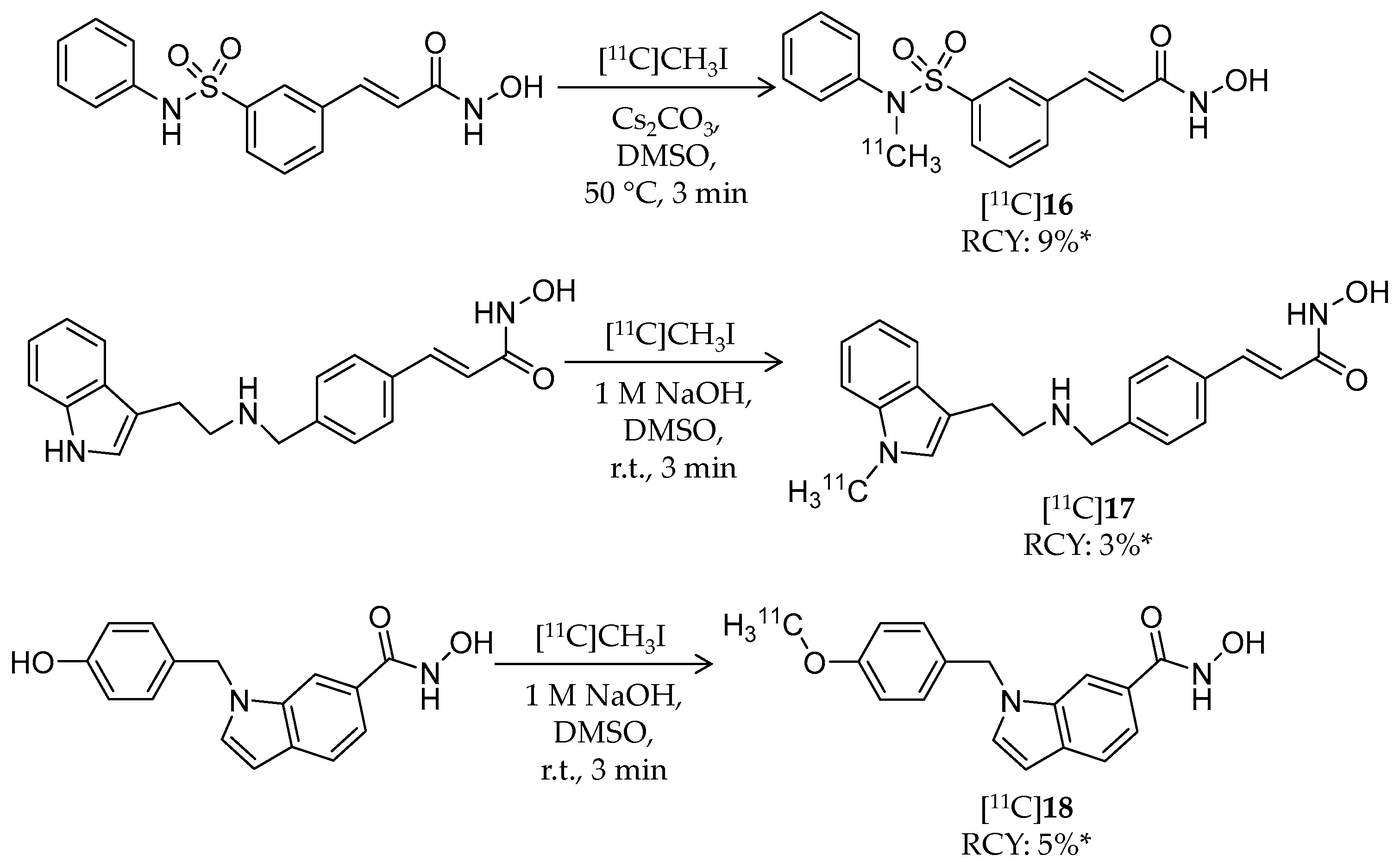
| [11C]16 | 331.37 | 2.03 1 | 9% 3 | HDAC1–3/6 | HDAC1: 5 nM HDAC2: 32 nM HDAC3: 3.4 nM HDAC6: 5 nM | Brain uptake: Around 0.1% ID/cc immediately after i.v. injection (rat) | [89] |
| [11C]17 | 348.43 | 2.66 1 | 3% 3 | HDAC1–3/6 | HDAC1: 0.2 nM HDAC2: 1.2 nM HDAC3: 0.4 nM HDAC6: 1.6 nM | Brain uptake: Around 0.15% ID/cc immediately after i.v. injection (rat) | [89] |
| [11C]18 | 295.33 | 2.61 1 | 5% 3 | HDAC8 | HDAC8: 18 nM | Brain uptake: Around 0.2% ID/cc immediately after i.v. injection (rat) | [89] |
