Enantioselective Michael Addition of Aldehydes to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide


 , and
, and
Abstract
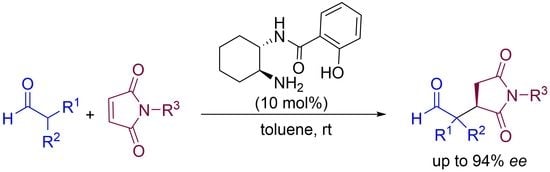
:
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Asymmetric Conjugate Addition Reaction
3.3. Computational Methods.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chauhan, P.; Kaur, J.; Chimni, S.S. Asymmetric organocatalytic addition reactions of maleimides: A promising approach towards the synthesis of chiral succinimide derivatives. Chem. Asian J. 2013, 8, 328–346. [Google Scholar] [CrossRef] [PubMed]
- Malochet-Grivois, C.; Roussakis, C.; Robillard, N.; Biard, J.F.; Riou, D.; Debitus, C.; Verbist, J.F. Effects in vitro of two marine substances, chlorolissoclimide and dichlorolissoclimide, on a non-small-cell bronchopulmonary carcinoma line (NSCLC-N6). Anticancer Drug Des. 1992, 7, 493–502. [Google Scholar] [PubMed]
- Ando, Y.; Fuse, E.; Figg, W.D. Thalidomide metabolism by the CYP2C subfamily. Clin. Cancer Res. 2002, 8, 1964–1973. [Google Scholar] [PubMed]
- Freiberg, C.; Brunner, N.A.; Schiffer, G.; Lampe, T.; Pohlmann, J.; Brands, M.; Raabe, M.; Haebich, D.; Ziegelbauer, K. Identification and characterization of the first class of potent bacterial Acetyl-CoA carboxylase inhibitors with antibacterial activity. J. Biol. Chem. 2004, 279, 26066–26073. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Rugseree, N.; Maithip, P.; Kongsaeree, P.; Prabpai, S.; Thebtaranonth, Y. Hirsutellones A-E, antimycobacterial alkaloids from the insect pathogenic fungus Hirsutella nivea body centered cubic 2594. Tetrahedron 2005, 61, 5577–5583. [Google Scholar] [CrossRef]
- Uddin, J.; Ueda, K.; Siwu, E.R.O.; Kita, M.; Uemura, D. Cytotoxic labdane alkaloids from an ascidian Lissoclinum sp.: isolation, structure elucidation, and structure-activity relationship. Bioorg. Med. Chem. 2006, 14, 6954–6961. [Google Scholar] [CrossRef] [PubMed]
- Nöth, J.; Frankowski, K.J.; Neuenswander, B.; Aubé, J.; Reiser, O. Efficient synthesis of γ-lactams by a tandem reductive amination/lactamization sequence. J. Comb. Chem. 2008, 10, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Fenster, E.; Hill, D.; Reiser, O.; Aube, J. Automated three-component synthesis of a library of γ-lactams. Beilstein J. Org. Chem. 2012, 8, 1804–1813. [Google Scholar] [CrossRef] [Green Version]
- Spaltenstein, A.; Almond, M.R.; Bock, W.J.; Cleary, D.G.; Furfine, E.S.; Hazen, R.J.; Kazmierski, W.M.; Salituro, F.G.; Tung, R.D.; Wright, L.L. Novel inhibitors of HIV protease: design, synthesis and biological evaluation of picomolar inhibitors containing cyclic P1/P2 scaffolds. Bioorg. Med. Chem. Lett. 2000, 10, 1159–1162. [Google Scholar] [CrossRef]
- Kazmierski, W.M.; Andrews, W.; Furfine, E.; Spaltenstein, A.; Wright, L. Discovery of potent pyrrolidone-based HIV-1 protease inhibitors with enhanced drug-like properties. Bioorg. Med. Chem. Lett. 2004, 14, 5689–5692. [Google Scholar] [CrossRef]
