Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes


1
Department of Chemistry, Georgia State University, 50 Decatur St., Atlanta, GA 30303, USA
2
Center for Diagnostics and Therapeutics, Georgia State University, Petit Science Center, 100 Piedmont Ave SE, Atlanta, GA 30303, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2018, 23(2), 226; https://doi.org/10.3390/molecules23020226
Submission received: 27 November 2017
/
Revised: 18 January 2018
/
Accepted: 20 January 2018
/
Published: 24 January 2018
(This article belongs to the Collection Heterocyclic Compounds)
Abstract
:Heptamethine cyanine dyes are a class of near infrared fluorescence (NIRF) probes of great interest in bioanalytical and imaging applications due to their modifiability, allowing them to be tailored for particular applications. Generally, modifications at the meso-position of these dyes are achieved through Suzuki-Miyaura C-C coupling and SRN1 nucleophilic substitution of the chlorine atom at the meso-position of the dye. Herein, a series of 15 meso phenyl-substituted heptamethine cyanines was synthesized utilizing a modified dianil linker. Their optical properties, including molar absorptivity, fluorescence, Stokes shift, and quantum yield were measured. The HSA binding affinities of two representative compounds were measured and compared to that of a series of trimethine cyanines previously synthesized by our lab. The results indicate that the binding of these compounds to HSA is not only dependent on hydrophobicity, but may also be dependent on steric interferences in the binding site and structural dynamics of the NIRF compounds.
1. Introduction
Heptamethine cyanine dyes are a class of near infared fluorescence (NIRF) probes that have shown great potential in numerous applications due to their versatility, low toxicity, narrow absorption band, and high extinction coefficients [1,2,3,4,5,6]. These dyes are comprised of two terminal nitrogen-containing heterocycles linked together by a conjugated polymethine chain. The heterocycles act as both electron donors and acceptors creating an electron deficient system throughout the molecule, allowing for long wavelength absorption [7,8,9,10]. Heptamethine cyanines have been used in medical imaging targeting of: cartilage, bone, endocrine gland, biomolecular labeling and much more all serving as contrast agents to aid in surgical application [5,11,12,13,14,15,16,17]. Modifications to the cyanine dye scaffold can alter optical properties, solubility, and allow for specific tailoring of dyes for their desired application.
A main contributing factor in the successful application of heptamethine cyanine dyes is their modifiability. Most commonly cyanines are modified by the use of different heterocycles and with different substituents on the nitrogen atom of the heterocycle. One point of modification that has not been thoroughly investigated is the central (meso) carbon of the methine bridge. Many derivatives described in literature have been made by replacing the chlorine atom at this position via a SRN1 mechanism [12,13,14]. The most common method of carbon-coupling at the meso-position thus far has been done by first synthesizing a heptamethine cyanine dye containing a chlorine atom at the meso-position followed by Suzuki-Miyaura coupling [18,19,20]. While this method is successful in synthesizing many scaffolds, it requires tedious purification and the use of an expensive palladium catalyst [12,13,14]. Although the phenyl-substituted dianil linker has been previously synthesized, it has not been thoroughly investigated for its effects on the optical properties on the NIRF dye [21,22].
Many of these cyanine dyes are administered via IV injection in which the dyes are transported to their target through the bloodstream [23]. Human serum albumin (HSA) is the most abundant protein in human blood plasma, serving an important role of transporting substances throughout the body [23,24,25]. It is synthesized in the liver and has great binding capacity for hydrophobic compounds [23,26]. HSA has been a widely studied protein because of its importance in drug delivery [15]. HSA contains four binding pockets and does not require biomolecular ligand specificity, increasing its versatility and usefulness in medical research [23,24,25,26]. It is well described in literature that HSA binds to hydrophobic entities. A unique attribute of HSA is that it forms reversible covalent bonds with the binding agent, this allows for stable complex formation; however, since the bonds are reversible also allows for localization and deposit [27].
Our lab has previously designed and synthesized a series of trimethine dyes and studied their hydrophobicity and its effect on their interactions with HSA [28]. In this paper, a series of heptamethine cyanines with varying degrees of hydrophobicity containing a meso-phenyl substituent were synthesized through the use of a phenyl-substituted dianil linker. This method not only allows for a more facile synthesis, but a wider array of dyes can be made and can serve for various applications. The effect of the phenyl ring on the dyes hydrophobicity, optical properties, and binding to HSA was studied and compared to the results from our previous study [28].
2. Results and Discussion
2.1. Synthesis
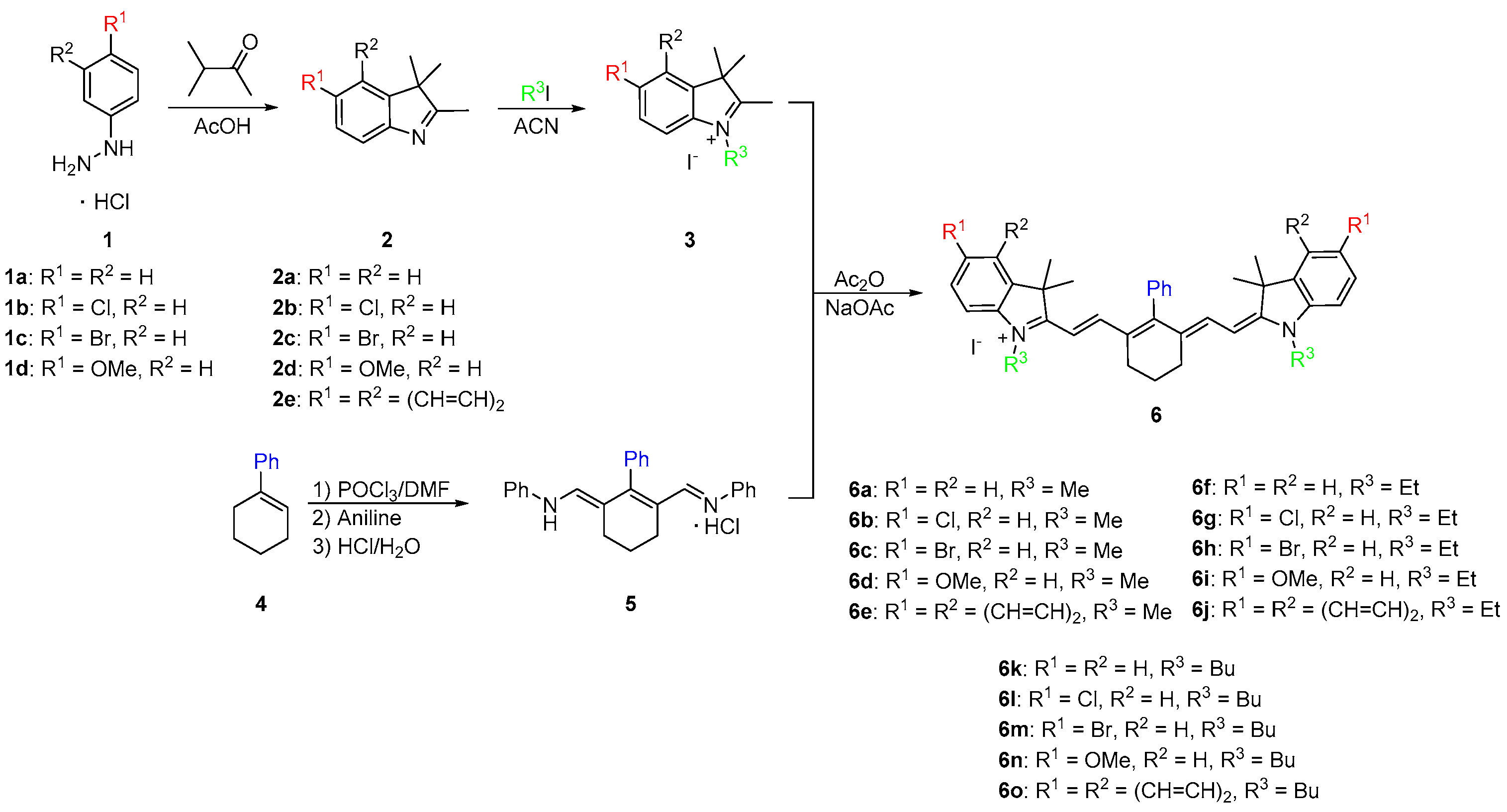
As shown in Scheme 1, the synthesis began with a Fischer indole cyclization by refluxing 4-substituted phenylhydrazines 1 overnight with 3-methyl-2-butanone in glacial acetic acid. After cooling to room temperature, the reaction mixture was neutralized and the substituted indoles 2 were extracted with dichloromethane to give brown oils. The oils were dissolved in acetonitrile and refluxed overnight with various alkyl halides to yield quaternary ammonium salts 3. In parallel to salt formation, a phenyl-substituted dianil compound was synthesized through a Vilsmeier Haack formylation with 1-phenylcyclohexene (4). The ends of the dianil linker were capped with aniline for stability to yield dianil compound 5 [21,22]. Various quaternary ammonium salts 3 and dianil compound 5 are then condensed in a 2:1 ratio in acetic anhydride to yield the final phenyl substituted heptamethine cyanines 6. Pure compounds were obtained in good yield by simply washing with methanol.
The synthetic route described in Scheme 1 provided a new carbon-carbon linked substituent position at the meso center adding to the versatility of heptamethine cyanine. The phenyl group was added before the dye was made. This allowed for an efficient method of preparation of the dye, which required no catalysts or complex purification methods and allowed for a wider array hydrophobic compounds to be made.
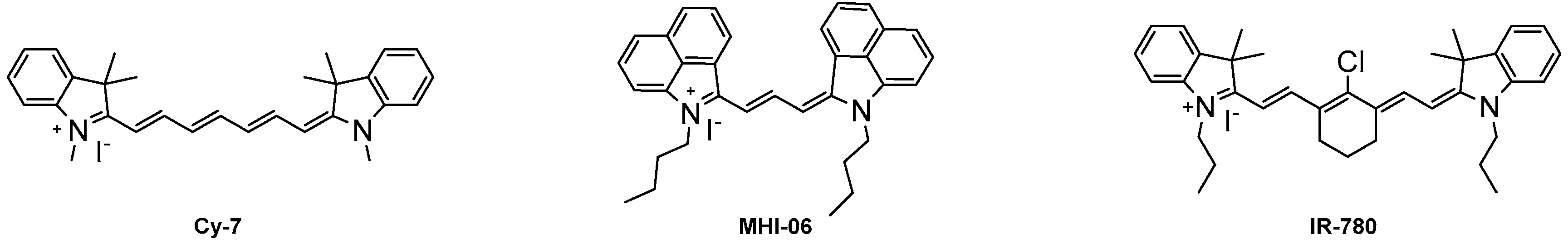
Once the dyes have been successfully synthesized, the optical properties were measured, and representative dyes 6a and 6k were studied for their binding affinity to HSA. The optical properties were compared to commercially available heptamethine dyes Cy-7 and IR-780 due to the similar absorbance and emission wavelengths. The binding studies of compounds 6a and 6k were compared to MHI-06, a dye previously reported as a strong HSA binding agent [28]. Figure 1 shows the three dye structures of the standards used in our study.
