
Chloro- and Dichloro-methylsulfonyl Nitrenes: Spectroscopic Characterization, Photoisomerization, and Thermal Decomposition

1
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China
2
Department of Chemistry, Graduate School of Science, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739-8526, Japan
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(12), 3312; https://doi.org/10.3390/molecules23123312
Submission received: 27 November 2018
/
Revised: 11 December 2018
/
Accepted: 12 December 2018
/
Published: 13 December 2018
(This article belongs to the Special Issue Matrix Infrared Spectra and Molecular Structures of Reactive Intermediates)
Abstract
:Chloro- and dichloro-methylsulfonyl nitrenes, CH2ClS(O)2N and CHCl2S(O)2N, have been generated from UV laser photolysis (193 and 266 nm) of the corresponding sulfonyl azides CH2ClS(O)2N3 and CHCl2S(O)2N3, respectively. Both nitrenes have been characterized with matrix-isolation IR and EPR spectroscopy in solid N2 (10 K) and glassy toluene (5 K) matrices. Triplet ground-state multiplicity of CH2ClS(O)2N (|D/hc| = 1.57 cm−1 and |E/hc| = 0.0026 cm−1) and CHCl2S(O)2N (|D/hc| = 1.56 cm−1 and |E/hc| = 0.0042 cm−1) has been confirmed. In addition, dichloromethylnitrene CHCl2N (|D/hc| = 1.57 cm−1 and |E/hc| = 0 cm−1), formed from SO2-elimination in CHCl2S(O)2N, has also been identified for the first time. Upon UV light irradiation (365 nm), the two sulfonyl nitrenes R–S(O)2N (R = CH2Cl and CHCl2) undergo concomitant 1,2-R shift to N-sulfonlyamines R–NSO2 and 1,2-oxygen shift to S-nitroso compounds R–S(O)NO, respectively. The identification of these new species with IR spectroscopy is supported by 15N labeling experiments and quantum chemical calculations at the B3LYP/6-311++G(3df,3pd) level. In contrast, the thermally-generated sulfonyl nitrenes CH2ClS(O)2N (600 K) and CHCl2S(O)2N (700 K) dissociate completely in the gas phase, and in both cases, HCN, SO2, HCl, HNSO, and CO form. Additionally, ClCN, OCCl2, HNSO2, •NSO2, and the atmospherically relevant radical •CHCl2 are also identified among the fragmentation products of CHCl2S(O)2N. The underlying mechanisms for the rearrangement and decomposition of CH2ClS(O)2N and CHCl2S(O)2N are discussed based on the experimentally-observed products and the calculated potential energy profile.
1. Introduction
Nitrenes R–N are neutral species containing monovalent nitrogen atoms [1]. Chemically, nitrenes are highly reactive intermediates that have been extensively used in chemical transformations such as the well-known aziridination and C–H amidation reactions, and also in the covalent functionalization of nanomaterials [2,3,4]. Typically, nitrenes R–N can be readily generated from the decomposition of azides R–N3 upon either photolysis or pyrolysis, in which molecular nitrogen is the only byproduct [5]. However, due to inherent instability and also high reactivity of nitrenes, the associated rapid intramolecular rearrangement and/or intermolecular reactions with solvent molecules in solution render their direct observations challenging. Therefore, conventional chemical trapping reactions have been frequently used to probe the structure and reactivity of nitrenes [6,7]. The direct characterization of nitrenes requires either ultrafast [8,9,10] or cryogenic matrix-isolation spectroscopic methods [11,12,13]. Recently, relatively more stable nitrenes stabilized by bulky organic ligands have also been synthesized as isolable compounds even at room temperatures [14].
Compared to the intensively studied alkyl [15], aryl [16,17], and carbonyl nitrenes [18,19,20], knowledge about the fundamental properties of sulfonyl nitrenes RS(O)2–N is limited, although this class of nitrenes have been frequently proposed as the key intermediate in the synthesis of N-sulfonyl-1,2,3-triazoles [21], biologically relevant sulfonamides [22], and diamines [23], where sulfonyl azides RS(O)2–N3 were usually used as the reagents. As the most frequently studied targets, naphthyl [8,10], phenyl [9], and methyl-substituted [9] sulfonyl nitrenes have already been directly observed in the photolytic decomposition of the corresponding azide precursors in solution by using ultrafast IR spectroscopy, and triplet ground-state multiplicity has been also established with EPR spectroscopy. Interestingly, experimental studies by chemical trapping [24] and ultrafast kinetics [8,9,10] in solutions have revealed that sulfonyl nitrenes are generally more rigid than carbonyl nitrenes RC(O)–N, since the latter may undergo facile rearrangement to isocyanate RNCO through 1,2-R shift [18]. In contrast, similar 1,2-R shift in sulfonyl nitrenes to N-sulfonlyamines R–NSO2, known as pseudo-Curtius rearrangement, has been only occasionally observed in the photochemistry of sulfonyl azides under matrix-isolation conditions [11,25,26]. Moreover, S-nitroso compounds R–S(O)NO, formally regarded as the 1,2-oxygen shifted isomers of sulfonyl nitrenes R–S(O)2N, have been identified among the photolysis products of fluorinated sulfonyl nitrenes such as CF3S(O)2–N [12] and FS(O)2–N [13].
In addition to the complex photochemistry of sulfonyl nitrenes in solution and cryogenic matrices, their thermal decompositions were found to yield diverse fragments, thus providing access to some highly reactive species in the gas phase. For instance, the thermolysis of FS(O)2–N [11] yields sulfonyl radical FSO2• and N2 via nitrene dimerization. Fragmentation of CH3S(O)2–N and CF3S(O)2–N [27] furnishes iminyl radical •NSO2 through homolytic C–S bond cleavage. Flash vacuum pyrolysis of CH3OS(O)2–N [26] and PhS(O)2–N [28] forms HNSO2 and Ph–N with concerted elimination of CH2O and SO2, respectively.
The distinct photolytic and thermal chemistry of various sulfonyl nitrenes prompted us to extend our studies to other sulfonyl nitrenes, including the barely investigated chlorinated methylsulfonyl nitrenes CHnX3-nClS(O)2N (n = 0–2), the key intermediates in the decomposition of the synthetically useful chloromethylsulfonyl azides [29]. Herein, we report the first generation and spectroscopic characterization of triplet sulfonyl nitrenes CH2ClS(O)2N and CHCl2S(O)2N in cryogenic matrices. In addition to the photolytic rearrangement products N-sulfonlyamines R–NSO2 and S-nitroso compounds R–S(O)NO, a novel triplet chloromethylnitrene species CHCl2N has also been identified. Furthermore, the complex decomposition of the two nitrenes in the gas phase has been presented. Unlike the dominant C–S bond cleavage in CH3S(O)2–N [27], thermal decompositions of CH2ClS(O)2N and CHCl2S(O)2N initiate mainly by the concomitant elimination of HCl and SO2.
2. Results and Discussion
2.1. Photolysis of CH2ClS(O)2N3
The photolysis of CH2ClS(O)2N3 in solid N2-matrix was performed by using an ArF excimer laser (193 nm). The IR difference absorption spectrum reflecting the decomposition of the azide is shown in Figure 1A. Upon irradiation, nearly 42% of the azide was depleted. As a result, new species with IR bands at 1354.1, 1155.5, and 500.8 cm−1 formed. These band positions are close to those of SO2 (1347.5, 1153.1, and 524.6 cm−1) [30]. In addition, several weak but distinguishable IR bands in the range of 900–700 cm−1 appear but partially overlap with those of the azide precursor.
Given the TD-B3LYP/6-311++G(3df,3pd) calculated vertical transition at 390 nm (oscillator strength f = 0.0084, Table S1) for the most likely candidate species CH2ClS(O)2N, the matrix containing the 193 nm laser photolysis products of CH2ClS(O)2N3 was further irradiated with UV-light (365 nm). The resulting IR difference absorption spectrum (Figure 1B) suggests the depletion of trace azide but mainly the carrier for the aforementioned new IR bands (1354.1, 1155.5, and 500.8 cm−1). The selective depletion enables the unambiguous identification of the remaining weak IR bands for this carrier at 3023.0, 2953.6, 1398.3, 1238.6, 1128.2, 870.1, 748.8, 718.9, and 688.1 cm−1 (Table 1). Most of these band positions agree with the calculated IR frequencies for the expected nitrene intermediate CH2ClS(O)2N in the triplet state (Figure 1C).
According to the calculated vibrational displacement vectors of triplet CH2ClS(O)2N, the two characteristic stretching vibrations of the SO2 moiety are located at 1354.1 (νasym(SO2)) and 1155.5 cm−1 (νsym(SO2)), which are very close to those of other triplet sulfonyl nitrenes such as CH3S(O)2N (1349.8 and 1156.6 cm−1, Ne-matrix) [31], CF3S(O)2N (1387.4 and 1171.8 cm−1, Ar-matrix) [12], and PhS(O)2N (1348.9 and 1168.2 cm−1, Ne-matrix) [28]. No noticeable shift occurs to both IR bands in the 15N-labeled nitrene. Only one band at 718.9 cm−1 displays a large 15N isotopic shift of 11.1 cm−1, which is in good agreement with the calculated shift of 11.2 cm−1 for the S–N stretching mode (Table 1). It is also close to those in CH3S(O)2N (11.0 cm−1) [31] and CF3S(O)2N (8.2 cm−1) [12]. It should be noted that the calculated S–N stretching mode in singlet CH2ClS(O)2N locates at 973 cm–1 (Δνcal(14/15N) = 11.5 cm−1). Moreover, the two SO2 stretching vibrations in the nitrene in the singlet state at 1400 and 1053 cm−1 are heavily mixed with the S–N stretching mode, as evidenced by the calculated 15N isotopic shifts of 2.5 and 9.4 cm−1, respectively. The absence of these bands in the IR spectrum of the photolysis products of CH2ClS(O)2N3 (Figure 1) suggests that the initially-generated singlet nitrene CH2ClS(O)2N from the N2-elimination in the azide relaxes to the triplet ground state through rapid intersystem crossing (ISC). According to the recent ultrafast spectroscopic studies on the kinetics of arylsulfonyl nitrenes in solutions, the ISC from singlet to triplet is extremely fast (700 ± 300 ps in CCl4) [8,10].
As can be seen in Figure 1B, the UV-light irradiation (365 nm) results in the depletion of the nitrene CH2ClS(O)2N and another species with a weak IR band at 1839.6 cm−1 (labeled with d in Figure 1B). It exhibits a large 15N-isotopic shift of 32.1 cm−1. Both the band position and isotopic shift are very close to those of the most prominent N=O stretching vibration in CF3S(O)NO (ν(NO) = 1832.3 cm−1, Δν(14/15N) = 32.0 cm−1, Ar-matrix) [12]. Furthermore, they also show good agreement with the calculated strongest IR band for the oxygen-shifted rearrangement product CH2ClS(O)NO (νcal(NO) = 1838 cm−1, Δνcal(14/15N) = 32.1 cm−1, Table S3). Nevertheless, the observation of only one band renders the identification of CH2ClS(O)NO tentative. In contrast, the formation of the 1,2-CH2Cl shifted rearrangement product CH2ClNSO2 from the 365 nm irradiation of nitrene CH2ClS(O)2N can be assured by the occurrence of one set of IR bands at 1470.3, 1387.0, 1324.7, 1278.0, 1219.7, 1113.1, 967.7, 849.4, 706.4, and 509.3 cm−1. Interestingly, each of these bands is accompanied with a weaker matrix-site band (Table 2), probably due to interactions of CH2ClNSO2 with the surrounding molecules in the matrix cages. The assignment is supported by the good agreement of the observed frequencies and 15N-isotopic shifts with the calculations (Figure 1D).
