
[2+2+2] Annulation of N-(1-Naphthyl)acetamide with Two Alkynoates via Cleavage of Adjacent C–H and C–N Bonds Catalyzed by an Electron-Deficient Rhodium(III) Complex


Abstract
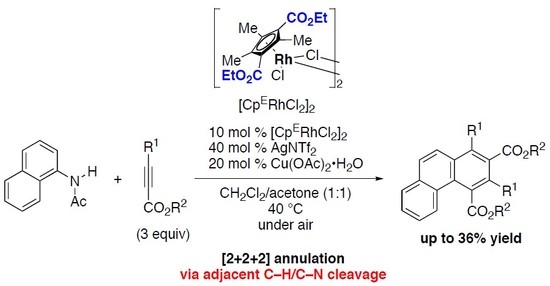
:
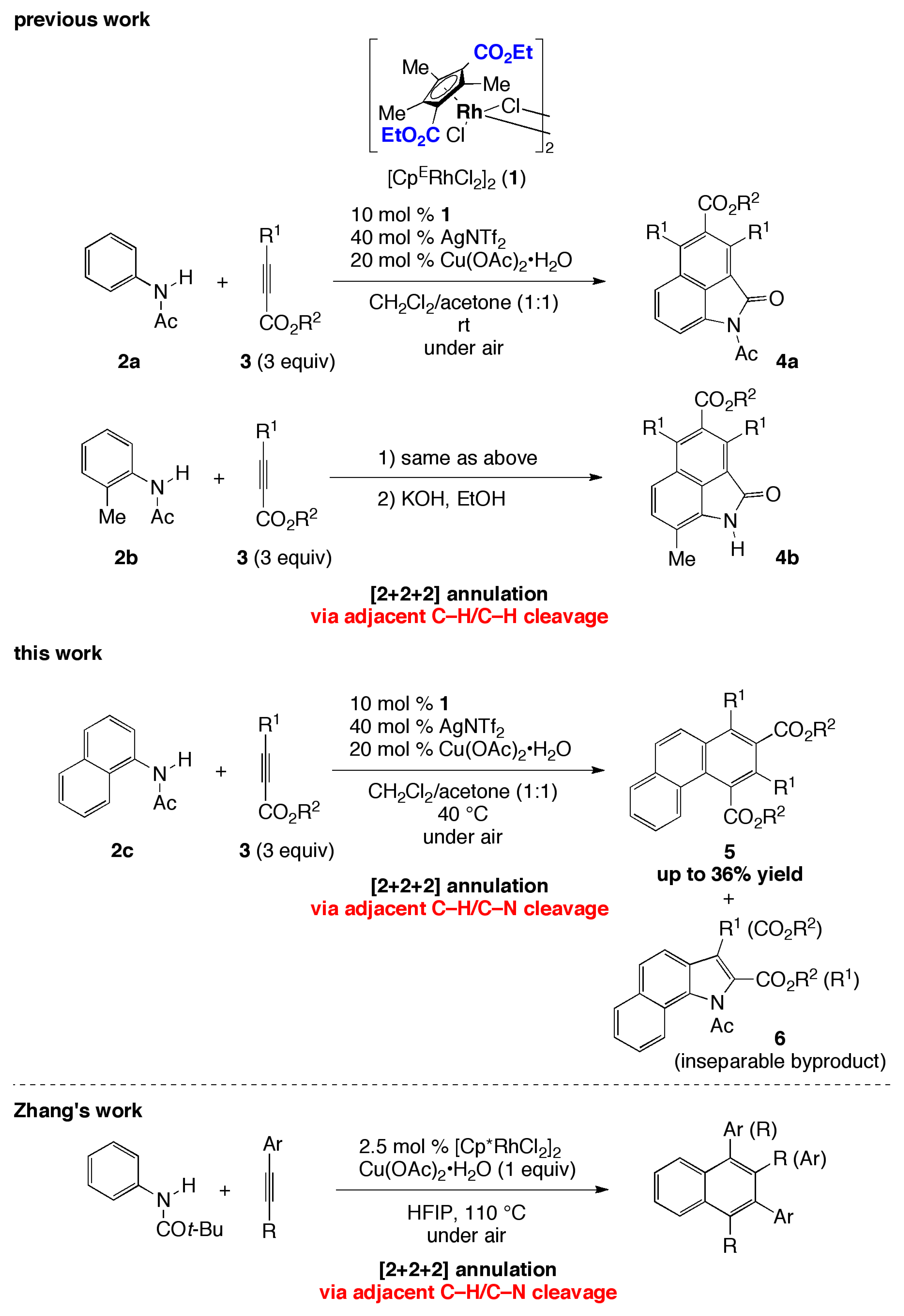
1. Introduction
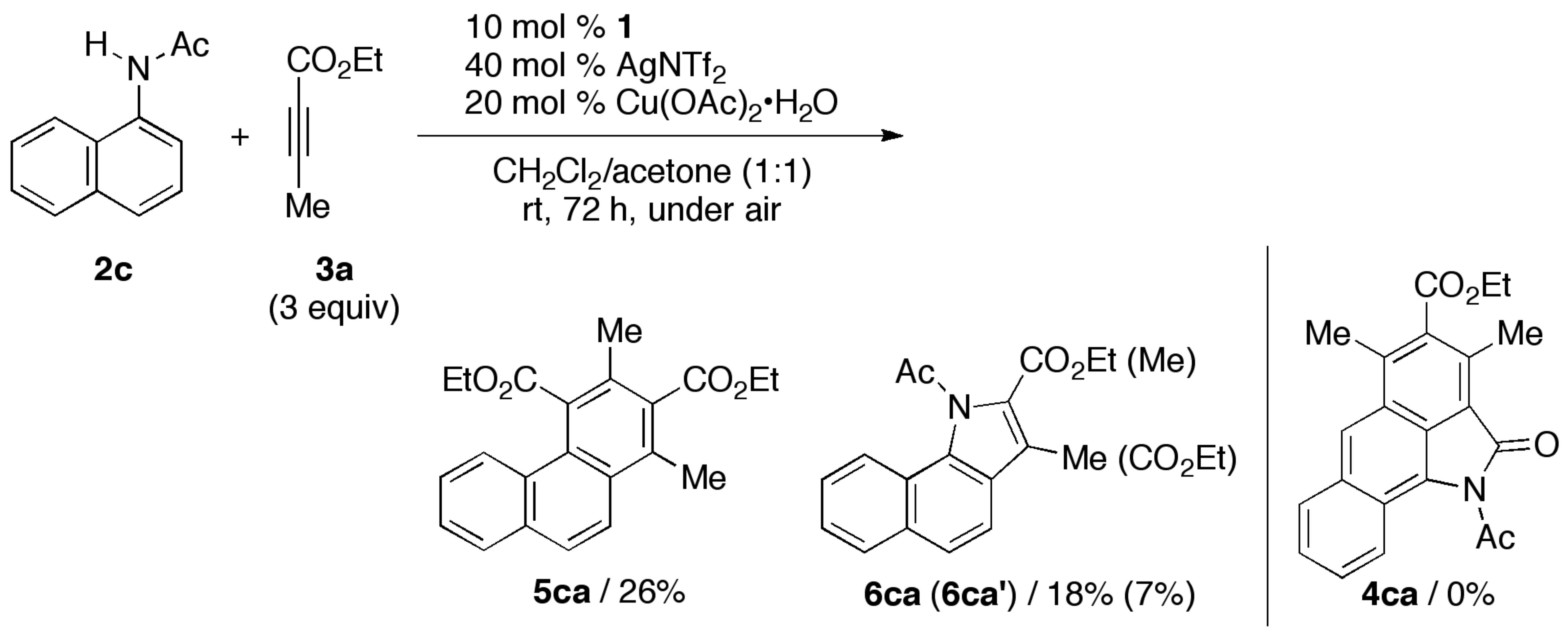
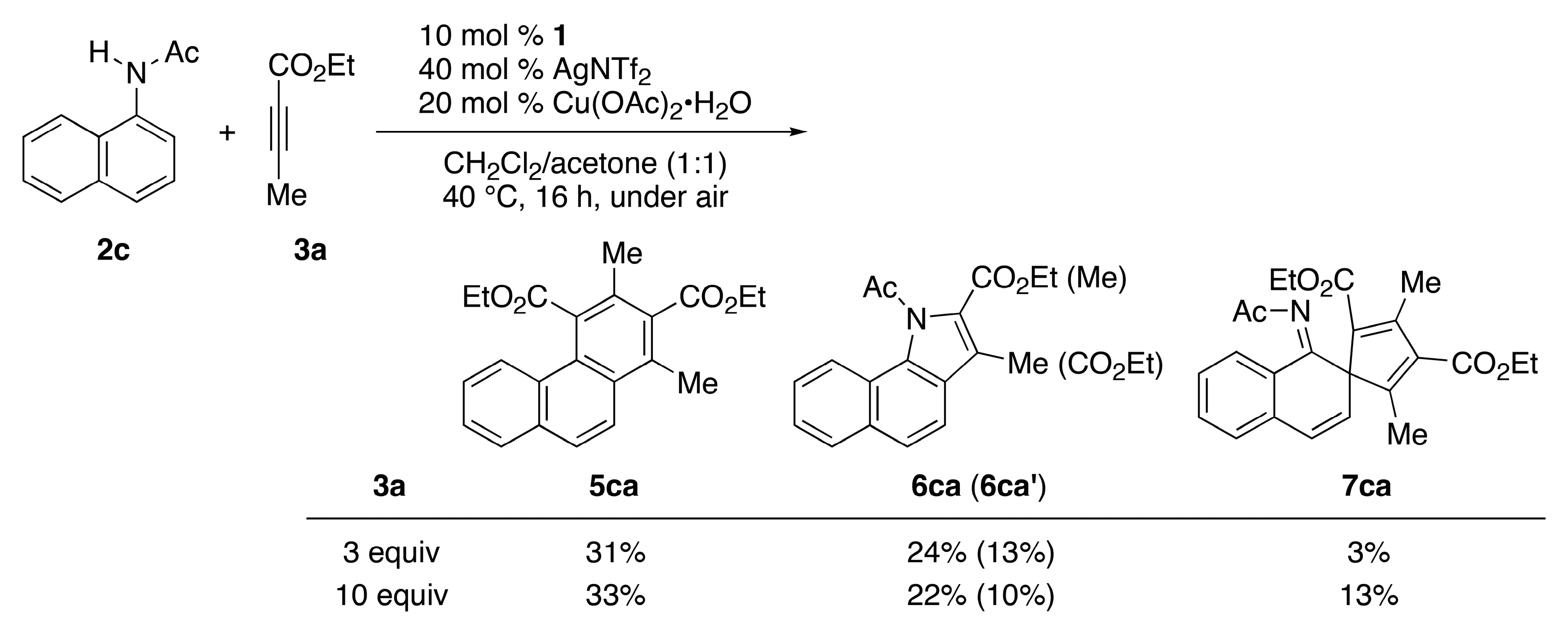
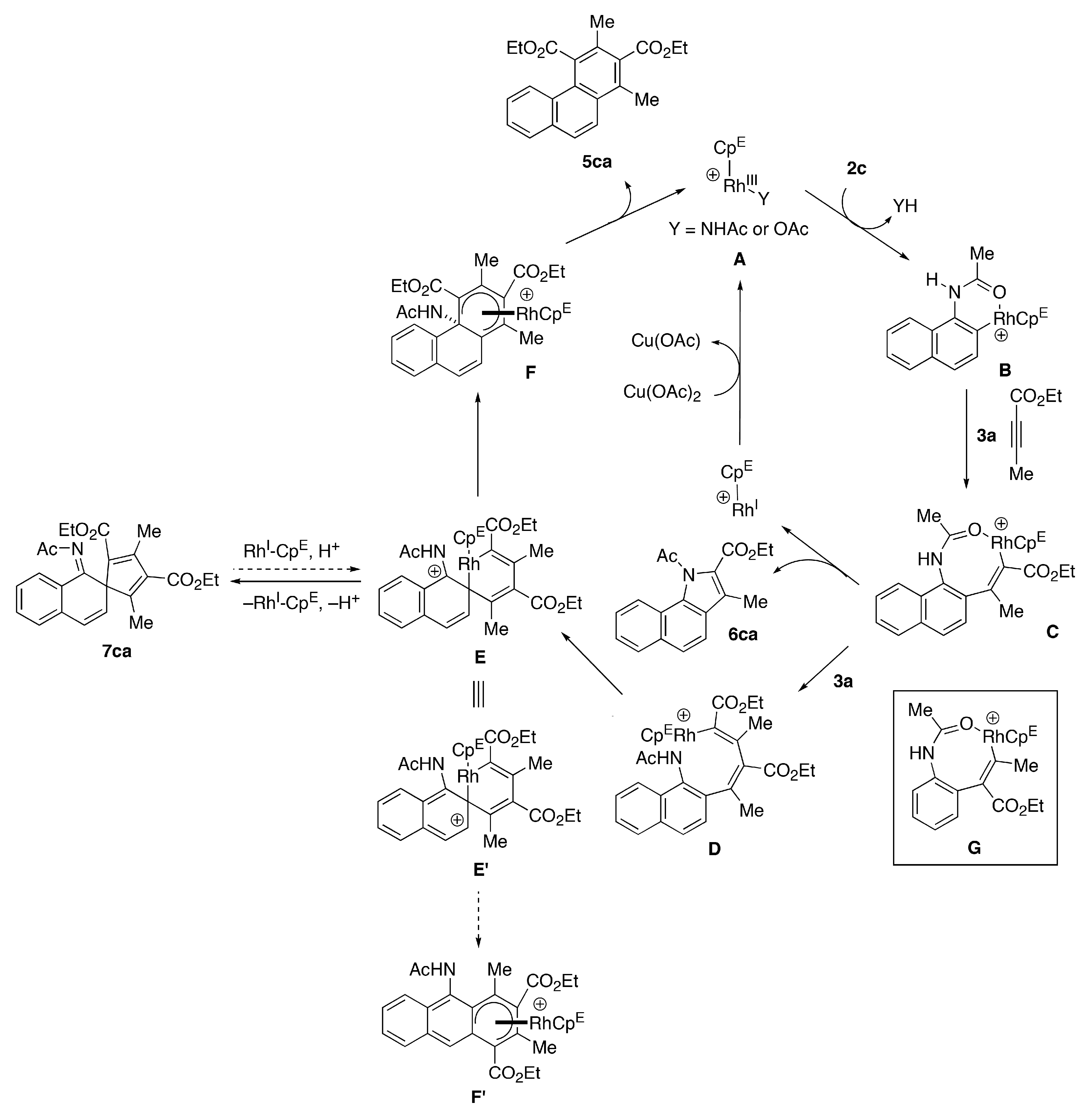
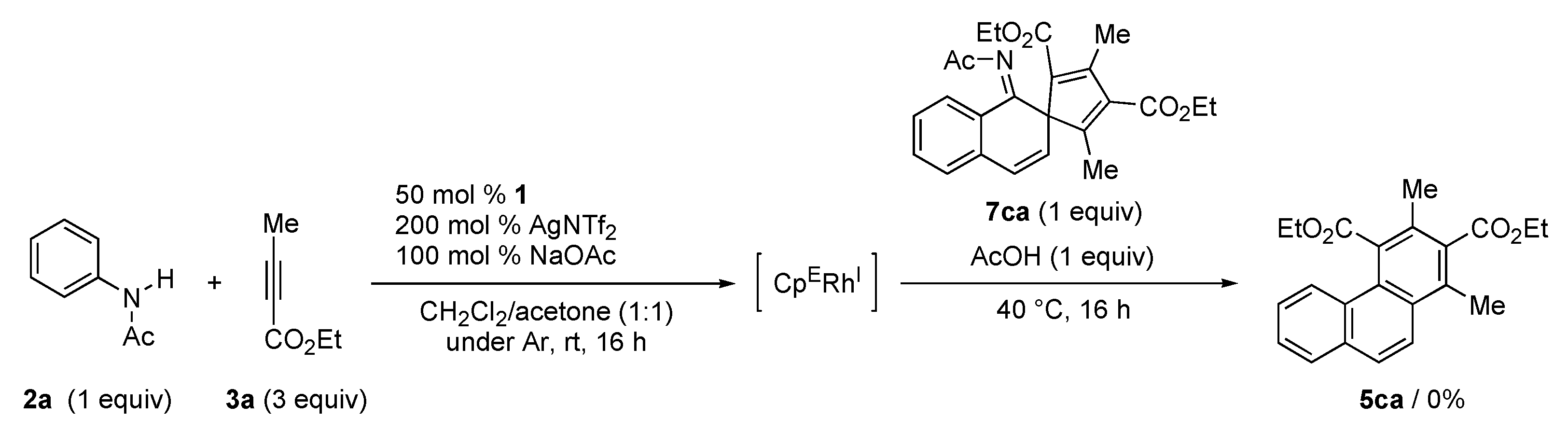
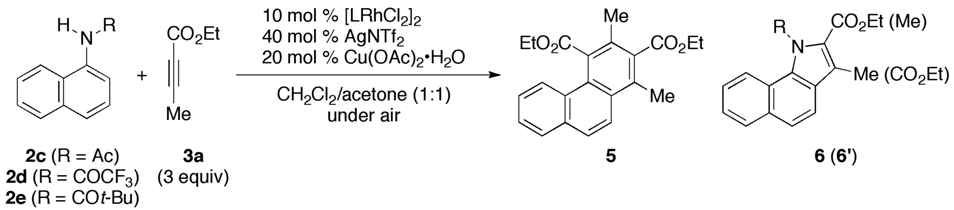
2. Results
3. Materials and Methods
3.1. General Information
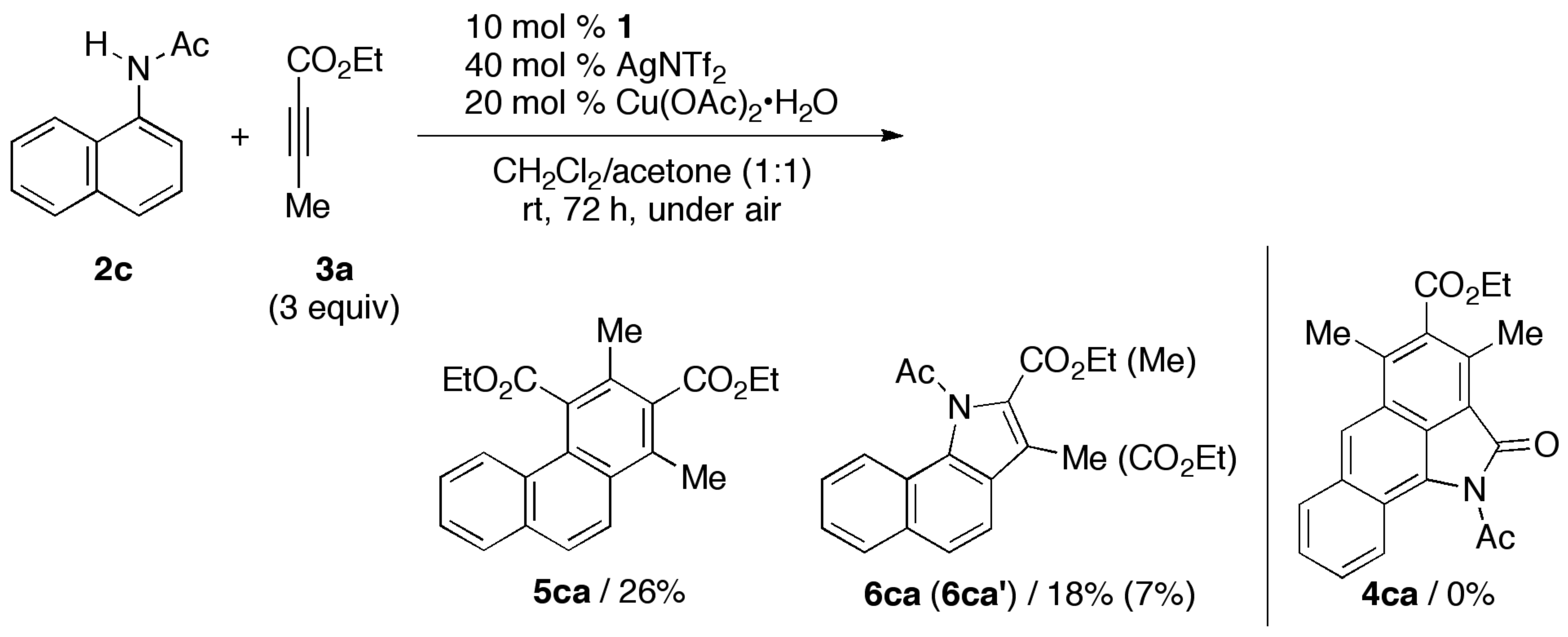
3.2. General Procedure for the Rhodium-Catalyzed Annulation of N-(1-Naphthyl)amides with Two Alkynoates (5ca, 6ca and 6ca′; Table 1, entry 2)
3.3. Product Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pia–Quintana, A.; Roglans, A. Chiral Induction in [2+2+2] Cycloaddition Reactions. Asian J. Org. Chem. 2018, 7, 1706–1718. [Google Scholar] [CrossRef]
- Beeck, S.; Wegner, H.A. Thieme Chemistry Journals Awardees—Where Are They Now? Aromaticity as a Source for Strain Energy? Synthesis of Curved Polycyclic Aromatic Structures via [2+2+2] Cycloaddition Reactions. Synlett 2017, 28, 1018–1027. [Google Scholar] [CrossRef]
- Domínguez, G.; Pérez–Castells, J. Alkens in [2+2+2] Cycloadditions. Chem. Eur. J. 2016, 22, 6720–6739. [Google Scholar] [CrossRef]