2.2. Optical Properties
As described in Scheme 1, fifteen final NIRF contrast agents were synthesized using the dianil linker to yield symmetrical heptamethine cyanines 6a–o. The compounds are broken down to three sets of five. Dyes 6a–e all contain a methyl substituent off the nitrogen of the heterocycle with varying substitutions at the six position of the heterocyclic ring.
Dyes 6f–j and 6k–o contain ethyl and butyl N-alkyl substituents, respectively. In comparison to Cy-7, the optical properties of the new compounds were found to be superior (Table 1). The addition of the cyclohexene ring provided rigidity to the compounds, increasing the molar absorptivity and quantum yield by 60,000 M−1·cm−1 and 5%, respectively, for 6a [29]. To determine the effects the phenyl ring had on the optical properties, the dyes were compared to IR-780 [30]. Although the molar absorptivity of the studied dyes were within the same range as the commercially available dye, the quantum yield was dramatically increased with the introduction of the electron rich phenyl ring, observing a 23%–47% increase in quantum yield. The chlorine atom at the meso-position of dye IR-780 promotes intersystem crossing due to the heavy atom effect, and allows for the molecule to relax in non-radiative means and decreases the fluorescence [31].
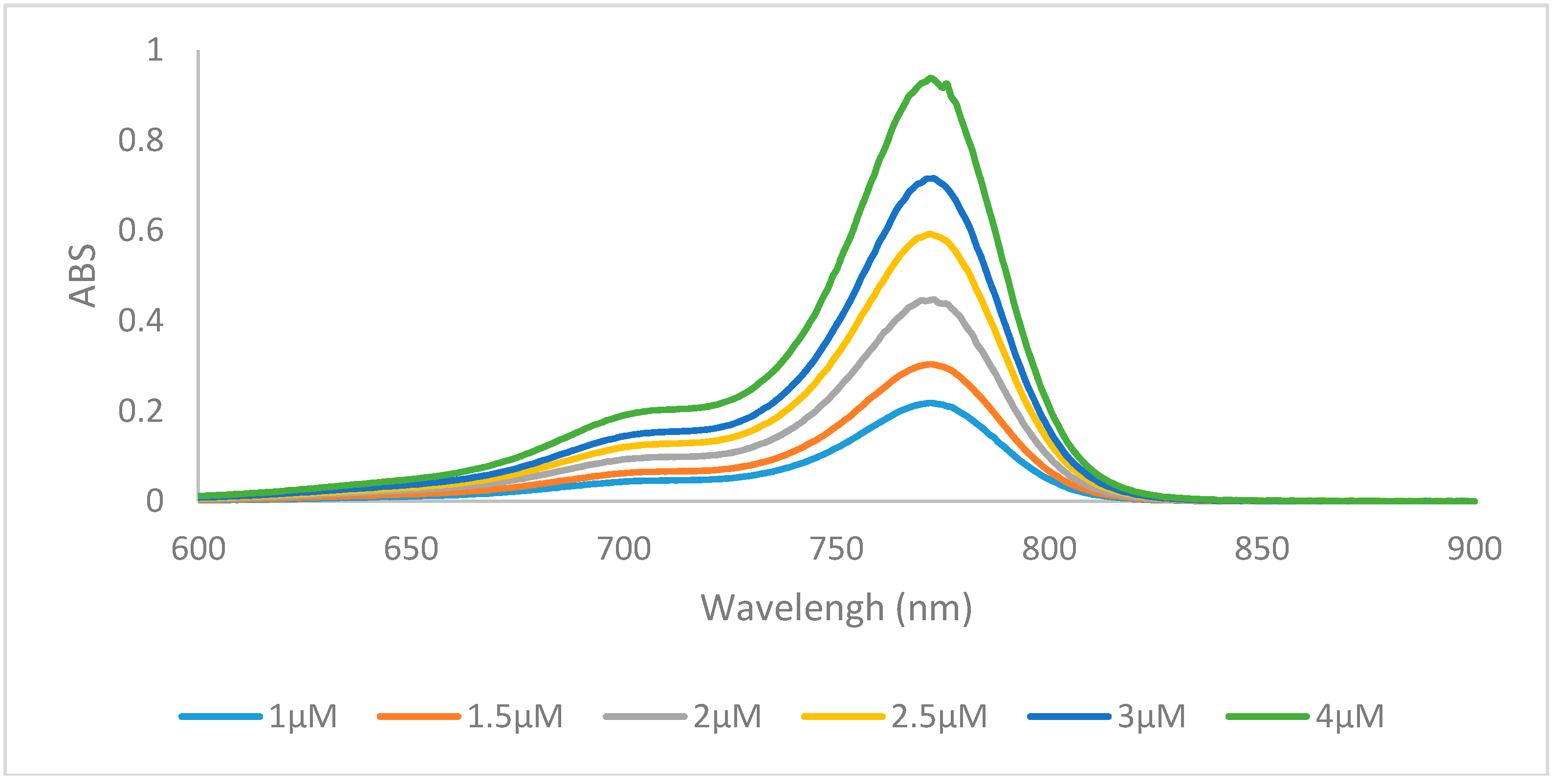
Increasing the length of N-alkyl substituents from a methyl to ethyl did not result in any significant changes in optical properties, but an increase in size to the butyl group generally lowered the molar absorptivity. Dyes containing hydrogen 6a, 6f, 6k, and halogens 6b, 6c, 6g, 6h, 6l, 6m at the 6 position of the heterocycle displayed absorption λmax values of 759–773 nm with redshifts from the hydrogen to the halogens from 6 nm to 10 nm which has previously been described [32]. Absorption spectra of representative compound 6m was shown in Figure 2. All 15 compounds have Stokes shifts ranging from 12 to 21 with the benz[e]indolenine containing compounds 6e, 6j, 6o having the shortest Stokes shifts. All three methoxy-substituted compounds 6d, 6i, 6n had redshifted absorption λmax values from 16 nm to 26 nm and lower quantum yields than the other compounds while dyes 6e, 6j, 6o were redshifted 37–39 nm around 800 nm due to the increased conjugation of the benz[e]indolenine heterocycle.
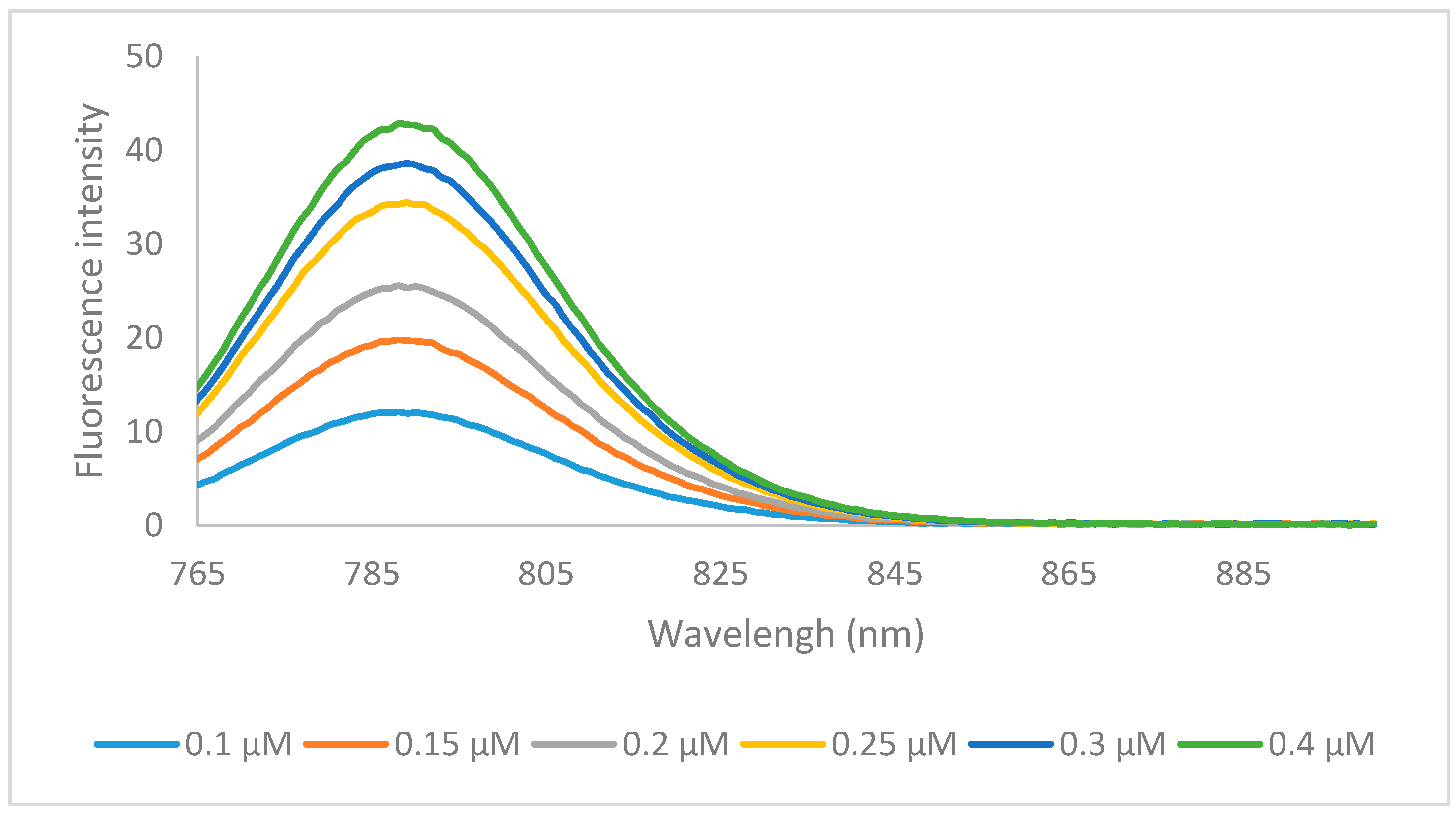
The emission data of all the dyes followed the same trends, whereby compounds containing the hydrogen 6a, 6f, 6k, and halogen 6b, 6c, 6g, 6h, 6l, 6m substituents had the highest quantum yields, and the longer N-alkyl chains increased the quantum yield by 4%–8% from methyl to ethyl and 5%–7% from ethyl to butyl. Emission spectra of representative compound 6m was shown in Figure 3. There was no significant difference in quantum yield between the hydrogen and halogens within the same set. Compounds 6e, 6j, 6o containing the benz[e]indolenine heterocycle and 6d, 6i, 6n containing the methoxy substituent displayed lower quantum yields at 16%–17% and 10%–12%, respectively, which is consistent with previous reports [12].