In CH2ClNSO2, the asymmetric and symmetric SO2 stretching vibration modes occur at 1387.0 and 1113.1 cm−1, respectively, and the latter strongly couples with the C–N stretching (ν(CN)), as indicated by the 15N-isotopic shift of 15.5 cm−1. The ν(N=S) stretching mode appears at 1278.0 cm−1 (Δν(14/15N) = 10.4 cm−1). It is slightly lower than that in CH3NSO2 (1294.9 cm−1, Ar-matrix), which has been very recently generated in the gas phase through flash vacuum pyrolysis of sulfamoyl chloride MeN(H)S(O)2Cl through HCl-elimination [32]. In addition to CH2ClNSO2, traces of SO2 were also produced. However, the counterpart fragmentation species CH2ClN or its isomer CHCl=NH was not observed due to weak IR intensities.
2.2. Flash Vacuum Pyrolysis of CH2ClS(O)2N3
Similar to the photochemistry, the thermal decomposition of sulfonyl azides (e.g., FS(O)2N3 [11] and PhS(O)2N3 [28]) should also initiate by extruding molecular nitrogen, followed by secondary fragmentation of the sulfonyl nitrene intermediates. To uncover the thermal behavior of CH2ClS(O)2N, flash vacuum pyrolysis (FVP, 600 K) of CH2ClS(O)2N3 in N2 dilution (1:1000) was performed. The IR spectrum of the pyrolysis products (Figure 2A) reveals complete dissociation of the azide, fragments SO2 (e) [33,34], HCN (g, 3287.8 and 735.5 cm−1) [35], HCl (h, 2854.5, 2803.8 cm−1) [32], HNSO (i, 3305.0, 1253.5, 1094.9, 925.6, and 775.5 cm−1) [36], H2CO (j, 1740.0 and 1499.7 cm−1) [26], CH2NH (k, 1637.4, 1450.9, and 1064.7 cm−1) [35], HNCO (l, 3489.2 and 2265.7 cm−1) [37], CO2 (m, 2348.9 and 662.3 cm−1) [38], CO (n, 2138.9 cm−1) [38], and N2 (IR inactive) form.
The absence of the IR band for nitrene CH2ClS(O)2N implies its immediate dissociation under the FVP conditions. Unlike the straightforward C–S bond cleavage in other alkylsulfonyl nitrenes CH3S(O)2N (→•CH3 + •NSO2) and CF3S(O)2N (→•CF3 + •NSO2), the absence of the IR bands for •CH2Cl and •NSO2 in the IR spectrum (Figure 2A), and the presence of very strong IR bands for SO2, mean that no C–S bond cleavage, but rather, SO2-elimination occurs for CH2ClS(O)2N. The identification of HCl and HCN strongly suggests that further fragmentation happens to the SO2-elimination product CH2ClN. The unexpected formation of a second pair of fragments HNSO/HCl/CO from CH2ClS(O)2N can be tentatively explained by first rearrangement to CH2ClNSO, as followed by further HCl-elimination via the intermediacy of a putative carbene species H–C–NSO2. As further proof of the identification of HNSO among the products, the previously-observed [36,39] isomerization to HOSN was repeated by irradiation with a 266 nm laser (Figure 2B).
2.3. Photolysis of CHCl2S(O)2N3
The photolysis of CHCl2S(O)2N3 in N2-matrix was also performed with a 193 nm laser. Compared to the photochemistry of CH2ClS(O)2N3, the depletion of CHCl2S(O)2N3 is more relatively efficient, since 52% of the azide vanishes in 11 min. In the corresponding IR difference absorption spectrum (Figure 3A), the IR bands of SO2 (1344.4, 1150.2, and 522.4 cm−1) and new species at 1835.1, 1777.3, 1366.5, 1153.2, and 512.6 cm–1 can be identified.
In order to distinguish these new IR bands, the matrix was irradiated with UV-light (365 nm), leading to the main depletion of the IR bands at 1835.1, 1366.5, 1153.2, 808.4, 747.7, 718.5, 675.8, and 512.6 cm−1 (Table 3). All these IR bands except the first one can be reasonably assigned to the nitrene intermediate CHCl2S(O)2N in the triplet state by comparing with the calculated IR data (Figure 3C). With the aid of the calculations, weaker bands at 3027.2, 1196.1, and 1163.7 cm−1 were also found to belong to the same carrier. This assignment is further supported by the observation of well-resolved 15N-isotopic shift of 9.6 cm−1 only for the S–N stretching mode (ν(SN)) at 718.5 cm−1. As expected, the band position is very close to that in triplet CH2ClS(O)2N at 718.9 cm−1. As for the IR band at 1835.1 cm−1, the accompanying large 15N-isotopic shift of 31.7 cm−1 suggests a tentative assignment to an NO-containing species (CHCl2S(O)NO, Table S5).
Upon the UV-light irradiation, CHCl2S(O)2N partially recombines molecular nitrogen and reforms CHCl2S(O)2N3 (Figure 3B). In the meantime, 1,2-CHCl2 shift occurs and furnishes CHCl2NSO2 (Figure 3D). Similar to the IR spectrum of CH2ClNSO2 (Table 2), most of the IR bands for CHCl2NSO2 (Table 4) split into doublets due to weak interactions with the surrounding molecules. Based on the distinct 15N-isotopic shifts, the IR bands at 1280.5/1262.7 and 1125.4/1117.0 cm−1 belong mainly to the ν(SN) and ν(CN) stretching modes, which are also very close to those in CHCl2NSO2 at 1283.7/1278.0 and 1114.6/1113.1 cm−1, respectively. Additionally, another species with IR bands at 1777.3, 1474.4, 964.5, 930.5, and 798.8 cm−1 appears from the UV-light photolysis of CHCl2S(O)2N, its identification remains unclear.
2.4. Flash Vacuum Pyrolysis of CHCl2S(O)2N3
The IR spectrum of the flash vacuum pyrolysis (700 K) products of CHCl2S(O)2N3 is depicted in Figure 4A. Traces of the azide survive, SO2, HCN, HCl, CO, HNSO, OCCl2 (m, 1817.9, 847.2 and 843.6 cm−1) [40], ClCN (k, 2208.0 cm−1) [41], •NSO2 (g, 1376.7, 1345.0, 1229.3 and 936.6 cm−1) [27], HNSO2 (l, 3329.6, 1387.9, 1300.3, and 672.1 cm−1) [26], and •CHCl2 (h, 1223.3 and 896.2 cm−1) [42] can be identified among the pyrolysis products. The identification of •NSO2 can be ascertained with the subsequent photoisomerization with OSNO• upon further UV-light irradiation (Figure 4B).
The presence of •NSO2 and •CHCl2 but no counterpart •N3 radicals among the FVP products of CHCl2S(O)2N3 clearly demonstrates that the azide decomposes by the first formation of CHCl2S(O)2N through N2-elimination. However, the initially generated nitrene CHCl2S(O)2N is thermally unstable, which dissociates by homolytic cleavage of the C–S bond. Alternatively, it may undergo heterolytic cleavage of the C–S bond with concerted H-migration to furnish HNSO2 and dichlorocarbene CCl2, and the latter reacts with oxygen-containing species and yields the experimentally observed OCCl2. Another possible pathway involves the isomerization of CHCl2S(O)2N to CHCl2NSO2, and the latter may break the C–N bond to a pair of radicals •NSO2/•CHCl2 and HNSO2/CCl2. It should be noted that the FVP of CHCl2S(O)2N3 provides an efficient and practical method for the gas-phase generation of the atmospherically-important radical •CHCl2 [43], since its production in the previous spectroscopic studies utilized either laser photolysis of CHCl2Br [44] or the reaction of chloroform with lithium atoms [45].
2.5. EPR Spectroscopy
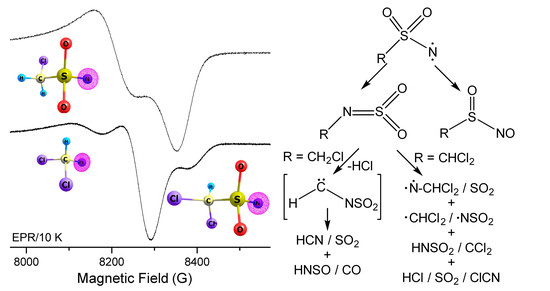
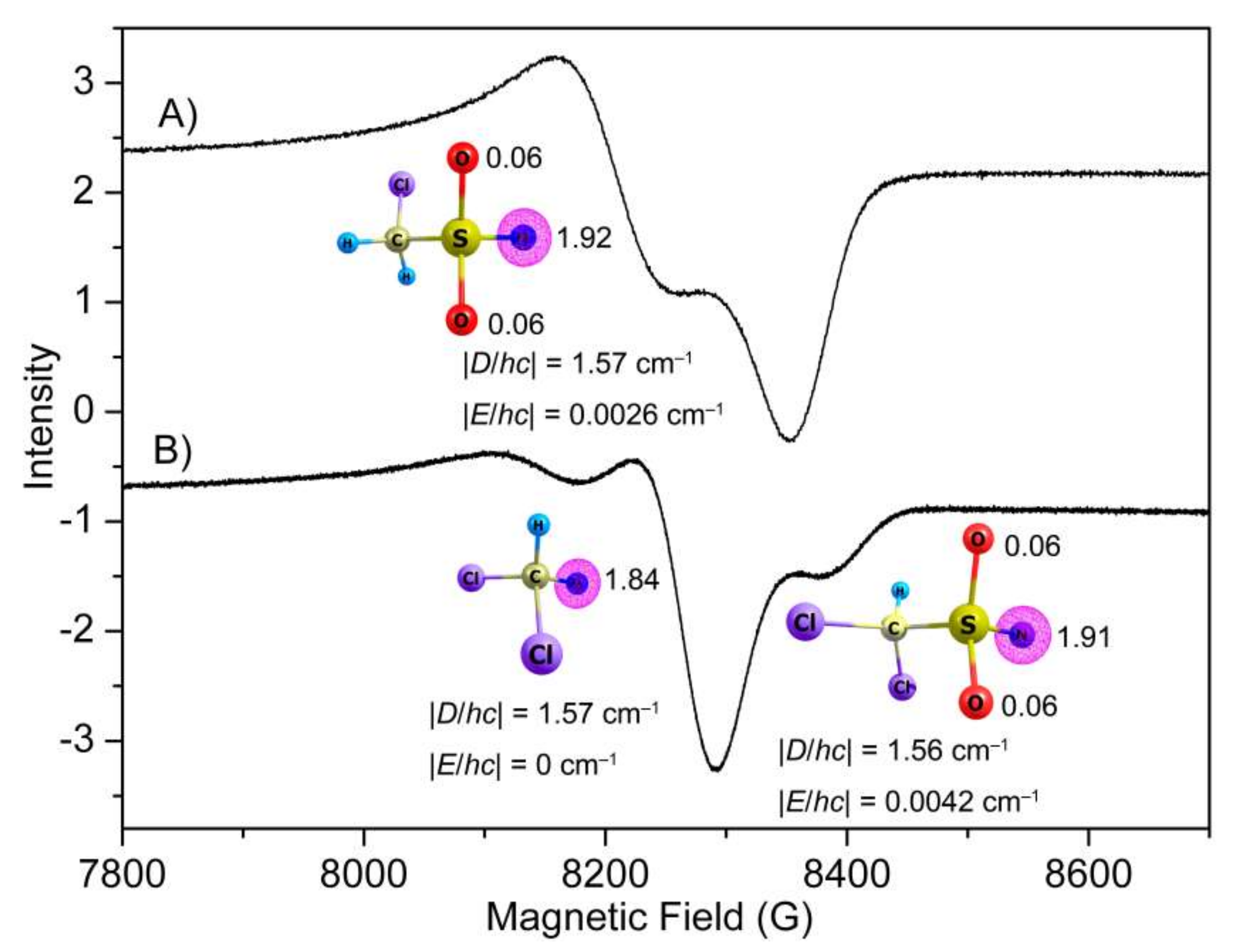
To capture the nitrene intermediates in the decomposition of CH2ClS(O)2N3 and CHCl2S(O)2N3, the photochemistry (266 nm) of both sulfonyl azides in glassy toluene matrices (5 K) was followed with EPR spectroscopy. The obtained EPR spectra (Figure 5) demonstrate typical triplet nitrene signals in the region of 7500–9000 G at about 9.40 GHz resonance frequency. The derived zero-field splitting parameters (ZFSP) for CH2ClS(O)2N (|D/hc| = 1.57 cm−1 and |E/hc| = 0.0026 cm−1) and CHCl2S(O)2N (|D/hc| = 1.56 cm−1 and |E/hc| = 0.0042 cm−1) are very similar to those of other sulfonyl nitrenes, such as Me2NS(O)2–N (|D/hc| = 1.57 cm−1 and |E/hc| = 0.0038 cm−1) [46], CF3S(O)2–N (|D/hc| = 1.741 cm−1 and |E/hc| = 0 cm−1) [12], and FS(O)2N (|D/hc| = 1.620 cm−1 and |E/hc| = 0.0055 cm−1) [13]. According to the linear correlation between the calculated spin densities (ρ, CH2ClS(O)2N: 1.92; CHCl2S(O)2N: 1.91, M06-2X/6-311++G(3df,3pd)) and zero-field D values [47,48], the predicted D values of 1.72 and 1.70 cm−1 agree with the experimental observations of 1.57 and 1.56 cm−1, respectively. Consistent with the observed nonzero E values in CH2ClS(O)2–N and CHCl2S(O)2–N, small spin densities of about 0.12 are equally distributed on the two neighboring O atoms.
