
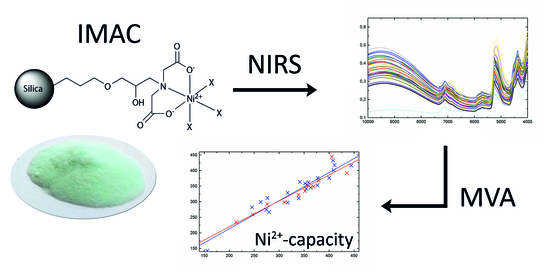
Direct Determination of Ni2+-Capacity of IMAC Materials Using Near-Infrared Spectroscopy

 , ,
, ,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
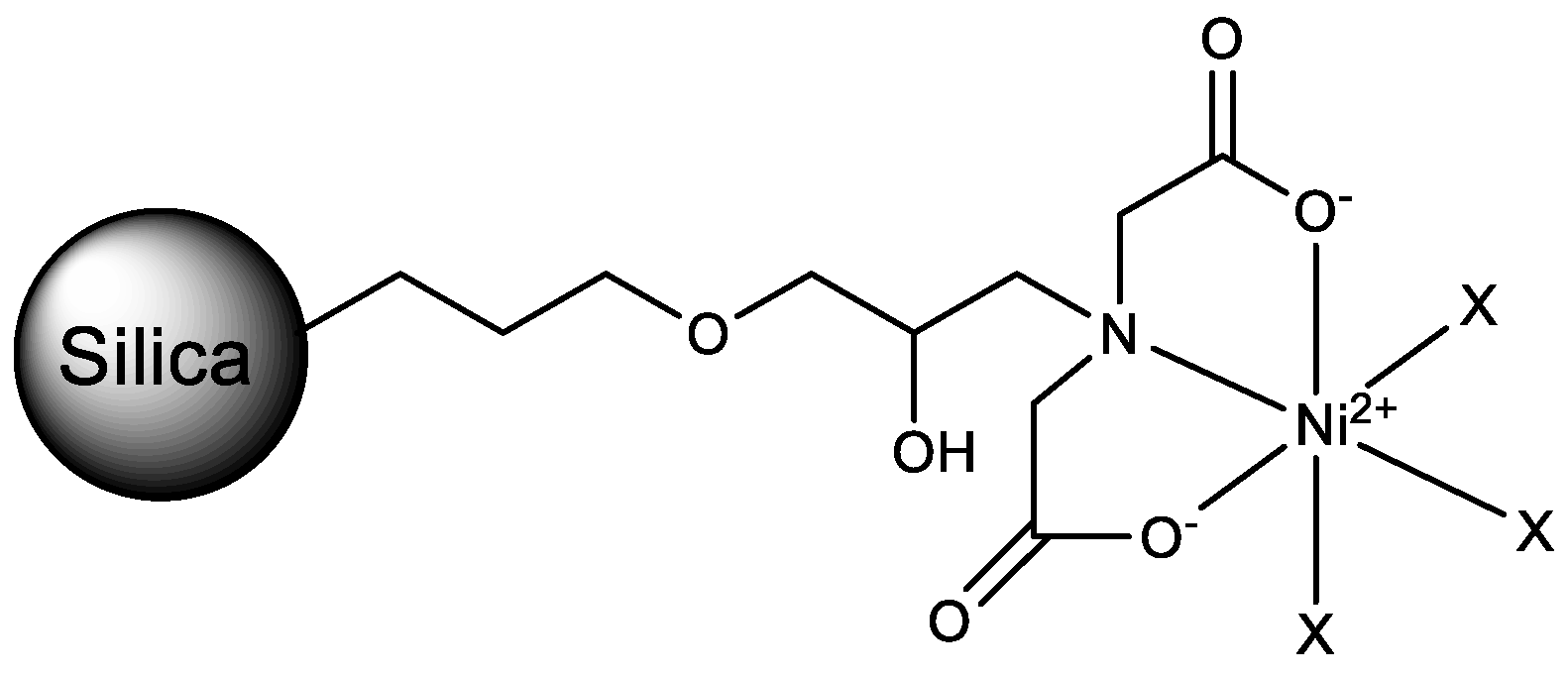
2.1. IMAC Materials
2.2. UV and AAS Method Validation
2.3. Reference Analysis Method Validation
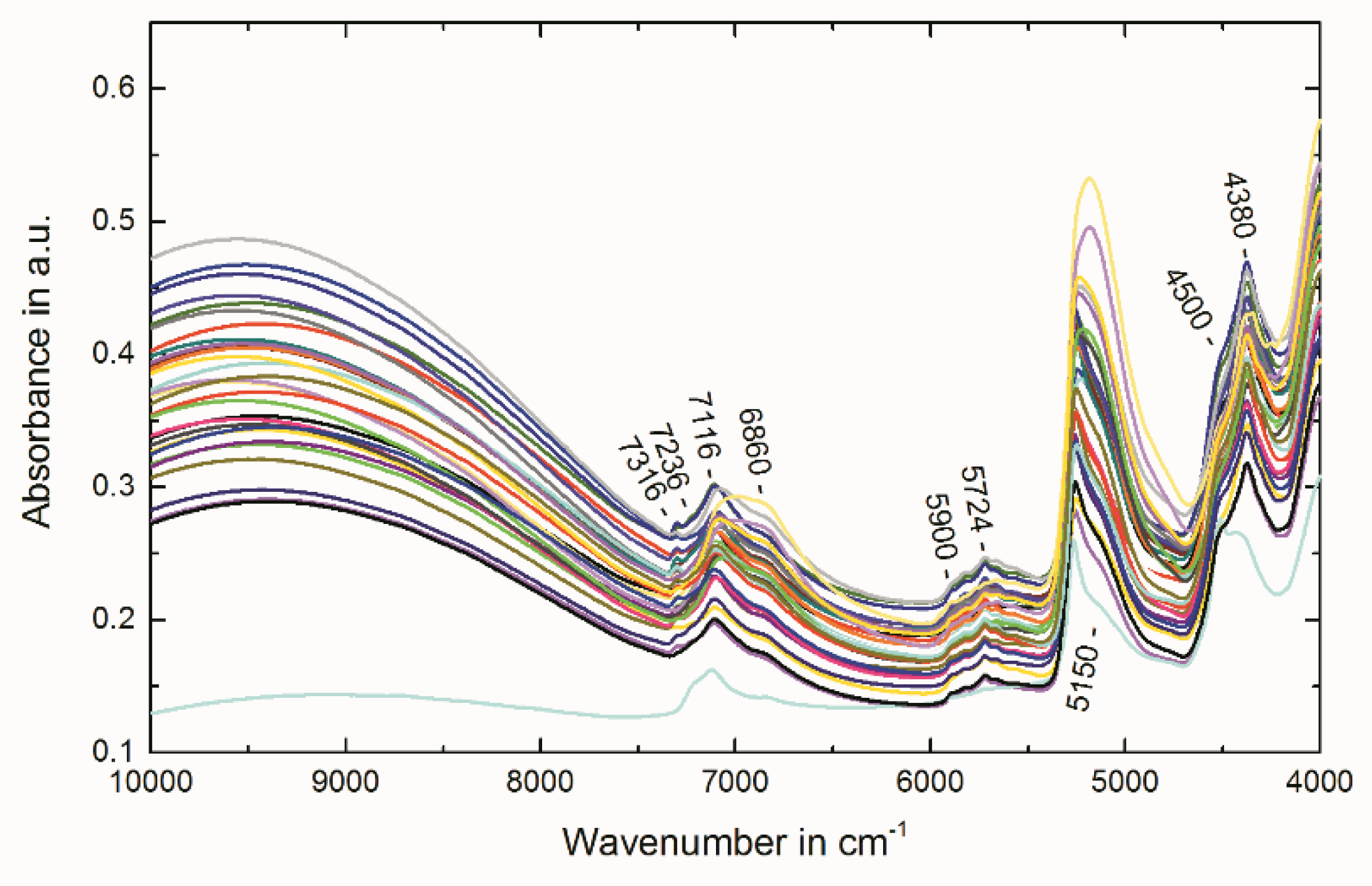
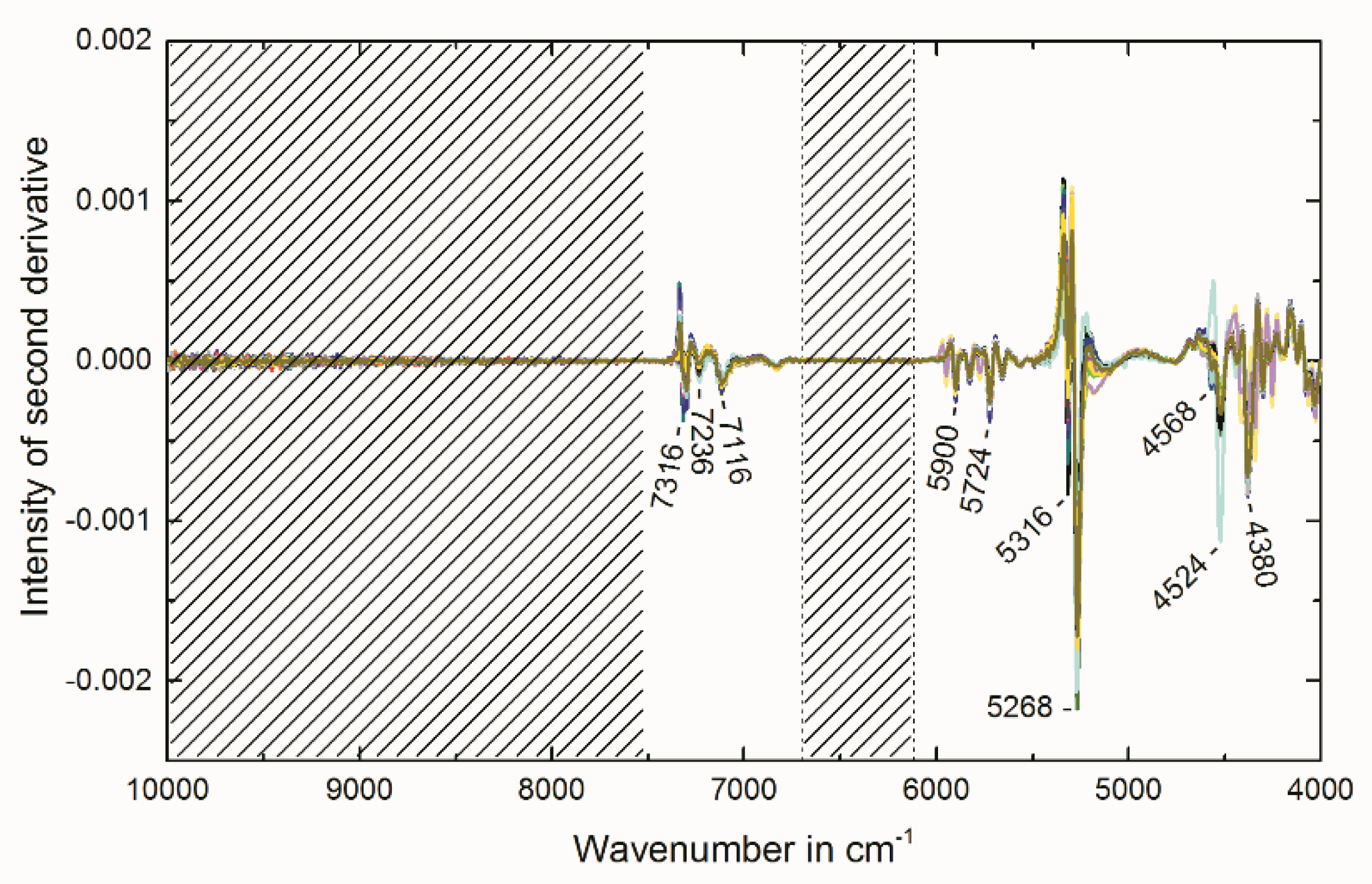
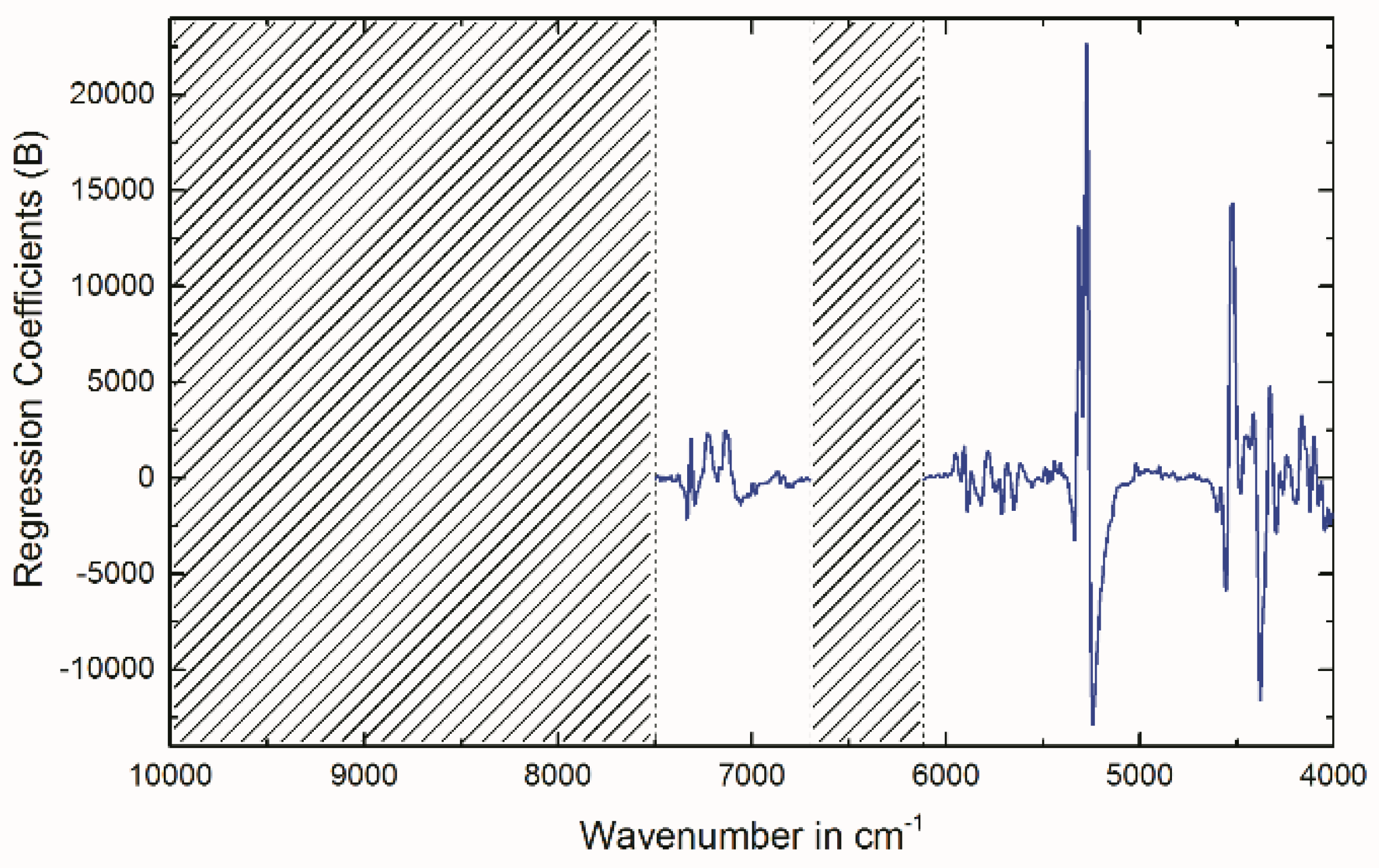
2.4. Multivariate Data Analysis of NIR Spectra
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis
3.2.1. Activation of Silica Gel
3.2.2. Preparation of GLYMO-IDA
3.2.3. Immobilization of GLYMO-IDA on the Silica Gel
3.2.4. Nickel Ion Immobilization
3.3. UV Absorption Spectroscopy
3.4. Atomic Absorption Spectrometry
3.5. Near-Infrared Spectroscopy
3.6. UV and AAS Method Validation
3.7. Reference Analysis Method Validation
3.8. Multivariate Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Porath, J.; Carlsson, J.; Olsson, I.; Belfrage, G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature 1975, 258, 598–599. [Google Scholar] [CrossRef] [PubMed]
- El Rassi, Z.; Horváth, C. Metal chelate-interaction chromatography of proteins with iminodiacetic acid-bonded stationary phases on silica support. J. Chromatogr. A 1986, 359, 241–253. [Google Scholar] [CrossRef]