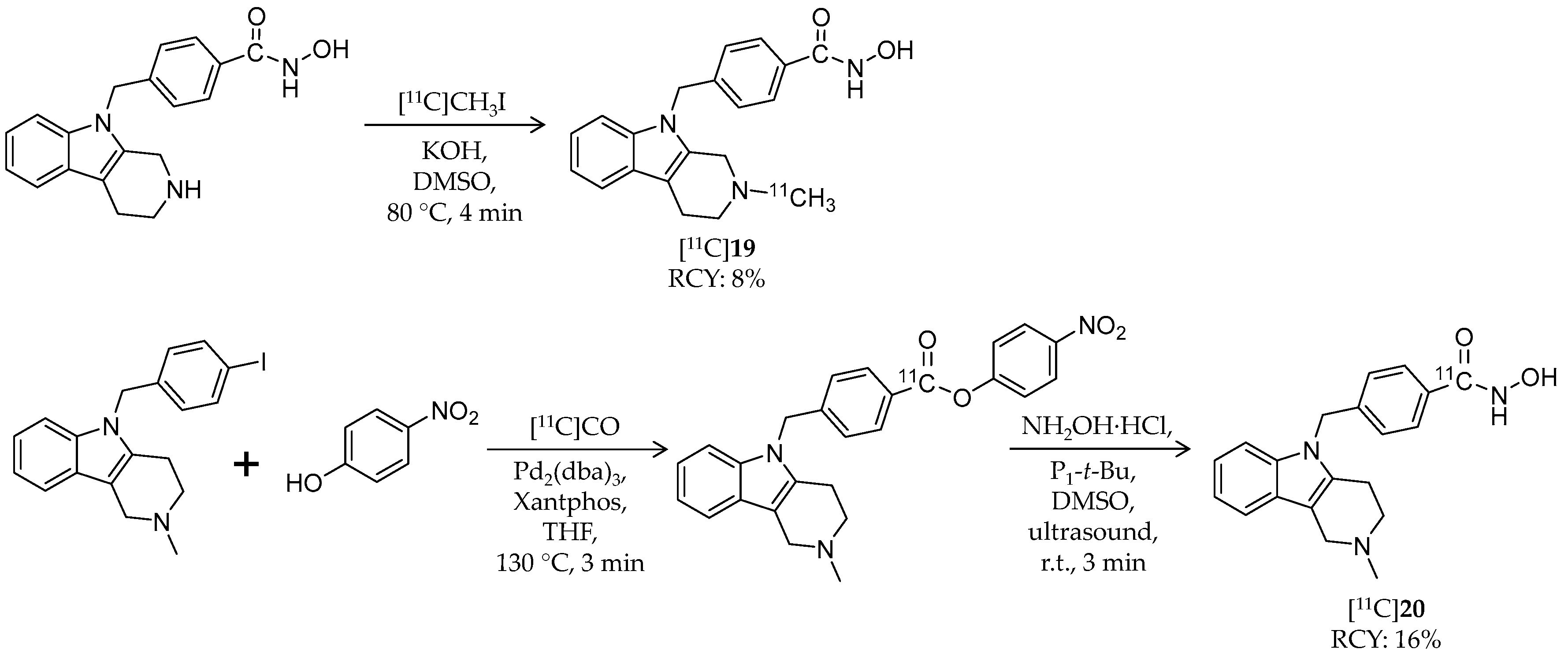
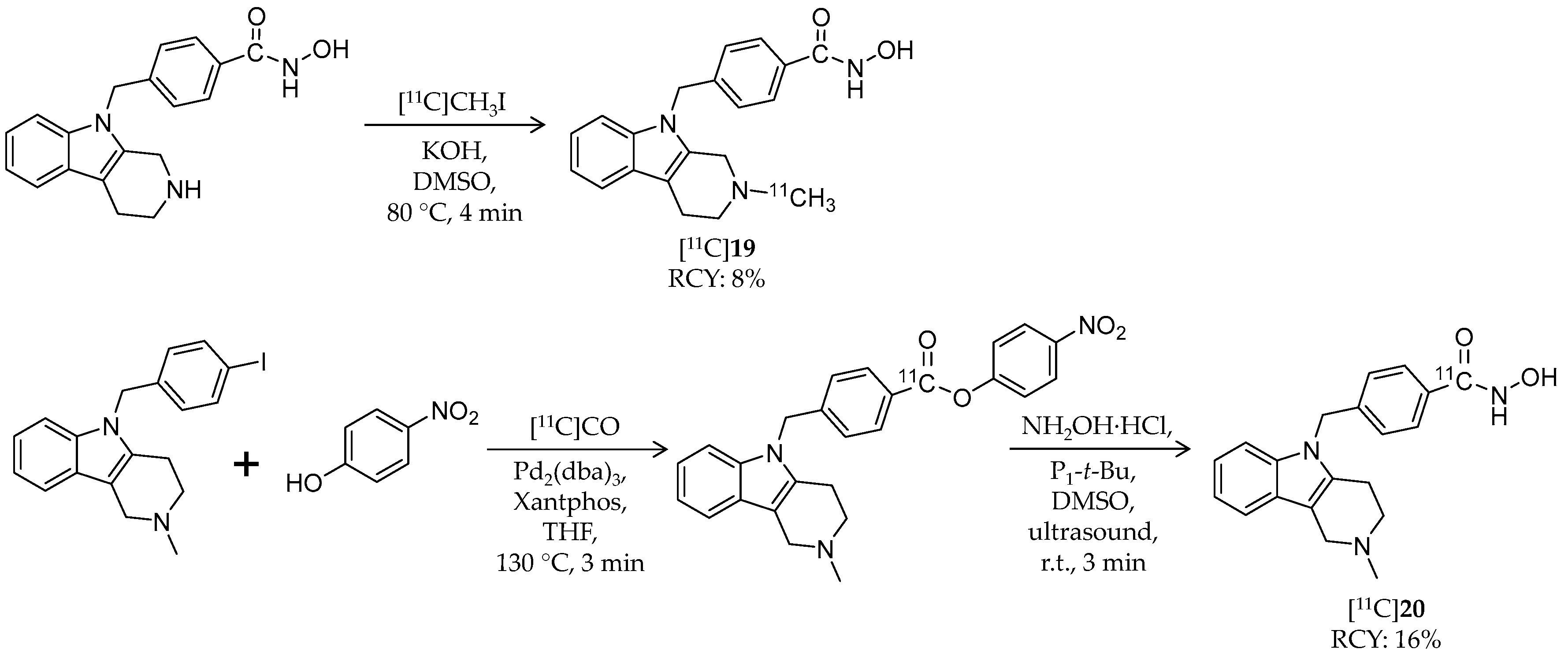
| [11C]19 | 334.41 | 1.33 2 | 8% | HDAC6 | HDAC1: 5.2 μM HDAC6: 1.4 nM | Brain uptake: Radioactivity peaked in forebrain at 0.44 SUV and in cerebellum at 0.48 SUV (rat) | [93,94] |
| [11C]20 | 334.41 | 1.33 2 | 16% | HDAC6 | HDAC6: 4 nM Others: ≥1.3 μM | – | [82,95] |
| [64Cu]21 | 1009.02 | – | ≥98% | Class I/IIb/III | Class I, IIb, III: 94 nM | Brain uptake: Biodistribution study was performed but brain uptake was not assessed (tumor-bearing mouse) | [96] |