In line with the IR spectroscopic observation of SO2-elimination during the photolysis of CHCl2S(O)2N3, a second triplet nitrene signal with ZFSP of |D/hc| = 1.57 cm−1 and |E/hc| = 0 cm−1 for dichloromethylnitrene CHCl2N was also observed in the EPR spectrum (Figure 5B). Its assignment is supported by the close similarity with other alkyl nitrenes such as CH3N (|D/hc| = 1.720 cm−1 and |E/hc| < 0.003 cm−1) [49] and CF3N (|D/hc| = 1.741 cm−1 and |E/hc| = 0 cm−1) [12]. In contrast, no EPR signal for chloromethylnitrene CH2ClN could be observed in the photolysis of CH2ClS(O)2N3, which is probably due to immediate isomerization to singlet species (CHCl=NH) under the photolysis conditions.
2.6. Quantum Chemical Calculations
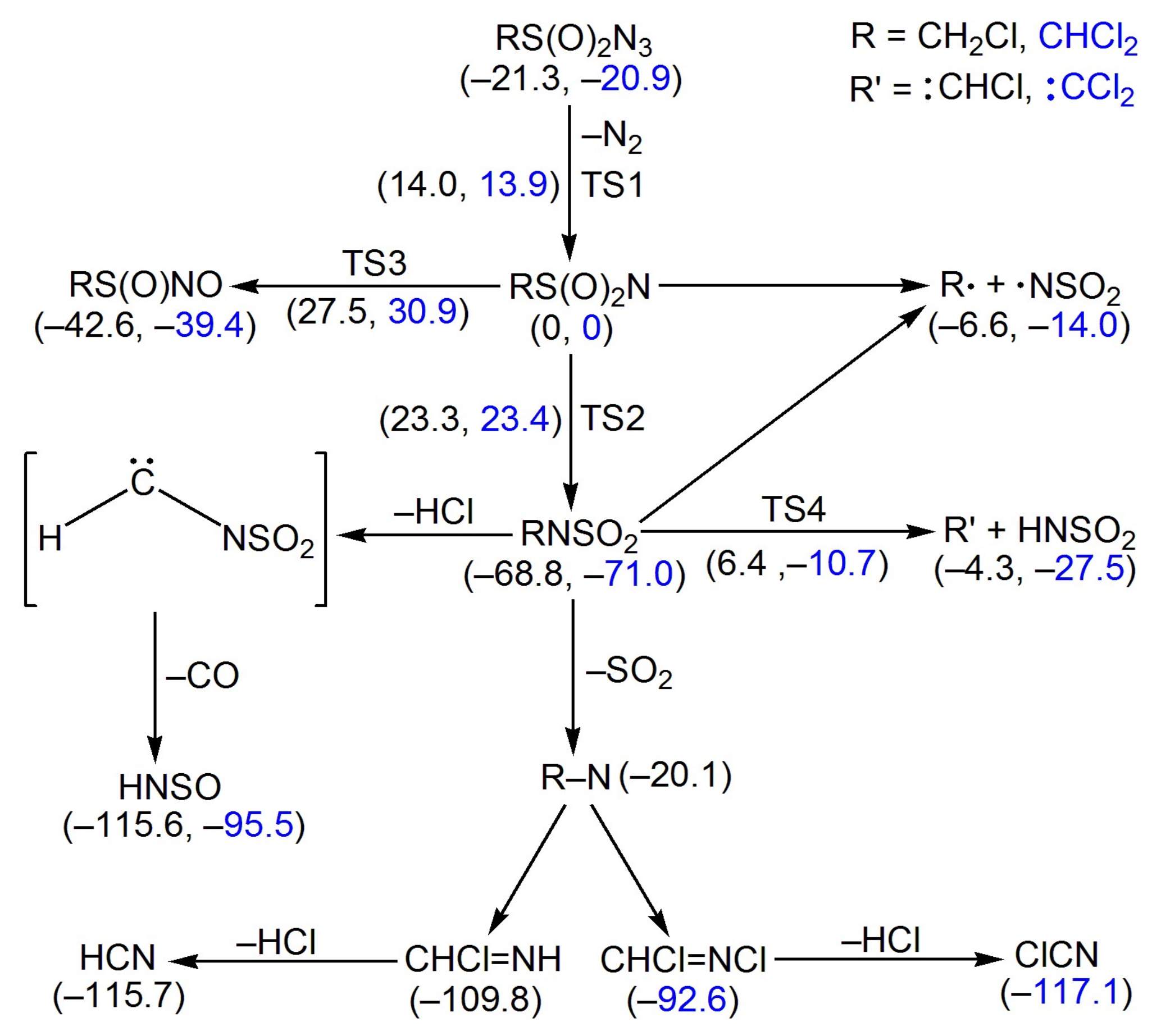
The energies for the species involving in the stepwise decomposition of CH2ClS(O)2N3 and CHCl2S(O)2N3 via the intermediacy of the corresponding nitrenes were calculated at the B3LYP/6-311++G(3df,3pd) level (Figure 6). The barriers (TS1) for the N2-elimination in CH2ClS(O)2N3 (35.3 kcal mol−1) and CHCl2S(O)2N3 (34.8 kcal mol−1) are comparable with those of other sulfonyl azides such as PhS(O)2N3 (35 kcal mol−1, CBS-QB3) [28] and CH3OS(O)2N3 (35 kcal mol−1, CCSD(T)/6-311++G(2df,2p)//UMP2/6-311++G(2df,2p)) [26], and they are also close to those for the nitrene formation in typical carbonyl azides such as FC(O)N3 (33 kcal mol−1, B3LYP/6-311+G(3df)) [50] and CH3OC(O)N3 (34 kcal mol–1, B3LYP/6-311++G(3df,3pd)) [51].
For the initially-generated singlet sulfonyl nitrenes RS(O)2N (R = CH2Cl and CHCl2), several competing processes might be responsible for their disappearance. Similar to the well-established Curtius-rearrangement of all carbonyl nitrenes (RC(O)N → RNCO) [52], RS(O)2N can undergo either 1,2-R shift to RNSO2 or 1,2-oxygen shift to RS(O)NO. Both pathways are highly exothermic, and the higher activation barriers for the latter (TS3, CH2ClS(O)2N: 27.5 kcal mol−1; CHCl2S(O)2N: 30.9 kcal mol−1) than the former (TS2, CH2ClS(O)2N: 23.3 kcal mol−1; CHCl2S(O)2N: 23.4 kcal mol−1) render their contribution in the gas phase reactions of the nitrenes unlikely. However, the excessive energy input from the ArF laser irradiation (193 nm, 148 kcal mol−1) [53] in the photochemistry can overcome the barrier and enable the formation of RS(O)NO as minor products. In addition to the intramolecular rearrangement, the sulfonyl nitrenes may decompose exothermically through the homolytic C–S bond cleavage to alkyl radicals R• (•CH2Cl and •CHCl2) and •NSO2 or heterolytic C–N bond fragmentation with concerted H-migration to carbenes HCCl and ClCCl and HNSO2. These two pairs of fragments may also be derived from the decomposition of RNSO2; however, the large C–N bond dissociation energies (CH2ClNSO2: 62.2 kcal mol−1; CHCl2NSO2: 57.0 kcal mol−1) and formidable barriers for the concerted H-migration (TS4, CH2ClNSO2: 75.2 kcal mol–1; CHCl2NSO2: 60.3 kcal mol−1) rule out these pathways under the pyrolysis conditions. In agreement with the IR detectable amounts of •CHCl2/•NSO2 and OCCl2/HNSO2 among the FVP products of CHCl2S(O)2N3, the energy release from the decomposition of CHCl2S(O)2N is larger than that of CHCl2NSO2.
The absence of RNSO2 (R = CH2Cl and CHCl2), but the presence of HCl, CO, and HNSO, among the pyrolysis products of the azides implies further dissociation through the thermodynamically favorable HCl- or Cl2-elimination (Figure 6). According to the B3LYP/6-311++G(3df,3pd) calculation, the putative carbene species H–C–NSO2 is highly unstable in the closed-shell singlet state and prefers either N–S bond breakage to HCN and SO2 or further CO-elimination to HNSO. The SO2 formation may also be attributed to the direct N–S bond breakage in RNSO2. By analogy to the thermal instability of other alkyl nitrenes (e.g., CH3N [54] and CF3N [12]), the thermally-generated nitrenes CH2ClN and CHCl2N isomerize to CHCl=NH and CHCl=NCl and then eliminate HCl to yield the observed HCN and ClCN, respectively.