- Belew, M.; Porath, J. Immobilized metal ion affinity chromatography: Effect of solute structure, ligand density and salt concentration on the retention of peptides. J. Chromatogr. A 1990, 516, 333–354. [Google Scholar] [CrossRef]
- Kato, Y.; Nakamura, K.; Hashimoto, T. High-performance metal chelate affinity chromatography of proteins. J. Chromatogr. A 1986, 354, 511–517. [Google Scholar] [CrossRef]
- Belew, M.; Yip, T.T.; Andersson, L.; Ehrnström, R. High-performance analytical applications of immobilized metal ion affinity chromatography. Anal. Biochem. 1987, 164, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, E. Purification of proteins by imac. Trends Biotechnol. 1985, 3, 1–7. [Google Scholar] [CrossRef]
- Hochuli, E.; Döbeli, H.; Schacher, A. New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. J. Chromatogr. A 1987, 411, 177–184. [Google Scholar] [CrossRef]
- Zachariou, M. Affinity Chromatography: Methods and Protocols; Humana Press: Totowa, ON, Canada, 2008; Volume 421, ISBN 978-1-58829-659-7. [Google Scholar]
- Bornhorst, J.A.; Falke, J.J. Purification of proteins using polyhistidine affinity tags. Methods Enzym. 2000, 326, 245–254. [Google Scholar] [CrossRef]
- Trojer, L.; Stecher, G.; Feuerstein, I.; Lubbad, S.; Bonn, G. Characterisation and evaluation of metal-loaded iminodiacetic acid–silica of different porosity for the selective enrichment of phosphopeptides. J. Chromatogr. A 2005, 