Proteome Investigation of Rat Lungs Subjected to Ex Vivo Perfusion (EVLP)

 ,
,  , , , , , , and
, , , , , , and 
Abstract
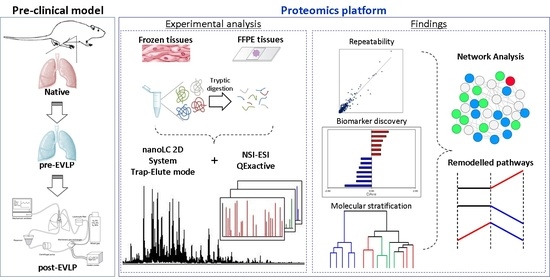
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
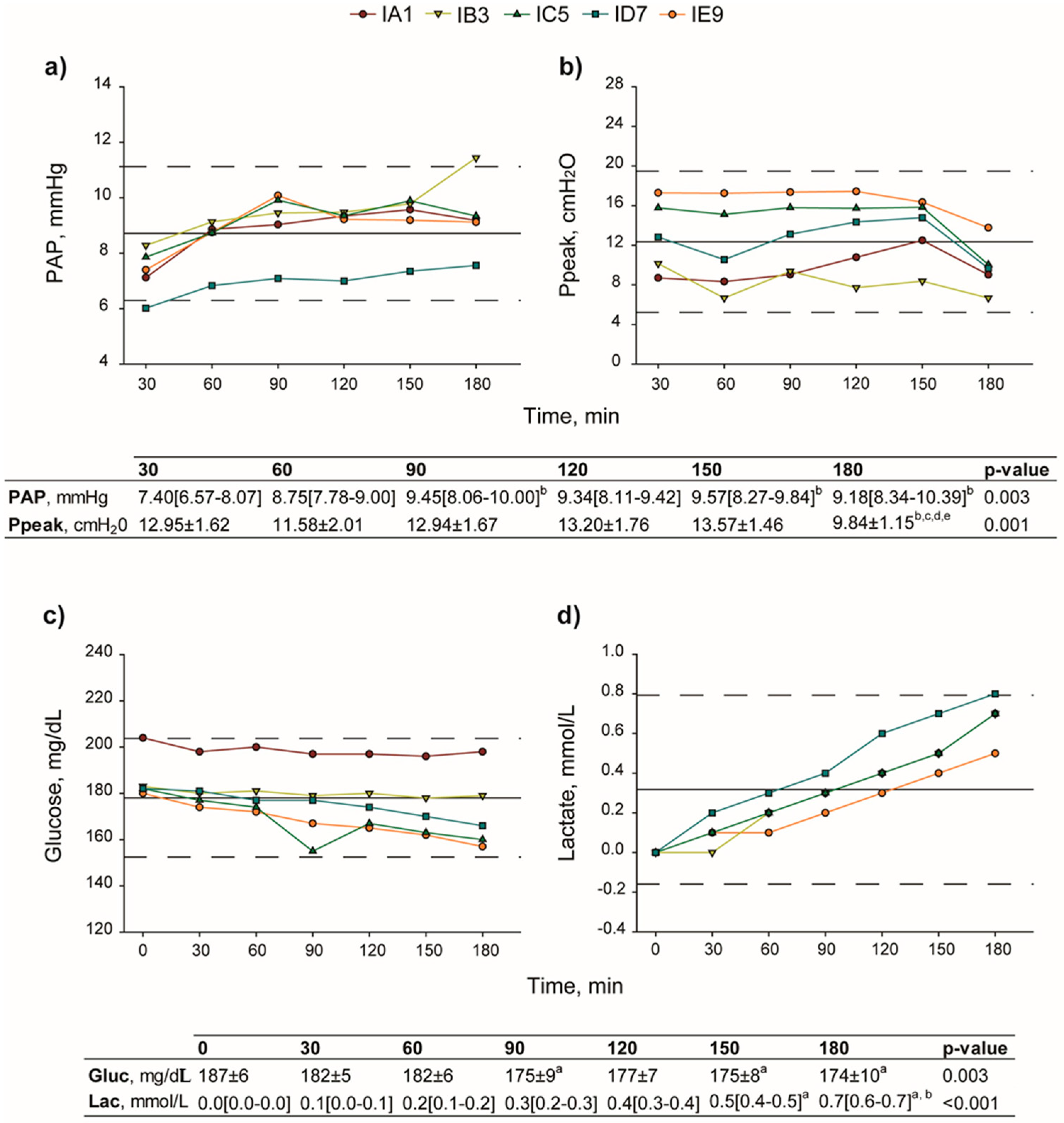
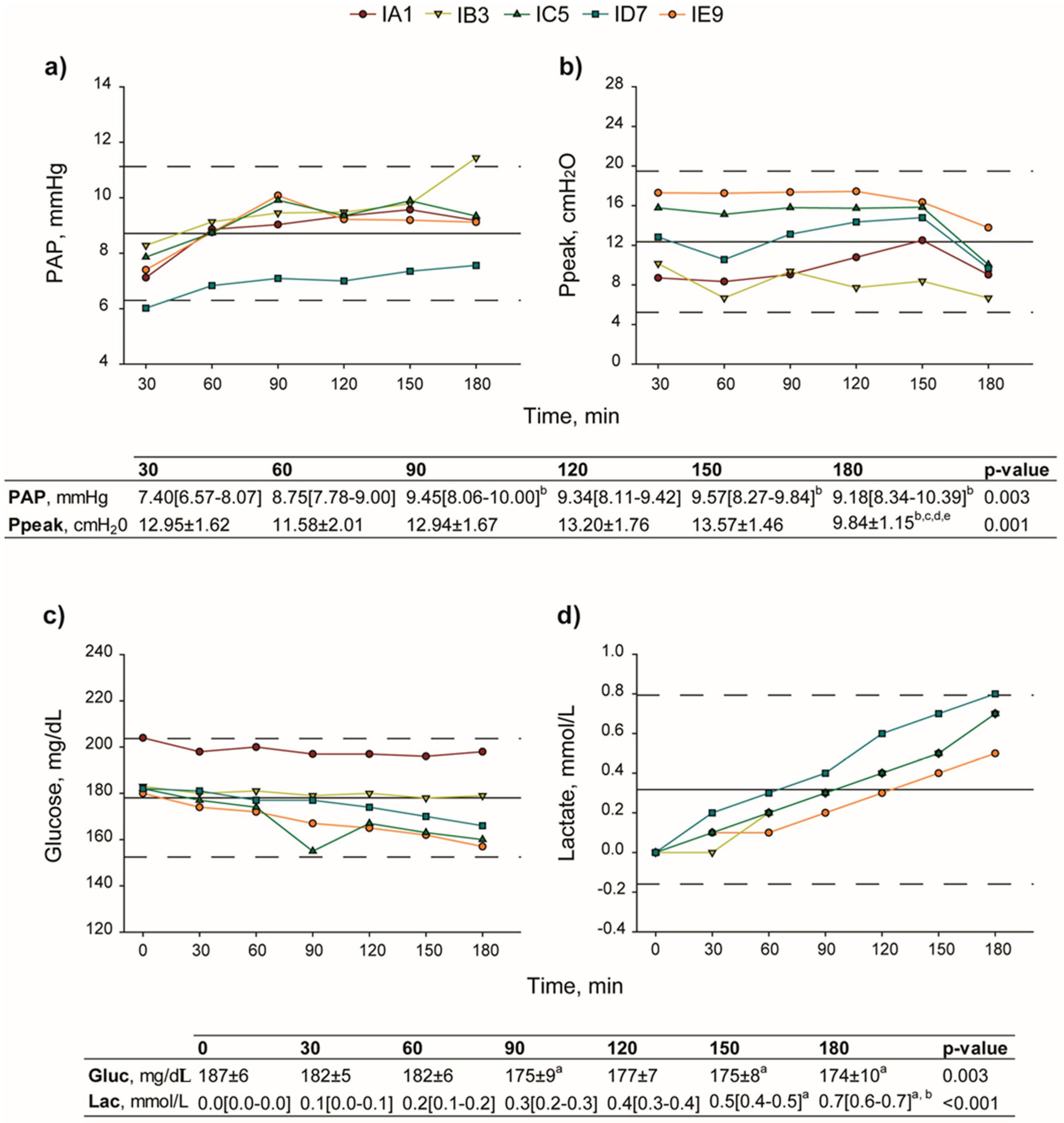