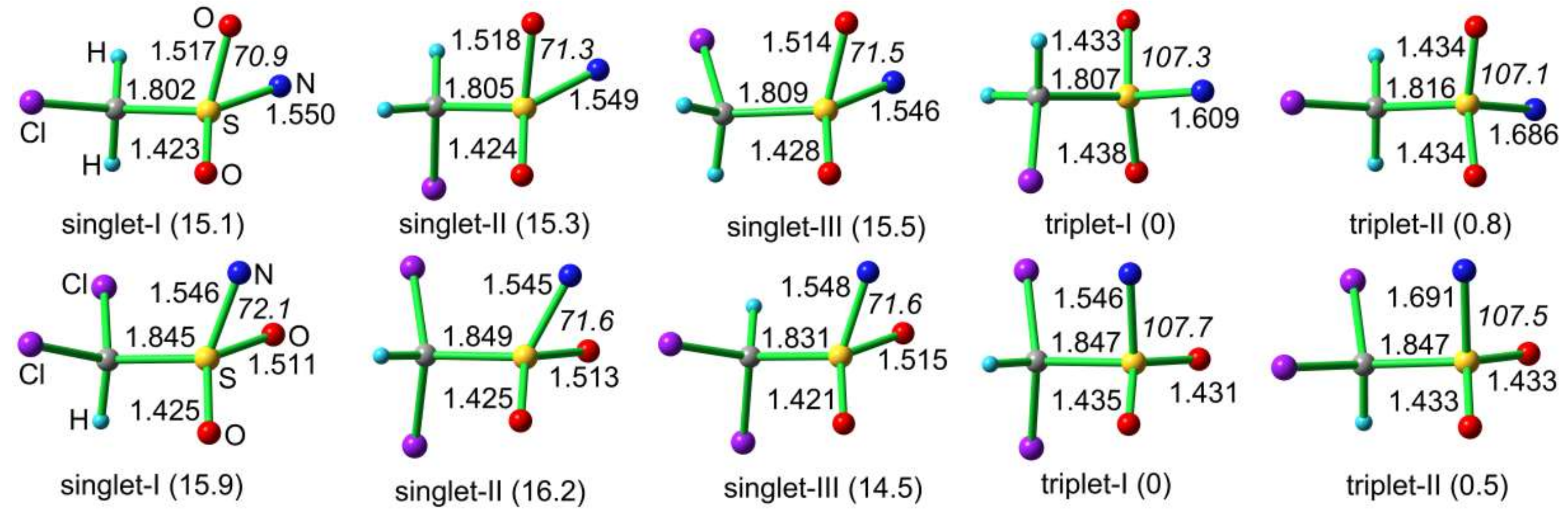
The calculated molecular structures and relative energies of CH2ClS(O)2N and CHCl2S(O)2N in the singlet and triplet states are depicted in Figure 7. For each nitrene in the singlet state, three rotamers differing in the staggered orientation of the methyl group with respect to the SO2N moiety are close-in-energy minima. Consistent with the IR and EPR spectroscopic observations, both sulfonyl nitrenes prefer triplet ground state, and the singlet state are about 15 kcal mol−1 higher in energy at the B3LYP/6-311++G(3df,3pd) level. The energy gaps between the singlet and triplet states (ΔEST) for CH2ClS(O)2N (15.1 kcal mol−1) and CHCl2S(O)2N (14.5 kcal mol−1) are close to those of CH3S(O)2N (13.6 kcal mol−1) and PhS(O)2N (14.7 kcal mol−1) at the same theoretical level [31].
Due to intramolecular interaction between the oxygen atom with the electron-deficient nitrogen center, the singlet sulfonyl nitrenes are distorted from Cs-symmetry. In the lowest-energy C1-symmetric rotamer (singlet-I) of CH2ClS(O)2N, the Cl–C and S–N bonds adopts an antiperiplanar configuration with a dihedral angle (φ(ClCSN)) of −158.8°. The φ(ClCSN) dihedral angles in singlet-II and singlet-III are 84.4 and −31.7°, respectively. Similarly, in the most stable rotamer of singlet CHCl2S(O)2N (singlet-III), the two S–N bond adopts an antiperiplanar configuration with the bisector of the ClSCl angle. In the triplet state of CH2ClS(O)2N, the more stable rotamer (triplet-I) adopts C1-symmetry with the S–N bond being synperiplanar with the Cl–C bond (φ(ClCSN) = −63.3°); however, the Cs-symmetric rotamer (triplet-II) exhibits an synperiplanar conformation between these two bonds (φ(ClCSN) = 180°). Structurally, sulfonyl nitrenes in the singlet state are stabilized by the intramolecular N–O interactions, as evidenced by the considerably smaller OSN angles in the singlet state (ca. 72°) than the triplet state (ca. 107°). Unlike the much stronger intramolecular N–N interactions in singlet ground-state sulfamoyl nitrenes [46], the stabilizing N–O interactions in alkylsulfonyl nitrenes can hardly switch the spin-state. In contrast, very recent studies on structurally-related carbonyl nitrenes RC(O)N have demonstrated that the spin multiplicity can be effectively switched by the stabilizing N–O interactions [18,52].
3. Materials and Methods
3.1. Sample Preparation
Caution! Covalent azides are explosive! Although no explosions occurred during this work, appropriate safety precautions (face shields, leather gloves, and protective leather clothing) should be taken, especially when working with pure CH2ClS(O)2N3 and CHCl2S(O)2N3 in the condensed phase.
Chloromethylsulfonyl azide, CH2ClS(O)2N3, was prepared by the reaction of chloromethylsulfonyl chloride with sodium azide. Briefly, acetonitrile (0.3 mL) and freshly purified chloromethylsulfonyl chloride (0.3 g, 2.0 mmol) were distilled into a glass vessel which contains dried NaN3 (0.2 g, 3.2 mmol). The mixture was stirred at room temperature for 18 h. The volatile crude products were separated by passing through three successive cold U-traps (−20, −80, and −196 °C). Pure azide CH2ClS(O)2N3 was retained in the first trap as white crystals. 1H-NMR (400MHz, CDCl3, 298 K): δ = 4.72 (s, 2H); 13C-NMR (400 MHz, CDCl3 298 K): δ = 57.9 ppm; IR (gas-phase): 2148, 1412, 1246, 1183, 1135, 868, 814, 750, 577, and 524 cm−1; Raman(solid): 3032, 2961, 2161, 1391, 1185, 1163, 1131, 869, 807, 747, 736, 575, 536, 515, 451, 407, 315, and 285 cm−1. 1-15N sodium azide (98 atom% 15N, EURISO-TOP GmbH) was used for the preparation of 15N-labeled sample.
Dichloromethylsulfonyl azide, CHCl2S(O)2N3, was synthesized in a similar manner by the reaction of freshly purified dichloromethylsulfonyl chloride (0.6 g, 3.0 mmol) with NaN3 (0.3 g, 4.6 mmol) in acetonitrile (0.5 mL). The mixture was stirred at room temperature for 24 h. Separation of the volatile products was carried out by using three cold traps of −16, −80, and −196 °C. The desired product CHCl2S(O)2N3 was retained in the first trap as colorless liquid. 1H-NMR (400 MHz, CDCl3, 298 K): δ = 6.41 ppm (s, 1H); 13C-NMR (400 MHz, CDCl3, 298 K): δ = 79.4 ppm; IR (gas-phase): 2148, 1422, 1355, 1222, 1179, 1150, 816, 751, 694, 608, 576, and 529 cm−1; Raman (liquid): 2993, 2961, 2155, 1382, 1203, 1156, 1144, 799, 746, 698, 603, 575, 536, 452, 410, 342, 295, 238, and 211 cm−1. 1-15N sodium azide (98 atom % 15N, EURISO-TOP GmbH) was used for the preparation of 15N labeled sample.
3.2. Spectroscopy
Gas-phase IR spectra were recorded on a Bruker spectrometer (Tensor 27) (Bruker Optik GmbH, Ettlingen, Germany). Raman spectra were recorded on a Horiba JY HR800 Raman spectroscopy (HORIBA, Lille, France).
3.3. Matrix IR Spectroscopy
Matrix IR spectra were recordedon a FT-IR spectrometer (Bruker 70 V) in a reflectance mode using a transfer optic. A KBr beam splitter and MCT detector were used in the mid-IR region (4000−500 cm−1). For each spectrum, 200 scans at a resolution of 0.5 cm−1 were coadded. The gas sample mixed by passing a flow of N2 gas through a cold U-trap (CH2ClS(O)2N3: −5 °C; CHCl2S(O)2N3: −10 °C) containing ca. 10 mg of the azide. The mixture (azide/dilution gas ≈ 1:1000 estimated) was passed through an aluminum oxide (o.d. 2.0 mm, i.d. 1.0 mm), which could be heated over a length of approximately 25 mm by a tantalum wire (o.d. 0.4 mm, resistance 0.4 Ω). Then, the mixture was immediately deposited (2 mmol/h) onto the Rh-plated copper block matrix support (15 K) in a high vacuum (~10−6 Pa). While not directly measured, the expected residence time of the mixture in the pyrolysis tube is about a few milliseconds, and the pressure inside the pyrolysis tube is about 10 mbar. The electric power (voltage/current) used in pyrolysis experiments was 4.0 V/1.9 A for CH2ClS(O)2N3 and 4.5 V/2.9 A for CHCl2S(O)2N3. Photolysis experiments were performed with ArF excimer laser (GAM LASER, Orlando, FL, USA) (193 nm, Gamlaser EX5/250, 5 mJ, 3 Hz), Nd3+: YAG laser (266 nm, MPL-F-266, 10 mW), and UV lamp (365 nm).
3.4. Computational Details
Geometry optimizations were performed using DFT-B3LYP [55] method combined with the 6-311++G(3df,3pd) basis set. Time-dependent (TD) DFT (B3LYP/6-311++G(3df,3pd)) [56,57] calculations were performed for the prediction of UV−vis transitions. Local minima were confirmed by vibrational frequency analysis, and transition states were further confirmed by intrinsic reaction coordinate (IRC) calculations [58,59]. All the calculations were performed using the Gaussian 09 software package (Gaussian, Inc., Wallingford, CT, USA) [60].
4. Conclusions
The UV laser photolytic (193 and 266 nm) and thermal decomposition of two alkylsulfonyl azides RS(O)2N3 (R = CH2Cl and CHCl2) have been studied by combining matrix-isolation IR and EPR spectroscopy and quantum chemical calculations. Two new sulfonyl nitrenes CH2ClS(O)2N and CHCl2S(O)2N in the triplet ground state have been directly observed and spectroscopically characterized. Upon subsequent UV light irradiations, both nitrenes undergo rearrangement reactions to the corresponding N-sulfonlyamines R–NSO2 and 1,2-oxygen shift to S-nitroso compounds R–S(O)NO in solid N2-matrices. In the gas phase, the monochloro-substituted sulfonyl nitrene CH2ClS(O)2N prefers rearrangement to CH2ClNSO2 with subsequent decomposition to HCl, HNSO, and CO, in which an intriguing carbene species H–C–NSO2 might be involved. The dichloro-substituted sulfonyl nitrene CHCl2S(O)2N partially undergoes homolytic C–S bond cleavage to a pair of radicals •CHCl2 and •NSO2. Its rearrangement product CHCl2NSO2 also decomposes to HNSO2/CCl2 and HCl/SO2/ClCN.
Supplementary Materials
The following are available online. Calculated vertical transitions, structures, energies and IR frequencies, and Calculated atomic coordinates for all optimized structures.
Author Contributions
Conceptualization, X.Z.; Methodology, Y.Y., X.C., M.A., and X.Z.; Software, X.C., Y.L., and M.A.; Formal Analysis, X.Z., Y.Y., Y.L., and X.C.; Writing, X.Z. and Y.Y.; Supervision, X.Z.