1079, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Trojer, L.; Stecher, G.; Feuerstein, I.; Bonn, G. Cu(ii)-loaded iminodiacetic acid-silica particles for protein profiling of human serum samples using surface-enhanced affinity capture: Support porosity effects. Rapid Commun. Mass Spectrom. 2005, 19, 3398–3404. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Suen, S.-Y.; Chen, S.-C.; Tzeng, J.-H. Analysis of protein adsorption on regenerated cellulose-based immobilized copper ion affinity membranes. J. Chromatogr. A 2003, 996, 53–70. [Google Scholar] [CrossRef]
- Burns, D.A.; Ciurczak, E.W. Handbook of Near-Infrared Analysis; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9780849373930. [Google Scholar]
- Ozaki, Y. Near-infrared spectroscopy—Its versatility in analytical chemistry. Anal. Sci. 2012, 28, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Siesler, H.W.; Ozaki, Y.; Satoshi, K.; Michael, H.H. Near-Infrared Spectroscopy: Principles, Instruments, Applications, 2nd ed.; Wiley-VCH Verlag: Weinheim, Germany, 2002; ISBN 3-527-30149-6. [Google Scholar]
- Chalmers, J.M.; Griffiths, P.R. Handbook of Vibrational Spectroscopy, 1st ed.; John Wiley & Sons: Chichester, UK, 2001; ISBN 0471988472. [Google Scholar]
- Kessler, W. Multivariate Datenanalyse für die pharma-, bio- und prozessanalytik: Ein lehrbuch; Wiley-VCH Verlag: Weinheim, Germany, 2007; ISBN 978-3-527-31262-7. [Google Scholar]