- Reddy, P.A.; Hsiang, B.C.H.; Latifi, T.N.; Hill, M.W.; Woodward, K.E.; Rothman, S.M.; Ferrendelli, J.A.; Covey, D.F. 3,3-Dialkyl- and 3-alkyl-3-benzyl-substituted 2-pyrrolidinones: A new class of anticonvulsant agents. J. Med. Chem. 1996, 39, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Das Sarma, K.; Zhang, J.; Huang, Y.; Davidson, J.G. Amino acid esters and amides for reductive amination of mucochloric acid: synthesis of novel γ-lactams, short peptides and antiseizure agent Levetiracetam (Keppra). Eur. J. Org. Chem. 2006, 3730–3737. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, J.-T. The effects of (-)-clausenamide on functional recovery in transient focal cerebral ischemia. Neurol. Res. 2002, 24, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-L.; Xu, Y.; Sundén, H.; Eriksson, L.; Sayah, M.; Cordova, A. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides. Chem. Commun. 2007, 7, 734–735. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Headley, A.D. A simple and highly effective water-compatible organocatalytic system for asymmetric direct Michael reactions of linear aldehydes to maleimides. Green Chem. 2013, 15, 2690–2694. [Google Scholar] [CrossRef]
- Xue, F.; Liu, L.; Zhang, S.; Duan, W.; Wang, W. A simple primary amine thiourea catalyzed highly enantioselective conjugate addition of α,α-disubstituted aldehydes to maleimides. Chem. Eur. J. 2010, 16, 7979–7982. [Google Scholar] [CrossRef]
- Bai, J.-F.; Peng, L.; Wang, L.-L.; Wang, L.-X.; Xu, X.-Y. Chiral primary amine thiourea promoted highly enantioselective Michael reactions of isobutylaldehyde with maleimides. Tetrahedron 2010, 66, 8928–8932. [Google Scholar] [CrossRef]
- Yu, F.; Jin, Z.; Huang, H.; Ye, T.; Liang, X.; Ye, J. A highly efficient asymmetric Michael addition of α,α-disubstituted aldehydes to maleimides catalyzed by primary amine thiourea salt. Org. Biomol. Chem. 2010, 8, 4767–4774. [Google Scholar] [CrossRef]
- Miura, T.; Masuda, A.; Ina, M.; Nakashima, K.; Nishida, S.; Tada, N.; Itoh, A. Asymmetric Michael reactions of α,α-disubstituted aldehydes with maleimides using a primary amine thiourea organocatalyst. Tetrahedron Asymmetry 2011, 22, 1605–1609. [Google Scholar] [CrossRef]
- Miura, T.; Nishida, S.; Masuda, A.; Tada, N.; Itoh, A. Asymmetric Michael additions of aldehydes to maleimides using a recyclable fluorous thiourea organocatalyst. Tetrahedron Lett. 2011, 52, 4158–4160. [Google Scholar] [CrossRef]
- Ma, Z.-W.; Liu, Y.-X.; Li, P.-L.; Ren, H.; Zhu, Y.; Tao, J.-C. A highly efficient large-scale asymmetric Michael addition of isobutyraldehyde to maleimides promoted by a novel multifunctional thiourea. Tetrahedron Asymmetry 2011, 22, 1740–1748. [Google Scholar] [CrossRef]
- Song, Z.T.; Zhang, T.; Du, H.L.; Ma, Z.W.; Zhang, C.H.; Tao, J.C. Highly enantioselective Michael addition promoted by a new diterpene-derived bifunctional thiourea catalyst: A doubly stereocontrolled approach to chiral succinimide derivatives. Chirality 2014, 26, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-W.; Liu, X.-F.; Liu, J.-T.; Liu, Z.-J.; Tao, J.-C. Highly enantioselective Michael addition of α,α-disubstituted aldehydes to maleimides catalyzed by new primary amine-squaramide bifunctional organocatalysts. Tetrahedron Lett. 2017, 58, 4487–4490. [Google Scholar] [CrossRef]
- Durmaz, M.; Sirit, A. Calixarene-based chiral primary amine thiourea promoted highly enantioselective asymmetric Michael reactions of α,α-disubstituted aldehydes with maleimides. Tetrahedron Asymmetry 2013, 24, 1443–1448. [Google Scholar] [CrossRef]
- De Simone, N.A.; Meninno, S.; Talotta, C.; Gaeta, C.; Neri, P.; Lattanzi, A. Solvent-free enantioselective Michael reactions catalyzed by a calixarene-based primary amine thiourea. J. Org. Chem. 2018, 83, 10318–10325. [Google Scholar] [CrossRef]