2.1. Surgical Procedures and Physiological Assessments
2.2. Protein Profiles of Frozen Tissue Lung Lobes
2.3. Data Handling
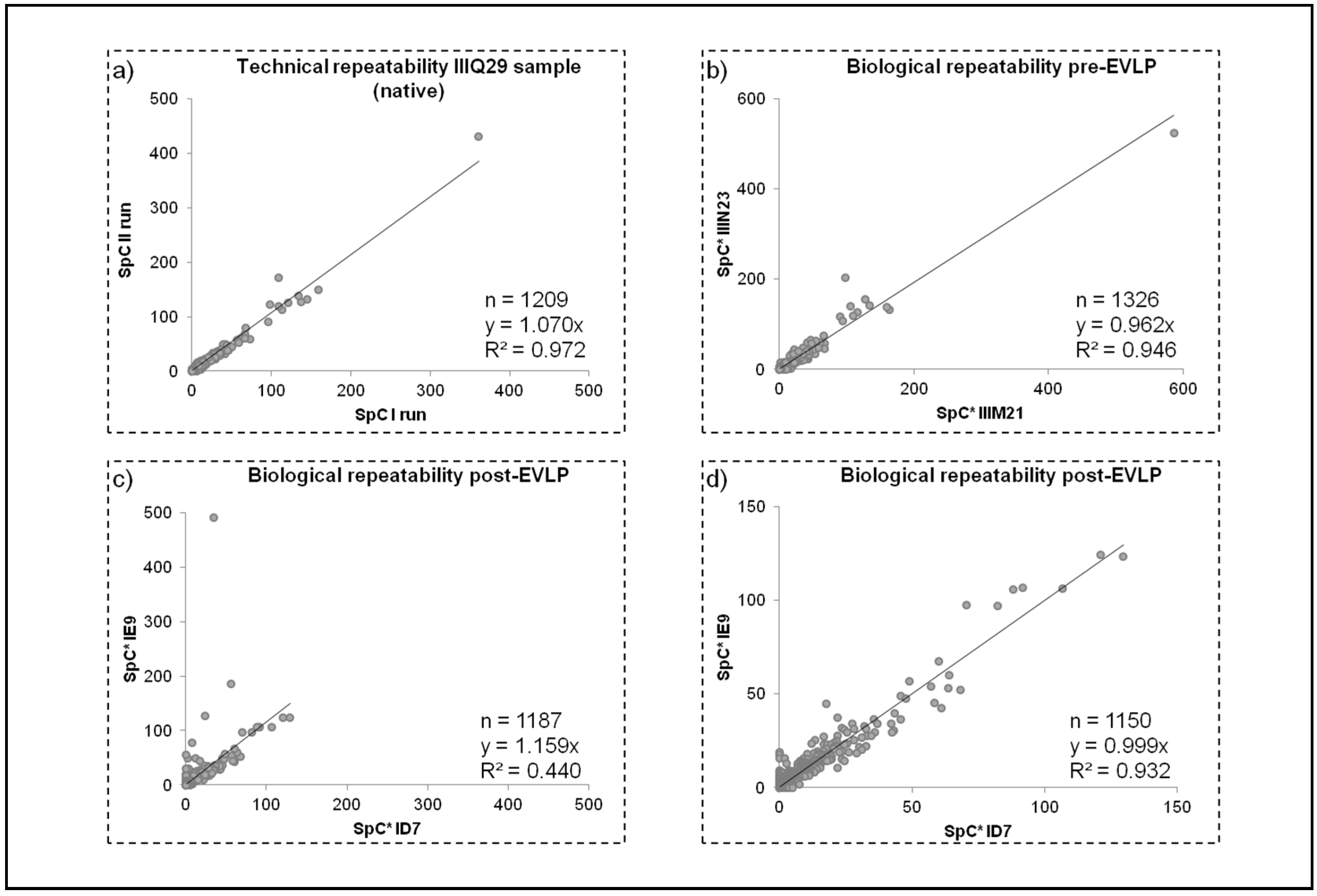
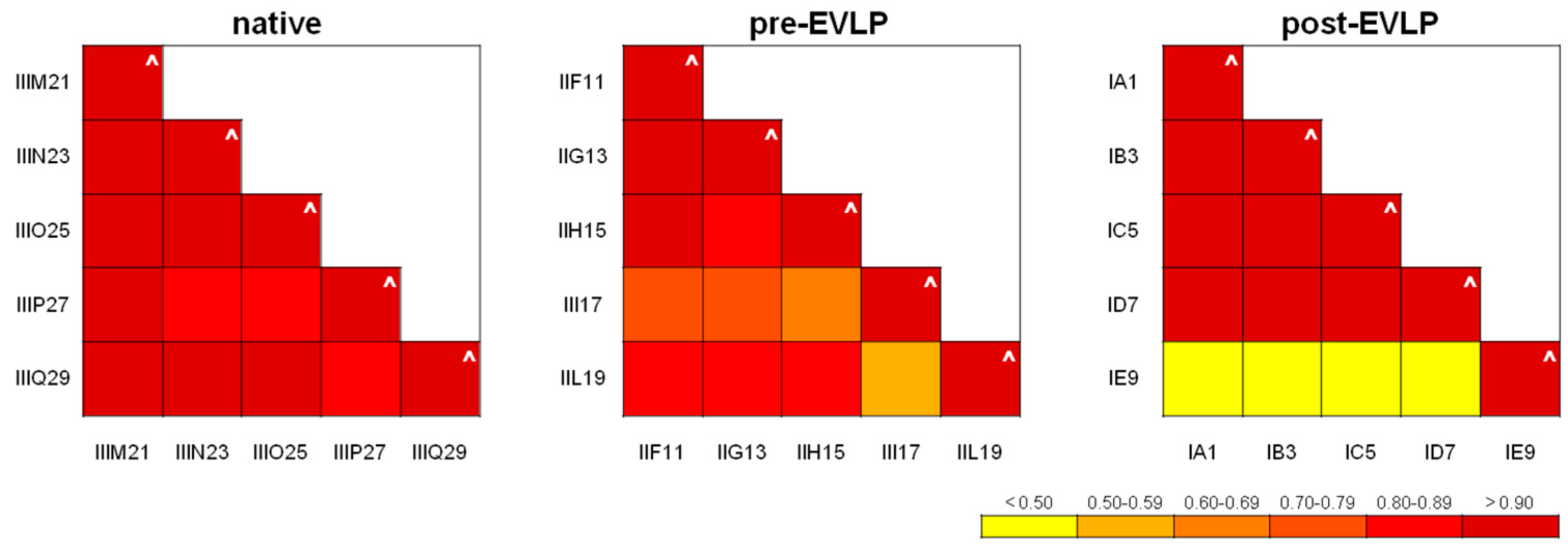
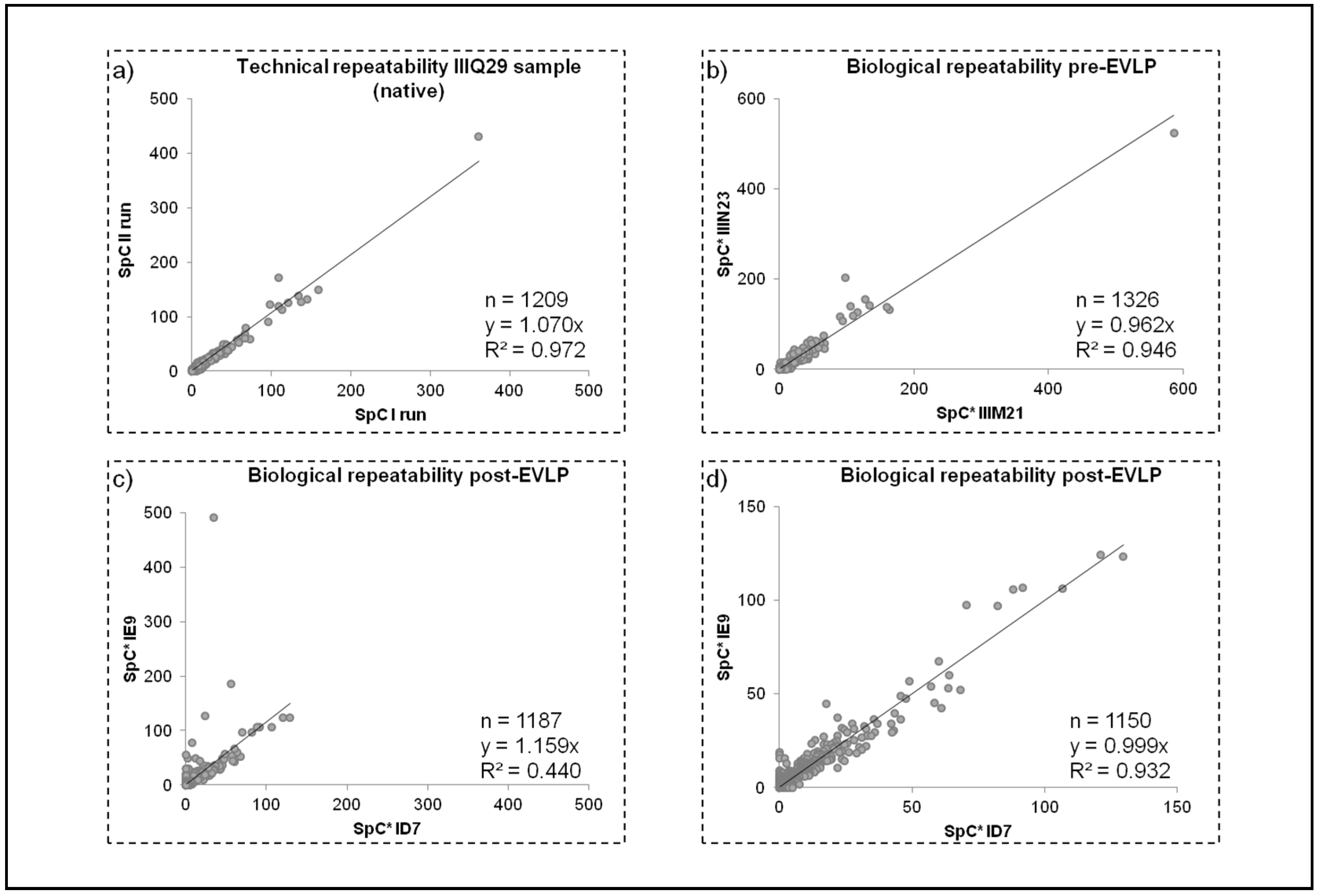
2.3.1. Repeatability
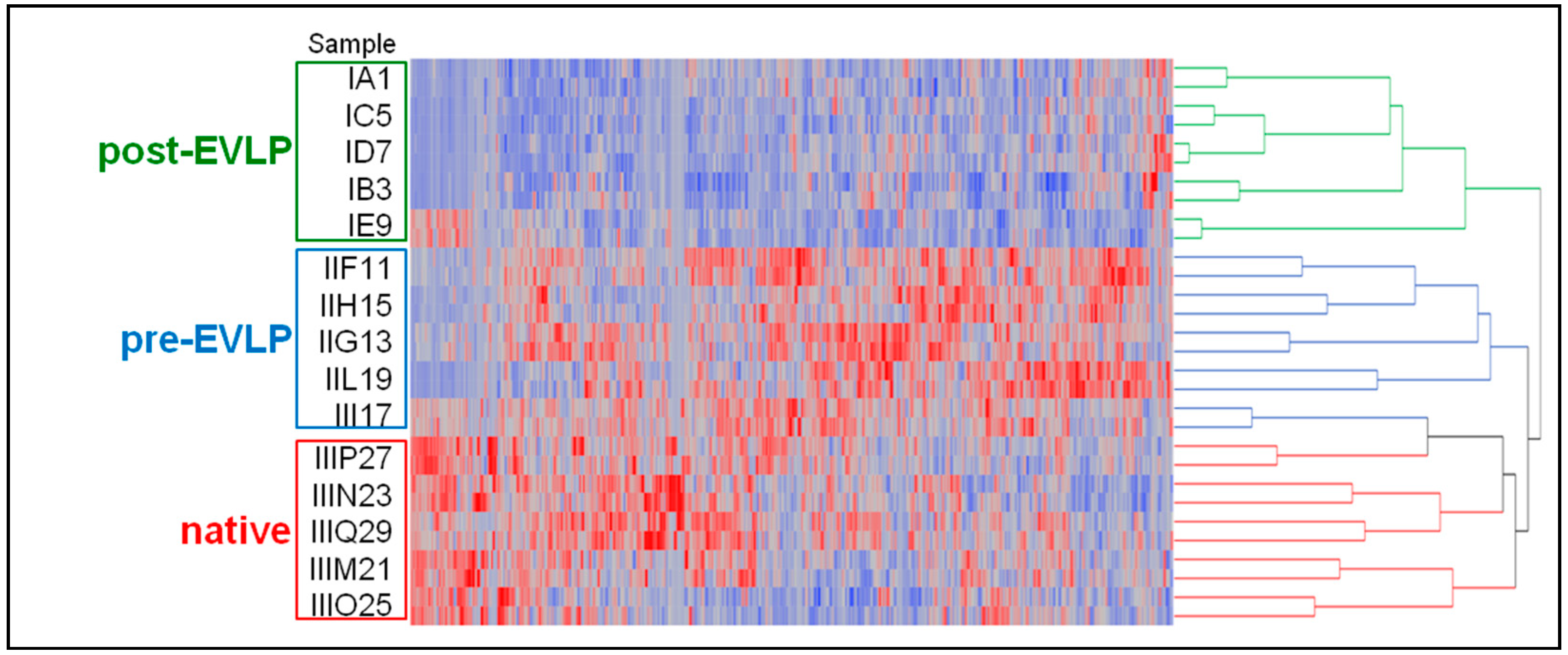