- Amatore, M.; Aubert, C. Recent Advances in Stereoselective [2+2+2] Cycloadditions. Eur. J. Org. Chem. 2015, 2015, 265–286. [Google Scholar] [CrossRef]
- Satoh, Y.; Obora, Y. Niobium Complexes in Organic Transformations: From Stoichiometric Reactions to Catalytic [2+2+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2015, 2015, 5041–5054. [Google Scholar] [CrossRef]
- Tanaka, K. (Ed.) Transition-Metal-Mediated Aromatic Ring Construction; John Wiley & Sons: Hoboken, NJ, USA, 2013; Part 1; ISBN 978-1-118-14892-1. [Google Scholar]
- Broere, D.L.J.; Ruijter, E. Recent Advances in Transition-Metal-Catalyzed [2+2+2]-Cyclo(co)trimerization Reactions. Synthesis 2012, 44, 2639–2672. [Google Scholar] [CrossRef]
- Dominguez, G.; Pérez-Castells, J. Recent advances in [2+2+2] cycloaddition reactions. Chem. Soc. Rev. 2011, 40, 3430–3444. [Google Scholar] [CrossRef]
- Weding, N.; Hapke, M. Preparation and synthetic applications of alkene complexes of group 9 transition metals in [2+2+2] cycloaddition reactions. Chem. Soc. Rev. 2011, 40, 4525–4538. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Sreevani, G. Design and Synthesis of Aromatics through [2+2+2] Cyclotrimerization. Synlett 2018, 29, 2342–2361. [Google Scholar] [CrossRef]