- Anspach, F.B. Silica-based metal chelate affinity sorbents i. Preparation and characterization of iminodiacetic acid affinity sorbents prepared via different immobilization techniques. J. Chromatogr. A 1994, 672, 35–49. [Google Scholar] [CrossRef]
- Pearson, K. Note on regression and inheritance in the case of two parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar] [CrossRef]
- Christy, A.A. New insights into the surface functionalities and adsorption evolution of water molecules on silica gel surface: A study by second derivative near infrared spectroscopy. Vib. Spectrosc. 2010, 54, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Blakeley, R.T.; Dixon, N.E.; Zerner, B. Jack bean urease vii. Light scattering and nickel(ii) spectrum thiolate → nickel(ii) charge-transfer peaks in the spectrum of the β-mercaptoethanol-urease complex. Biochim. Et Biophys. Acta (BBA)—Protein Struct. Mol. Enzym. 1983, 744, 219–229. [Google Scholar] [CrossRef]
- Jørgensen, C.K. Comparative crystal field studies of some ligands and the lowest singlet state of paramagnetic nickel(ii) complexes. Acta Chem. Scand. 1955, 9, 1362–1377. [Google Scholar] [CrossRef]
- Christy, A.A. The nature of silanol groups on the surfaces of silica, modified silica and some silica based materials. Adv. Mater. Res. 2014, 998–999, 3–10. [Google Scholar] [CrossRef]