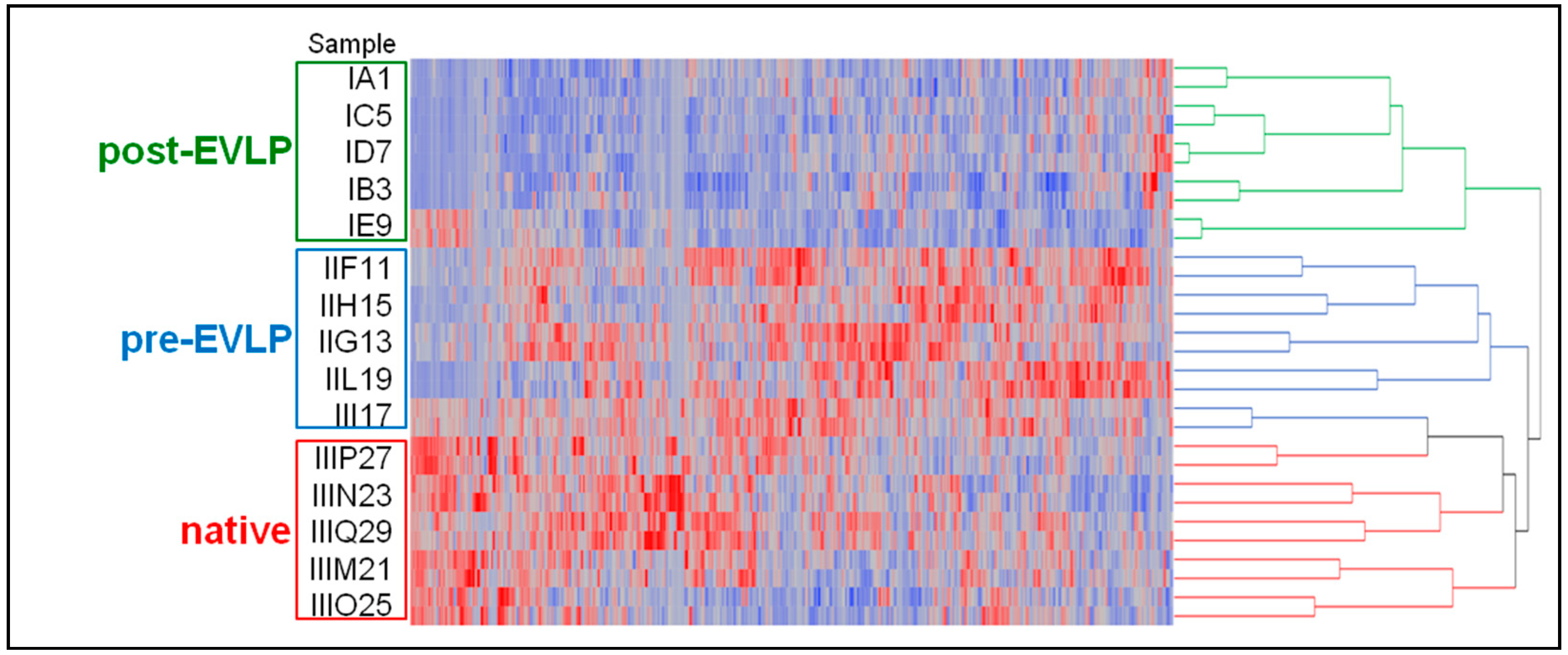
2.3.2. Clustering and Linear Discriminant Analyses
2.3.3. Differential Analysis
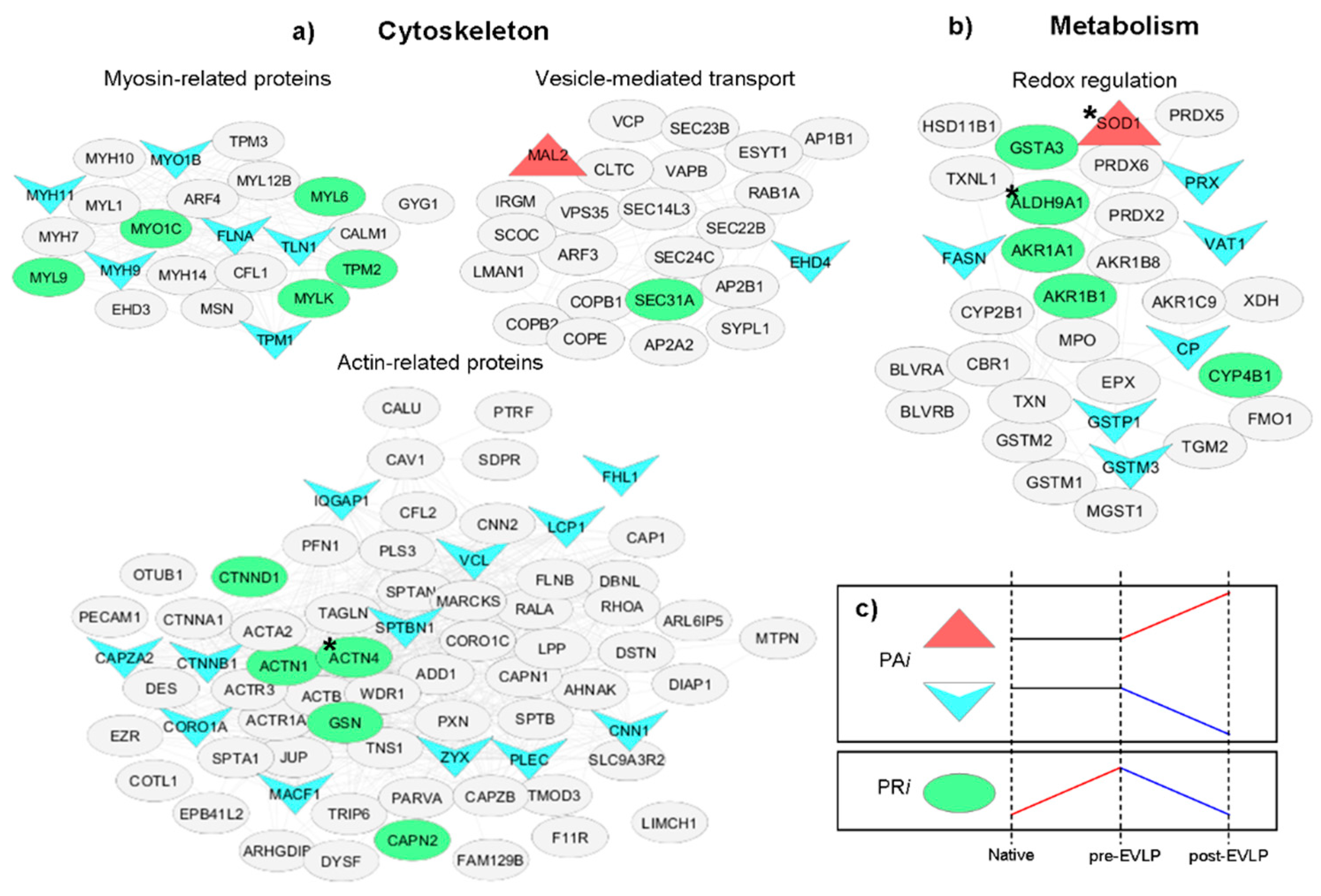
2.4. Network Analysis
2.5. Comparison between Frozen and FFPE Tissue Lung Lobes
3. Discussion
4. Materials and Methods
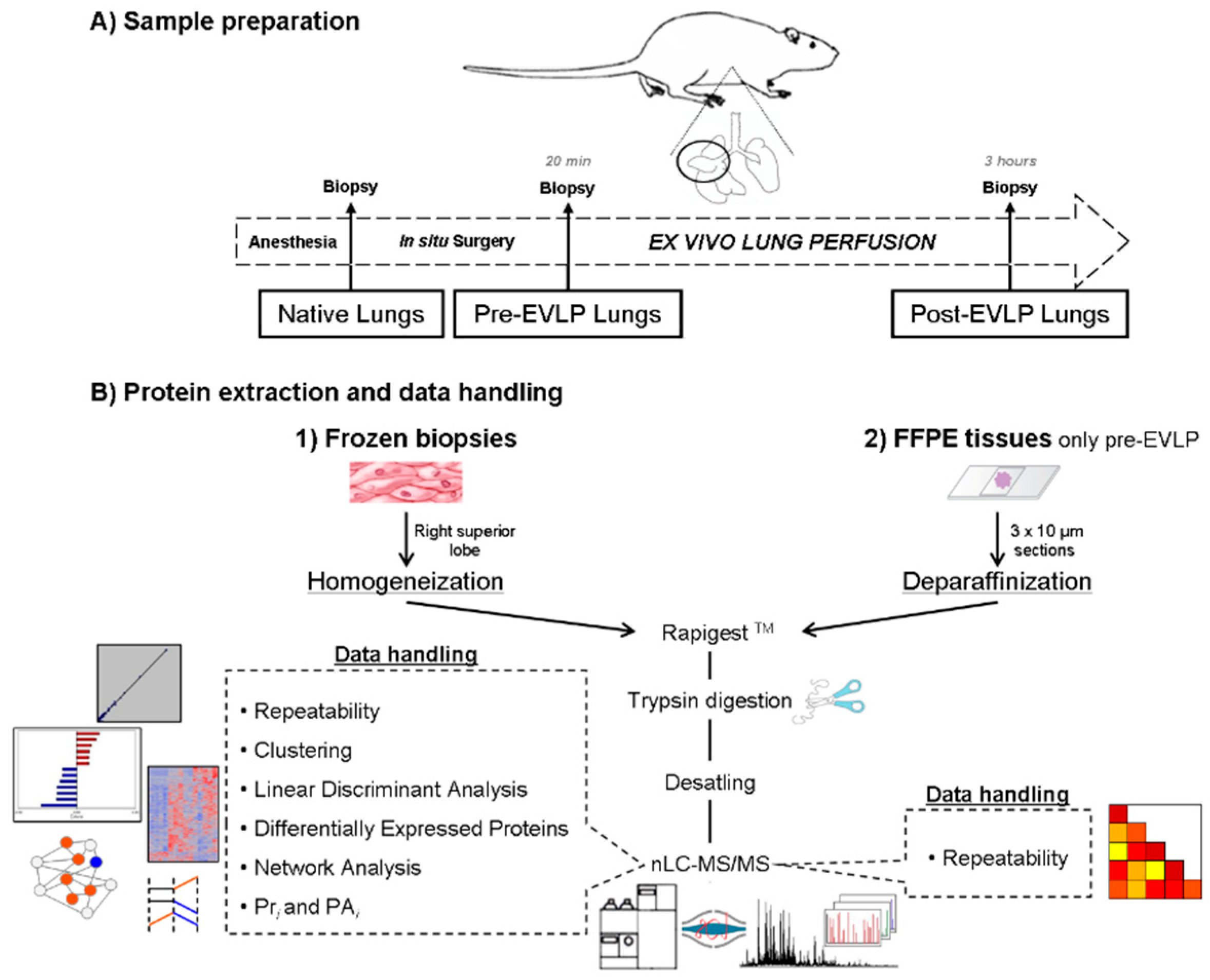