- Tanaka, K.; Kimura, Y.; Murayama, K. Enantioselective Helicene Synthesis by Rhodium-Catalyzed [2+2+2] Cycloadditions. Bull. Chem. Soc. Jpn. 2015, 88, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Tanaka, K. Rhodium-Catalyzed [2+2+2] Cycloaddition of Alkynes for the Synthesis of Substituted Benzenes: Catalysts, Reaction Scope, and Synthetic Applications. Synthesis 2012, 3, 323–350. [Google Scholar] [CrossRef]
- Li, S.; Zhou, L.; Kanno, K.; Takahashi, T. Recent development for enantioselective synthesis of aromatic compounds from alkynes via metallacyclopentadienes. J. Heterocycl. Chem. 2011, 48, 517–528. [Google Scholar] [CrossRef]
- Galan, B.R.; Rovis, T. Beyond Reppe: Building Substituted Arenes by [2+2+2] Cycloadditions of Alkynes. Angew. Chem. Int. Ed. 2009, 48, 2830–2834. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.; Cheng, C. O-Dihaloarenes as aryne precursors for nickel-catalyzed [2+2+2] cycloaddition with alkynes and nitriles. Chem. Commun. 2008, 0, 2992–2994. [Google Scholar] [CrossRef]
- Hsieh, J.; Cheng, C. Nickel-catalyzed cocyclotrimerization of arynes with diynes; a novel method for synthesis of naphthalene derivatives. Chem. Commun. 2005, 0, 2459–2461. [Google Scholar] [CrossRef]
- Yoshikawa, E.; Radhakrishnan, K.V.; Yamamoto, Y. Palladium-Catalyzed Controlled Carbopalladation of Benzyne. J. Am. Chem. Soc. 2000, 122, 7280–7286. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, X.; Feng, X.; Yamamoto, Y.; Almansour, A.I.; Arumugam, N.; Kumar, R.S.; Bao, M. Rhodium-Catalyzed Oxidative Benzannulation of N-Pivaloylanilines with Internal Alkynes through Dual C–H Bond Activation: Synthesis of Highly Substituted Naphthalenes. Chem. Asian J. 2016, 11, 3241–3250. [Google Scholar] [CrossRef]
- He, Z.; Huang, Y. Diverting C–H Annulation Pathways: Nickel-Catalyzed Dehydrogenative Homologation of Aromatic Amides. ACS Catal. 2016, 6, 7814–7823. [Google Scholar] [CrossRef]
- Pham, M.V.; Cramer, N. Aromatic Homologation by Non-Chelate-Assisted RhIII-Catalyzed C–H Functionalization of Arenes with Alkynes. Angew. Chem. Int. Ed. 2014, 53, 3484–3487. [Google Scholar] [CrossRef]
- Gao, M.; Lam, J.W.Y.; Liu, Y.; Li, J.; Tang, B.Z. A new route to functional polymers: Atom-economical synthesis of poly(pyrazolylnaphthalene)s by rhodium-catalyzed oxidative polycoupling of phenylpyrazole and internal diynes. Polym. Chem. 2013, 4, 2841–2849. [Google Scholar] [CrossRef]
- Song, G.; Gong, X.; Li, X. Synthesis of Quinolines via Rh(III)-Catalyzed Oxidative Annulation of Pyridines. J. Org. Chem. 2011, 76, 7583–7589. [Google Scholar] [CrossRef]
- Fukui, M.; Shibata, Y.; Hoshino, Y.; Sugiyama, H.; Teraoka, K.; Uekusa, H.; Noguchi, K.; Tanaka, K. Rhodium(III)-Catalyzed Tandem [2+2+2] Annulation–Lactamization of Anilides with Two Alkynoates via Cleavage of Two Adjacent C–H or C–H/C–O bonds. Chem. Asian J. 2016, 11, 2260–2264. [Google Scholar] [CrossRef]