Although molar absorptivity and quantum yield are important properties of fluorophores, in regards to application, the molecular brightness gives a more useful indication of the dye utility. Molecular brightness takes into account both molar absorptivity and quantum yield [33,34]. Dyes that have high quantum yield, but do not absorb light efficiently (low molar absorptivity) are still not emitting as many photons and are less useful for fluorescent applications. N-alkyl substituents from a methyl to ethyl increased the molecular brightness by approximately 20,000 M−1·cm−1 for the hydrogen-6a, 6f, 6k, and halogen-6b, 6c, 6g, 6h, 6l, 6m substituted compounds while the butyl compounds showed lower molecular brightness. Due to the low quantum yield of the benz[e]indolenine heterocycle 6e, 6j, 6o and the methoxy 6d, 6i, 6n substituted compounds, the two sets showed the lowest molecular brightness of 38,000–40,000 M−1·cm−1 and 17,000–20,000 M−1·cm−1, respectively.
2.3. Physiochemical Properties
In our previous study of trimethine cyanine dyes it was shown that MHI-06 (Figure 1) bound HSA with an affinity of 1.0 × 106 M−1 [28]. In that study of the binding affinity of trimethine cyanines, a trend was observed correlating hydrophobicity to the binding affinity. As the dyes became more hydrophobic greater binding affinity was observed. However, the correlation did not hold when the large N-phenylpropyl side chain was introduced in the trimethine series [28]. It was hypothesized that the binding affinity decreased due to the increased size of the N-phenylpropyl side chain hindering the dye from entering the HSA binding pocket. The heptamethine cyanines 6a–o synthesized in this work were tailored to be hydrophobic in order to observe if binding increases due to increase of hydrophobicity or decreases due to the size and steric hindrance.
The physicochemical properties were calculated using ChemAxon for the 15 heptamethine dyes synthesized and compared to our pervious reported compound MHI-06 (Table 2) [28]. Physicochemical trends were observed in each series with the same heterocyclic substituents as well as with the same N-alkyl side chain. Compounds 6d, 6i, 6n with the methoxy substituent showed the lowest logD, due to its ability to form hydrogen bonds. Slightly higher values were observed for compounds with the hydrogen-substitued 6a, 6f, 6k. A 1.2 and 1.5 increase of logD was observed from the hydrogen to the chloro-and bromo-substituted compounds, respectively. As expected, the series with the benz[e]indolenine heterocycle 6e, 6j, 6o had the highest logD values due to the addition of another phenyl ring. All heptamethine cyanines 6a–o had significantly higher logD values compared to that of MHI-06 due to the presence of the phenyl substituent at the meso-position and the increased size of the hydrocarbon skeleton. The dipole moments decreased as the length of the alkyl chain increased from methyl to butyl, with methoxy-substituted dyes having significantly higher values. In comparison to MHI-06 most of the compounds, especially that of the methyl series 6a–e, had a higher dipole moment, but for 6k which had fairly similar results to MHI-06 at 2.85 and 2.48, respectively. The number of rotatable bonds increased by 2 from methyl to ethyl and by 4 from ethyl to butyl. Only the methoxy heterocyclic substitution affected the number of rotatable bonds with an additional rotatable bond for each methoxy in compounds 6d, 6i, 6n. MHI-06 had eight rotatable bonds. All of the dyes with the exception of those containing the methoxy substituent had a total polar surface area (TPSA) of 6.25, 0 hydrogen bond donors, and 1 hydrogen bond acceptor (ChemAxon). Dyes 6d, 6i, 6n containing the methoxy substituent had higher TPSA at 24.71 and three hydrogen bond acceptors. The presence of the polar oxygen increases the TPSA, and increases the number of hydrogen bond that can form by two.
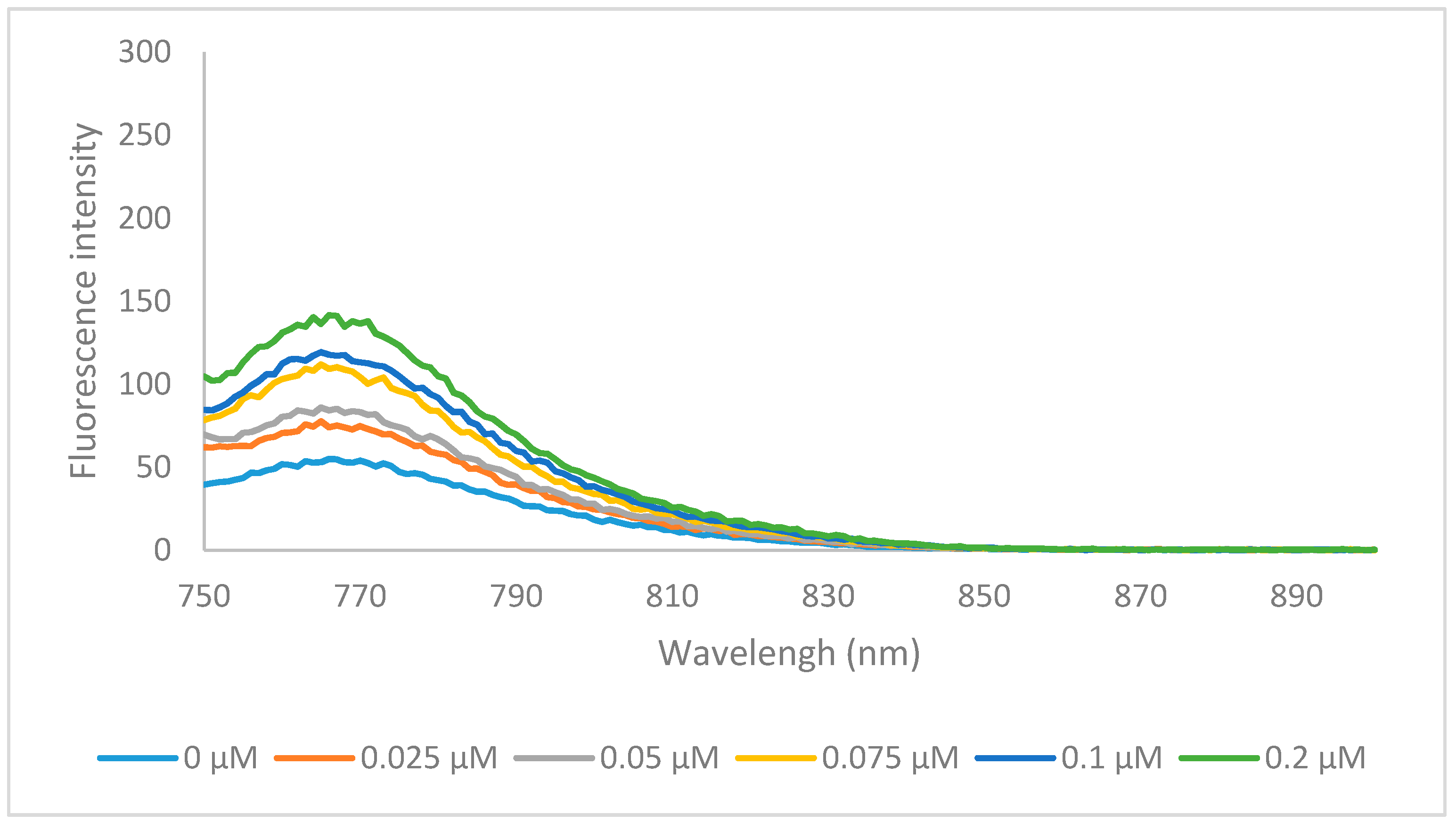
In summary, the newly synthesized heptamethine dyes 6a–o were significantly more hydrophobic then MHI-06, but had a much larger volume. Although hydrophobicity plays a key role in binding to HSA, reversibly the size of the compound could inhibit the ability of its binding to HSA pocket. Compounds 6a and 6k were tested for their ability to bind to HSA, to further determine if other factors than hydrophobicity, play an important role in HSA binding allowing a better understanding of binding nature of the larger heptamethine cyanines to HSA. The HSA binding spectra for 6a is shown below (Figure 4) and binding data of 6k is shown in the supplemental information (6k HSA binding).
2.4. HSA Binding
The formation of a dye/substrate conjugate was studied with HSA in phosphate buffered saline (PBS), pH 7.4. Previous research by Kim et al. suggested that cyanine dyes bind HSA in with a 1:1 stoichiometry which was confirmed by our and other research groups using trimethine cyanine dyes [3,28,35,36,37]. The binding interactions were studied by measuring the changes in emission intensities at a fixed concentration of dye with varying micromolar concentrations of HSA and using a double reciprocal plot of [HSA]−1 vs. ΔF−1, where ΔF is the change in emission intensity of the Dye/HSA conjugate, that should give a linear relationship. The binding affinity is then calculated by dividing the intercept by the slope of the line. Our lab has previously shown that both the N-alkyl substituents and the heterocyclic ring of the cyanines have profound effects on overall conjugation with the biomolecule [20]. It has also previously been shown that cyanine dyes aggregate in polar solvents such as PBS buffer due to strong intermolecular van der Waals interactions between the heterocycles that cause the dyes to form H-aggregates [38]. Generally, organic solvents are used to disrupt this aggregate formation, but organic solvents cannot be used in the presence of HSA due to their ability to denature the biomolecule. Conjugate formation with HSA disrupts the aggregation increasing monomer formation and thereby increasing fluorescence emission of the dyes. It was determined that heptamethine dye 6a binds HSA with an affinity of 8 × 101 M−1. This is 5 orders of magnitude lower than the previously tested trimethine cyanines, MHI-06, which bound on the order of 1 × 106 M−1 [28]. This further confirms our previous hypothesis that the binding affinity of the dyes is not only hydrophobicity dependent, but dependent on steric interferences in the HSA binding site. It also confirms that the delocalized cationic nature of the dyes may has no electrostatic interference with the binding cavity as this heptamethine cyanine displays increased delocalization over the previously tested trimethine cyanines. Albumin is known to bind a variety of compounds including fatty acids, nucleic acids, and oligoproteins therefore this information on the steric specificity of these binding sites is of potential interest when developing methods to study them [28].