4.1. Experimental Proteomic Pipeline
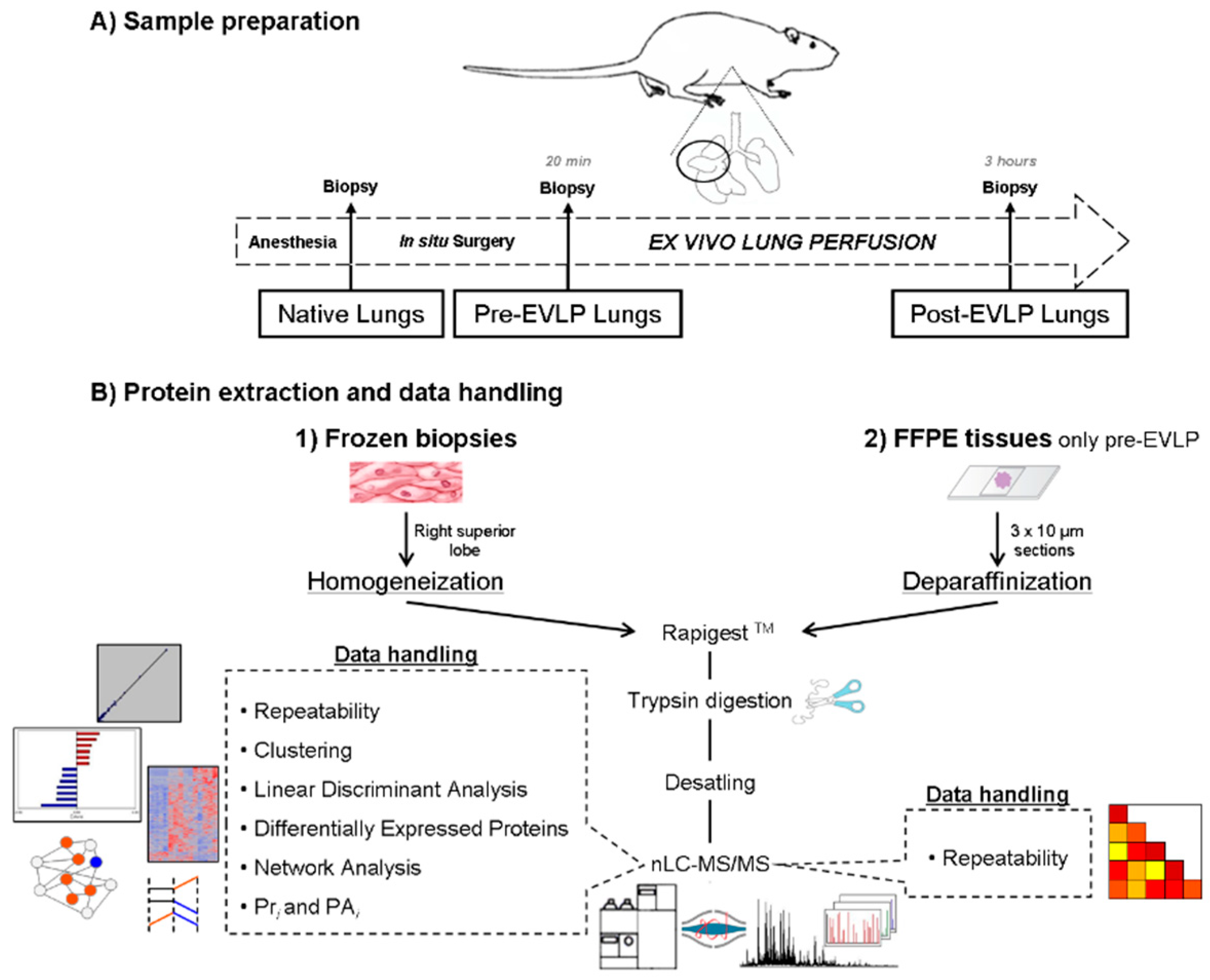
4.2. Surgical Procedure and Frozen and FFPE Tissue Collection
4.2.1. Animals and Experimental Groups
4.2.2. Ex Vivo Lung Perfusion (EVLP)
4.2.3. Lung Function
- Pulmonary artery and airway pressure assessment.PAP and Ppeak were continuously recorded, and digitally stored via an amplifier (data acquisition software Colligo Elekton, Milan, Italy).
- Gas analysis and determination of glucose and lactate concentration.Gas analysis, glucose, and lactate concentration evaluation were performed on perfusate samples using a gas analyzer (ABL 800 FLEX, A. De Mori Strumenti, Milan, Italy).
- Lung edema index.At the end of the procedure, the right superior lobe was weighed (KERN & SOHN GmbH, Balingen, Germany), before (wet) and after (dry) storage at 50 °C for 24 h (LTE Scientific, Oldham, UK). Wet-to-dry ratio (W/D) was then calculated and used as index for pulmonary edema.