- Nugent, T.C.; Sadiq, A.; Bibi, A.; Heine, T.; Zeonjuk, L.L.; Vankova, N.; Bassil, B.S. Noncovalent bifunctional organocatalysts: Powerful tools for contiguous quaternary-tertiary stereogenic carbon formation, scope, and origin of enantioselectivity. Chem. Eur. J. 2012, 18, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, C.G. An asymmetric Michael addition of α,α-disubstituted aldehydes to maleimides leading to a one-pot enantioselective synthesis of lactones catalyzed by amino acids. Org. Lett. 2013, 15, 2406–2409. [Google Scholar] [CrossRef]
- Avila-Ortiz, C.G.; Diaz-Corona, L.; Jimenez-Gonzalez, E.; Juaristi, E. Asymmetric Michael addition organocatalyzed by α,β-dipeptides under solvent-free reaction conditions. Molecules 2017, 22, 1328. [Google Scholar] [CrossRef]
- Muramulla, S.; Ma, J.-A.; Zhao, J.C.-G. Michael addition of ketones and aldehydes to maleimides catalyzed by modularly designed organocatalysts. Adv. Synth. Catal. 2013, 355, 1260–1264. [Google Scholar] [CrossRef]
- Yang, W.; Jiang, K.-Z.; Lu, X.; Yang, H.-M.; Li, L.; Lu, Y.; Xu, L.-W. Molecular assembly of an achiral phosphine and a chiral primary amine: A highly efficient supramolecular catalyst for the enantioselective Michael reaction of aldehydes with maleimides. Chem. Asian J. 2013, 8, 1182–1190. [Google Scholar] [CrossRef]
- Nakashima, K.; Kawada, M.; Hirashima, S.-I.; Kosugi, A.; Kato, M.; Yoshida, A.; Koseki, Y.; Miura, T. Stereoselective conjugate addition of carbonyl compounds to maleimides using a diaminomethyleneindenedione organocatalyst. Tetrahedron Asymmetry 2016, 27, 888–895. [Google Scholar] [CrossRef]
- Kochetkov, S.V.; Kucherenko, A.S.; Zlotin, S.G. Asymmetric Michael addition of aldehydes to maleimides in primary amine-based aqueous ionic liquid-supported recyclable catalytic system. Mendeleev Commun. 2017, 27, 473–475. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Nájera, C. Enantioselective Michael addition of α,α-disubstituted aldehydes to maleimides organocatalyzed by chiral primary amine-guanidines. Tetrahedron Asymmetry 2012, 23, 1625–1627. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Gómez-Bengoa, E.; Nájera, C. Enantioselective synthesis of succinimides by Michael addition of aldehydes to maleimides organocatalyzed by chiral primary amine-guanidines. Eur. J. Org. Chem. 2013, 23, 5085–5092. [Google Scholar] [CrossRef]
- Vizcaíno-Milla, P.; Sansano, J.M.; Nájera, C.; Fiser, B.; Gómez-Bengoa, E. Primary amine-2-aminopyrimidine chiral organocatalysts for the enantioselective conjugate addition of branched aldehydes to maleimides. Synthesis 2015, 47, 2199–2206. [Google Scholar] [CrossRef]
- Fernandes, T.d.A.; Vizcaino-Milla, P.; Ravasco, J.M.J.M.; Ortega-Martinez, A.; Sansano, J.M.; Nájera, C.; Costa, P.R.R.; Fiser, B.; Gomez-Bengoa, E. Bifunctional primary amine 2-aminobenzimidazole organocatalyst anchored to trans-cyclohexane-1,2-diamine in enantioselective conjugate additions of aldehydes. Tetrahedron Asymmetry 2016, 27, 118–122. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Solvent-dependent enantioswitching in the Michael addition of α,α-disubstituted aldehydes to maleimides organocatalyzed by mono-N-BOC-protected cyclohexa-1,2-diamines. Tetrahedron Asymmetry 2014, 25, 1091–1094. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Fiser, B.; Gómez-Bengoa, E.; Chinchilla, R. Solvent-induced reversal of enantioselectivity in the synthesis of succinimides by the addition of aldehydes to maleimides catalysed by carbamate-monoprotected 1,2-diamines. Eur. J. Org. Chem. 2015, 1218–1225. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides in deep eutectic solvents. Tetrahedron Asymmetry 2017, 28, 302–306. [Google Scholar] [CrossRef]