3. Experimental Section
3.1. General Information
All chemicals and solvents were of American Chemical Society grade or HPLC purity and were used as received. HPLC grade ethanol were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were purchased from Fisher Scientific (Pittsburgh, PA, USA) or Acros Organics (Pittsburgh, PA, USA). The reactions were followed using silica gel 60 F254 thin layer chromatography plates (Merck EMD Millipore, Darmstadt, Germany). The 1H NMR and 13C NMR spectra were obtained using high quality Kontes NMR tubes (Kimble Chase, Vineland, NJ, USA) rated to 500 MHz and were recorded on an Avance spectrometer (Bruker, Billerica, MA; 400 MHz for 1H and 100 MHz for 13C) in DMSO-d6, acetone-d6 CD3Cl-d3. High-resolution accurate mass spectra (HRMS) were obtained at the Georgia State University Mass Spectrometry Facility using a Q-TOF micro (ESI-Q-TOF) mass spectrometer (Waters, Milford, MA, USA). All compounds tested were >95% pure.
A solution of POCl3 (11 mL, 117.66 mmol) in dichloromethane (10 mL) was added dropwise to a solution of DMF (13 mL, 167.89 mmol) in dichloromethane (13 mL) at 0 °C for 30 min under inert conditions. Then 1-phenylcyclohexene (4, 5.5 mL, 32.81 mmol) was dissolved in dry dichloromethane (5 mL) and added dropwise to the solution which was then refluxed for 3 h. The solution was allowed to cool to room temperature and then poured over 500 mL of ice/water. Aniline (9 mL, 98.57 mmol) in ethanol (9 mL) was added to cap the ends. The crude solid was collected and washed with diethyl ether and hexanes. Resulting in the dianil linker 5 as a pure compound and used without further purification.
In parallel, substituted hydrazines 1 (4.0 g, 22.25 mmol) were reacted with 3-methylbutanone (3 mL, 28.04 mmol) in acetic acid and heated to a 100 °C for 24 h. The solution was then neutralized using sodium bicarbonate and extracted using dichloromethane; affording substituted indolenine heterocycles 2 which was dried under reduced pressure. The heterocycles 2 were then reacted with an alkyl halide in acetonitrile at 100 °C for 12–18 h. The quaternary ammonium salts 3 were precipitated with diethyl ether, and collected.
The various salts 3 (2 molar eq), the dianil linker 5 (1 molar eq), and sodium acetate (2 molar eq) were dissolved in acetic anhydride and heated to 60 °C for 2–3 h. The crude product was then precipitated with diethyl ether, collected, and washed with methanol to yield heptamethine dyes 6 as pure sample.
3.2. Characterization
1,3,3-Trimethyl-2-((E)-2-((E)-6-(2-((E)-1,3,3-trimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-3H-indol-1-ium iodide, 6a: Yield 77%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.11 (s, 12H), 1.95 (m, 2H), 2.68 (t, J = 6.0 Hz, 4H), 3.58 (s, 6H), 6.17 (d, J = 14.0 Hz, 2H), 7.16 (m, 4H), 7.25 (d, J = 8.4 Hz, 2H), 7.34 (m, 4H), 7.46 (d, J = 7.6 Hz, 2H), 7.62 (m, 3H). 13C NMR (CDCl3) δ 21.3, 24.5, 27.3, 31.5, 48.5, 100.7, 111.3, 122.7, 124.9, 128.5, 128.8, 129.0, 129.6, 131.0, 139.1, 140.9, 143.3, 147.3, 161.5, 172.1. HRMS (ESI) m/z: calcd. for C38H41N2+ 525.3264, obsd 525.3241.
5-Chloro-2-((E)-2-((E)-6-(2-((E)-5-chloro-1,3,3-trimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium iodide, 6b: Yield 78%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.17 (s, 12H), 2.07 (m, 2H), 2.74 (t, J = 6.4 Hz, 4H), 3.65 (s, 6H), 6.12 (d, J = 14.0 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 7.17 (m, 6H), 7.30 (t, J = 8.4 Hz, 2H), 7.56 (m, 3H); 13C NMR (CDCl3) δ 21.1, 25.0, 27.5, 32.5, 48.5, 100.8, 111.3, 122.5, 128.2, 128.5, 128.7, 129.4, 130.2, 133.0, 138.7, 141.5, 142.1, 148.2, 163.0, 171.4. HRMS (ESI) m/z: calcd. for C38H39Cl2N2+ 593.2485, obsd 593.2475.
5-Bromo-2-((E)-2-((E)-6-(2-((E)-5-bromo-1,3,3-trimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium iodide, 6c: Yield 75%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.12 (s, 12H), 2.08 (m, 2H), 2.76 (t, J = 6.4 Hz, 4H), 3.66 (s, 6H), 6.15 (d, J = 14.0 Hz, 2H), 6.99 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 14.0 Hz, 2H), 7.21 (dd, J = 7.6 Hz, 2H), 7.46 (dd, J = 8.4 Hz, 2H), 7.56 (m, 3H). 13C NMR (CDCl3) δ 21.0, 25.0, 27.5, 32.5, 48.4, 100.9, 111.7, 117.7, 125.3, 128.2, 128.5, 129.4, 131.5, 133.2, 138.7, 142.0, 142.4, 148.1, 162.9, 171.2. HRMS (ESI) m/z: calcd. for C38H39Br2N2+ 681.1475, obsd 681.1475.
5-Methoxy-2-((E)-2-((E)-6-(2-((E)-5-methoxy-1,3,3-trimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1,3,3-trimethyl-3H-indol-1-ium iodide, 6d: Yield 73%; m.p. 238–240 °C; 1H NMR (CDCl3) δ 1.17 (s, 12H), 2.06 (m, 2H), 2.70 (t, J = 6.4 Hz, 4H), 3.61 (s, 6H), 3.82 (s, 6H), 6.03 (d, J = 14.0 Hz, 2H), 6.76 (s, 2H), 6.86 (d, J = 8.8 Hz, 2H), 7.04 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 14.0 Hz, 2H), 7.21 (d, J = 6.8 Hz, 2H), 7.55 (m, 3H); 13C NMR (CDCl3) δ 21.2, 24.9, 27.5, 32.2, 48.5, 55.9, 99.8, 109.1, 110.9, 112.8, 128.0, 128.4, 129.5, 131.4, 136.5, 139.1, 142.2, 146.8, 157.8, 161.3, 171.0. HRMS (ESI) m/z: calcd. for C40H75N2O2+ 585.3476, obsd 585.3469.
1,1,3-Trimethyl-2-((E)-2-((E)-6-((E)-2-(1,1,3-trimethyl-1,3-dihydro-2H-benzo[e]indol-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1H-benzo[e]indol-3-ium iodide, 6e: Yield 63%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.50 (s, 12H), 2.11 (m, 2H), 2.79 (t, J = 6.4 Hz, 4H), 3.77 (s, 6H), 6.16 (d, J = 14.0 Hz, 2H), 7.30 (m, 2H), 7.44 (m, 4H), 7.55 (t, J = 7.6 Hz, 2H), 7.66 (m, 3H), 7.92 (m, 6H); 13C NMR (CDCl3) δ 21.2, 25.0, 27.1, 32.5, 50.2, 99.9, 110.5, 121.9, 124.7, 127.5, 128.0, 128.3, 128.6, 129.5, 130.1, 130.5, 131.7, 132.2, 132.9, 139.0, 140.2, 147.1, 162.0, 173.3. HRMS (ESI) m/z: calcd. for C46H45N2+ 625.3577, obsd 625.3570.
1-Ethyl-2-((E)-2-((E)-6-(2-((E)-1-ethyl-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-3,3-dimethyl-3H-indol-1-ium iodide, 6f; Yield: 70%; m.p. > 260 °C; 1H NMR (acetone-d6) δ 1.11 (s, 12H), 1.24 (t, J = 7.2 Hz, 6H), 1.96 (m, 2H), 2.97 (t, 4H), 4.14 ( m, 4H) 6.20 (d, J = 14 Hz, 2H), 7.18 (m, 4H), 7.35 (m, 4H), 7.47 (d, J = 7.6 Hz, 2H), 7.61 (m, 3H); 13C NMR (acetone-d6) δ 12.5, 21.3, 24.6, 27.4, 39.8, 40.0, 40.2, 48.7, 100.2, 111.2, 123.0, 125.0, 128.6, 129.0, 129.1, 129.6, 131.1, 141.1, 142.1, 147.7, 171.2. HRMS (ESI) m/z: calcd. for C40H45N2+ 553.3577, obsd 553.1566.
5-Chloro-2-((E)-2-((E)-6-(2-((E)-5-chloro-1-ethyl-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1-ethyl-3,3-dimethyl-3H-indol-1-ium iodide, 6g; Yield 64%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.16 (s, 12H), 1.38 (t, J = 7.2 Hz, 6H), 2.07 (t, J = 5.6, 2H), 2.73 (t, J = 6.0 Hz, 4H), 4.142 (m, 4H), 6.10 (d, J = 14.0 Hz, 2H), 7.07 (d, J = 8.4 Hz, 2H), 7.28 (m, 8H) 7.59 (m, 3H); 13C NMR (CDCl3) δ 12.2, 21.1, 25.0, 27.5, 40.7, 48.6, 100.1, 111.4, 122.6, 128.3, 128.7, 128.8, 129.4, 130.3, 132.6, 138.7, 140.5, 142.4, 148.3, 163.1, 170.5. HRMS (ESI) m/z: calcd. for C40H43Cl2N2+ 621.2798, obsd 621.2788.
5-Bromo-2-((E)-2-((E)-6-(2-((E)-5-chloro-1-ethyl-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1-ethyl-3,3-dimethyl-3H-indol-1-ium iodide, 6h; Yield 71%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.18 (s, 12H), 1.41 (t, J = 7.2 Hz, 6H), 2.10 (t, J = 6.4 Hz, 2H), 2.75 (t, 5.6 Hz, 4H), 4.16 (m, 4H), 6.13 (d, J = 14.4 Hz, 2H), 7.22 (m, 10H), 7.59 (m, 3H); 13C NMR (CDCl3) δ 12.2, 21.1, 25.1, 27.5, 40.1, 48.6, 100.3, 111.7, 117.7, 125.5, 128.3, 128.6, 129.4, 131.6, 133.0, 138.8, 141.0, 142.7, 148.2, 162.9, 170.3. HRMS (ESI) m/z: calcd. for C40H43Br2N2+ 709.1788, obsd 709.1780.