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
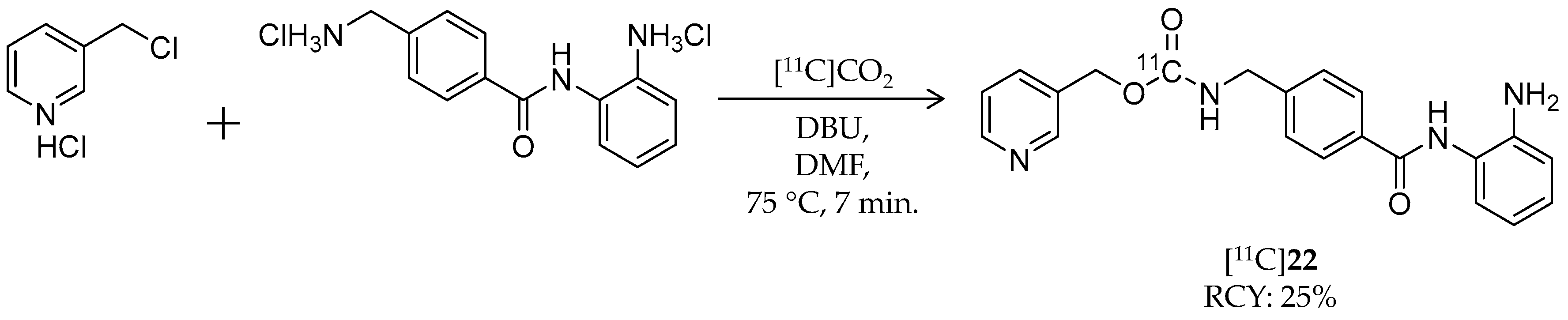
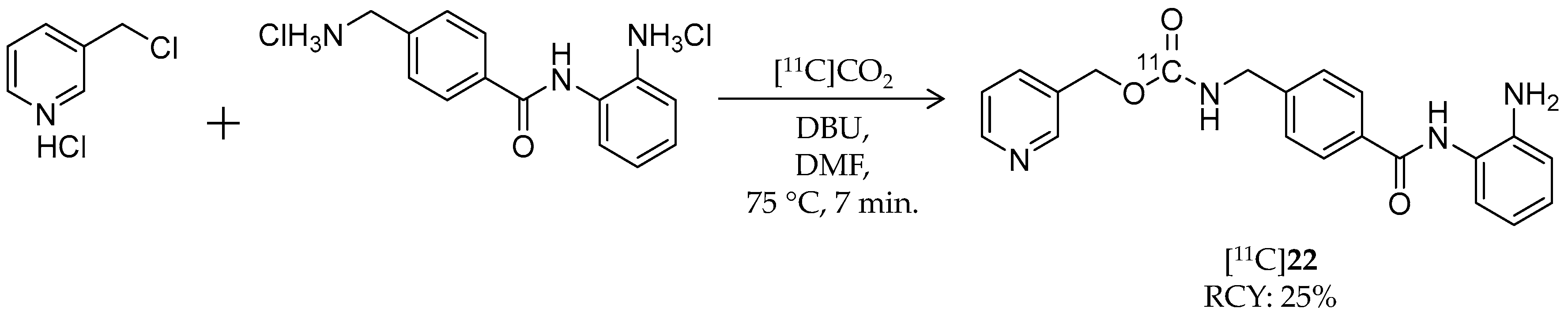
| [11C]22 | 375.42 | 1.8 | 25% | Class I | HDAC1: 60 nM HDAC2: 153 nM | Brain uptake: <0.10% ID/cm3 after 3 min Metabolism: 80% of radioactivity in the brain was unchanged [11C]22 (rat) | [101,105] |
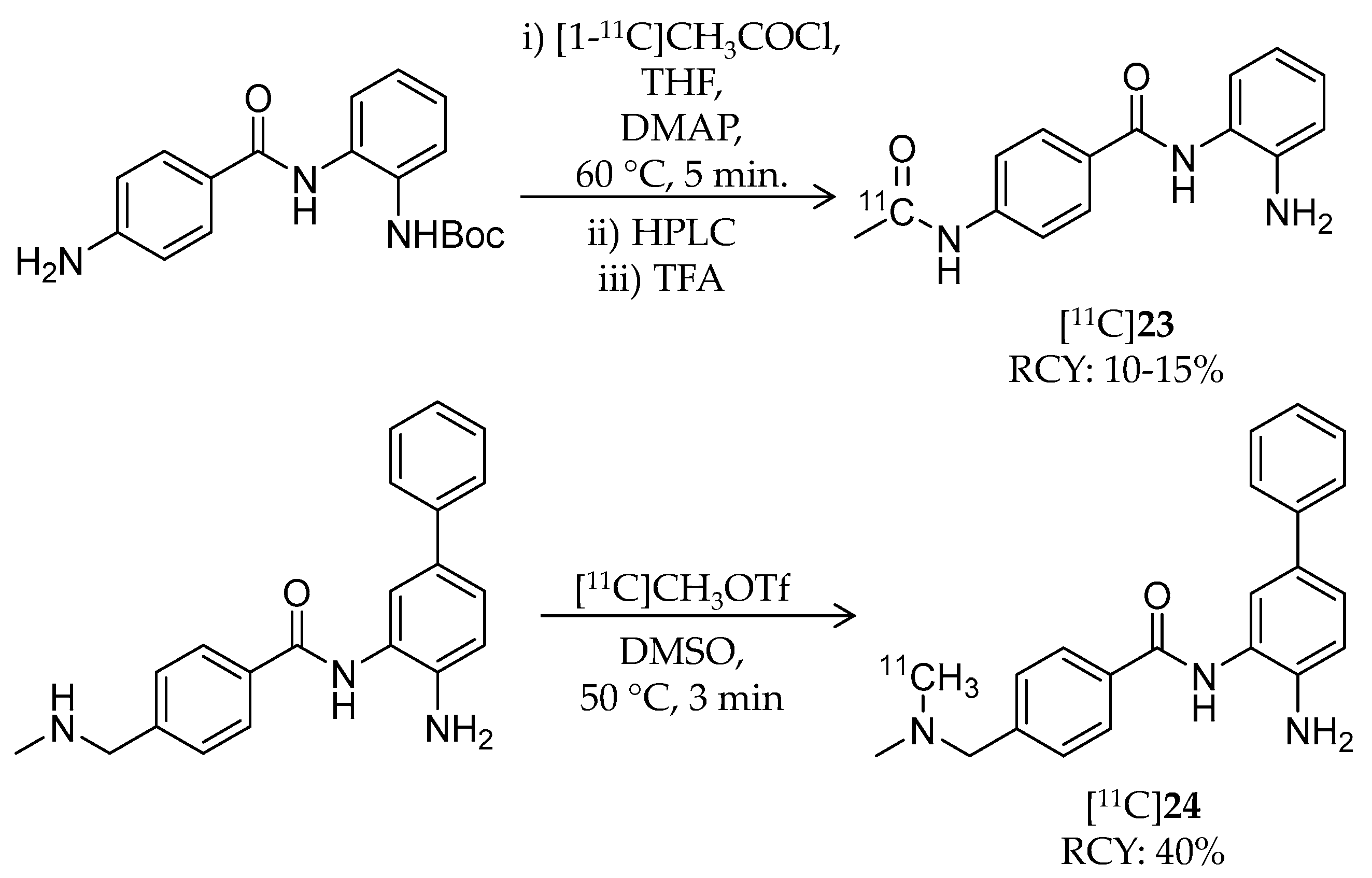
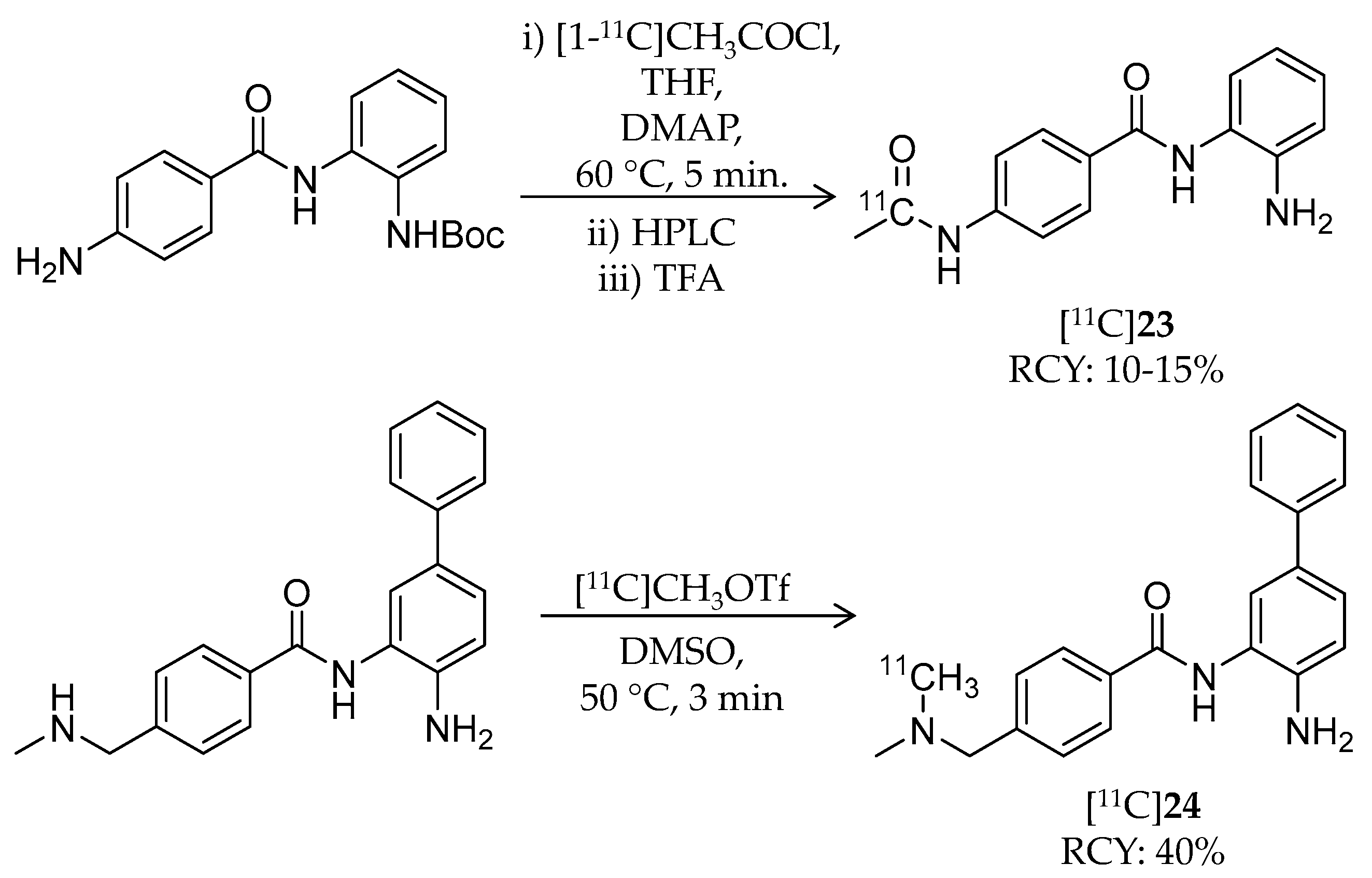
| [11C]23 | 268.30 | 1.0 | 10–15% | Class I | HDAC1: 45 nM HDAC2: 31 nM HDAC3: 20 nM | – | [105] |
| [11C]24 | 344.45 | 2.1 | 40% | Class I | HDAC1: 10 nM HDAC2: 20 nM | – | [105] |