- Honjo, Y.; Shibata, Y.; Kudo, E.; Namba, T.; Masutomi, K.; Tanaka, K. Room Temperature Decarboxylative and Oxidative [2+2+2] Annulation of Benzoic Acids with Alkynes Catalyzed by an Electron-Deficient Rhodium(III) Complex. Chem. Eur. J. 2018, 24, 317–321. [Google Scholar] [CrossRef]
- Loginov, D.A.; Muratov, D.V.; Nelyubina, Y.V.; Laskova, J.; Kudinov, A.R. μ-Borole triple-decker complexes as catalysts for oxidative coupling of benzoic acid with alkynes. Structure of a hybrid rhodacyclopentadienyl/borole triple-decker complex. J. Mol. Catal. 2017, 426, 393–397. [Google Scholar] [CrossRef]
- Loginov, D.A.; Belova, A.O.; Kudinov, M.R. Rhodacarboranes as catalysts for oxidative coupling of benzoic acid with diphenylacetylene. Russ. Chem. Bull. 2014, 63, 983–986. [Google Scholar] [CrossRef]
- Ueura, K.; Satoh, T.; Miura, M. Rhodium- and Iridium-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes via Regioselective C–H Bond Cleavage. J. Org. Chem. 2007, 72, 5362–5367. [Google Scholar] [CrossRef]
- Geng, K.; Fan, Z.; Zhang, A. Available Rhodium-catalyzed oxidative coupling of N-acyl anilines with alkynes using an acylamino moiety as the traceless directing group. Org. Chem. Front. 2016, 3, 349–353. [Google Scholar] [CrossRef]