1-Ethyl-2-((E)-2-((E)-6-(2-((E)-1-ethyl-5-methoxy-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-5-methoxy-3,3-dimethyl-3H-indol-1-iumiodide, 6i; Yield 61%; m.p. > 260 °C; 1H NMR (acetone-d6) δ 1.22 (s; 12H), 1.34 (t, J = 7.2 Hz, 6H), 2.02 (m, 2H), 2.73 (t, J = 6.0 Hz, 4H), 3.84 (s, 6H), 4.21 (m, 4H), 6.23 (d, J = 14 Hz, 2H), 6.23 (d, J = 2.8 Hz, 2H) 6.94 (d, J = 2.4 Hz 2H) 7.22 (s, 2H) 7.28 (m, 6H) 7.66 (m, 3H); 13C NMR (acetone-d6) δ 11.6, 21.3 24.5, 26.8, 39.0, 48.8, 55.4, 99.1, 109.0, 111.2, 113.4, 128.0, 128.7, 129.5, 130.3, 135.5, 139.4, 142.8, 145.0, 158.1, 161.0, 170.6. HRMS (ESI) m/z: calcd. for C42H49N2O2+ 613.3789, obsd 613.3777.
3-Ethyl-2-((E)-2-((E)-6-((E)-2-(3-ethyl-1,1-dimethyl-1,3-dihydro-2H-benzo[e]indol-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1,1-dimethyl-1H-benzo[e]indol-3-ium iodide, 6j; Yield 65%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.46 (t, J = 6.8 Hz, 6H), 1.51 (s, 12H), 2.13 (t, J = 5.6 Hz, 2H), 2.79 (t, J = 6.0 Hz, 4H), 4.28 (m, 4H), 6.17 (d, J = 14.0 Hz, 2H), 7.45 (m, 13H), 7.94 (m, 6H); 13C NMR (CDCl3) δ 99.3, 110.4, 122.0, 124.8, 127.6, 128.1, 128.3, 128.7, 129.5, 130.1, 130.7, 131.7, 132.0, 133.3, 139.1, 139.3, 147.3, 162.0, 172.4. HRMS (ESI) m/z: calcd. for C48H49Cl2N2+ 593.2485, obsd 593.2475.
1,3,3-Trimethyl-2-((E)-2-((E)-6-(2-((E)-1,3,3-trimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-3H-indol-1-ium iodide, 6k; yield 68%; m.p. > 260 °C; 1H-NMR (CDCl3) δ 1.04 (t, J = 7.2 Hz, 6H), 1.23 (s, 12H), 1.49 (m, 4H), 1.80 (m, 4H), 2.11 (t, J = 6.0 Hz, 2H), 2.745 (s, 4H), 4.04 (t, J = 7.2 Hz, 4H), 6.10 (d, J = 14 Hz, 2H), 7.07 (d, J = 11.6 Hz, 2H), 7.22 (m, 6H), 7.33 (t, J = 7.2 Hz, 2H), 7.61 (m, 3H); 13C NMR (CDCl3) 14.0, 20.5, 21.3, 25.0, 27.7, 29.4, 110.4, 122.1, 124.8, 128.2, 128.6, 129.6, 140.8, 142.3. HRMS (ESI) m/z: calcd. for C44H53N2+ 609.4203, obsd 609.4198.
1-Butyl-2-((E)-2-((E)-6-(2-((E)-1-butyl-5-chloro-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetra-hydro-[1,1′-biphenyl]-2-yl)vinyl)-5-chloro-3,3-dimethyl-3H-indol-1-ium iodide, 6l: Yield 67%; m.p. > 260; 1H NMR (CDCl3) δ 1.03 (t, J = 7.2 Hz, 6H), 1.191 (s, 12H), 1.47 (m, 4H), 1.78 (t, J = 6.8 Hz, 4H), 2.12 (s, 2H), 2.75 (s, 4H), 4.02 (s, 4H), 6.09 (d, J = 12.4 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 7.28 (m, 8H), 7.86 (m, 3H); 13C NMR (CDCl3) δ 14.0, 20.4, 21.3, 25.0, 27.7, 29.3, 44.7, 48.6, 100.4, 111.3, 122.7, 128.3, 128.6, 129.6, 130.2, 138.7, 141.0, 142.4, 148.5, 171.0. HRMS (ESI) m/z: calcd. for C44H51Cl2N2+ 677.3424, obsd 677.3421.
1-Butyl-2-((E)-2-((E)-6-(2-((E)-1-butyl-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-3,3-dimethyl-3H-indol-1-ium iodide, 6m: Yield 65%; m.p. > 260 °C; 1H NMR (CDCl3), δ 1.04 (t, J = 7.2 Hz, 6H), 1.20 (s, 12H), 1.51 (m, 4H), 1.81 (m, 4H), 2.13 (t, J = 5.2 Hz, 2H), 2.75 (s, 4H), 4.04 (t, J =7.2 Hz, 4H), 6.10 (d, J = 14.0 Hz, 2H), 7.07 (d, J = 8 Hz, 2H), 7.20 (m, 8H), 7.61 (m, 3H); 13C NMR (CDCl3) δ 14.0, 20.5, 21.3, 25.0, 27.7, 29.4, 44.4, 48.6, 100.0, 110.3, 122.1, 124.7, 128.2, 128.6, 129.6, 132.1, 138.8, 140.8, 142.3, 148.4, 162.9, 171.4. HRMS (ESI) m/z: calcd. for C44H51Br2N2+ 765.2414, obsd 765.2422.
1-Butyl-2-((E)-2-((E)-6-(2-((E)-1-butyl-5-methoxy-3,3-dimethylindolin-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-5-methoxy-3,3-dimethyl-3H-indol-1-ium iodide, 6n: Yield 66%; m.p. > 260 °C; 1H NMR (CDCl3) δ 1.01 (t, J = 6.8 Hz, 6H), 1.17 (s, 12H), 1.46 (m, 4H), 1.80 (m, 4H), 2.07 (s, 2H), 2.68 (s, 4H), 3.82 (s, 6H), 4.03 (t, J = 7.2 Hz, 4H), 6.00 (d, J = 14.0 Hz, 2H) 6.78 (s, 2H), 6.88 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 7.11 (d, J = 14.0 Hz, 2H), 7.21 (d, J = 6.8 Hz, 2H), 7.57 (m, 3H); 13C NMR (CDCl3) δ 13.9, 20.4, 21.3, 24.8, 27.7, 29.4, 44.6, 48.7, 56.0, 99.4, 109.1, 111.1, 113.1, 128.1, 128.6, 129.4, 130.9, 135.9, 139.0, 142.4, 146.9, 157.9, 161.4, 170.6. HRMS (ESI) m/z: calcd. for C46H57N2O2+ 669.4415, obsd 669.4404.
3-Butyl-2-((E)-2-((E)-6-((E)-2-(3-butyl-1,1-dimethyl-1,3-dihydro-2H-benzo[e]indol-2-ylidene)ethylidene)-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)vinyl)-1,1-dimethyl-1H-benzo[e]indol-3-ium iodide, 6o: Yield 70%; m.p. >260 °C; 1H NMR (CDCl3) δ 1.03 (t, J = 7.2 Hz, 6H), 1.52 (m, 16H), 1.86 (m, 4H), 2.16 (t, J = 6.0 Hz, 2H), 2.79 (s, 4H), 4.17 (s, 4H), 6.15 (d, J = 14.4 Hz, 2H), 7.34 (m, 6H), 7.45 (d, J = 7.6 Hz, 2H), 7.53 (m, 2H), 7.68 (t, J = 2.8 Hz, 3H), 7.93 (m, 6H); 13C NMR (CDCl3) δ 14.0, 20.4, 21.4, 25.0, 27.2, 29.7, 44.6, 50.4, 99.6, 110.6, 122.0, 124.8, 127.6, 128.1, 128.4, 128.7, 129.6, 130.1, 130.6, 131.7, 133.2, 139.0, 139.7, 147.3, 172.8. HRMS (ESI) m/z: calcd. for C52H57N2+ 709.4516, obsd 709.4540.
3.3. Stock Solutions
Stock solutions of the dyes and standard were prepared by weighing the solid on a 5-digit analytical balance in an amber vial and adding solvent via a class A volumetric pipette to a final concentration of 1.0 mM. The vials were vortexed for 20 s and then sonicated for 15 min to ensure complete dissolution. The stock solutions were stored in a dark freezer at 4 °C when not in use. Working solutions were prepared just prior to use by dilution of the stock to final concentrations.
3.4. Method of Determining Molar Absorptivity and Fluorescence Quantum Yield
Stock solutions were used to prepare six dilutions of dyes in ethanol and the standard with concentrations ranging from 1 μM to 4 μM using a class A volumetric pipette in order to maintain absorption between 0.1 and 1.0. The dye solutions were diluted ten-fold for fluorescence in order to minimize inner filter effect. The absorbance spectra of each sample was measured in triplicate from 400 nm to 900 nm. The emission spectrum of each sample was measured in triplicate with a 750 nm excitation wavelength.
For molar absorptivity, the absorbance at the wavelength of maximum absorbance (λmax) was determined and the absorbance of each sample at λmax was plotted as a function of dye concentration. The linear regression equation was computed using Microsoft Excel.
The fluorescence quantum yields were determined relative to the indocyanine green standard utilizing the gradient from the plot of integrated fluorescence intensity vs. absorbance (Grad) and the published quantum yield of the standard (φS, 13.2% [29]) as per Equation (1):
φD = φS * GradD/GradS ∗ η2S/η2D
3.5. HSA Binding Study
A stock solution of 6a (4 × 10−5 M) and HSA (4 × 10−5 M; Sigma Aldrich, St. Louis, MO, USA) were prepared in PBS buffer. Fluorescence titration with HSA concentrations (0–2 μM) were made by mixing 35 μL dye solution with PBS buffer solution with and without HSA to a total volume of 4000 μL in a fluorescence cuvette to make working solutions of 2 μM dye. Fluorescence spectra were measured in duplicate with excitation at 740 nm and slit widths of 5 nm for both excitation and emission.