Funding
This work was supported by the National Natural Science Foundation of China (21673147, 21422304) and the project of scientific and technologic infrastructure of Suzhou (SZS201708).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gritsan, N.P. Properties of Carbonyl Nitrenes and Related Acyl Nitrenes. Nitrenes and Nitrenium Ions, 1st ed.; Falvey, D.E., Gudmundsdottir, A.D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 6, pp. 481–548. ISBN 978-0-470-39059-7. [Google Scholar]
- Jiang, H.L.; Lang, K.; Lu, H.J.; Wojtas, L.; Zhang, X.P. Intramolecular radical aziridination of allylic sulfamoyl azides by cobalt(II)-based metalloradical catalysis: Effective construction of strained heterobicyclic structures. Angew. Chem. Int. Ed. 2016, 55, 11604–11608. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.J.; Li, C.Q.; Jiang, H.L.; Lizardi, C.L.; Zhang, X.P. Chemoselective amination of propargylic C(sp3)‒H bonds by cobalt(II)-based metalloradical catalysis. Angew. Chem. Int. Ed. 2014, 126, 7148–7152. [Google Scholar] [CrossRef]
- Liu, L.-H.; Yan, M. Perfluorophenyl azides: New applications in surface functionalization and nanomaterial synthesis. Acc. Chem. Res. 2010, 43, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Lwowski, W. Nitrenes and the decomposition of carbonylazides. Angew. Chem. Int. Ed. 1967, 6, 897–1012. [Google Scholar] [CrossRef]
- Kundu, S.; Miceli, E.; Farquhar, E.; Pfaff, F.F.; Kuhlmann, U.; Hildebrandt, P.; Braun, B.; Greco, C.; Ray, K. Lewis acid trapping of an elusive copper−tosylnitrene intermediate using scandium triflate. J. Am. Chem. Soc. 2012, 134, 14710–14713. [Google Scholar] [CrossRef] [PubMed]
- Gritsan, N.P.; Likhotvorik, I.; Tsao, M.-L.; Celebi, N.; Platz, M.S.; Karney, W.L.; Kemnitz, C.R.; Borden, W.T. Ring-expansion reaction of cyano-substituted singlet phenyl nitrenes: Theoretical predictions and kinetic results from laser flash photolysis and chemical trapping experiments. J. Am. Chem. Soc. 2001, 123, 1425–1433. [Google Scholar] [CrossRef]
- Kubicki, J.; Luk, H.L.; Zhang, Y.; Vyas, S.; Peng, H.-L.; Hadad, C.M.; Platz, M.S. Direct observation of a sulfonyl azide excited state and its decay processes by ultrafast time-resolved IR Spectroscopy. J. Am. Chem. Soc. 2012, 134, 7036–7044. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, A.V.; Neumann, C.; van Wilderen, L.J.G.W.; Shainyanb, B.A.; Bredenbeck, J. Exploring photochemistry of p-bromophenylsulfonyl, p-toylsulfonyl and methylsulfonyl azides by ultrafast UV-Pump-IR-Probe spectroscopy and computations. Phys. Chem. Chem. Phys. 2016, 18, 8662–8672. [Google Scholar] [CrossRef] [PubMed]
- Kubicki, J.; Zhang, Y.; Xue, J.; Luk, H.L.; Platz, M.S. Ultrafast time resolved studies of the photochemistry of acyl and sulfonyl azides. Phys. Chem. Chem. Phys. 2012, 14, 10377–10390. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Beckers, H.; Willner, H. Thermally persistent fluorosulfonyl nitrene and unexpected formation of the fluorosulfonyl radical. J. Am. Chem. Soc. 2013, 135, 2096–2099. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Beckers, H.; Willner, H.; Neuhaus, P.; Grote, D.; Sander, W. Photochemistry of matrix isolated (trifluoromethyl)sulfonyl azide, CF3SO2N3. J. Phys. Chem. A 2015, 119, 2281–2288. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Beckers, H.; Neuhaus, P.; Grote, D.; Sander, W. Elusive fluoro sulfinyl nitrite, FS(O)NO, produced by photolysis of matrix-isolated FS(O)2N. Z. Anorg. Allg. Chem. 2012, 638, 526–533. [Google Scholar] [CrossRef]
- Obenhuber, A.H.; Gianetti, T.L.; Berrebi, X.; Bergman, R.G.; Arnold, J. Reaction of (bisimido)niobium(V) complexes with organic azides: [3+2] cycloaddition and reversible cleavage of β-diketiminato ligands involving nitrene transfer. J. Am. Chem. Soc. 2014, 136, 2994–2997. [Google Scholar] [CrossRef] [PubMed]
- Klima, R.F.; Gudmundsdottir, A.D. Intermolecular triplet-sensitized photolysis of alkyl azides trapping of triplet alkyl nitrenes. J. Photochem. Photobiol. A. 2004, 162, 239–247. [Google Scholar] [CrossRef]
- Hayes, J.C.; Sheridan, R.S. Infrared spectrum of triplet phenylnitrene. On the origin of didehydroazepine in low-temperature matrices. J. Am. Chem. Soc. 1990, 112, 5879–5881. [Google Scholar] [CrossRef]
- Leyva, E.; Platz, M.S.; Persy, G.; Wirz, J. Photochemistry of phenyl azide: The role of singlet and triplet phenylnitrene as transient intermediates. J. Am. Chem. Soc. 1986, 108, 3783–3790. [Google Scholar] [CrossRef]
- Kubicki, J.; Zhang, Y.; Vyas, S.; Burdzinski, G.; Luk, H.L.; Wang, J.; Xue, J.; Peng, H.-L.; Pritchina, E.A.; Sliwa, M.; et al. Photochemistry of 2-naphthoyl azide. An ultrafast time-resolved UV-vis and IR spectroscopic and computational Study. J. Am. Chem. Soc. 2011, 133, 9751–9761. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Kubicki, J.; Luk, H.L.; Zhang, Y.; Gritsan, N.P.; Hadad, C.M.; Platz, M.S. An ultrafast time-resolved infrared and UV-vis spectroscopic and computational study of the photochemistry of acyl azides. J. Phys. Org. Chem. 2012, 25, 693–703. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Beckers, H.; Willner, H.; Grote, D.; Sander, W. The missing link: Triplet fluorocarbonyl nitrene FC(O)N. Chem. Eur. J. 2011, 17, 3977–3984. [Google Scholar] [CrossRef]
- Chuprakov, S.; Worrell, B.T.; Selander, N.; Sit, R.K.; Fokin, V.V. Stereoselective 1,3-insertions of rhodium(II) azavinyl carbenes. J. Am. Chem. Soc. 2014, 136, 195–202. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Coelho, P.S.; Farwell, C.C.; Wang, Z.J.; Lewis, J.C.; Brown, T.R.; Arnold, F.H. Enantioselective intramolecular C‒H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem. Int. Ed. 2013, 125, 9479–9482. [Google Scholar] [CrossRef]
- Lu, H.J.; Jiang, H.L.; Wojtas, L.; Zhang, X.P. Selective intramolecular C‒H amination through the metalloradical activation of azides: Synthesis of 1,3-diamines under neutral and nonoxidative conditions. Angew. Chem. Int. Ed. 2010, 122, 10390–10394. [Google Scholar] [CrossRef]
- Lwowski, W.; Scheiffele, E. Curtius and lossen rearrangements. I. The benzenesulfonyl system. J. Am. Chem. Soc. 1965, 87, 4359–4365. [Google Scholar] [CrossRef]
- Sheridan, R.S.; Rempala, P. Books of Abstracts. In Proceedings of the 217th ACS National Meeting, Anaheim, CA, USA, 21–25 March 1999; American Chemical Society: Washington, DC, USA, 1999. [Google Scholar]