| Compound | Target | Findings in PET Studies in NHPs | Reference |
|---|---|---|---|
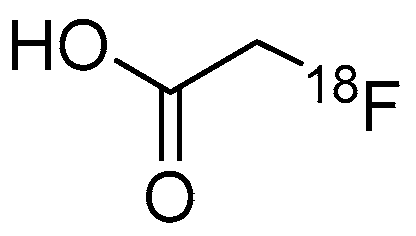
| [18F]1 | Class IIa | Radioactivity accumulation was brain region specific in rhesus macaques (~0.03% ID/g) By 30 min p.i., almost all [18F]1 was metabolized to [18F]FACE Radioactivity in the brain was decreased by pre-treatment with SAHA in a dose-dependent manner | [26,46] |
| [11C]6 [11C]7 | – | Brain uptake of both ligands was very low in baboons (~0.004% ID/cc) The unchanged fraction of both ligands in baboon plasma was less than 20% at 30 min p.i. | [60] |
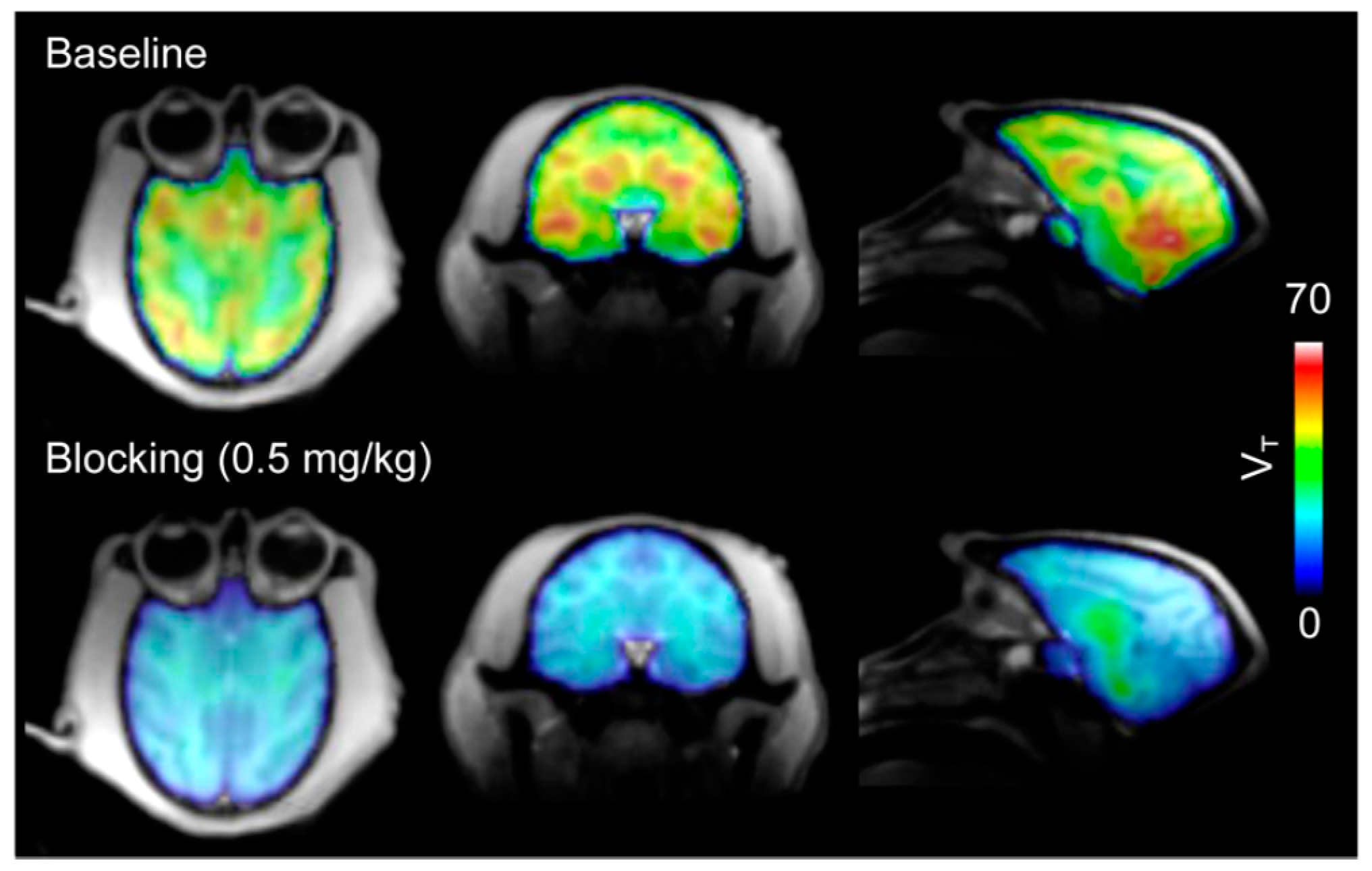
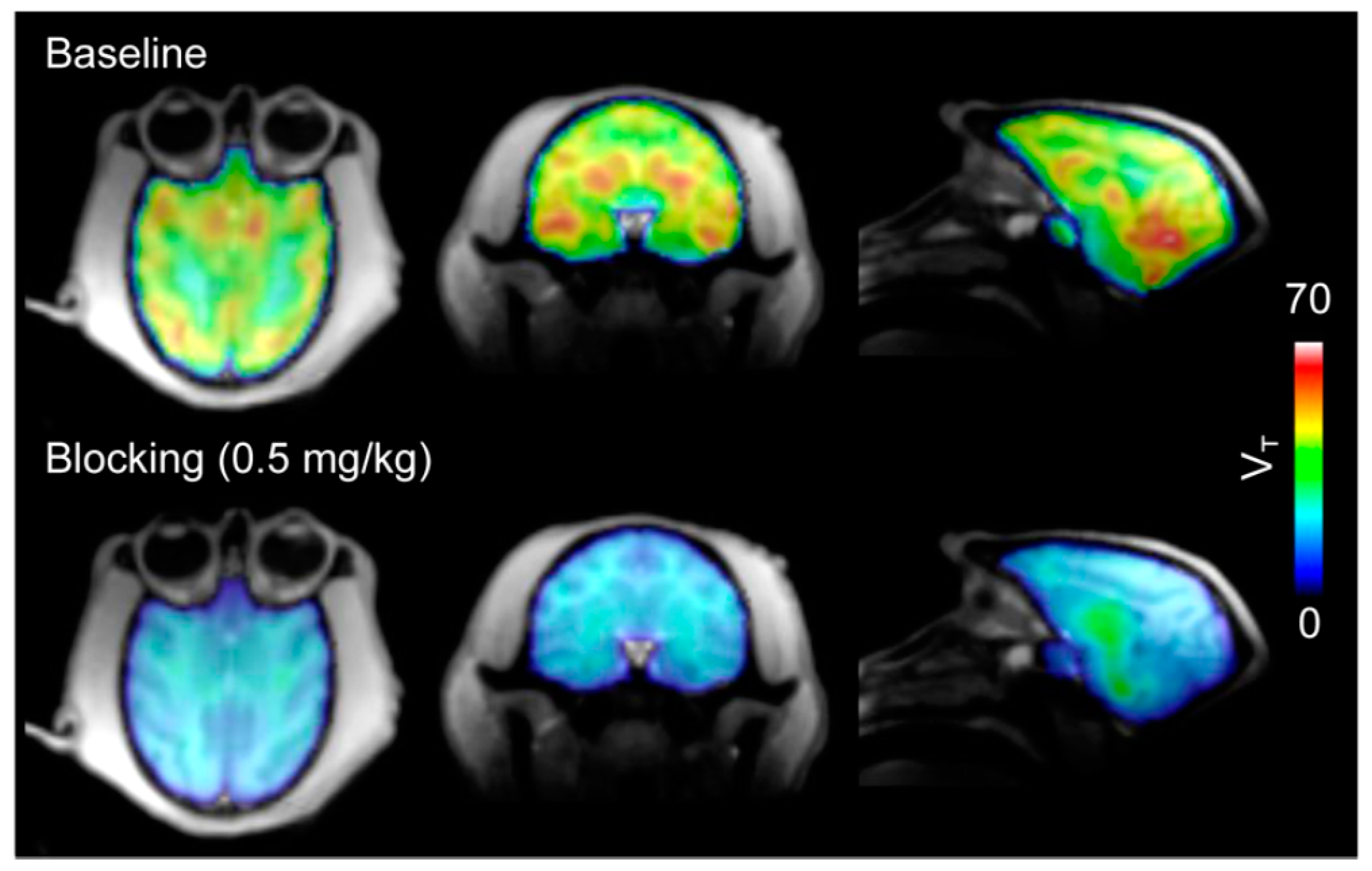
| [11C]8 | Class I/IIb | Regional VT (90-min scan) in the baboon brain ranged from 29.9 to 54.4 mL/cm3 Parent fraction in plasma decreased gradually (50% at 30 min p.i. and 40% at 60 min p.i.) The mean VT in the brain decreased by 82.3 ± 5.5% with a 1-mg/kg blocking dose | [28,68,78] |
| [18F]9 [18F]10 [18F]11 | Class I/IIb | Whole brain SUV30–60 min of [18F]9, [18F]10, and [18F]11 in baboons were 0.57, 1.2, and 