
The Antitumor Activity of a Lead Thioxanthone is Associated with Alterations in Cholesterol Localization

 ,
,  , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
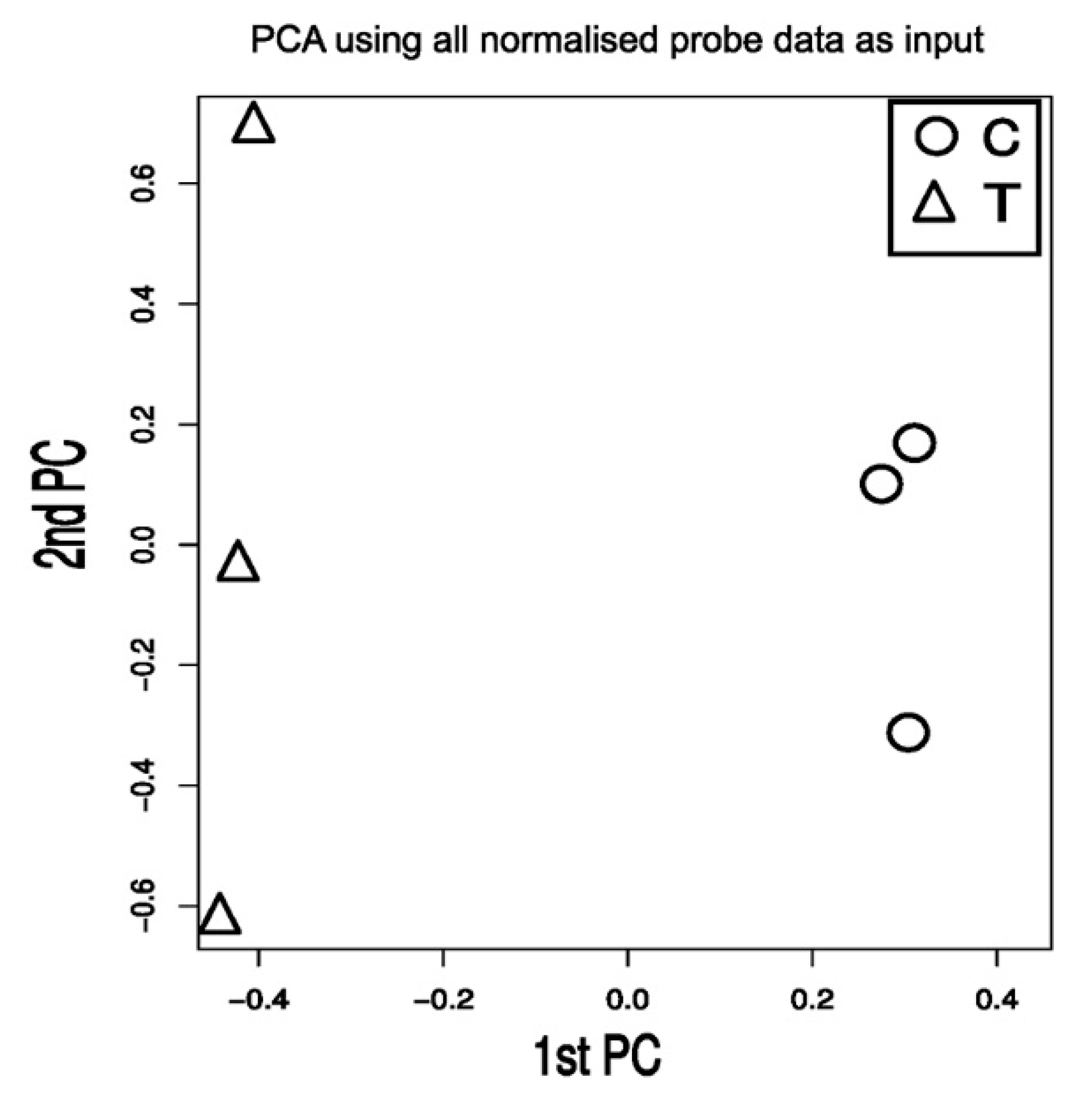
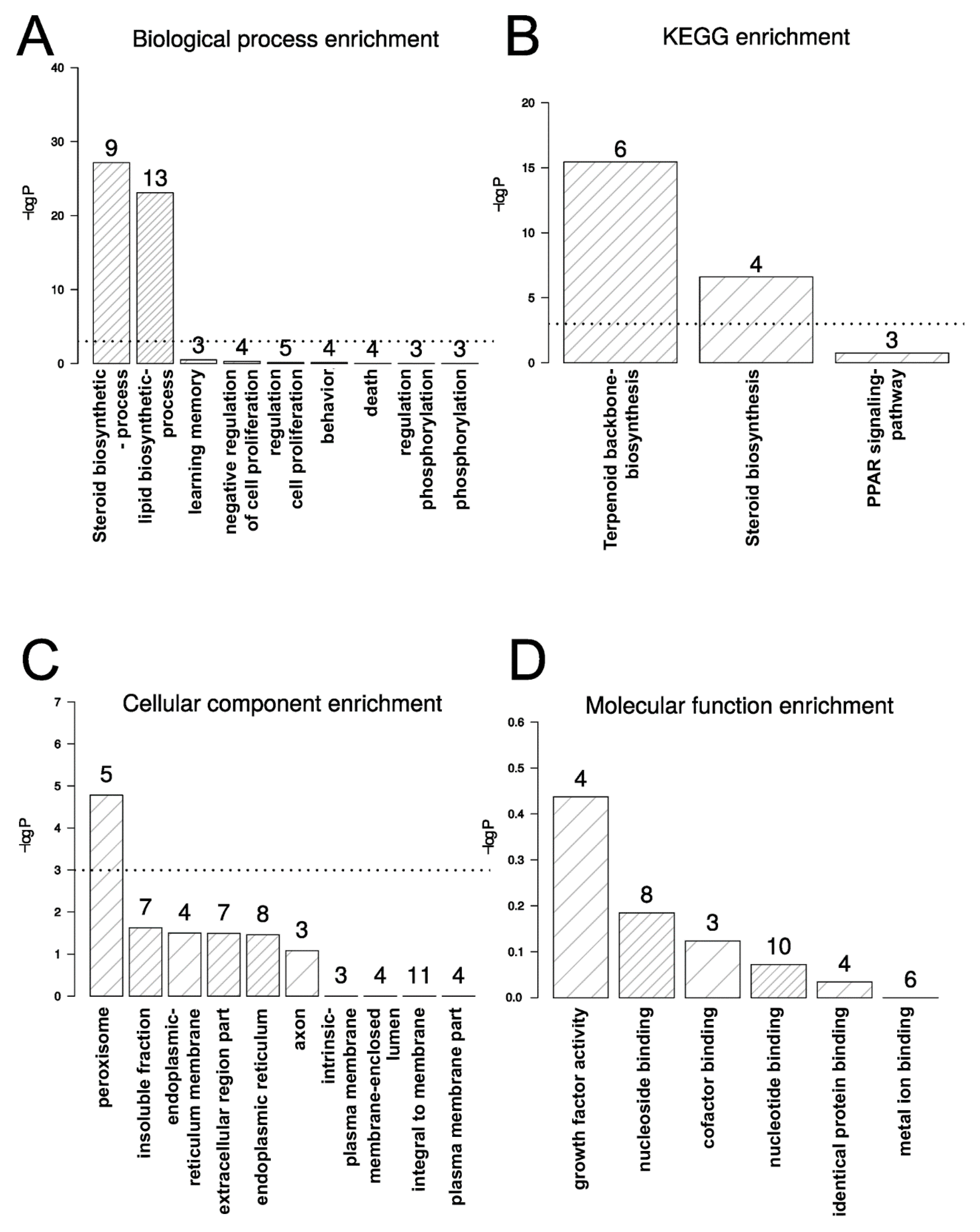
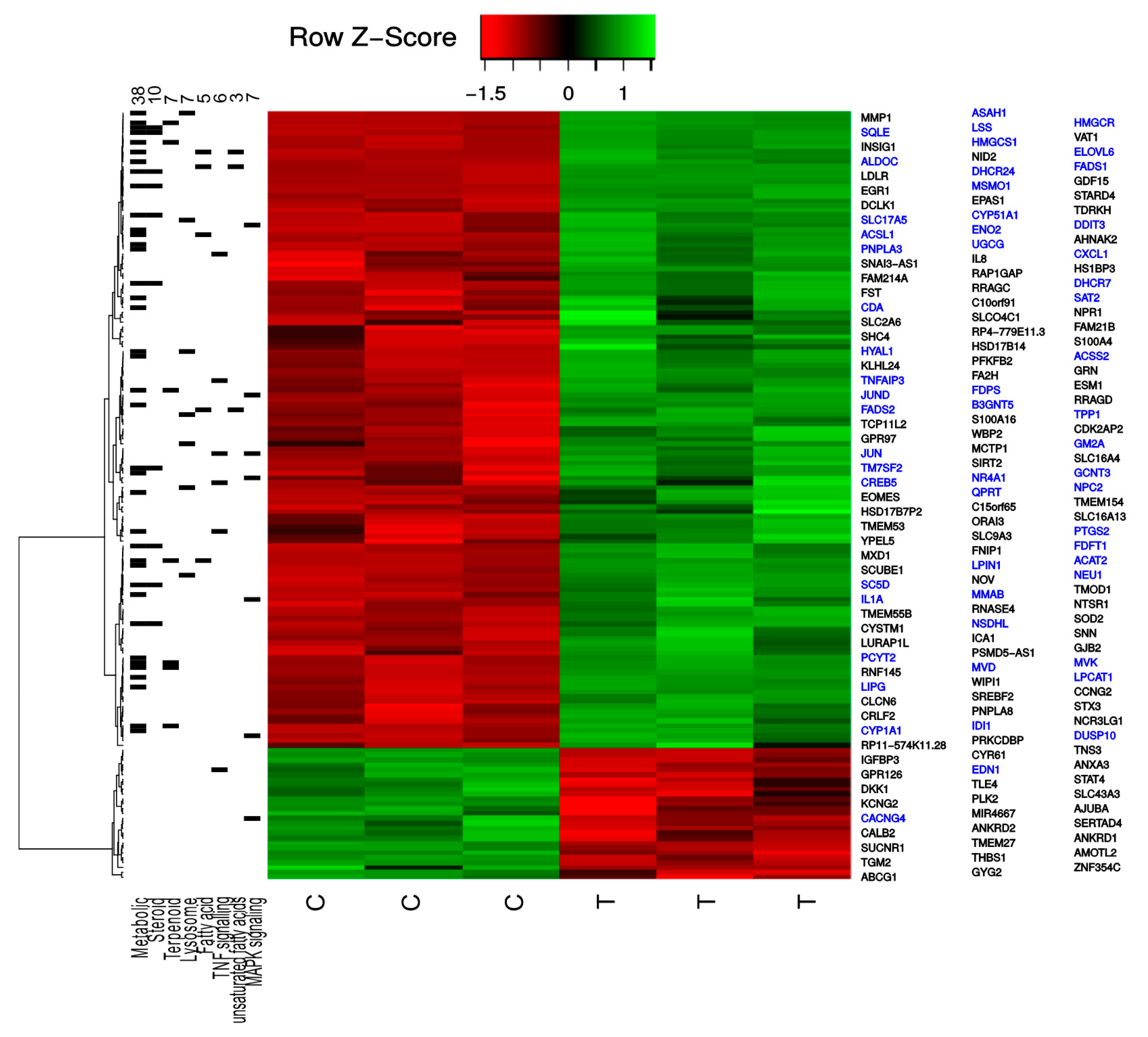
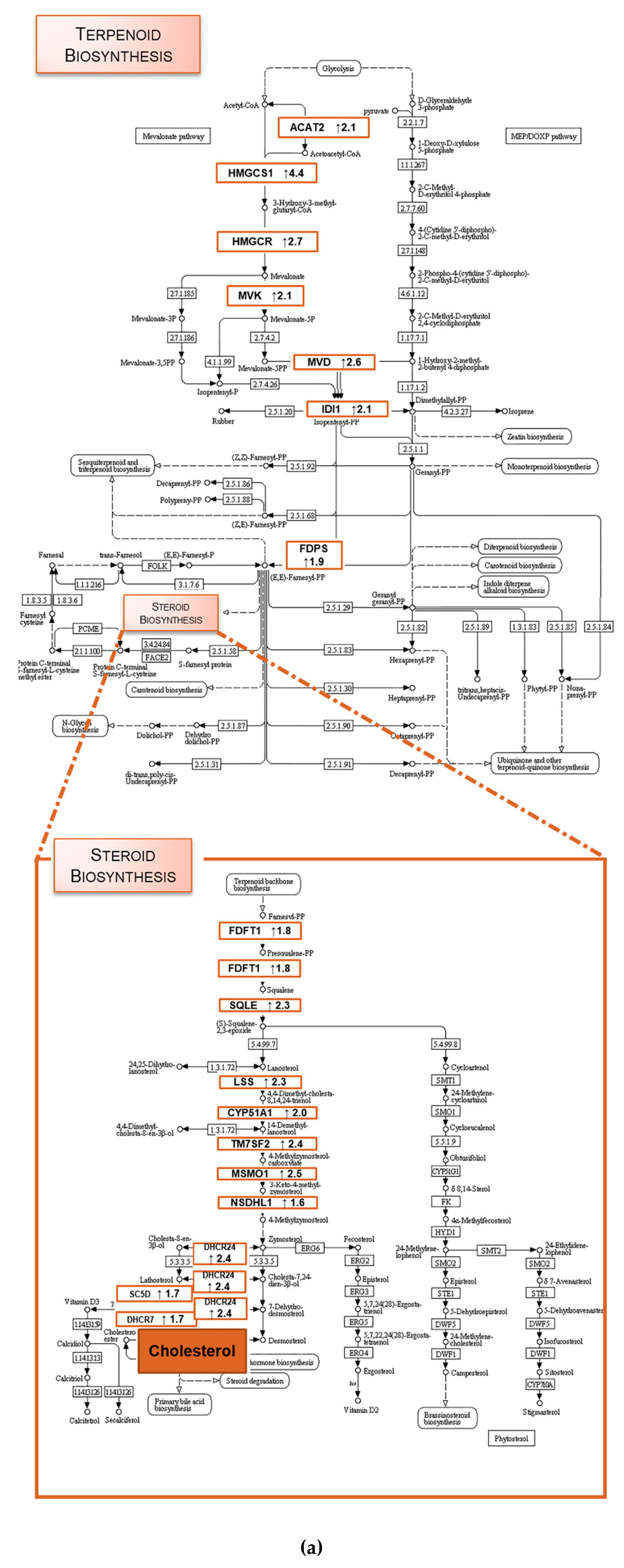
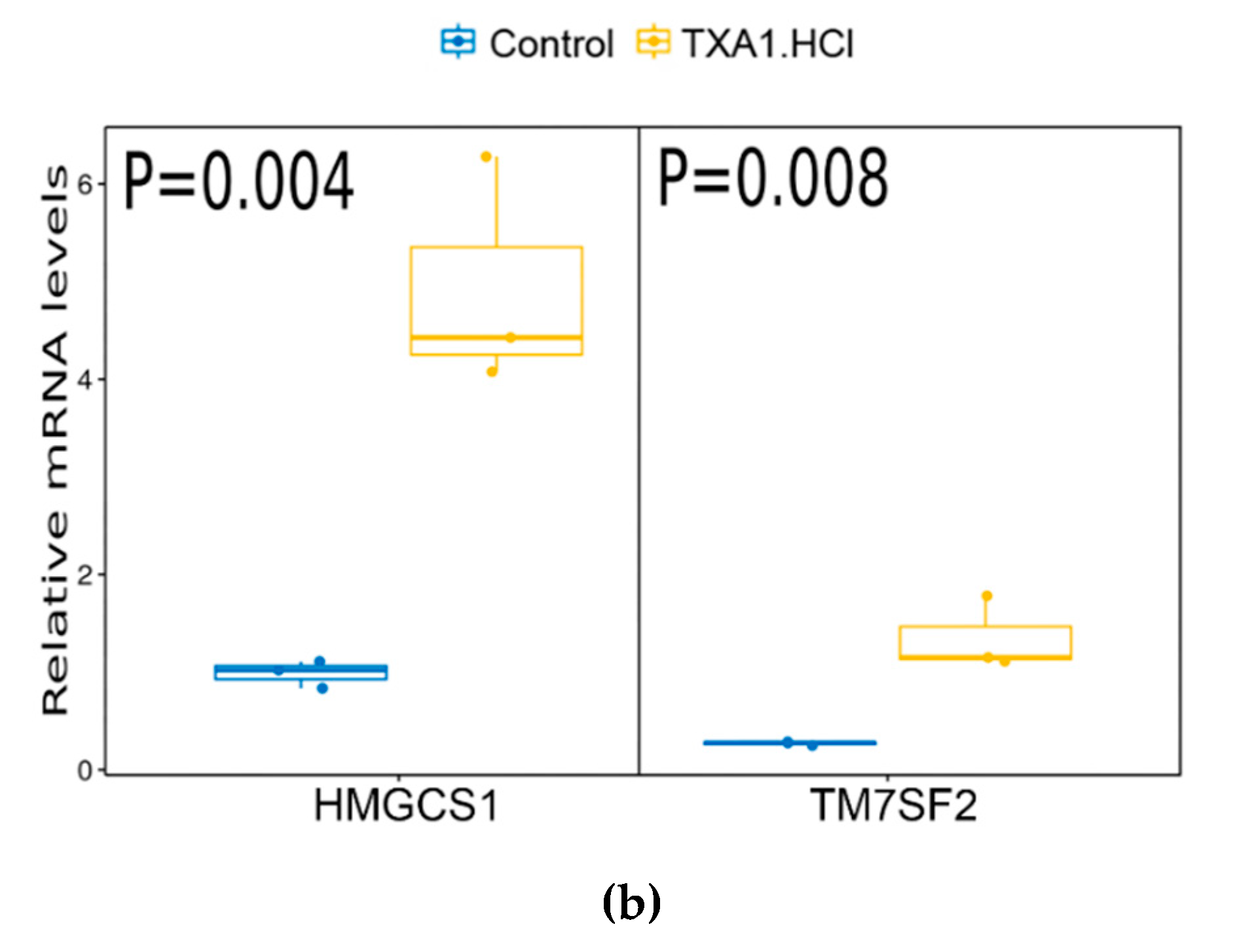