- Deng, G.H.; Wu, Z.; Li, D.Q.; Linguerri, R.; Francisco, J.S.; Zeng, X.Q. Simplest N-sulfonylamine HNSO2. J. Am. Chem. Soc. 2016, 138, 11509–11512. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.Q.; Beckers, H.; Willner, W. The iminyl radical O2SN. Angew. Chem. Int. Ed. 2013, 52, 7981–7984. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.H.; Dong, X.L.; Liu, Q.F.; Li, D.Q.; Li, H.M.; Sun, Q.; Zeng, X.Q. The decomposition of benzenesulfonyl azide: A matrix isolation and computational study. Phys. Chem. Chem. Phys. 2017, 19, 3792–3799. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.; Giroult, A.; Panchaud, P.; Renaud, P. Efficient carboazidation of alkenes using a radical desulfonylative azide transfer process. J. Am. Chem. Soc. 2010, 132, 17511–17515. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wan, H.B.; Xu, J.; Lu, B.; Lu, Y.; Eckhardt, A.K.; Schreiner, P.R.; Xie, C.J.; Guo, H.; Zeng, X.Q. The near-UV absorber OSSO and its isomers. Chem. Commun. 2018, 54, 4517–4520. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.H.; Li, D.Q.; Wu, Z.; Li, H.M.; Bernhardt, E.; Zeng, X.Q. Methanesulfonyl azide: Molecular structure and photolysis in solid noble gas matrices. J. Phys. Chem. A 2016, 120, 5590–5597. [Google Scholar] [CrossRef]
- Lu, Y.; Li, H.M.; Wan, H.B.; Liu, Q.; Deng, G.H.; Zeng, X.Q. Flash vacuum pyrolysis of sulfamoyl azides and chlorides: Facile gas-hhase generation of transient N-sulfonylamines. J. Anal. Appl. Pyrolysis. 2018, 134, 476–483. [Google Scholar] [CrossRef]
- Schriver-Mazzuoli, L.; Chaabouni, H.; Schriver, A. Infrared spectra of SO2 and SO2: H2O ices at low temperature. J. Mol. Struct. 2003, 644, 151–164. [Google Scholar] [CrossRef]
- Allavena, M.; Rysnik, R.; White, D.; Calder, V.; Mann, D.E. Infrared spectra and geometry of SO2 isotopes in solid krypton matrices. J. Chem. Phys. 1969, 50, 3399–3410. [Google Scholar] [CrossRef]
- Hooper, N.; Beeching, L.J.; Dyke, J.M.; Morris, A.; Ogden, J.S. A study of the thermal decomposition of 2-azidoethanol and 2-azidoethyl acetate by ultraviolet photoelectron spectroscopy and matrix isolation infrared spectroscopy. J. Phys. Chem. A 2002, 106, 9968–9975. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, Q.F.; Li, D.Q.; Feng, R.J.; Zeng, X.Q. Flash vacuum pyrolysis of methoxysulfinyl azide: Stepwise decomposition via methoxysulfinyl nitrene. J. Anal. Appl. Pyrolysis. 2017, 124, 610–617. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Bernhardt, E.; Beckers, H.; Banert, K.; Hagedorn, M.; Liu, H.L. Formyl azide: Properties and solid-state structure. Angew. Chem. Int. Ed. 2013, 52, 3503–3506. [Google Scholar] [CrossRef] [PubMed]
- d’Hendecourt, L.B.; Grim, R.J.A. Time-dependent chemistry in dense molecular clouds. Astron. Astrophys. 1986, 167, 161–165. [Google Scholar]
- Nonella, M.; Huber, J.R.; Ha, T.K. Photolytic preparation and isomerization of HNSO, HOSN, HSNO, and HONS in an argon matrix. An experimental and theoretical study. J. Phys. Chem. 1987, 91, 5203–5209. [Google Scholar] [CrossRef]
- Tobón, Y.A.; Nieto, L.I.; Romano, R.M.; Della Védova, C.O.; Downs, A.J. Photochemical reaction channels of OCS with Cl2, ICl, or IBr isolated together in an argon matrix: Isolation of syn-iodocarbonylsulfenyl bromide. J. Phys. Chem. A 2006, 110, 2674–2681. [Google Scholar] [CrossRef]
- Ramos, L.A.; Zeng, X.Q.; Ulic, S.E.; Beckers, H.; Willner, H.; Della Védova, C.O. Chlorodifluoroacetyl azide, ClF2CC(O)N3: Preparation, Properties, and Decomposition. J. Org. Chem. 2012, 77, 6456–6462. [Google Scholar] [CrossRef]
- Fridgen, T.D.; Zhang, X.K.; Parnis, J.M.; March, R.E. Isomerization and fragmentation products of CH2Cl2 and other dihalomethanes in rare-gas matrices: An electron bombardment matrix-isolation FTIR spectroscopic study. J. Phys. Chem. A 2000, 104, 3487–3497. [Google Scholar] [CrossRef]
- Tsang, W. Mechanisms for the formation and destruction of chlorinated organic products of incomplete combustion. Combust. Sci. Technol. 1990, 74, 99–116. [Google Scholar] [CrossRef]
- Yang, S.X.; Hou, G.Y.; Dai, J.H.; Chang, C.H.; Chang, B.C. Spectroscopic investigation of the multiphoton photolysis reactions of bromomethanes (CHBr3, CHBr2Cl, CHBrCl2, and CH2Br2) at near-ultraviolet wavelengths. J. Phys. Chem. A 2010, 114, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.G.; Andrews, L. Matrix infrared spectrum and bonding in the dichloromethyl radical. J. Chem. Phys. 1969, 50, 4235–4245. [Google Scholar] [CrossRef]
- Lu, Y.; Li, H.M.; Abe, M.; Bégué, D.; Wan, H.B.; Deng, G.H.; Xu, J.; Liu, K.; Zeng, X.Q. Sulfamoyl nitrenes: Singlet or triplet ground state? Chem. Commun. 2018, 54, 6136–6139. [Google Scholar] [CrossRef] [PubMed]
- Wentrup, C. Carbenes and nitrenes: Recent developments in fundamental chemistry. Angew. Chem. Int. Ed. 2018, 57, 11508–11521. [Google Scholar] [CrossRef] [PubMed]
- Wentrup, C. Flash vacuum pyrolysis of azides, triazoles, and tetrazoles. Chem. Rev. 2017, 117, 4562–4623. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, W.A.; Kebede, N.; Showalter, B.M.; Matsunaga, N.; Miceli, A.P.; Liu, Y.; Ryzhkov, L.R.; Hadad, C.M.; Toscano, J.P. Generation of oxynitrenes and confirmation of their triplet ground states. J. Am. Chem. Soc. 2006, 128, 13142–13150. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; Zhu, B.F.; Wu, Z.; Zeng, X.Q.; Beckers, H.; Jenks, W.S. Thermally persistent carbonyl nitrene: FC(O)N. J. Org. Chem. 2015, 80, 2006–2009. [Google Scholar] [CrossRef]
- Li, H.M.; Wu, Z.; Li, D.Q.; Wan, H.B.; Xu, J.; Abe, M.; Zeng, X.Q. Direct observation of methoxycarbonylnitrene. Chem. Commun. 2017, 53, 4783–4786. [Google Scholar] [CrossRef]
- Feng, R.J.; Lu, Y.; Deng, G.H.; Xu, J.; Wu, Z.; Li, H.M.; Liu, Q.; Kadowaki, N.; Abe, M.; Zeng, X.Q. Magnetically bistable nitrenes: Matrix isolation of furoylnitrenes in both singlet and triplet states and triplet 3-furylnitrene. J. Am. Chem. Soc. 2018, 140, 10–13. [Google Scholar] [CrossRef]
- Yeh, P.-S.; Leu, G.-H.; Lee, Y.-P.; Chen, I.-C. Photodissociation of HNO3 at 193 nm: Near-infrared emission of NO detected by time-resolved fourier transform spectroscopy. J. Chem. Phys. 1995, 103, 4879–4886. [Google Scholar] [CrossRef]
- Tsao, M.-L.; Hadad, C.M.; Platz, M.S. A computational study of cyclopropylnitrene. Tetrahedron Lett. 2002, 43, 745–748. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Foresman, J.B.; Head-Gordon, M.; Pople, J.A.; Frisch, M.J. Toward a systematic molecular orbital theory for excited states. J. Phys. Chem. 1992, 96, 135–149. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions – The IRC approach. Acc. Chem. Res. 1981, 4, 363–368. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Using hessian updating to increase the efficiency of a hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian, 09; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
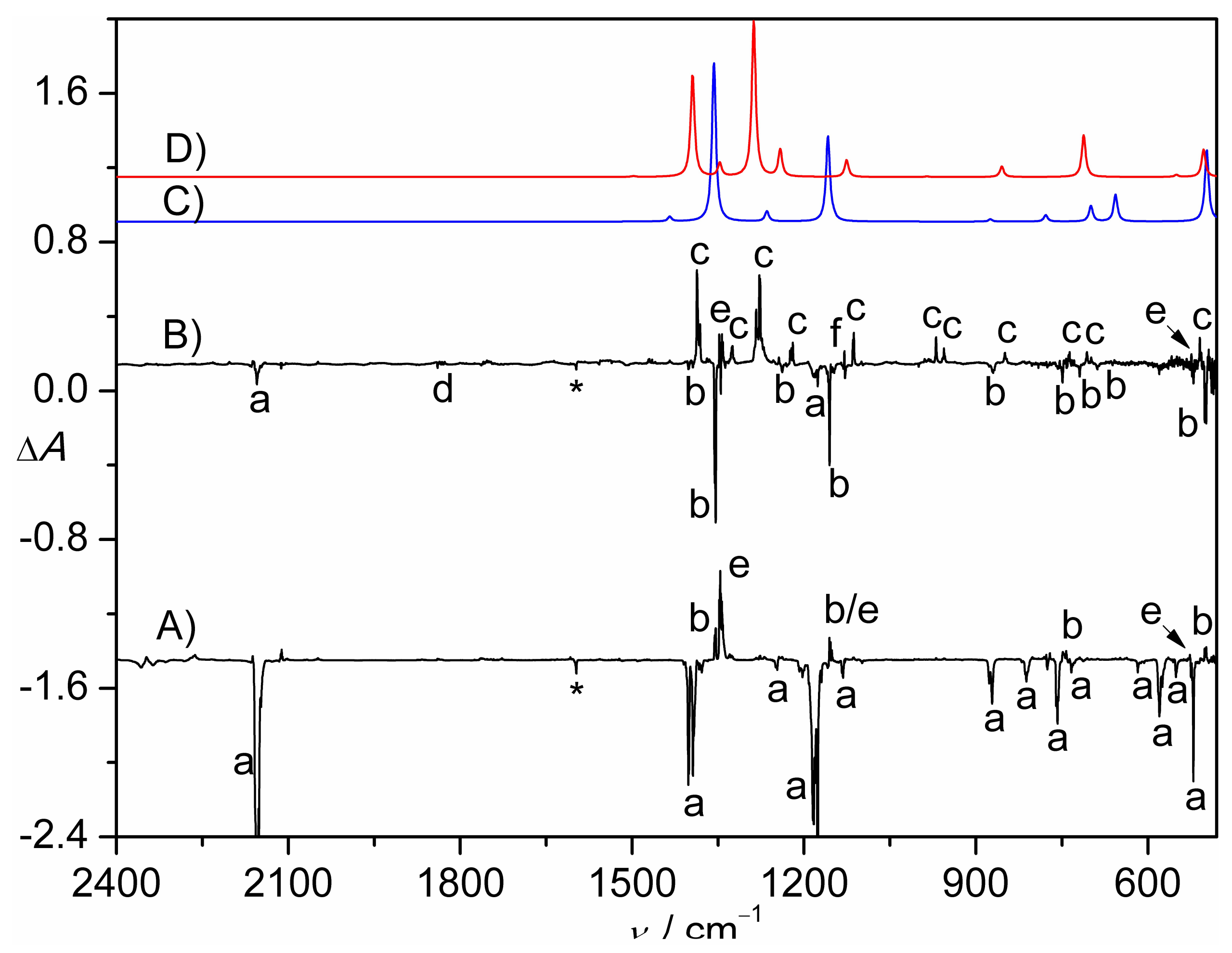
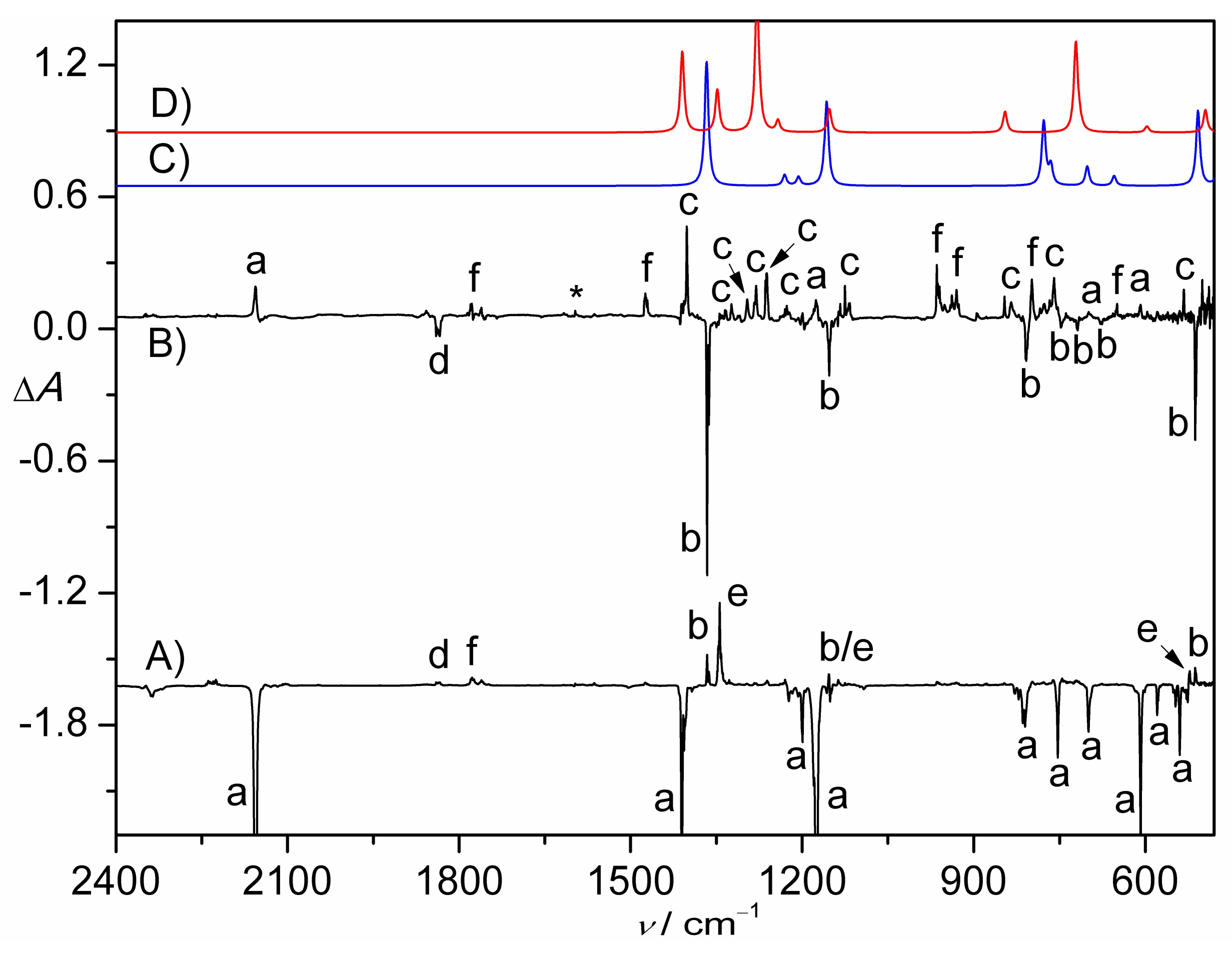
Figure 1.
(A) IR difference spectrum (Absorbance, ΔA) showing the decomposition of CH2ClS(O)2N3 in N2-matrix upon a 193 nm laser photolysis (30 min); (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent UV-light photolysis (365 nm, 8 min). For clarity, spectrum B is 12-fold expanded along the ΔA axis. (C) Calculated IR spectrum of triplet CH2ClS(O)2N at the B3LYP/6-311++G(3df,3pd) level. (D) Calculated IR spectrum of CH2ClNSO2 at the B3LYP/6-311++G(3df,3pd) level. The IR bands of CH2ClS(O)2N3 (a), CH2ClS(O)2N (b), CH2ClNSO2 (c), CH2ClS(O)NO (d), SO2 (e), unknown species (f), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.