- Saxena, P.; Thirupathi, N.; Nethaji, M. Depalladation of Neutral Monoalkyne- and Dialkyne-Inserted Palladacycles and Alkyne Insertion/Depalladation Reactions of Cationic Palladacycles Derived from N,N′,N″-Triarylguanidines as Facile Routes for Guanidine-Containing Heterocycles/Carbocycles: Synthetic, Structural, and Mechanistic Aspects. Organometallics 2014, 33, 5554–5565. [Google Scholar] [CrossRef]
- Aparna, P.S.; Prabha, B.; Prakash, P.; Jijy, E.; Varma, R.L.; Radhakrishnan, K.V. Ruthenium catalyzed desymmetrization of diazabicyclic olefins to access heteroaryl substituted cyclopentenes through C–H activation of phenylazoles. Tetrahedron Lett. 2014, 55, 865–868. [Google Scholar] [CrossRef]
- Qi, Z.; Li, X. Rhodium(III)-Catalyzed Coupling of Arenes with 7-Oxa/Azabenzonorbornadienes by C–H Activation. Angew. Chem. Int. Ed. 2013, 52, 8995–9000. [Google Scholar] [CrossRef]
- Ohtaka, J.; Sakamoto, T.; Kikugawa, Y. A one-pot procedure for trifluoroacetylation of arylamines using trifluoroacetic acid as a trifluoroacetylating reagent. Tetrahedron Lett. 2009, 50, 1681–1683. [Google Scholar] [CrossRef]
- Li, X.; Gong, X.; Zhao, M.; Song, G.; Deng, J.; Li, X. Rh(III)-Catalyzed Oxidative Olefination of N-(1-Naphthyl)sulfonamides Using Activated and Unactivated Alkenes. Org. Lett. 2011, 13, 5808–5811. [Google Scholar] [CrossRef] [PubMed]