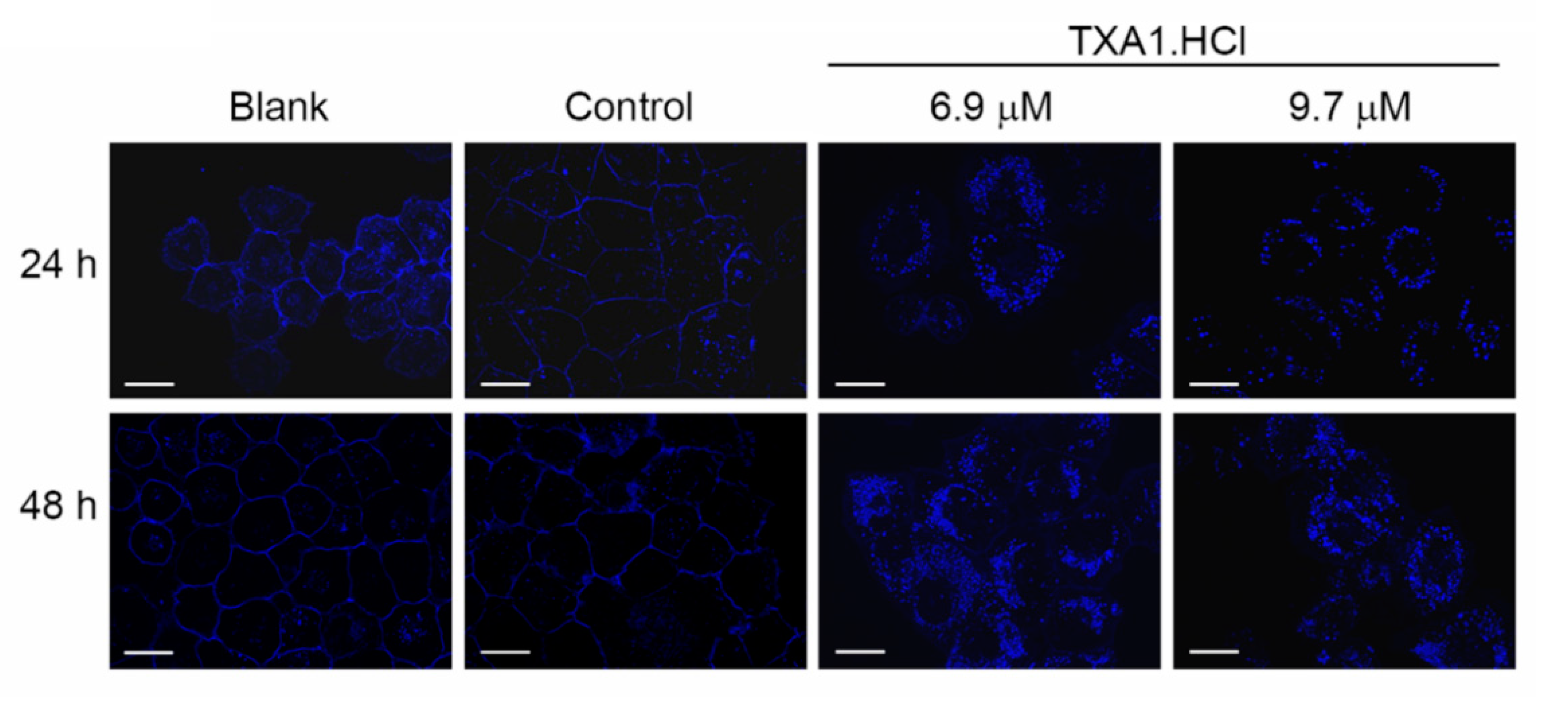
2.1. TXA1.HCl Affects Steroid Biosynthesis and Cholesterol Localization
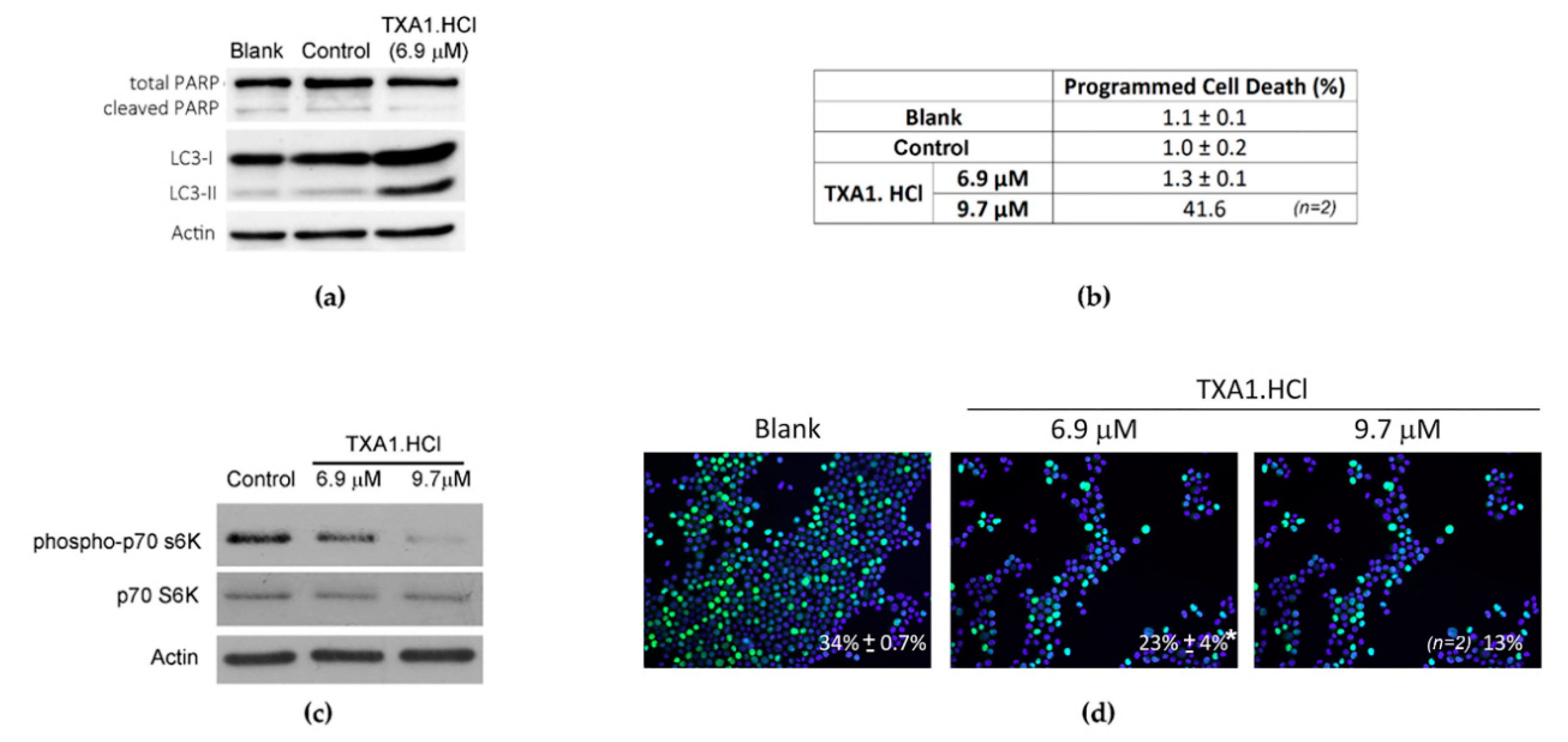
2.2. TXA1.HCl Induces Autophagy