4.3. Protein Extraction
4.3.1. Frozen Lobes
4.3.2. Formalin-Fixed Paraffin Embedded (FFPE) tissues
4.4. Proteomic Analysis
4.4.1. Liquid Chromatography
4.4.2. Mass Spectrometry
4.4.3. Data Handling
4.4.4. Label-Free Differential Analysis
4.4.5. Hierarchical Clustering and Linear Discriminant Analysis
4.4.6. Network Analysis
4.4.7. Proteome Recovery and Proteome Activation Index
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Briot, R.; Gennai, S.; Maignan, M.; Souilamas, R.; Pison, C. Ex vivo lung graft perfusion. Anaesth. Crit. Care Pain Med. 2016, 35, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, A.S.I.; Dark, J.H.; Fisher, A.J. Ex vivo lung perfusion in clinical lung transplantation—State of the art. Eur. J. Cardio-Thorac. Surg. 2014, 46, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.F.; Sabashnikov, A.; Patil, N.P.; Zeriouh, M.; Mohite, P.N.; Zych, B.; Saez, D.G.; Schmack, B.; Ruhparwar, A.; Dohmen, P.M.; et al. Ex vivo lung perfusion—State of the art in lung donor pool expansion. Med. Sci. Monit. Basic Res. 2015, 21, 9–14. [Google Scholar] [PubMed]
- Levin, K.; Kotecha, S.; Westall, G.; Snell, G. How can we improve the quality of transplantable lungs? Expert Rev. Respir. Med. 2016, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Valenza, F.; Rosso, L.; Coppola, S.; Froio, S.; Palleschi, A.; Tosi, D.; Mendogni, P.; Salice, V.; Ruggeri, G.M.; Fumagalli, J.; et al. Ex vivo lung perfusion to improve donor lung function and increase the number of organs available for transplantation. Transpl. Int. 2014, 27, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raemdonck, D.; Neyrinck, A.; Cypel, M.; Keshavjee, S. Ex-vivo lung perfusion. Transpl. Int. 2015, 28, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Makdisi, G.; Makdisi, T.; Jarmi, T.; Caldeira, C.C. Ex vivo lung perfusion review of a revolutionary technology. Ann. Transl. Med. 2017, 5, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Friend, S.H. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat. Rev. Clin. Oncol. 2011, 8, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.L.; Longo, D.L. Precision medicine--personalized, problematic, and promising. N. Engl. J. Med. 2015, 372, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Thompson, K.E.; Tabary, O.; le Rouzic, P.; Guillot, L. Translating the genetics of cystic fibrosis to personalized medicine. Transl Res. 2016, 168, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Kaminski, N. Personalized medicine: Applying ‘omics’ to lung fibrosis. Biomark Med. 2012, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Bai, X.Y.; Li, Y.; Bai, J.Z.; Yuan, L.R.; Ren, S.A.; Hu, X.Y.; Zhang, X.R.; Yu, B.F.; Guo, R.; et al. Label-free LC-MS/MS shotgun proteomics to investigate the anti-inflammatory effect of rCC16. Mol. Med. Rep. 2016, 1, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; De Palma, A.; Benazzi, L.; Riccio, A.M.; Canonica, G.W.; Mauri, P. Biomarker discovery in asthma and COPD by proteomic approaches. Proteomics Clin. Appl. 2014, 8, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Gupta, V.; Balch, W.E.; Yates, J.R. Quantitative proteomic profiling reveals differentially regulated proteins in cystic fibrosis cells. J. Proteome Res. 2014, 13, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Lässer, C.; O’Neil, S.E.; Shelke, G.V.; Sihlbom, C.; Hansson, S.F.; Gho, Y.S.; Lundbäck, B.; Lötvall, J. Exosomes in the nose induce immune cell trafficking and harbour an altered protein cargo in chronic airway inflammation. J. Transl. Med. 2016, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Mauri, P.; Riccio, A.M.; Rossi, R.; Di Silvestre, D.; Benazzi, L.; De Ferrari, L.; Dal Negro, R.W.; Holgate, S.T.; Canonica, G.W. Proteomics of bronchial biopsies: Galectin-3 as a predictive biomarker of airway remodelling modulation in omalizumab-treated severe asthma patients. Immunol. Lett. 2014, 162, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.S.A. Ex Vivo Lung Perfusion and Transplant: State of the Art and View to the Future. Exp. Clin. Transplant. 2015, 13, 493–499. [Google Scholar] [PubMed]
- Mohamed, M.S.A. Translational Insights on Lung Transplantation: Learning from Immunology. Iran. J. Immunol. 2015, 12, 156–165. [Google Scholar] [PubMed]
- Reeb, J.; Cypel, M. Ex vivo lung perfusion. Clin. Transplant. 2016, 30, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bassani, G.A.; Lonati, C.; Brambilla, D.; Rapido, F.; Valenza, F.; Gatti, S. Ex Vivo Lung Perfusion in the Rat: Detailed Procedure and Videos. PLoS ONE 2016, 11, e0167898. [Google Scholar] [CrossRef] [PubMed]
- Lonati, C.; Bassani, G.A.; Brambilla, D.; Leonardi, P.; Carlin, A.; Faversani, A.; Gatti, S.; Valenza, F. Influence of ex vivo perfusion on the biomolecular profile of rat lungs. FASEB J. 2018, 32, 5532–5549. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef] [PubMed]
- Mauri, P.; Scarpa, A.; Nascimbeni, A.C.; Benazzi, L.; Parmagnani, E.; Mafficini, A.; Della Peruta, M.; Bassi, C.; Miyazaki, K.; Sorio, C. Identification of proteins released by pancreatic cancer cells by multidimensional protein identification technology: A strategy for identification of novel cancer markers. FASEB J. 2005, 19, 1125–1127. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.H.; Yates, J.R. Shotgun proteomics: Integrating technologies to answer biological questions. Curr. Opin. Mol. Ther. 2003, 5, 302–309. [Google Scholar] [PubMed]
- Chait, BT. Chemistry. Mass spectrometry: Bottom-up or top-down? Science 2006, 314, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Rossi, R.; Palladini, G.; Obici, L.; Verga, L.; Mauri, P.; Merlini, G. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood 2012, 119, 1844–1847. [Google Scholar] [CrossRef] [PubMed]
- Mauri, P.; Dehò, G. A proteomic approach to the analysis of RNA degradosome composition in Escherichia coli. Methods Enzymol. 2008, 447, 99–117. [Google Scholar] [PubMed]