4. Conclusions
A series of 15 phenyl-substituted heptamethine cyanines was synthesized in good yields and characterized by 1H and 13C NMR. Their optical properties including molar absorptivity, fluorescence, Stokes shift, and quantum yield were measured. The optical properties followed similar trends to previously published cyanine dye MHI-06. The binding affinity of one of these heptamethine cyanine dyes to HSA was measured to be 5 orders of magnitude lower than our previous synthesized trimethine cyanines further confirming the hypothesis that the binding affinity of the dyes is not only hydrophobicity dependent, but dependent on steric interferences in the binding site [28]. Because albumin is known to bind a variety of compounds, this information on the steric specificity of these binding sites is of potential interest when developing methods to study them.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
M.H., A.L. and F.M. would like to thank the Department of Chemistry at Georgia State University for their support and the funds provided for the Ph.D. program for A.L. and Master’s program for F.M. M.H., A.L. and F.M. would like to thank Ariana Laskey for her contribution in the synthesis of some the compounds. M.H. wishes to thank the NIH/NIBI R01EB022230, the Brains and Behavior Seed Grant, the Atlanta Clinical & Translational Science Institute for the Healthcare Innovation Program Grant, and the Georgia Research Alliance for the Ventures Phase 1 Grant.
Author Contributions
M.H. designed the research and all authors wrote the paper. F.M. and A.L. performed experiments. All authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Flanagan, J.H., Jr.; Khan, S.H.; Menchen, S.; Soper, S.A.; Hammer, R.P. Functionalized tricarbocyanine dyes as near-infrared fluorescent probes for biomolecules. Bioconjug. Chem. 1997, 8, 751–756. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Patonay, G. Fluorescence studies of carbocyanines using AOTF. Talanta 1997, 45, 285–289. [Google Scholar] [CrossRef]
- Patonay, G.; Salon, J.; Sowell, J.; Strekowski, L. Noncovalent labeling of biomolecules with red and near-infrared dyes. Molecules 2004, 9, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shi, C.; Tong, R.; Qian, W.; Zhau, H.E.; Wang, R.; Zhu, G.; Cheng, J.; Yang, V.W.; Cheng, T.; et al. Near IR heptamethine cyanine dye-mediated cancer imaging. Clin. Cancer Res. 2010, 16, 2833–2844. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wu, J.B.; Pan, D. Review on near-infrared heptamethine cyanine dyes as theranostic agents for tumor imaging, targeting, and photodynamic therapy. J. Biomed. Opt. 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Li, Y.L.M.; Chen, H.; Li, H. Near-infrared chemodosimetric probes based on heptamethine cyanine dyes for the “naked-eye” detection of cyanide in aqueous media. J. Lumin. 2017, 185, 286–291. [Google Scholar] [CrossRef]
- Soriano, E.; Holder, C.; Levitz, A.; Henary, M. Benz[c,d]indolium-containing monomethine cyanine dyes: Synthesis and photophysical properties. Molecules 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Kurutosa, A.; Tarabara, O.R.U.; Trusovab, V.; Gorbenko, G.; Gadjev, N.; Deligeorgiev, T. Novel synthetic approach to near-infrared heptamethine cyanine dyes and spectroscopic characterization in presence of biological molecules. J. Photochem. Photobiol. A 2016, 328, 87–96. [Google Scholar] [CrossRef]
- Ernst, L.A.; Gupta, R.K.; Mujumdar, R.B.; Waggoner, A.S. Cyanine dye labeling reagents for sulfhydryl groups. Cytometry 1989, 10, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Honig, B.; Greenberg, A.D.; Dinur, U.; Ebrey, T.G. Visual-pigment spectra: Implications of the protonation of the retinal Schiff base. Biochemistry 1976, 15, 4593–4599. [Google Scholar] [CrossRef] [PubMed]
- Ashitate, Y.; Levitz, A.; Park, M.H.; Hyun, H.; Venugopal, V.; Park, G.; El Fakhri, G.; Henary, M.; Gioux, S.; Frangioni, J.V.; et al. Endocrine-specific NIR fluorophores for adrenal gland targeting. Chem. Commun. 2016, 52, 10305–10308. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Owens, E.A.; Wada, H.; Levitz, A.; Park, G.; Park, M.H.; Frangioni, J.V.; Henary, M.; Choi, H.S. Cartilage-specific near-infrared fluorophores for biomedical imaging. Angew. Chem. Int. Ed. Engl. 2015, 54, 8648–8652. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Park, M.H.; Owens, E.A.; Wada, H.; Henary, M.; Handgraaf, H.J.; Vahrmeijer, A.L.; Frangioni, J.V.; Choi, H.S. Structure-inherent targeting of near-infrared fluorophores for parathyroid and thyroid gland imaging. Nat. Med. 2015, 21, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Wada, H.; Bao, K.; Gravier, J.; Yadav, Y.; Laramie, M.; Henary, M.; Frangioni, J.V.; Choi, H.S. Phosphonated near-infrared fluorophores for biomedical imaging of bone. Angew. Chem. Int. Ed. Engl. 2014, 53, 10668–10672. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, G.; Hyun, H.; Lee, J.H.; Ashitate, Y.; Choi, J.; Hong, G.H.; Owens, E.A.; Henary, M.; Choi, H.S. Near-infrared lipophilic fluorophores for tracing tissue growth. Biomed. Mater. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Owens, E.A.; Henary, M.; El Fakhri, G.; Choi, H.S. Tissue-specific near-infrared fluorescence imaging. Acc. Chem. Res. 2016, 49, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Shershov, V.E.; Spitsyn, M.A.; Kuznetsova, V.E.; Timofeev, E.N.; Ivashkina, O.A.; Abramov, I.S.; Nasedkina, T.V.; Zasedatelev, A.S.; Chudinov, A.V. Near-infrared heptamethine cyanine dyes. Synthesis, spectroscopic characterization, thermal properties and photostability. Dyes Pigment. 2013, 97, 353–360. [Google Scholar] [CrossRef]
- Lee, H.; Berezin, M.Y.; Tang, R.; Zhegalova, N.; Achilefu, S. Pyrazole-substituted near-infrared cyanine dyes exhibit pH-dependent fluorescence lifetime properties. Photochem. Photobiol. 2013, 89, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mason, J.C.; Achilefu, S. Heptamethine cyanine dyes with a robust C-C bond at the central position of the chromophore. J. Org. Chem. 2006, 71, 7862–7865. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mason, J.C.; Achilefu, S. Synthesis and spectral properties of near-infrared aminophenyl-, hydroxyphenyl-, and phenyl-substituted heptamethine cyanines. J. Org. Chem. 2008, 73, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.; Stanford, C.; Morton, M.D.; Zhu, Q.; Smith, M.B. Structurally modified indocyanine green dyes. Modification of the polyene linker. Dyes Pigment. 2013, 99, 275–283. [Google Scholar] [CrossRef]
- Jozef Salon, E.W.; Raszkiewicz, A.; Patonay, G.; Strekowski, L. Synthesis of Benz[e]indolium Heptamethine Cyanines Containing C-Substituents at the Central Portion of the Heptamethine Moiety. J. Heterocycl. Chem. 2005, 42, 959–961. [Google Scholar] [CrossRef]
- Theodore Peters, J. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic: San Diego, CA, USA, 1995. [Google Scholar]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, M.; Maggioli, C.; Zaccherini, G. Human albumin in the management of complications of liver cirrhosis. Crit. Care 2012, 16. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Beckford, G.; Owens, E.; Henary, M.; Patonay, G. The solvatochromic effects of side chain substitution on the binding interaction of novel tricarbocyanine dyes with human serum albumin. Talanta 2012, 92, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Rurack, K.; Spieles, M. Fluorescence quantum yields of a series of red and near-infrared dyes emitting at 600–1000 nm. Anal. Chem. 2011, 83, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Chapman, G.; Henary, M.; Patonay, G. The effect of varying short-chain alkyl substitution on the molar absorptivity and quantum yield of cyanine dyes. Anal. Chem. Insights 2011, 6, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.A.; Jacobs, L.D.; Furlani, T.R.; Nalley, E.A.; Laboy, J. Conformationally dependent heavy atom effect of chlorine on alkene triplet lifetimes. experimental and ab initio calculations. J. Am. Chem. Soc. 1991, 144, 1623–1625. [Google Scholar]
- Dost, T.L.; Gressel, M.T.; Henary, M. Synthesis and optical properties of pentamethine cyanine dyes with carboxylic acid moieties. Anal. Chem. Insights 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Toseland, C.P. Fluorescent labeling and modification of proteins. J. Chem. Biol. 2013, 6, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, A. Covalent labeling of proteins and nucleic acids with fluorophores. Methods Enzymol. 1995, 246, 362–373. [Google Scholar] [PubMed]
- Patonay, G.; Kim, J.S.; Kodagahally, R.; Strekowski, L. Spectroscopic study of a novel bis(heptamethine cyanine) dye and its interaction with human serum albumin. Appl. Spectrosc. 2005, 59, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, H.; Tang, Y.; Xu, G.; Yan, W. Spectroscopic investigation on the interaction of J-aggregate with human serum albumin. Biophys Chem. 2007, 128, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Berezin, M.Y.; Lee, H.; Akers, W.; Nikiforovich, G.; Achilefu, S. Ratiometric analysis of fluorescence lifetime for probing binding sites in albumin with near-infrared fluorescent molecular probes. Photochem. Photobiol. 2007, 83, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kodagahally, R.; Strekowski, L.; Patonay, G. A study of intramolecular H-complexes of novel bis(heptamethine cyanine) dyes. Talanta 2005, 67, 947–954. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
Scheme 1.
Synthetic routes of heptamethine dyes containing a phenyl ring at the meso-position.