and Decreases Proliferation of NSCLC Cells
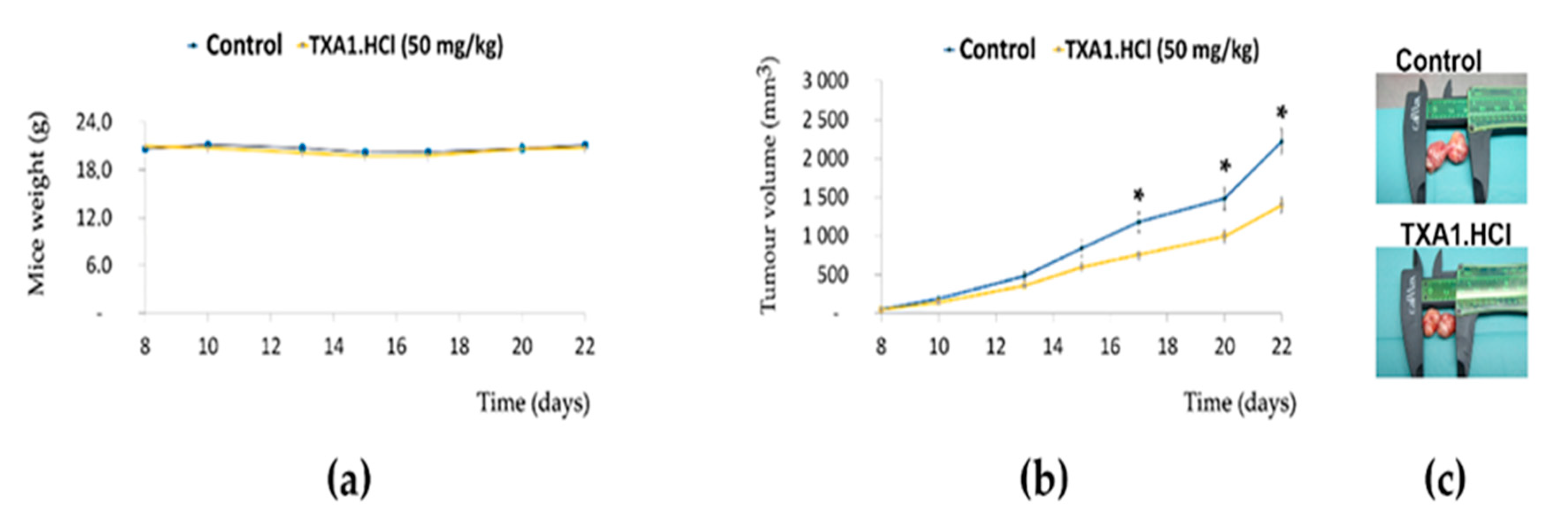
2.3. TXA1.HCl Reduces the Growth of NCI-H460 Xenografts in Nude Mice
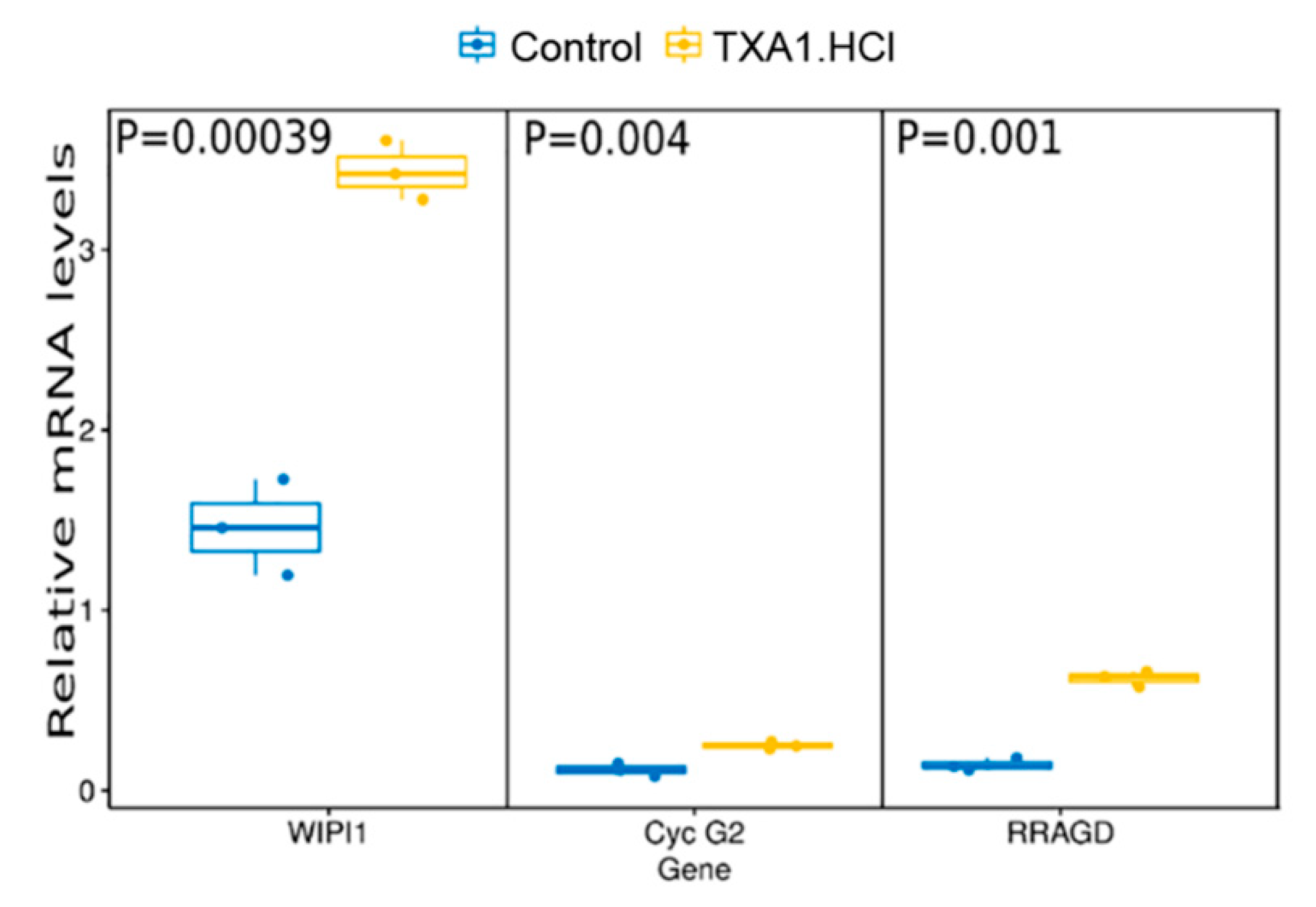