Figure 1.
(A) IR difference spectrum (Absorbance, ΔA) showing the decomposition of CH2ClS(O)2N3 in N2-matrix upon a 193 nm laser photolysis (30 min); (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent UV-light photolysis (365 nm, 8 min). For clarity, spectrum B is 12-fold expanded along the ΔA axis. (C) Calculated IR spectrum of triplet CH2ClS(O)2N at the B3LYP/6-311++G(3df,3pd) level. (D) Calculated IR spectrum of CH2ClNSO2 at the B3LYP/6-311++G(3df,3pd) level. The IR bands of CH2ClS(O)2N3 (a), CH2ClS(O)2N (b), CH2ClNSO2 (c), CH2ClS(O)NO (d), SO2 (e), unknown species (f), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.

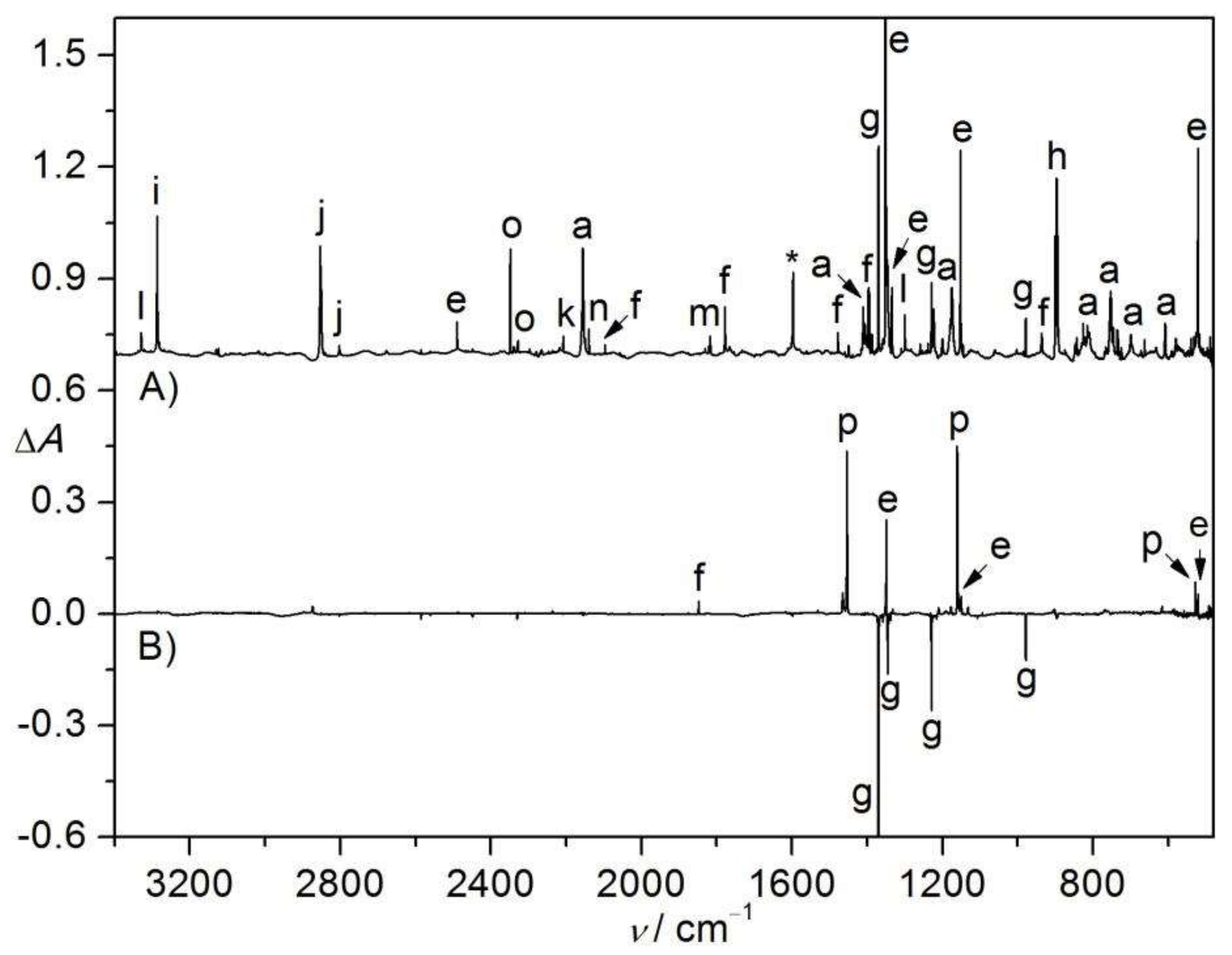
Figure 2.
(A) IR spectrum (Absorbance, A) of the N2-matrix isolated flash vacuum pyrolysis (600 K) products of CH2ClS(O)2N3. (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent 266 nm laser photolysis (25 min). For clarity, spectrum B is 12-fold expanded along the ΔA axis. The IR bands of SO2 (e), unknown species (f), HCN (g), HCl (h), HNSO (i), H2CO (j), CH2NH (k), HNCO (l), CO2 (m), CO (n), HOSN (o), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.
Figure 2.
(A) IR spectrum (Absorbance, A) of the N2-matrix isolated flash vacuum pyrolysis (600 K) products of CH2ClS(O)2N3. (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent 266 nm laser photolysis (25 min). For clarity, spectrum B is 12-fold expanded along the ΔA axis. The IR bands of SO2 (e), unknown species (f), HCN (g), HCl (h), HNSO (i), H2CO (j), CH2NH (k), HNCO (l), CO2 (m), CO (n), HOSN (o), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.

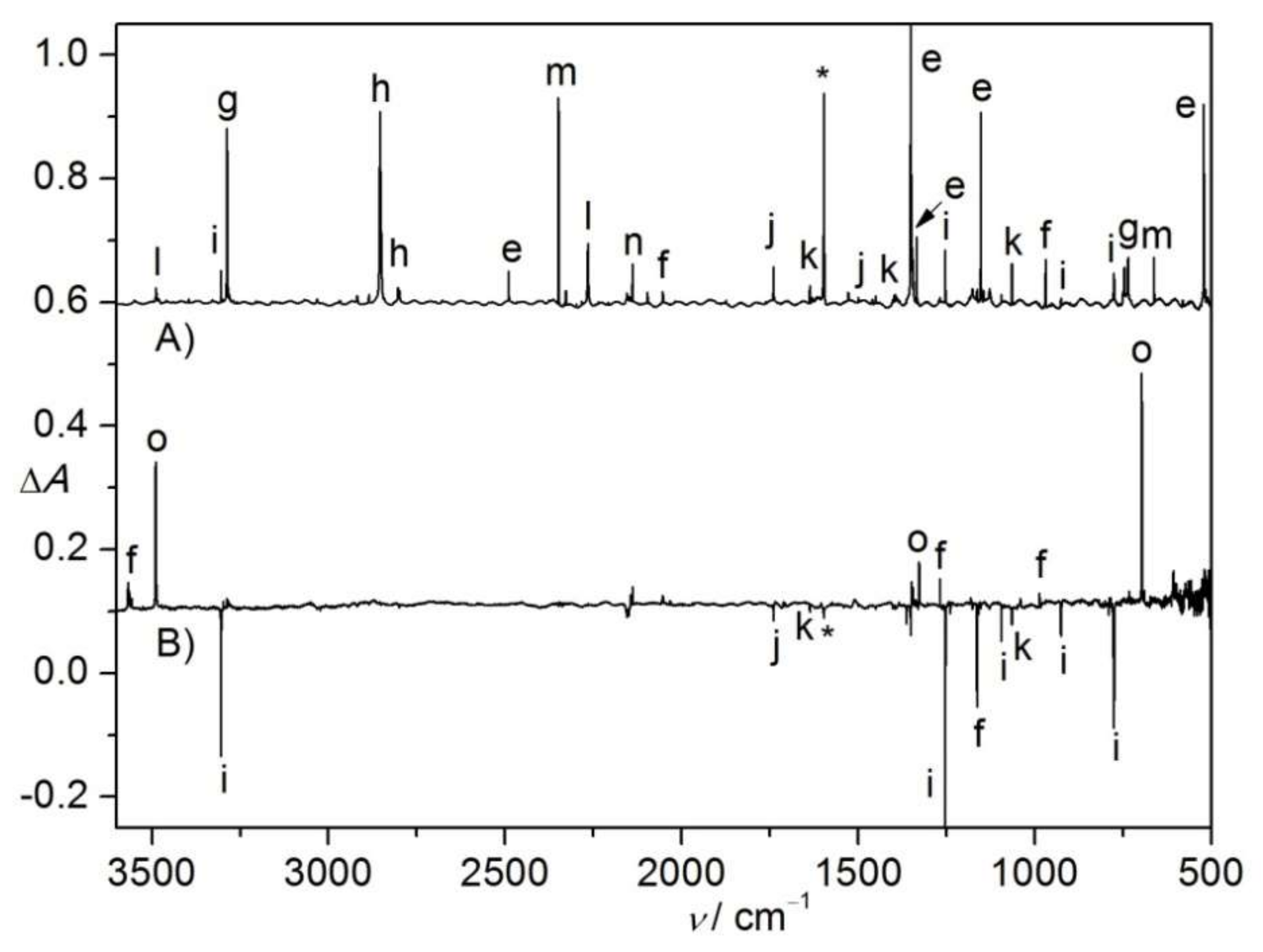
Figure 3.
(A) IR difference spectrum (Absorbance, ΔA) showing the decomposition of CHCl2S(O)2N3 in N2-matrix upon a 193 nm laser photolysis (11 min); (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent UV-light photolysis (365 nm, 10 min). (C) Calculated IR spectrum of triplet CHCl2S(O)2N at the B3LYP/6-311++G(3df,3pd) level. (D) Calculated IR spectrum of CHCl2NSO2 (singlet-II) at the B3LYP/6-311++G(3df,3pd) level. The IR bands of CHCl2SO2N3 (a), CHCl2SO2N (b), CHCl2NSO2 (c), CHCl2S(O)NO (d), SO2 (e), unknown species (f), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.
Figure 3.
(A) IR difference spectrum (Absorbance, ΔA) showing the decomposition of CHCl2S(O)2N3 in N2-matrix upon a 193 nm laser photolysis (11 min); (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent UV-light photolysis (365 nm, 10 min). (C) Calculated IR spectrum of triplet CHCl2S(O)2N at the B3LYP/6-311++G(3df,3pd) level. (D) Calculated IR spectrum of CHCl2NSO2 (singlet-II) at the B3LYP/6-311++G(3df,3pd) level. The IR bands of CHCl2SO2N3 (a), CHCl2SO2N (b), CHCl2NSO2 (c), CHCl2S(O)NO (d), SO2 (e), unknown species (f), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.

Figure 4.
(A) IR spectrum (Absorbance, A) of the N2-matrix isolated flash vacuum pyrolysis (700 K) products of CHCl2S(O)2N3. (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent UV-light photolysis (365 nm, 10 min). The IR bands of CHCl2SO2N3 (a), SO2 (e), unknown species (f), •NSO2 (g), CHCl2 (h), HCN (i), HCl (j), ClCN (k), HNSO2 (l), OCCl2 (m), CO (n), CO2 (o), OSNO (p), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.
Figure 4.
(A) IR spectrum (Absorbance, A) of the N2-matrix isolated flash vacuum pyrolysis (700 K) products of CHCl2S(O)2N3. (B) IR difference spectrum (Absorbance, ΔA) showing the change of the N2-matrix upon subsequent UV-light photolysis (365 nm, 10 min). The IR bands of CHCl2SO2N3 (a), SO2 (e), unknown species (f), •NSO2 (g), CHCl2 (h), HCN (i), HCl (j), ClCN (k), HNSO2 (l), OCCl2 (m), CO (n), CO2 (o), OSNO (p), and impurity H2O (*) are labeled. For clarity, the spectra are arbitrarily shifted along the ΔA axis.