- Comunian, C.; Rusconi, F.; De Palma, A.; Brunetti, P.; Catalucci, D.; Mauri, P.L. A comparative MudPIT analysis identifies different expression profiles in heart compartments. Proteomics 2011, 11, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Di Silvestre, D.; Brunetti, P.; Vella, D.; Brambilla, F.; De Palma, A.; Mauri, P. Automated Extraction of Proteotypic Peptides by Shotgun Proteomic Experiments: A New Computational Tool and Two Actual Cases. Curr. Biotechnol. 2015, 4, 39–45. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, O.J.; Arentz, G.; Hoffmann, P. Proteomic developments in the analysis of formalin-fixed tissue. Biochim. Biophys. Acta 2015, 559–580. [Google Scholar] [CrossRef] [PubMed]
- Hilario, M.; Kalousis, A. Approaches to dimensionality reduction in proteomic biomarker studies. Brief. Bioinform. 2008, 9, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Pawłowski, K.; Muszewska, A.; Lenart, A.; Szczepińska, T.; Godzik, A.; Grynberg, M. A widespread peroxiredoxin-like domain present in tumor suppression- and progression-implicated proteins. BMC Genom. 2010, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Kosieradzki, M.; Rowiński, W. Ischemia/reperfusion injury in kidney transplantation: Mechanisms and prevention. Transplant. Proc. 2008, 40, 3279–3288. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Hearth mitochondria: Gates of life and death. Cardiovasc Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Kako, K.; Kato, M.; Matsuoka, T.; Mustapha, A. Depression of membrane-bound Na+-K+-ATPase activity induced by free radicals and by ischemia of kidney. Am. J. Physiol. 1988, 254, C330–C337. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kako, K.J. Effects of N-(2-mercaptopropionyl)glycine on ischemic-reperfused dog kidney in vivo and membrane preparation in vitro. Mol. Cell. Biochem. 1987, 78, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.N.; Christini, D.J. NHE inhibition does not improve Na(+) or Ca(2+) overload during reperfusion: Using modeling to illuminate the mechanisms underlying a therapeutic failure. PLoS Comput. Biol. 2011, 7, e1002241. [Google Scholar] [CrossRef] [PubMed]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol Heart Circ. Physiol. 2011, 301, 1723–1741. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, L.; Simonsen, U.; Eskildsen-Helmond, Y.; Vorum, H.; Uldbjerg, N.; Honoré, B.; Mulvany, M.J. Proteomics reveals lowering oxygen alters cytoskeletal and endoplasmatic stress proteins in human endothelial cells. Proteomics 2009, 9, 4457–4467. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Schmitt, S.; Veit, F.; Dahal, B.K.; Wilhelm, J.; Klepetko, W.; Marta, G.; Seeger, W.; Schermuly, R.T.; Grimminger, F.; et al. Cofilin, a hypoxia-regulated protein in murine lungs identified by 2DE: Role of the cytoskeletal protein cofilin in pulmonary hypertension. Proteomics 2013, 13, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.A.; Freundlich, B.; Adams, S.; Rosenbloom, J. Regulation of human lung fibroblast collagen production by recombinant interleukin-1, tumor necrosis factor, and interferon-gamma. Ann. N. Y. Acad. Sci. 1990, 580, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, A.; Hosokawa, T.; Mochitate, K. Interleukin-1beta and tumor necrosis factor-alpha have opposite effects on fibroblasts and epithelial cells during basement membrane formation. Matrix Biol. 2008, 27, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004, 68, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Pai, H.I.; Liu, S.L. Ventilator-induced lung injury (VILI) promotes ischemia/reperfusion lung injury (I/R) and NF-kappaB antibody attenuates both injuries. Resuscitation 2008, 79, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, R.G.; Dodia, C.; Tan, Z.; Fisher, A.B. Oxygen-dependent reperfusion injury in the isolated rat lung. J. Appl. Physiol. 1992, 72, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Puskas, J.D.; Hirai, T.; Christie, N.; Mayer, E.; Slutsky, A.S.; Patterson, G.A. Reliable thirty-hour lung preservation by donor lung hyperinflation. J. Thorac. Cardiovasc. Surg. 1992, 104, 1075–1083. [Google Scholar] [PubMed]
- Date, H.; Matsumura, A.; Manchester, J.K.; Obo, H.; Lima, O.; Cooper, J.M.; Sundaresan, S.; Lowry, O.H.; Cooper, J.D. Evaluation of lung metabolism during successful twenty-four-hour canine lung preservation. J. Thorac. Cardiovasc. Surg. 1993, 105, 480–491. [Google Scholar] [PubMed]
- Vallabhajosyula, P.; Korutla, L.; Habertheuer, A.; Reddy, S.; Schaufler, C.; Lasky, J.; Diamond, J.; Cantu, E. Ex Vivo Lung Perfusion Model to Study Pulmonary Tissue Extracellular Microvesicle Profiles. Ann. Thorac. Surg. 2017, 103, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Nomura, E.; Katsuta, K.; Ueda, T.; Toriyama, M.; Mori, T.; Inagaki, N. Acid-labile surfactant improves in-sodium dodecyl sulfate polyacrylamide gel protein digestion for matrix-assisted laser desorption/ionization mass spectrometric peptide mapping. J. Mass Spectrom. 2004, 39, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Karypis, G. Data clustering in life sciences. Mol. Biotechnol. 2005, 31, 55–80. [Google Scholar] [CrossRef]
- Jain, A.K.; Murty, M.N.; Flynn, P.J. Data Clustering: A Review. ACM Comput. Surv. 1999, 31, 264–323. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Di Silvestre, D.; Brambilla, F.; Scardoni, G.; Brunetti, P.; Motta, S.; Matteucci, M.; Laudanna, C.; Recchia, F.A.; Lionetti, V.; Mauri, P. Proteomics-based network analysis characterizes biological processes and pathways activated by preconditioned mesenchymal stem cells in cardiac repair mechanisms. Biochim. Biophys. Acta 2017, 1861, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roffia, V.; De Palma, A.; Lonati, C.; Di Silvestre, D.; Rossi, R.; Mantero, M.; Gatti, S.; Dondossola, D.; Valenza, F.; Mauri, P.; et al. Proteome Investigation of Rat Lungs Subjected to Ex Vivo Perfusion (EVLP). Molecules 2018, 23, 3061. https://doi.org/10.3390/molecules23123061
Roffia V, De Palma A, Lonati C, Di Silvestre D, Rossi R, Mantero M, Gatti S, Dondossola D, Valenza F, Mauri P, et al. Proteome Investigation of Rat Lungs Subjected to Ex Vivo Perfusion (EVLP). Molecules. 2018; 23(12):3061. https://doi.org/10.3390/molecules23123061
Chicago/Turabian StyleRoffia, Valentina, Antonella De Palma, Caterina Lonati, Dario Di Silvestre, Rossana Rossi, Marco Mantero, Stefano Gatti, Daniele Dondossola, Franco Valenza, Pierluigi Mauri, and et al. 2018. "Proteome Investigation of Rat Lungs Subjected to Ex Vivo Perfusion (EVLP)" Molecules 23, no. 12: 3061. https://doi.org/10.3390/molecules23123061