1.8, respectively (2.3 for [11C]8) [18F]11 showed the highest correlation in regional brain distribution with [11C]8 | [79] |
| [18F]12 | HDAC6 | Excellent brain uptake (SUV ≈ 3 around 30 min p.i.) was observed in baboons Nonspecific binding in the brain determined with 1 mg/kg unlabeled 12 was low (<1 SUV) | [80] |
| [11C]13 [11C]14 [11C]15 | Class I | In the baboon brain, uptake of the three ligands was low (~0.006% ID/cc) | [86] |
| [11C]16 [11C]17 | HDAC1–3/6 | Brain uptake in baboons was very low over the 80-min scan time | [89] |
| [11C]18 | HDAC8 | Brain uptake in baboons was very low over the 80-min scan time | [89] |
| [11C]22 | Class I | Brain uptake in baboons was very low (<0.001% ID/cc) over the 90-min scan time Approximately 60% of the plasma radioactivity was unchanged ligand at 40 min p.i. | [101] |
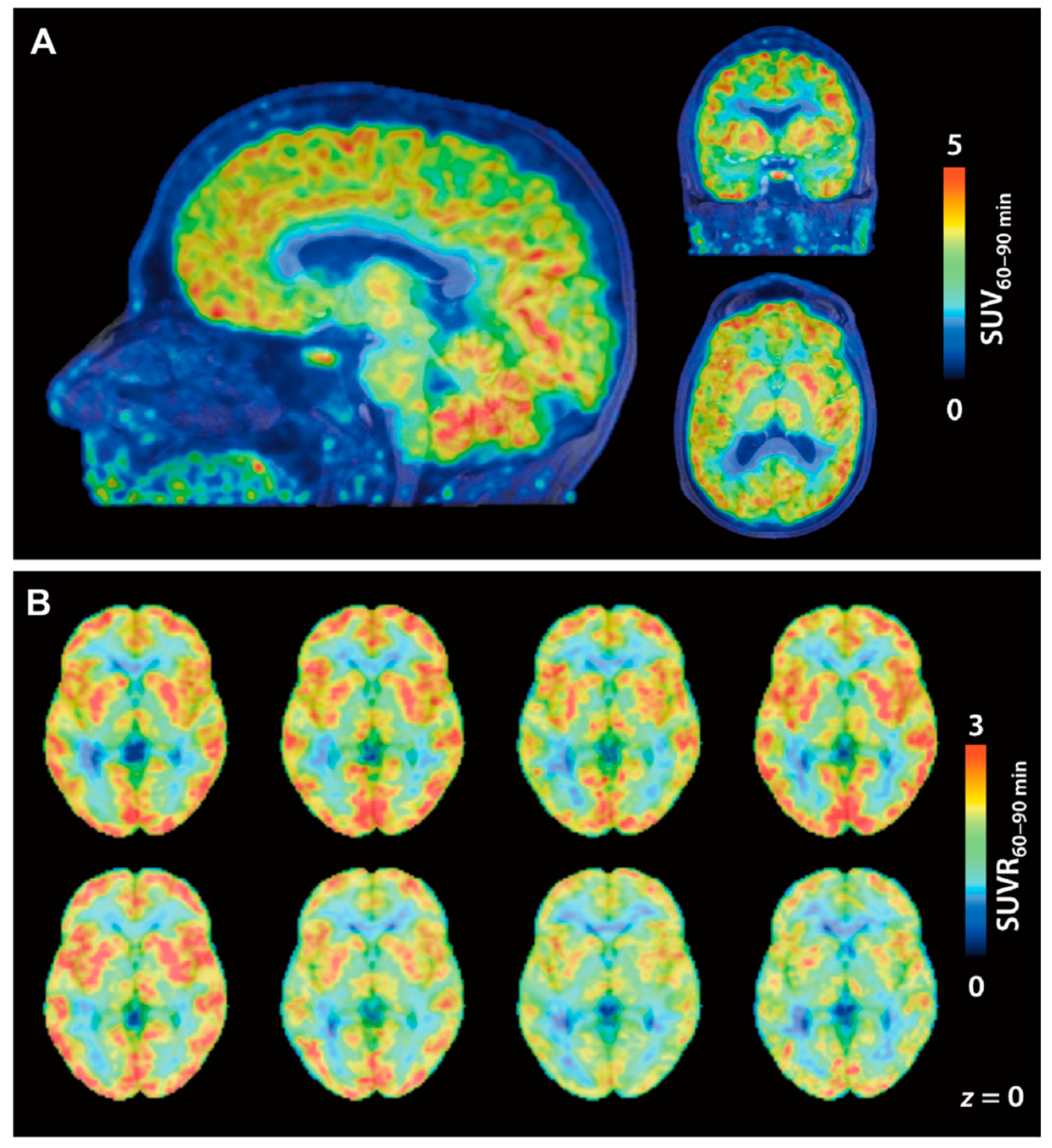
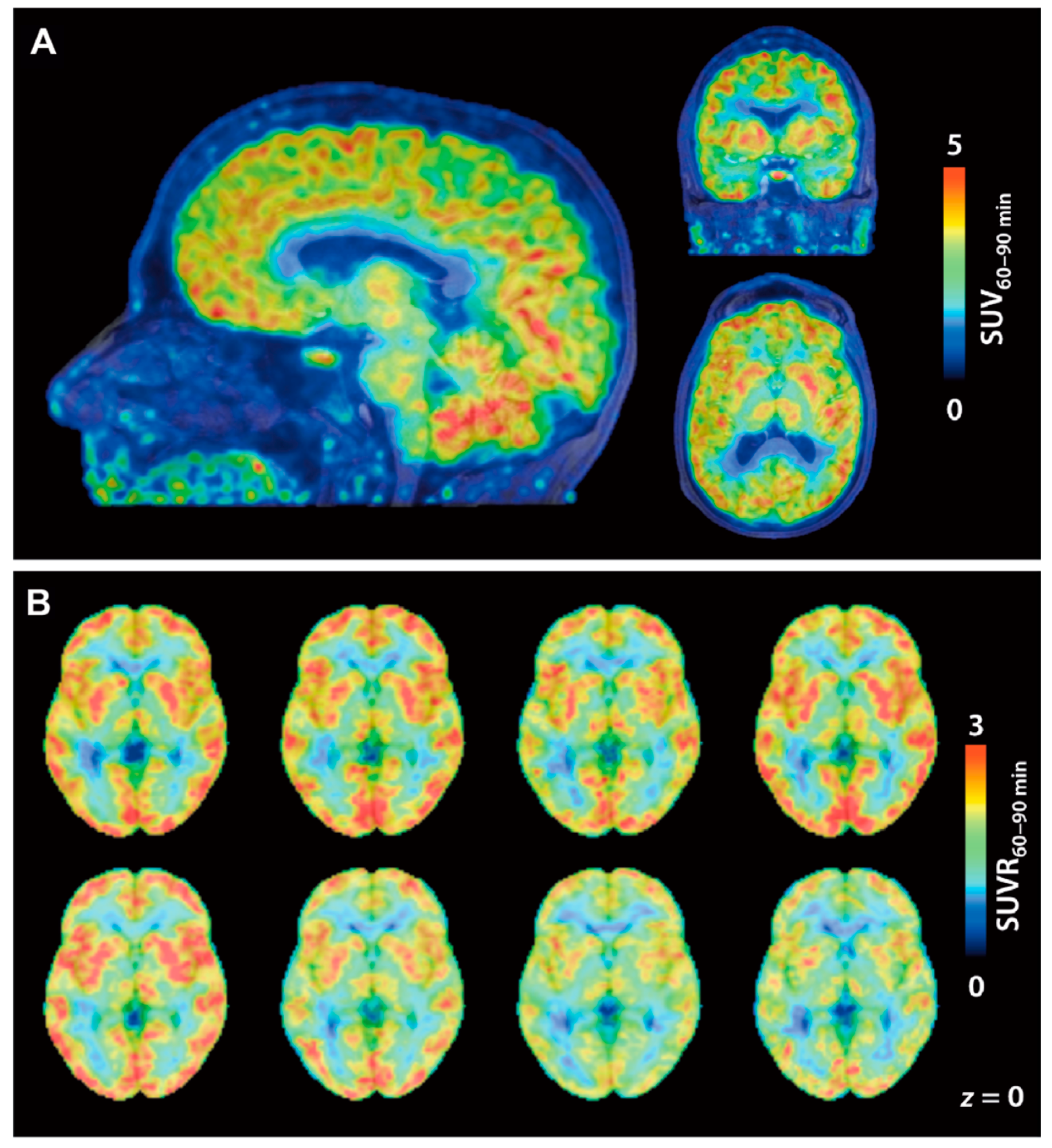
| [11C]23 [11C]24 | Class I | Total VT values of [11C]23 and [11C]24 in the baboon brain were 0.41 and 12 mL/cm3, respectively The degree of VT reduction of [11C]24 by unlabeled 24 or SAHA (1 mg/kg) ranged from 8–24% in various brain regions | [105] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tago, T.; Toyohara, J. Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules 2018, 23, 300. https://doi.org/10.3390/molecules23020300
Tago T, Toyohara J. Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules. 2018; 23(2):300. https://doi.org/10.3390/molecules23020300
Chicago/Turabian StyleTago, Tetsuro, and Jun Toyohara. 2018. "Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases" Molecules 23, no. 2: 300. https://doi.org/10.3390/molecules23020300