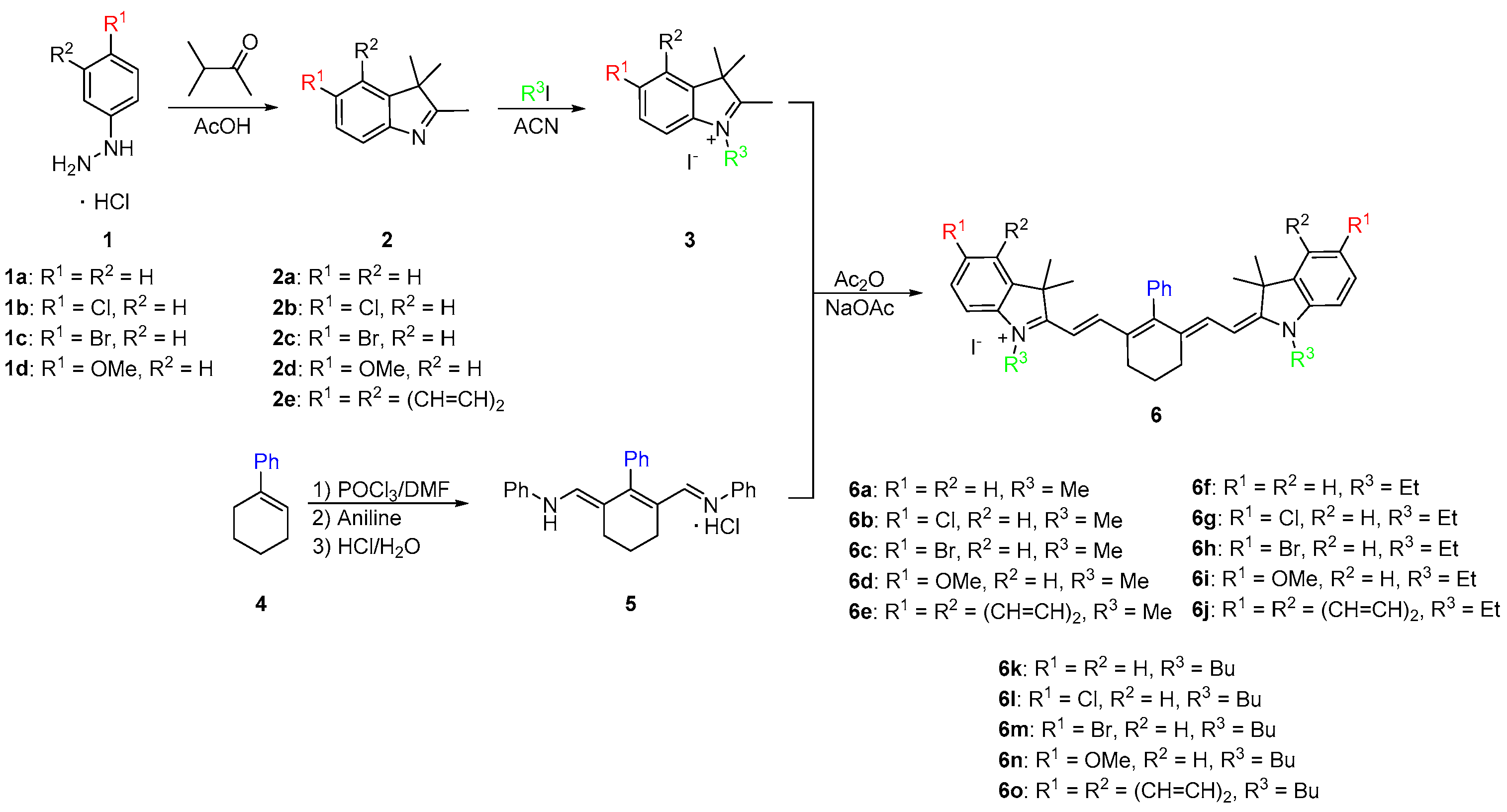
Figure 1.
The structures of the three NIR standards used for the study.
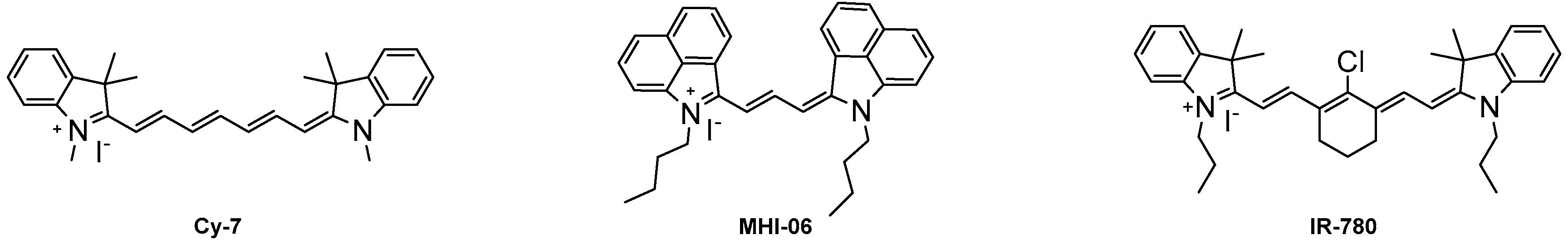
Figure 2.
Absorption spectra of dye 6m in ethanol.
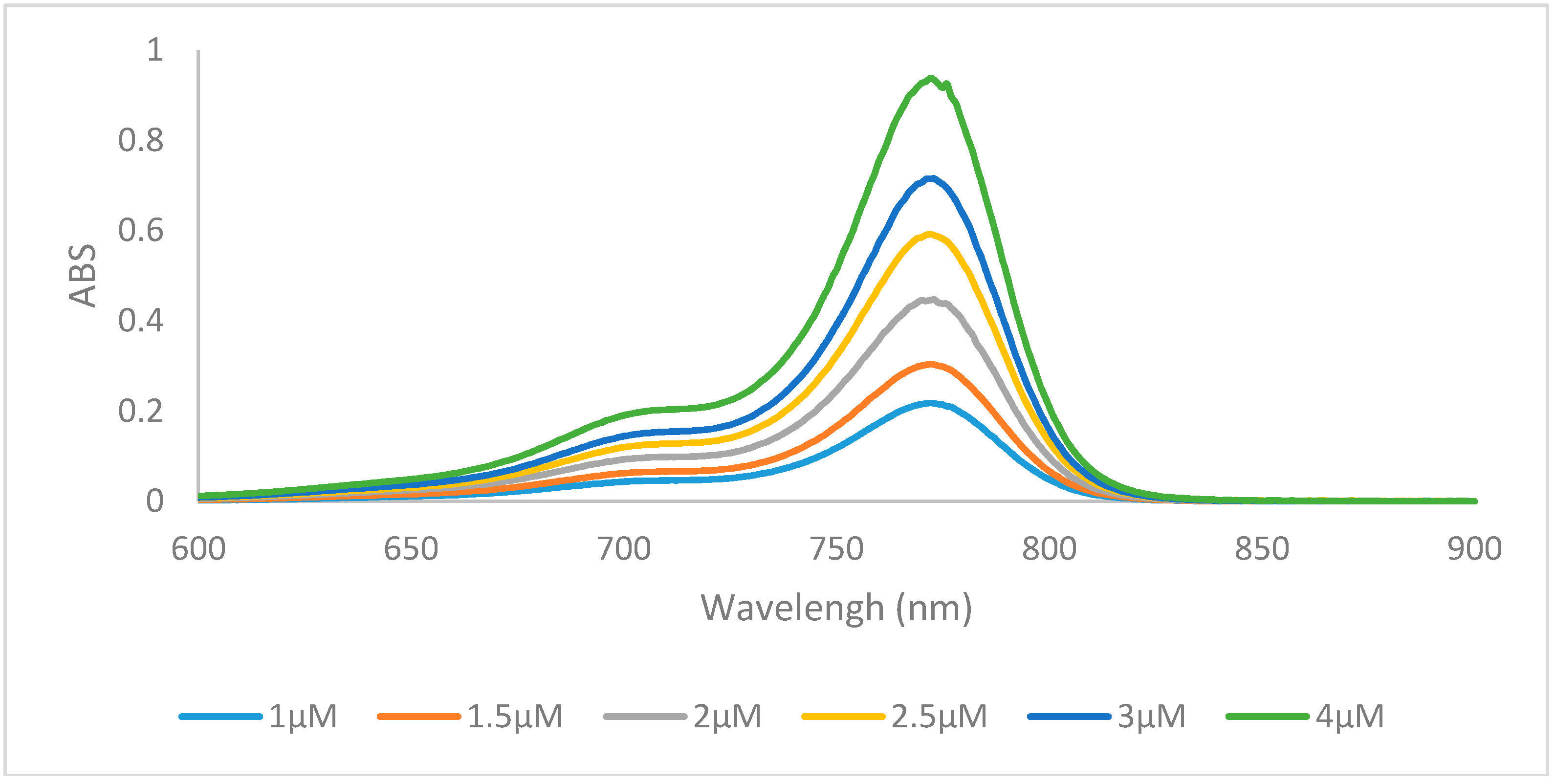
Figure 3.
Emission spectra of dye 6m in ethanol with excitation wavelength of 750 nm.