- Anderson, J.H.; Wickersheim, K.A. Near infrared characterization of water and hydroxyl groups on silica surfaces. Surf. Sci. 1964, 2, 252–260. [Google Scholar] [CrossRef]
- Klier, K.; Shen, J.H.; Zettlemoyer, A.C. Water on silica and silicate surfaces. I. Partially hydrophobic silicas. J. Phys. Chem. 1973, 77, 1458–1465. [Google Scholar] [CrossRef]
- Workman, J.; Weyer, L. Practical Guide and Spectral Atlas for Interpretive Near-infrared Spectroscopy, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012; ISBN 9781439875261. [Google Scholar]
- Guidance for Industry: Q2b Validation of Analytical Procedures: Methodology. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073384.pdf (accessed on 24 May 2018).
- Shabir, G.A. A practical approach to validation of hplc methods under current good manufacturing practices. J. Valid. Technol. 2004, 10, 210–218. [Google Scholar]
- Kromidas, S. Handbuch Validierung in der analytik; Wiley-VCH: Weinheim, Germany, 2011; ISBN 9783527329380. [Google Scholar]
- Geladi, P.; MacDougall, D.; Martens, H. Linearization and scatter-correction for near-infrared reflectance spectra of meat. Appl. Spectrosc. 1985, 39, 491–500. [Google Scholar] [CrossRef]
- Barnes, R.J.; Dhanoa, M.S.; Lister, S.J. Standard normal variate transformation and de-trending of near-infrared diffuse reflectance spectra. Appl. Spectrosc. 1989, 43, 772–777. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Haaland, D.M.; Thomas, E.V. Partial least-squares methods for spectral analyses. 