- Szőllősi, G.; Kozma, V. Design of heterogeneous organocatalyst for the asymmetric Michael addition of aldehydes to maleimides. ChemCatChem 2018. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Michael addition of aldehydes to maleimides organocatalyzed by chiral 1,2-diamines: an experimental and theoretical study. Tetrahedron Asymmetry 2013, 24, 1531–1535. [Google Scholar] [CrossRef]
- Martínez-Guillén, J.R.; Flores-Ferrándiz, J.; Gómez, C.; Gómez-Bengoa, E.; Chinchilla, R. Asymmetric conjugate addition of α,α-disubstituted aldehydes to nitroalkenes organocatalyzed by chiral monosalicylamides from trans-cyclohexane-1,2-diamines. Molecules 2018, 23, 141. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 15 and 19 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Additive | Solvent | Yield (%) a | ee (%) b |
|---|---|---|---|---|---|
| 1 | 15 | - | PhMe | 98 | 94 (R) |
| 2 | 15 | - | CH2Cl2 | 96 | 74 (R) |
| 3 | 15 | - | CHCl3 | 98 | 75 (R) |
| 4 | 15 | - | DMF/H2O c | 91 | 79 (S) |
| 5 | 15 | PhCO2H | PhMe | 94 | 77 (R) |
| 6 | 15 | LiCl | PhMe | 88 | 85 (R) |
| 7 | 15 | DMAP | PhMe | 45 | 83 (R) |
| 8 | 19 | - | PhMe | 98 | 87 (S) |

| Entry | Aldehyde | Maleimide | Michael Adduct | ||||
|---|---|---|---|---|---|---|---|
| R1, R2 | No. | R3 | No. | No. | Yield (%) a | eeb (%) b | |
| 1 | Me,Me | 16a | Ph | 17a | (R)-18aa | 98 | 94 |
| 2 | Me,Me | 16a | 4-MeC6H4 | 17b | (R)-18ab | 87 | 88 |
| 3 | Me,Me | 16a | 4-MeOC6H4 | 17c | (R)-18ac | 77 | 89 |
| 4 | Me,Me | 16a | 4-ClC6H4 | 17d | (R)-18ad | 92 | 88 |
| 5 | Me,Me | 16a | 4-AcC6H4 | 17e | (R)-18ae | 73 | 13 |
| 6 | Me,Me | 16a | 4-O2NC6H4 | 17f | (R)-18af | 78 | 70 |
| 7 | Me,Me | 16a | Me | 17g | (R)-18ag | 91 | 82 |
| 8 | Me,Me | 16a | H | 17h | (R)-18ah | 71 | 56 |
| 9 | -(CH2)4- | 16b | Ph | 17a | (R)-18ba | 93 | 82 |
| 10 | -(CH2)5- | 16c | Ph | 17a | (R)-18ca | 84 | 36 |
| 11 | Me,H | 16d | Ph | 17a | (2S,3R)-,(2R,3R)-18da c | 98 d | 79,89 e |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torregrosa-Chinillach, A.; Moragues, A.; Pérez-Furundarena, H.; Chinchilla, R.; Gómez-Bengoa, E.; Guillena, G. Enantioselective Michael Addition of Aldehydes to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide. Molecules 2018, 23, 3299. https://doi.org/10.3390/molecules23123299
Torregrosa-Chinillach A, Moragues A, Pérez-Furundarena H, Chinchilla R, Gómez-Bengoa E, Guillena G. Enantioselective Michael Addition of Aldehydes to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide. Molecules. 2018; 23(12):3299. https://doi.org/10.3390/molecules23123299
Chicago/Turabian StyleTorregrosa-Chinillach, Alejandro, Adrien Moragues, Haritz Pérez-Furundarena, Rafael Chinchilla, Enrique Gómez-Bengoa, and Gabriela Guillena. 2018. "Enantioselective Michael Addition of Aldehydes to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide" Molecules 23, no. 12: 3299. https://doi.org/10.3390/molecules23123299