- Mezaki, N.; Yagi, S.; Yoshigami, R.; Maeda, J.; Suzuki, T.; Ohsawa, S.; Tsukamoto, K.; Tanaka, T. Pd-Catalyzed Sulfinylzincation of Activated Alkynes with 1-Alkynyl Sulfoxides as a Sulfinyl Source. J. Org. Chem. 2003, 68, 5550–5558. [Google Scholar] [CrossRef] [PubMed]
- Dieter, R.N.; Lu, K. α-(N-Carbamoyl)alkylcuprate Chemistry in the Synthesis of Nitrogen Heterocycles. J. Org. Chem. 2002, 67, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Borah, G. Direct Access to Indoles by IrIII-Catalyzed C–H Functionalization of Acetanilides with Diazo Compounds. Eur. J. Org. Chem. 2017, 2271–2279. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds not are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | L | 2 (R) | Conditions | 3a (Equiv.) | Yield (%) b | |
|---|---|---|---|---|---|---|
| 5 | 6 (6′) | |||||
| 1 | CpE | 2c (Ac) | rt, 72 h | 3 | 5ca/26 | 6ca (6ca′)/18 (7) |
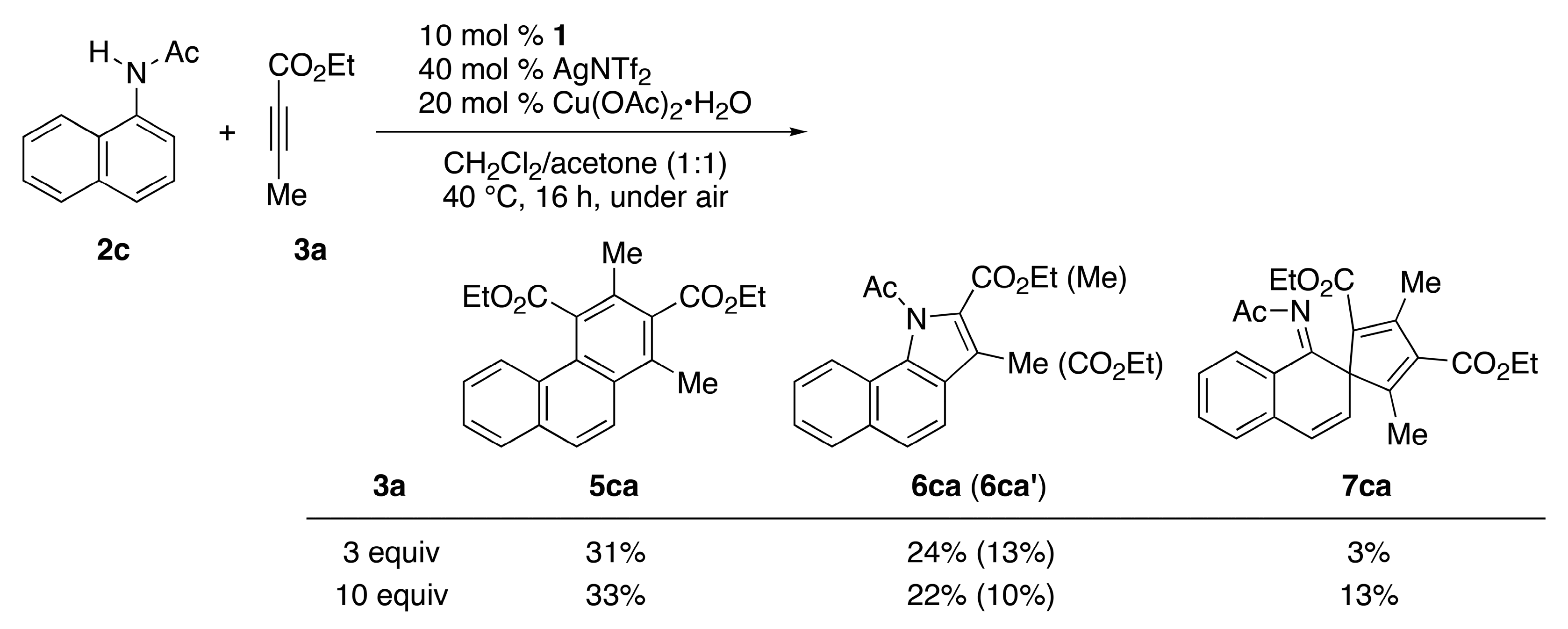
| 2 | CpE | 2c (Ac) | 40 °C, 16 h | 3 | 5ca/31 | 6ca (6ca′)/24 (13) |
| 3 | CpE | 2c (Ac) | 40 °C, 16 h | 10 | 5ca/33 | 6ca (6ca′)/22 (10) |
| 4 c | CpE | 2d (COCF3) | 80 °C, 16 h | 3 | 5da/0 | 6da (6da′)/0 (0) |
| 5 | CpE | 2e (COt-Bu) | 40 °C, 16 h | 3 | 5ea/0 | 6ea (6ea′)/55 (3) |
| 6 | Cp* | 2c (Ac) | 40 °C, 16 h | 3 | 5ca/13 | 6ca (6ca′)/3 (<1) |
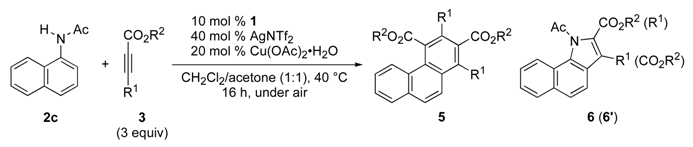