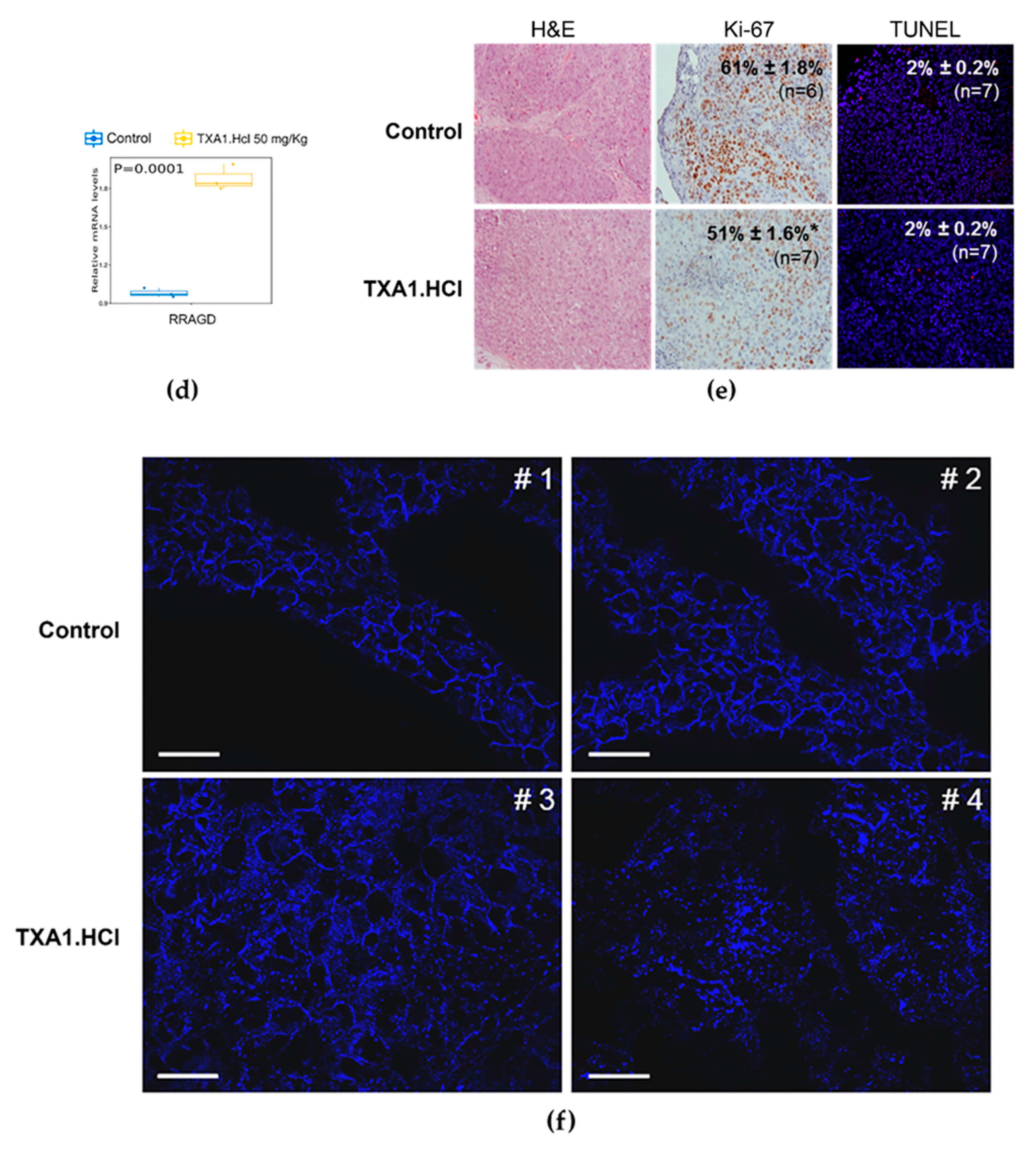
2.4. TXA1.HCl Treatment Reduces Proliferation, Increases RagD Expression and Affects Cholesterol Localization in NSCLC Xenografted Nude Mice