Figure 5.
EPR spectra of the 266 nm laser photolysis products of CH2ClS(O)2N3 (A) and CHCl2S(O)2N3 (B) in glassy toluene matrices (0.4 mmol mL−1) at 5 K. Natural spin densities of the nitrenes computed at the M06-2X/6-311++G(3df,3pd) level are depicted.
Figure 5.
EPR spectra of the 266 nm laser photolysis products of CH2ClS(O)2N3 (A) and CHCl2S(O)2N3 (B) in glassy toluene matrices (0.4 mmol mL−1) at 5 K. Natural spin densities of the nitrenes computed at the M06-2X/6-311++G(3df,3pd) level are depicted.

Figure 6.
Calculated energy profile for the decomposition of RS(O)2N3 (R = CH2Cl and CHCl2) in the singlet state at the B3LYP/6-311++G(3df,3pd) level of theory. The calculated molecules structures (bond lengths in Å and angles in °) for the selected species are also depicted.
Figure 6.
Calculated energy profile for the decomposition of RS(O)2N3 (R = CH2Cl and CHCl2) in the singlet state at the B3LYP/6-311++G(3df,3pd) level of theory. The calculated molecules structures (bond lengths in Å and angles in °) for the selected species are also depicted.

Figure 7.
Calculated molecular structures and relative energies (in parentheses, kcal mol−1) of CH2ClS(O)2N and CHCl2S(O)2N at the B3LYP/6-311++G(3df,3pd) level. Selected bond lengths in Å and angles in ° (in italics) are given.
Figure 7.
Calculated molecular structures and relative energies (in parentheses, kcal mol−1) of CH2ClS(O)2N and CHCl2S(O)2N at the B3LYP/6-311++G(3df,3pd) level. Selected bond lengths in Å and angles in ° (in italics) are given.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated and observed IR frequencies (>500 cm−1) of CH2ClS(O)2N.
| Calculated a | Observed b | Assignment c | ||||
|---|---|---|---|---|---|---|
| Singlet | Triplet | N2-matrix | ||||
| ν | Δν | ν | Δν | ν | Δν | |
| 3183 (4) | 0.0 | 3176 (2) | 0.0 | 3023.0 (4) | <0.5 | νasym(CH2) |
| 3098 (5) | 0.0 | 3097 (3) | 0.0 | 2953.6 (7) | <0.5 | νsym(CH2) |
| 1431 (5) | 0.0 | 1435 (4) | 0.0 | 1398.3 (3) | <0.5 | δ(CH2) |
| 1400 (164) | 2.5 | 1357 (155) | 0.0 | 1354.1 (100) | <0.5 | νasym(SO2) |
| 1268 (13) | 0.1 | 1265 (10) | 0.0 | 1238.6 (6) | <0.5 | ω(CH2) |
| 1161 (1) | 0.0 | 1158 (83) | 0.0 | 1155.5 (40) | <0.5 | νsym(SO2) |
| 1053 (67) | 9.4 | 1149 (2) | 0.0 | 1128.2 (5) | <0.5 | τ(CH2) |
| 973 (12) | 11.5 | 875 (2) | 0.0 | 870.1 (8) | <0.5 | ρ(CH2) |
| 870 (<1) | 0.1 | 778 (6) | 0.3 | 748.8 (9) | <0.5 | ν(CCl) |
| 761 (21) | 0.5 | 699 (15) | 11.2 | 718.9 (6) | 11.1 | ν(SN) |
| 686 (16) | 0.9 | 656 (26) | 0.2 | 688.1 (7) | <0.5 | ν(SC) |
| 515 (83) | 3.0 | 497 (69) | 4.0 | 500.8 (42) | <0.5 | δ(SO2) |
a The calculated IR harmonic frequencies (ν, unscaled), 15N-isotopic shifts (Δν), and band intensities (km mol−1, in parentheses) for the singlet and triplet states at the B3LYP/6-311++G(3df,3pd) level. Full list of the calculated IR frequencies for all the conformers are given in Table S2. b The observed band positions (ν), 15N-isotopic shifts (Δν), and relative intensities (in parentheses) in N2-matrix. c Tentative assignment based on the calculated vibrational displacement vectors for the triplet state.
Table 2.
Calculated and observed IR frequencies (>500 cm–1) of CH2ClNSO2.
| Calculated a | Observed (N2-matrix) b | Assignment c | ||
|---|---|---|---|---|
| ν | Δν | ν | Δν | |
| 3166 (<1) | 0.0 | νasym (CH2) | ||
| 3096 (8) | 0.0 | νsym (CH2) | ||
| 1497 (1) | 0.3 | 1470.3/1465.3 (2) | <0.5 | δ (CH2) |
| 1394 (198) | 0.3 | 1387.0/1381.6 (57) | <0.5 | νasym (SO2) |
| 1347 (26) | 4.2 | 1326.0/1324.7 (10) | 4.4 | ω (CH2) + ν (N=S) |
| 1287 (303) | 11.6 | 1283.7/1278.0 (100) | 10.4 | ν (N=S) |
| 1241 (52) | 0.8 | 1224.2/1219.7 (14) | <0.5 | τ (CH2) |
| 1125 (33) | 15.5 | 1114.6/1113.1 (13) | 15.5 | ν (CN) + νsym (SO2) |
| 985 (1) | 2.6 | 967.7/955.9 (12) | <0.5 | ρ (CH2) |
| 854 (20) | 9.1 | 849.4/846.7 (7) | 8.8 | ν (C–N–S) |
| 712 (81) | 0.8 | 706.4/699.0 (11) | 2.3 | ν (CCl) |
| 550 (4) | 6.0 | δ (S–O–N) | ||
| 502 (53) | 2.0 | 509.3/506.7 (8) | 2.2 | δ (SO2) |
a The calculated IR harmonic frequencies (ν, unscaled), 15N-isotopic shifts (Δν), and intensities (km mol−1, in parentheses) at the B3LYP/6-311++G(3df,3pd) level. Full list of the calculated IR frequencies for all the conformers are given in Table S4. b The observed band positions (ν), 15N-isotopic shifts (Δν), and relative band intensities (in parentheses) in N2-matrix. c Tentative assignment based on the calculated vibrational displacement vectors.
Table 3.
Calculated and observed IR frequencies (>500 cm−1) of CHCl2S(O)2N.
| Calculated a | Observed b | Assignment c | ||||
|---|---|---|---|---|---|---|
| Singlet | Triplet | N2-matrix | ||||
| ν | Δν | ν | Δν | ν | Δν | |
| 3167 (8) | 0.0 | 3152 (6) | 0.0 | 3027.2 (4) | <0.5 | ν(CH) |
| 1409 (139) | 2.7 | 1367 (137) | 0.0 | 1366.5 (100) | <0.5 | νasym(SO2) |
| 1225 (15) | 0.0 | 1230 (11) | 0.0 | 1196.1 (7) | <0.5 | ρ(CH) |
| 1213 (5) | 0.1 | 1206 (9) | 0.0 | 1163.7 (4) | <0.5 | ω(CH) |
| 1048 (74) | 10.0 | 1157 (93) | 0.0 | 1153.2 (32) | <0.5 | νsym(SO2) + ω(CH) |
| 978 (13) | 11.1 | 777 (67) | 0.2 | 808.4 (35) | <0.5 | νasym(CCl2) |
| 762 (100) | 0.1 | 764 (24) | 0.4 | 747.7 (16) | <0.5 | νsym(CCl2) |
| 760 (17) | 0.6 | 701 (21) | 10.7 | 718.5 (7) | 9.6 | ν(SN) |
| 681 (13) | 0.4 | 654 (10) | 0.4 | 675.8 (5) | <0.5 | ν(SC) |
| 520 (101) | 2.7 | 507 (82) | 1.3 | 512.6 (50) | <0.5 | δ(SO2) |
a The calculated IR harmonic frequencies (ν, unscaled), 15N-isotopic shifts (Δν), and band intensities (km mol–1, in parentheses) for the singlet and triplet states at the B3LYP/6-311++G(3df,3pd) level. Full list of the calculated IR frequencies for all the conformers are given in Table S6. b The observed band positions (ν), 15N-isotopic shifts (Δν), and relative intensities (in parentheses) in N2-matrix. c Tentative assignment based on the calculated vibrational displacement vectors for the triplet state.
Table 4.
Calculated and observed IR frequencies (> 500 cm–1) of CHCl2NSO2.
| Calculated a | Observed b | Assignment c | ||||
|---|---|---|---|---|---|---|
| Singlet-I | Singlet-II | N2-matrix | ||||
| ν | Δν | ν | Δν | ν | Δν | |
| 3151 (2) | 0.0 | 3175 (1) | 0.0 | ν(CH) | ||
| 1403 (190) | 0.2 | 1409 (163) | 0.0 | 1410.2/1401.7 (100) | <0.5 | νasym(SO2) |
| 1340 (7) | 1.7 | 1348 (97) | 9.1 | 1335.0/1323.8 (22) | <0.5 | ρ(CH) |
| 1299 (431) | 15.0 | 1278 (326) | 7.8 | 1280.5/1262.7 (98) | 16.0 | ν(SN) + δ(SO2) |
| 1252 (27) | 0.0 | 1243 (25) | 0.9 | 1230.2/1227.1 (7) | <0.5 | ω(CH) |
| 1129 (32) | 15.3 | 1152 (52) | 14.3 | 1125.4/1117.0 (35) | 11.8 | ν(CN) + νsym(SO2) |
| 890 (44) | 12.1 | 845 (47) | 8.1 | 846.2/834.4 (33) | 9.0 | ν(CN) |
| 758 (58) | 1.4 | 721 (201) | 1.0 | 783.9/776.9 (56) | <0.5 | νsym(CCl2) |
| 745 (169) | 0.9 | 714 (18) | 2.5 | 767.2/759.7 (40) | <0.5 | νasym(CCl2) |
| 526 (70) | 1.8 | 597 (13) | 8.5 | 532.5 (13) | 2.2 | δ(SO2) |
a The calculated IR harmonic frequencies (ν, unscaled), 15N-isotopic shifts (Δν), and band intensities (km mol–1, in parentheses) at the B3LYP/6-311++G(3df,3pd) level. Full list of the calculated IR frequencies for all the conformers are given in Table S4. b The observed band positions (ν), 15N-isotopic shifts (Δν), and relative intensities (in parentheses) in N2-matrix. c Tentative assignment based on the calculated vibrational displacement vectors.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, Y.; Chu, X.; Lu, Y.; Abe, M.; Zeng, X. Chloro- and Dichloro-methylsulfonyl Nitrenes: Spectroscopic Characterization, Photoisomerization, and Thermal Decomposition. Molecules 2018, 23, 3312. https://doi.org/10.3390/molecules23123312
AMA Style
Yang Y, Chu X, Lu Y, Abe M, Zeng X. Chloro- and Dichloro-methylsulfonyl Nitrenes: Spectroscopic Characterization, Photoisomerization, and Thermal Decomposition. Molecules. 2018; 23(12):3312. https://doi.org/10.3390/molecules23123312
Chicago/Turabian StyleYang, Yang, Xianxu Chu, Yan Lu, Manabu Abe, and Xiaoqing Zeng. 2018. "Chloro- and Dichloro-methylsulfonyl Nitrenes: Spectroscopic Characterization, Photoisomerization, and Thermal Decomposition" Molecules 23, no. 12: 3312. https://doi.org/10.3390/molecules23123312