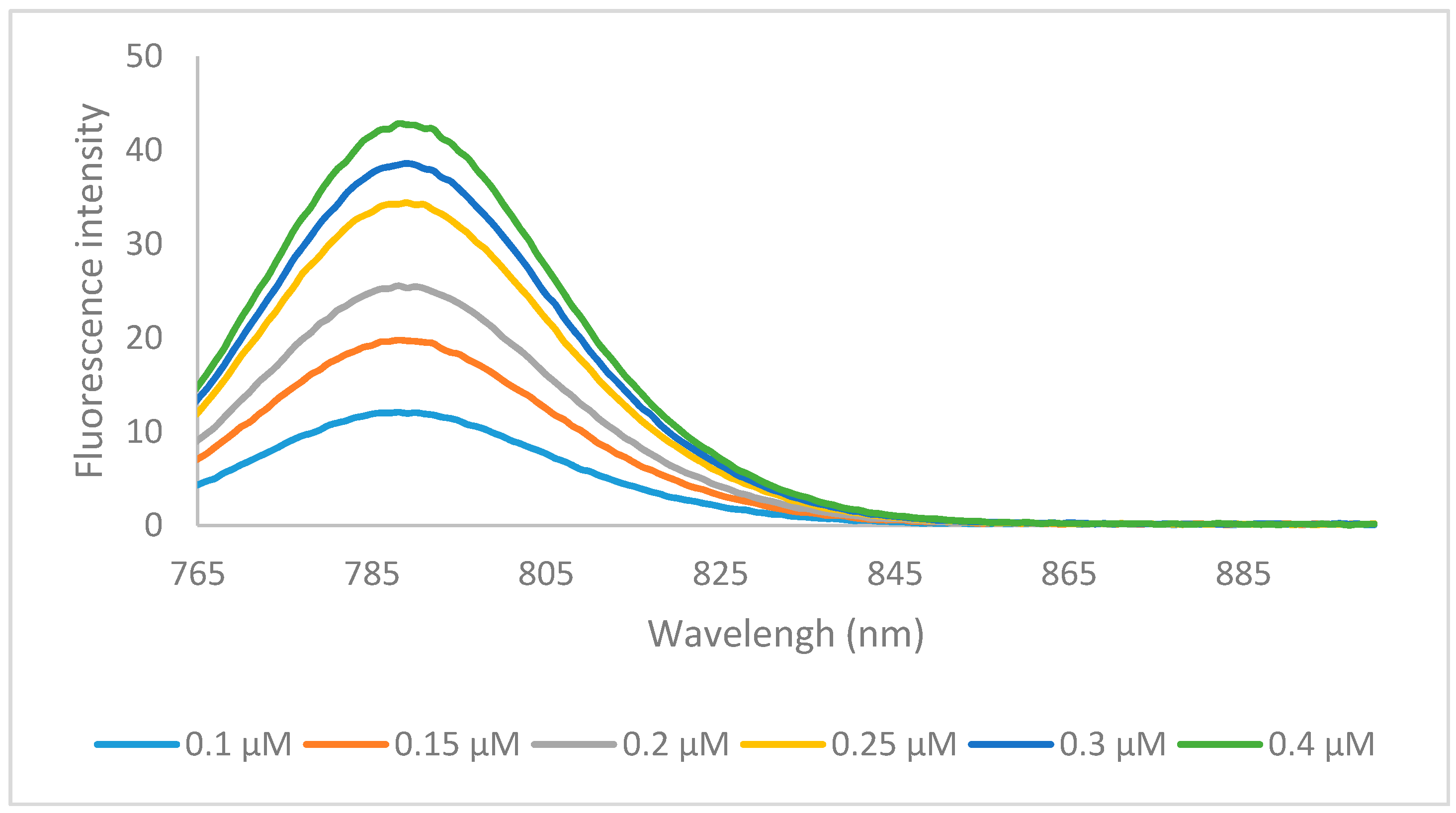
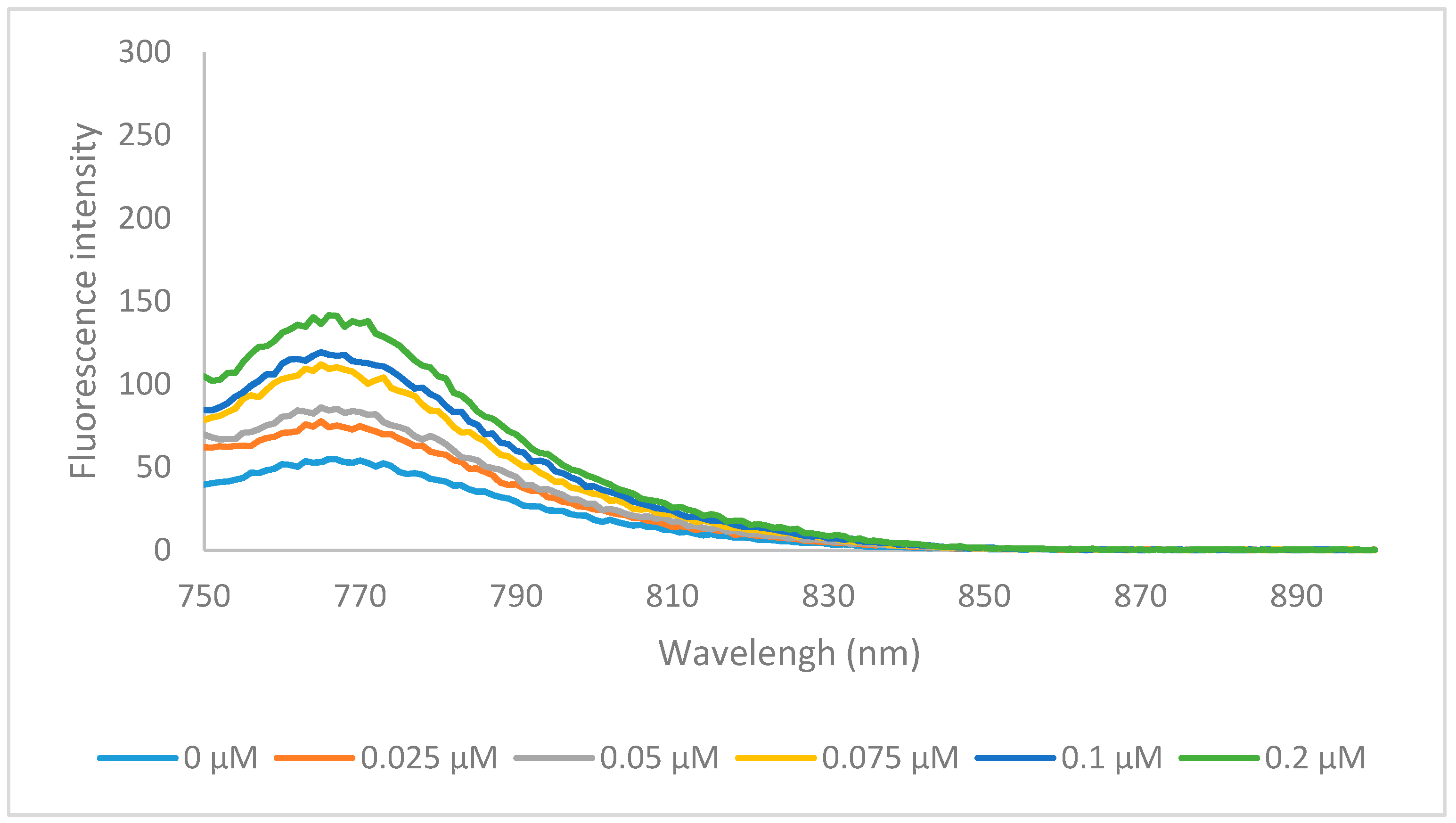
Figure 4.
The emission specra of 6a (0.2 μM) binding with various concentration of HSA, in PBS buffer at excitation wavelength of 740 nm.
Figure 4.
The emission specra of 6a (0.2 μM) binding with various concentration of HSA, in PBS buffer at excitation wavelength of 740 nm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Spectral Characteristics of dyes Cy-7, IR-780 and 6a–o. All optical properties of the dyes were measured in ethanol.
Table 1.
Spectral Characteristics of dyes Cy-7, IR-780 and 6a–o. All optical properties of the dyes were measured in ethanol.
| Dye | λmax (nm) | λemission (nm) | Stokes Shift (nm) | ε (L·mol−1·cm−1) | Φ (%) | Molecular Brightness (M−1·cm−1) |
|---|---|---|---|---|---|---|
| Cy-7 | 753 | 775 | 22 | 200,000 | 28 | 56,000 |
| IR-780 | 779 | 799 | 20 | 274,000 | 8.0 | 20,800 |
| 6a | 759 | 774 | 15 | 265,700 | 31 | 82,000 |
| 6b | 765 | 780 | 15 | 261,000 | 34 | 88,700 |
| 6c | 767 | 783 | 16 | 275,600 | 35 | 96,500 |
| 6d | 782 | 802 | 20 | 249,500 | 10 | 25,000 |
| 6e | 798 | 810 | 12 | 255,400 | 16 | 40,900 |
| 6f | 760 | 781 | 21 | 263,900 | 39 | 102,300 |
| 6g | 769 | 785 | 16 | 286,600 | 38 | 109,300 |
| 6h | 770 | 786 | 16 | 282,900 | 42 | 119,200 |
| 6i | 786 | 804 | 18 | 143,500 | 12 | 17,200 |
| 6j | 797 | 810 | 13 | 231,800 | 17 | 39,400 |
| 6k | 763 | 780 | 17 | 198,500 | 45 | 89,300 |
| 6l | 772 | 787 | 15 | 123,400 | 47 | 58,000 |
| 6m | 773 | 788 | 15 | 239,200 | 48 | 113,600 |
| 6n | 789 | 805 | 16 | 249,900 | 11 | 27,100 |
| 6o | 800 | 812 | 12 | 226,600 | 17 | 38,500 |
Table 2.
Physicochemical properties (in silico) of dyes MHI-06 and 6a–o calculated using ChemAxon. The data calculated (at pH 7.4) include: logD, polarizability, dipole moment (debye), number of rotatable bonds, volume (Å3), molecular surface area (Å3), and molar mass (g/mol).
Table 2.
Physicochemical properties (in silico) of dyes MHI-06 and 6a–o calculated using ChemAxon. The data calculated (at pH 7.4) include: logD, polarizability, dipole moment (debye), number of rotatable bonds, volume (Å3), molecular surface area (Å3), and molar mass (g/mol).
| Dye | logD | Polarizability | Dipole Moment | Rot. Bonds | Volume | Molec. Surface Area | Molar Mass |
|---|---|---|---|---|---|---|---|
| MHI-06 | 4.97 | 59.45 | 2.48 | 8 | 441.44 | 693.216 | 584.54 |
| 6a | 6.07 | 65.57 | 8.35 | 4 | 519.85 | 814.001 | 652.66 |
| 6b | 7.28 | 69.15 | 12.3 | 4 | 547.45 | 845.982 | 721.55 |
| 6c | 7.61 | 70.67 | 13.02 | 4 | 556.15 | 854.732 | 810.46 |
| 6d | 5.75 | 70.51 | 27.66 | 6 | 569.71 | 907.778 | 712.72 |
| 6e | 8.05 | 80.82 | 11.87 | 4 | 604.27 | 936.392 | 752.78 |
| 6f | 6.75 | 69.26 | 3.2 | 6 | 552.14 | 874.685 | 680.72 |
| 6g | 7.99 | 72.84 | 4.81 | 6 | 579.48 | 906.491 | 749.6 |
| 6h | 8.32 | 74.35 | 4.97 | 6 | 588.47 | 915.629 | 838.51 |
| 6i | 6.47 | 74.2 | 25.29 | 8 | 603.83 | 969.908 | 740.77 |
| 6j | 8.76 | 84.51 | 4.59 | 6 | 638.63 | 999.272 | 7803.84 |
| 6k | 8.72 | 76.65 | 2.85 | 10 | 620.01 | 996.744 | 736.81 |
| 6l | 9.92 | 80.23 | 4.63 | 10 | 647.91 | 1029.597 | 805.71 |
| 6m | 10.25 | 81.71 | 4.96 | 10 | 656.64 | 1038.143 | 894.62 |
| 6n | 8.40 | 81.59 | 26.66 | 12 | 672.63 | 1094.66 | 796.88 |
| 6o | 10.69 | 91.9 | 3.96 | 10 | 708.29 | 1124.8 | 836.95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Levitz, A.; Marmarchi, F.; Henary, M. Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes. Molecules 2018, 23, 226. https://doi.org/10.3390/molecules23020226
AMA Style
Levitz A, Marmarchi F, Henary M. Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes. Molecules. 2018; 23(2):226. https://doi.org/10.3390/molecules23020226
Chicago/Turabian StyleLevitz, Andrew, Fahad Marmarchi, and Maged Henary. 2018. "Synthesis and Optical Properties of Near-Infrared meso-Phenyl-Substituted Symmetric Heptamethine Cyanine Dyes" Molecules 23, no. 2: 226. https://doi.org/10.3390/molecules23020226