3. Materials and Methods
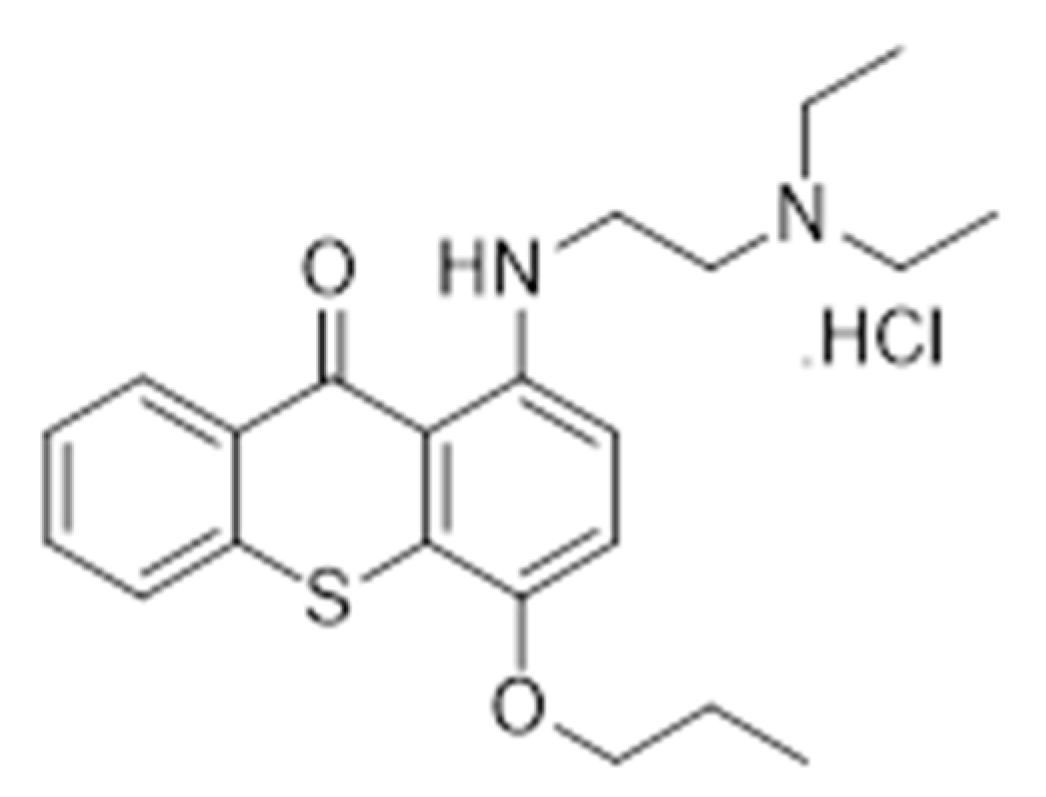
3.1. Synthesis of TXA1 Hydrochloride (TXA1.HCl).
3.2. Cell Culture and Treatment with TXA1.HCl
3.3. Mouse Xenograft Model
3.4. RNA Extraction
3.5. Microarray cDNA Analysis
3.6. Quantitative Real-time PCR
3.7. Tumor Histology and Immunohistochemical Analysis
3.8. TUNEL Assay
3.9. BrdU Incorporation Assay
3.10. Filipin Staining
3.11. Protein Expression Analysis
3.12. Biochemical and Toxicological Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Funding
Conflicts of Interest
References
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell. Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiøtt, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Z.; Shen, W.-J.; Azhar, S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. 2010, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.; Mo, H.; McConathy, W.; Sabnis, N.; Lacko, A. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Silvente-Poirot, S.; Poirot, M. Cholesterol and cancer, in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The role of cholesterol in cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Gu, D.; Bjorklund, C.C.; Kuiatse, I.; Remaley, A.T.; Bashir, T.; Vreys, V.; Orlowski, R.Z. The novel anticancer agent jnj-26854165 induces cell death through inhibition of cholesterol transport and degradation of abca1. J. Pharmacol. Exp. Ther. 2013, 346, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br. J. Cancer 2017, 117, 513–524. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Gowda, R.; Sharma, A.; Robertson, G.P. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol. Cancer Ther. 2014, 13, 1690–1703. [Google Scholar] [CrossRef]
- Lee, J.-K.; Nam, D.-H.; Lee, J. Repurposing antipsychotics as glioblastoma therapeutics: Potentials and challenges. Oncol. Lett. 2016, 11, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lafrasse, C.; Rousson, R.; Bonnet, J.; Pentchev, P.G.; Louisot, P.; Vanier, M.T. Abnormal cholesterol metabolism in imipramine-treated fibroblast cultures. Similarities with niemann-pick type c disease. Biochim. Biophys. Acta, Lipids Lipid Metab. 1990, 1043, 123–128. [Google Scholar] [CrossRef]
- Kristiana, I.; Sharpe, L.J.; Catts, V.S.; Lutze-Mann, L.H.; Brown, A.J. Antipsychotic drugs upregulate lipogenic gene expression by disrupting intracellular trafficking of lipoprotein-derived cholesterol. Pharmacogenomics J. 2010, 10, 396–407. [Google Scholar] [CrossRef]
- Wiklund, E.D.; Catts, V.S.; Catts, S.V.; Ng, T.F.; Whitaker, N.J.; Brown, A.J.; Lutze-Mann, L.H. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer 2010, 126, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (redo)-itraconazole as an anti-cancer agent. Ecancermedicalscience 2015, 9, 521. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef]
- Barbosa, J.; Lima, R.T.; Sousa, D.; Gomes, A.S.; Palmeira, A.; Seca, H.; Choosang, K.; Pakkong, P.; Bousbaa, H.; Pinto, M.M.; et al. Screening a small library of xanthones for antitumor activity and identification of a hit compound which induces apoptosis. Molecules 2016, 21, 81. [Google Scholar] [CrossRef]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of p-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef]
- Lima, R.T.; Sousa, D.; Paiva, A.M.; Palmeira, A.; Barbosa, J.; Pedro, M.; Pinto, M.M.; Sousa, E.; Vasconcelos, M.H. Modulation of autophagy by a thioxanthone decreases the viability of melanoma cells. Molecules 2016, 21. [Google Scholar] [CrossRef]
- Kanehisa, F.M.; Tanabe, M.S.Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carvalho, A.S.; Molina, H.; Matthiesen, R. New insights into functional regulation in ms-based drug profiling. Sci. Rep. 2016, 6, 18826. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The ldl receptor. Arterioscl. Throm. Vas. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Models Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Ohsaki, Y.; Tauchi-Sato, K.; Fujita, A.; Fujimoto, T. Cholesterol depletion induces autophagy. Biochem. Biophys. Res. Commun. 2006, 351, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dang, Y.; Ren, Y.R.; Liu, J.O. Cholesterol trafficking is required for mtor activation in endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTor regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. Mtor: A pharmacologic target for autophagy regulation. J. Clin. Invest. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Npc2 facilitates bidirectional transfer of cholesterol between npc1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Matsumoto, Y.; Inagaki, Y.; Sekimizu, K. Niemann-pick disease type c2 protein induces autophagy and inhibits growth in fm3a breast cancer cells. Drug Discoveries Ther. 2015, 9, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhao, M.; Qiu, X.; Deis, J.A.; Huang, H.; Tang, Q.Q.; Chen, X. Niemann-pick type c2 deficiency impairs autophagy-lysosomal activity, mitochondrial function, and tlr signaling in adipocytes. J. Lipid Res. 2016, 57, 1644–1658. [Google Scholar] [CrossRef] [PubMed]