1. Relation to other quantitative calibration methods and the extraction of qualitative information. Anal. Chem. 1988, 60, 1193–1202. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CV | TSV | |
|---|---|---|
| RMSEC in µmol/g | 21 | 21 |
| RMSECV or RMSEP in µmol/g | 24 | 22 |
| R2calibration | 0.90 | 0.90 |
| R2validation | 0.88 | 0.88 |
| % RSDPLSR | 8.0 | 7.5 |
| Factor | 2 | 2 |
| Calibration range in µmol/g | 156–444 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirchler, C.G.; Henn, R.; Modl, J.; Münzker, F.; Baumgartner, T.H.; Meischl, F.; Kehle, A.; Bonn, G.K.; Huck, C.W. Direct Determination of Ni2+-Capacity of IMAC Materials Using Near-Infrared Spectroscopy. Molecules 2018, 23, 3072. https://doi.org/10.3390/molecules23123072
Kirchler CG, Henn R, Modl J, Münzker F, Baumgartner TH, Meischl F, Kehle A, Bonn GK, Huck CW. Direct Determination of Ni2+-Capacity of IMAC Materials Using Near-Infrared Spectroscopy. Molecules. 2018; 23(12):3072. https://doi.org/10.3390/molecules23123072
Chicago/Turabian StyleKirchler, Christian G., Raphael Henn, Julia Modl, Felix Münzker, Tanja H. Baumgartner, Florian Meischl, Alexander Kehle, Günther K. Bonn, and Christian W. Huck. 2018. "Direct Determination of Ni2+-Capacity of IMAC Materials Using Near-Infrared Spectroscopy" Molecules 23, no. 12: 3072. https://doi.org/10.3390/molecules23123072