| Entry | 3 | R1 | R2 | Yield (%) b | |
|---|---|---|---|---|---|
| 5 | 6 (6′) | ||||
| 1 | 3a | Me | Et | 5ca/33 | 6ca (6ca′)/22 (10) |
| 2 | 3b | Me | Me | 5cb/30 | 6cb (6bb′)/22 (8) |
| 3 | 3c | n-Bu | Et | 5cc/32 | 6cc (6cc′)/37 (7) |
| 4 | 3d | Ph(CH2)3 | Me | 5cd/35 | 6cd (6cd′)/38 (5) |
| 5 | 3e | Cl(CH2)3 | Me | 5ce/36 | 6ce (6ce′)/31 (5) |
| 6 | 3f | Ph | Et | 5cf/0 | 6cf (6cf′)/0 (69) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terasawa, J.; Shibata, Y.; Fukui, M.; Tanaka, K. [2+2+2] Annulation of N-(1-Naphthyl)acetamide with Two Alkynoates via Cleavage of Adjacent C–H and C–N Bonds Catalyzed by an Electron-Deficient Rhodium(III) Complex. Molecules 2018, 23, 3325. https://doi.org/10.3390/molecules23123325
Terasawa J, Shibata Y, Fukui M, Tanaka K. [2+2+2] Annulation of N-(1-Naphthyl)acetamide with Two Alkynoates via Cleavage of Adjacent C–H and C–N Bonds Catalyzed by an Electron-Deficient Rhodium(III) Complex. Molecules. 2018; 23(12):3325. https://doi.org/10.3390/molecules23123325
Chicago/Turabian StyleTerasawa, Jyunichi, Yu Shibata, Miho Fukui, and Ken Tanaka. 2018. "[2+2+2] Annulation of N-(1-Naphthyl)acetamide with Two Alkynoates via Cleavage of Adjacent C–H and C–N Bonds Catalyzed by an Electron-Deficient Rhodium(III) Complex" Molecules 23, no. 12: 3325. https://doi.org/10.3390/molecules23123325