- Tsuyuki, S.; Takabayashi, M.; Kawazu, M.; Kudo, K.; Watanabe, A.; Nagata, Y.; Kusama, Y.; Yoshida, K. Detection of wipi1 mrna as an indicator of autophagosome formation. Autophagy 2014, 10, 497–513. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Pfisterer, S.G. Assessing mammalian autophagy by wipi-1/atg18 puncta formation. Methods Enzymol. 2009, 452, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Grolleau, A.; Bowman, J.; Pradet-Balade, B.; Puravs, E.; Hanash, S.; Garcia-Sanz, J.A.; Beretta, L. Global and specific translational control by rapamycin in t cells uncovered by microarrays and proteomics. J. Biol. Chem. 2002, 277, 22175–22184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Su, P.; Wang, L.; Chen, J.; Zimmermann, M.; Genbacev, O.; Afonja, O.; Horne, M.C.; Tanaka, T.; Duan, E.; et al. Mtor supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7840–7845. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, L.; Imbert, V.; Nebout, M.; Colosetti, P.; Neffati, Z.; Lagadec, P.; Verhoeyen, E.; Peng, C.; Duprez, E.; Legros, L.; et al. The bmi1 polycomb protein represses cyclin g2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015, 29, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of torc1 in response to amino acid starvation via lysosomal recruitment of tsc2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef]
- Shaw, R.J. Mtor signaling: Rag gtpases transmit the amino acid signal. Trends Biochem. Sci. 2008, 33, 565–568. [Google Scholar] [CrossRef]
- Yuan, H.X.; Xiong, Y.; Guan, K.L. Nutrient sensing, metabolism, and cell growth control. Mol. Cell 2013, 49, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Groenewoud, M.J.; Zwartkruis, F.J. Rheb and rags come together at the lysosome to activate mtorc1. Biochem. Soc. Trans. 2013, 41, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Liang, C. Negative regulation of autophagy. Cell Death Differ. 2010, 17, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mtor: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.N.; Sy, L.K.; Liu, F.; Che, C.M. Activation of autophagy of aggregation-prone ubiquitinated proteins by timosaponin a-iii. J. Biol. Chem. 2011, 286, 31684–31696. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A. Transcriptional activation of ragd gtpase controls mtorc1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mtor and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef]
- Meijer, A.J.; Codogno, P. Nutrient sensing: Tor’s ragtime. Nat. Cell Biol. 2008, 10, 881–883. [Google Scholar] [CrossRef]
- Narita, M.; Inoki, K. Rags connect mtor and autophagy. Small GTPases 2012, 3, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, E. Rag gtpase in amino acid signaling. Amino Acids 2016, 48, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-S.; Choi, C.S. The role of amino acid-induced mammalian target of rapamycin complex 1 (mtorc1) signaling in insulin resistance. Exp. Mol. Med. 2017, 48, e201. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Sabatini, D.M. Rag proteins regulate amino-acid-induced mtorc1 signalling. Biochem. Soc. Trans. 2009, 37, 289–290. [Google Scholar] [CrossRef]
- Nicastro, R.; Sardu, A.; Panchaud, N.; De Virgilio, C. The architecture of the rag gtpase signaling network. Biomolecules 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of mtorc1 by the rag gtpases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mtorc1 via an slc38a9–niemann-pick c1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Azar, H.A.; Hansen, C.T.; Costa, J. N:Nih (s) ii-nu/nu mice with combined immunodeficiency: A new model for human tumor heterotransplantation. J. Natl. Cancer Inst. 1980, 65, 421–430. [Google Scholar] [CrossRef]
- Wettenhall, J.M.; Simpson, K.M.; Satterley, K.; Smyth, G.K. Affylmgui: A graphical user interface for linear modeling of single channel microarray data. Bioinformatics 2006, 22, 897–899. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2008, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Iwase, A.; Bayasula, B.; Nagatomo, Y.; Kondo, M.; Nakahara, T.; Takikawa, S.; Goto, M.; Kotani, T.; Kiyono, T.; et al. Cyp51a1 induced by growth differentiation factor 9 and follicle-stimulating hormone in granulosa cells is a possible predictor for unfertilization. Reprod. Sci. 2015, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Almeida, G.M.; Duarte, T.L.; Farmer, P.B.; Steward, W.P.; Jones, G.D. Multiple end-point analysis reveals cisplatin damage tolerance to be a chemoresistance mechanism in a nsclc model: Implications for predictive testing. Int. J. Cancer 2008, 122, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Seca, H.; Soares, P.; Nascimento, M.S.; Vasconcelos, M.H. Ebv interferes with the sensitivity of burkitt lymphoma akata cells to etoposide. J. Cell. Biochem. 2011, 112, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.J.; Sousa, D.; Lima, R.T.; Carvalho, A.M.; Ferreira, I.C.F.R.; Vasconcelos, M.H. Flower extracts of filipendula ulmaria (l.) maxim inhibit the proliferation of the nci-h460 tumour cell line. Ind. Crop. Prod. 2014, 59, 149–153. [Google Scholar] [CrossRef]
- Borralho, P.M.; Simoes, A.E.; Gomes, S.E.; Lima, R.T.; Carvalho, T.; Ferreira, D.M.; Vasconcelos, M.H.; Castro, R.E.; Rodrigues, C.M. Mir-143 overexpression impairs growth of human colon carcinoma xenografts in mice with induction of apoptosis and inhibition of proliferation. PLoS ONE 2011, 6, e23787. [Google Scholar] [CrossRef]
- Preto, A.; Goncalves, J.; Rebocho, A.P.; Figueiredo, J.; Meireles, A.M.; Rocha, A.S.; Vasconcelos, H.M.; Seca, H.; Seruca, R.; Soares, P.; et al. Proliferation and survival molecules implicated in the inhibition of braf pathway in thyroid cancer cells harbouring different genetic mutations. BMC Cancer 2009, 9, 387. [Google Scholar] [CrossRef]
- Pereira, D.; Lima, R.T.; Palmeira, A.; Seca, H.; Soares, J.; Gomes, S.; Raimundo, L.; Maciel, C.; Pinto, M.; Sousa, E.; et al. Design and synthesis of new inhibitors of p53–mdm2 interaction with a chalcone scaffold. Arab. J. Chem. 2016. [Google Scholar] [CrossRef]
- Seca, H.; Lima, R.T.; Almeida, G.M.; Sobrinho-Simoes, M.; Bergantim, R.; Guimaraes, J.E.; Vasconcelos, M.H. Effect of mir-128 in DNA damage of hl-60 acute myeloid leukemia cells. Curr. Pharm. Biotechnol. 2014, 15, 492–502. [Google Scholar] [CrossRef]
- Lima, R.T.; Barron, G.A.; Grabowska, J.A.; Bermano, G.; Kaur, S.; Roy, N.; Vasconcelos, M.H.; Lin, P.K. Cytotoxicity and cell death mechanisms induced by a novel bisnaphthalimidopropyl derivative against the nci-h460 non-small lung cancer cell line. Anti-Cancer Agents Med. Chem. 2013, 13, 414–421. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds TXA1 and TXA1.HCl are available from the authors (E.S.). |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Creatinine (mg/dL) | Urea (mg/dL) | Total Protein (g/dL) | CKMB (U/L) | AST (U/L) | ALT (U/L) | |
|---|---|---|---|---|---|---|
| Control | 0.17 ± 0.03 | 48.63 ± 2.17 | 4.86 ± 0.30 | 128.00 ± 15.27 | 221.50 ± 30.85 | 29.67 ± 0.88 |
| TXA1.HCl (50 mg/kg) | 0.27 ± 0.10 | 49.58 ± 5.28 | 5.65 ± 0.69 | 151.50 ± 28.60 | 230.20 ± 57.9 | 38.25 ± 6.05 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, R.T.; Sousa, D.; Gomes, A.S.; Mendes, N.; Matthiesen, R.; Pedro, M.; Marques, F.; Pinto, M.M.; Sousa, E.; Vasconcelos, M.H. The Antitumor Activity of a Lead Thioxanthone is Associated with Alterations in Cholesterol Localization. Molecules 2018, 23, 3301. https://doi.org/10.3390/molecules23123301
Lima RT, Sousa D, Gomes AS, Mendes N, Matthiesen R, Pedro M, Marques F, Pinto MM, Sousa E, Vasconcelos MH. The Antitumor Activity of a Lead Thioxanthone is Associated with Alterations in Cholesterol Localization. Molecules. 2018; 23(12):3301. https://doi.org/10.3390/molecules23123301
Chicago/Turabian StyleLima, Raquel T., Diana Sousa, Ana Sara Gomes, Nuno Mendes, Rune Matthiesen, Madalena Pedro, Franklim Marques, Madalena M. Pinto, Emília Sousa, and M. Helena Vasconcelos. 2018. "The Antitumor Activity of a Lead Thioxanthone is Associated with Alterations in Cholesterol Localization" Molecules 23, no. 12: 3301. https://doi.org/10.3390/molecules23123301
APA StyleLima, R. T., Sousa, D., Gomes, A. S., Mendes, N., Matthiesen, R., Pedro, M., Marques, F., Pinto, M. M., Sousa, E., & Vasconcelos, M. H. (2018). The Antitumor Activity of a Lead Thioxanthone is Associated with Alterations in Cholesterol Localization. Molecules, 23(12), 3301. https://doi.org/10.3390/molecules23123301