Simulated and Experimental Time-Resolved Photoelectron Spectra of the Intersystem Crossing Dynamics in 2-Thiouracil

 , , and
, , and
Abstract
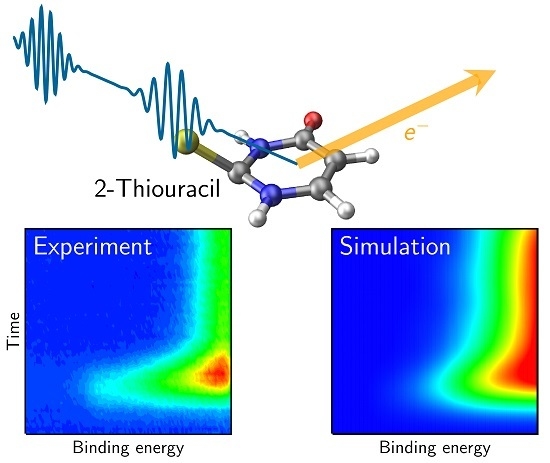
:
1. Introduction
2. Experimental Details
3. Computational Details
3.1. Excited-State Dynamics Simulations
3.2. TRPES Simulations
4. Results and Discussion
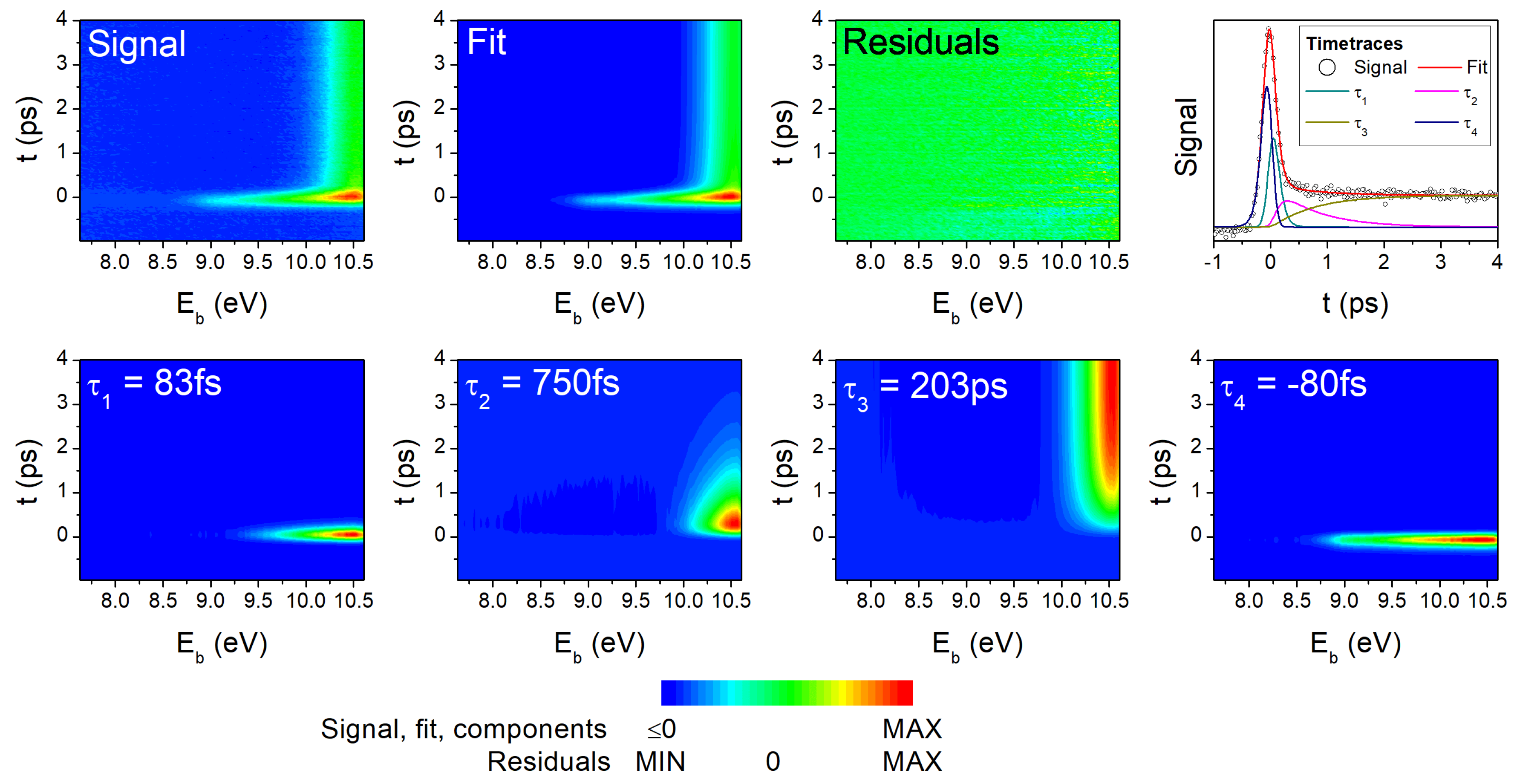
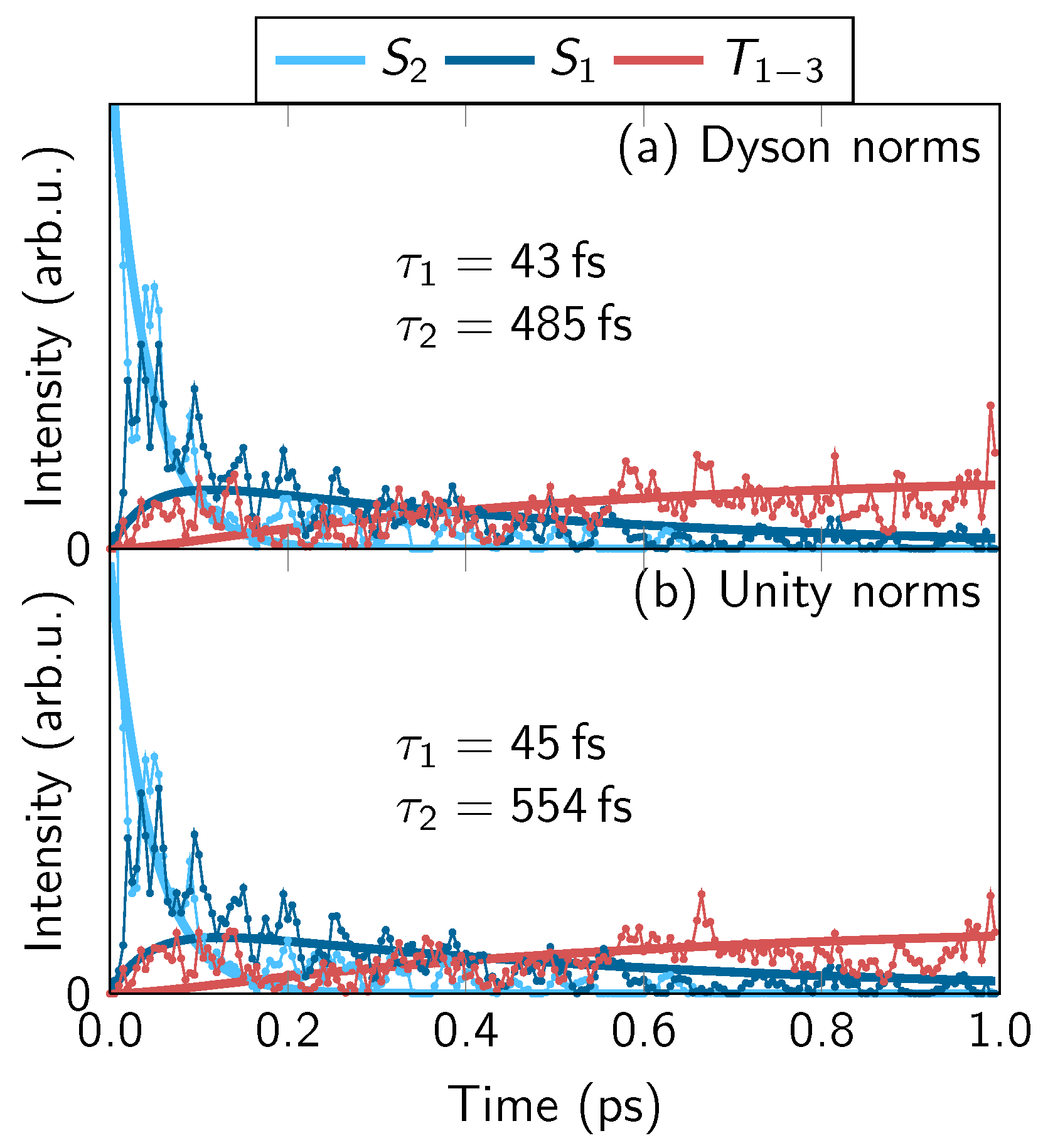
4.1. Experimental Results
4.2. Comparison of One- and Two-Photon TRPES Data
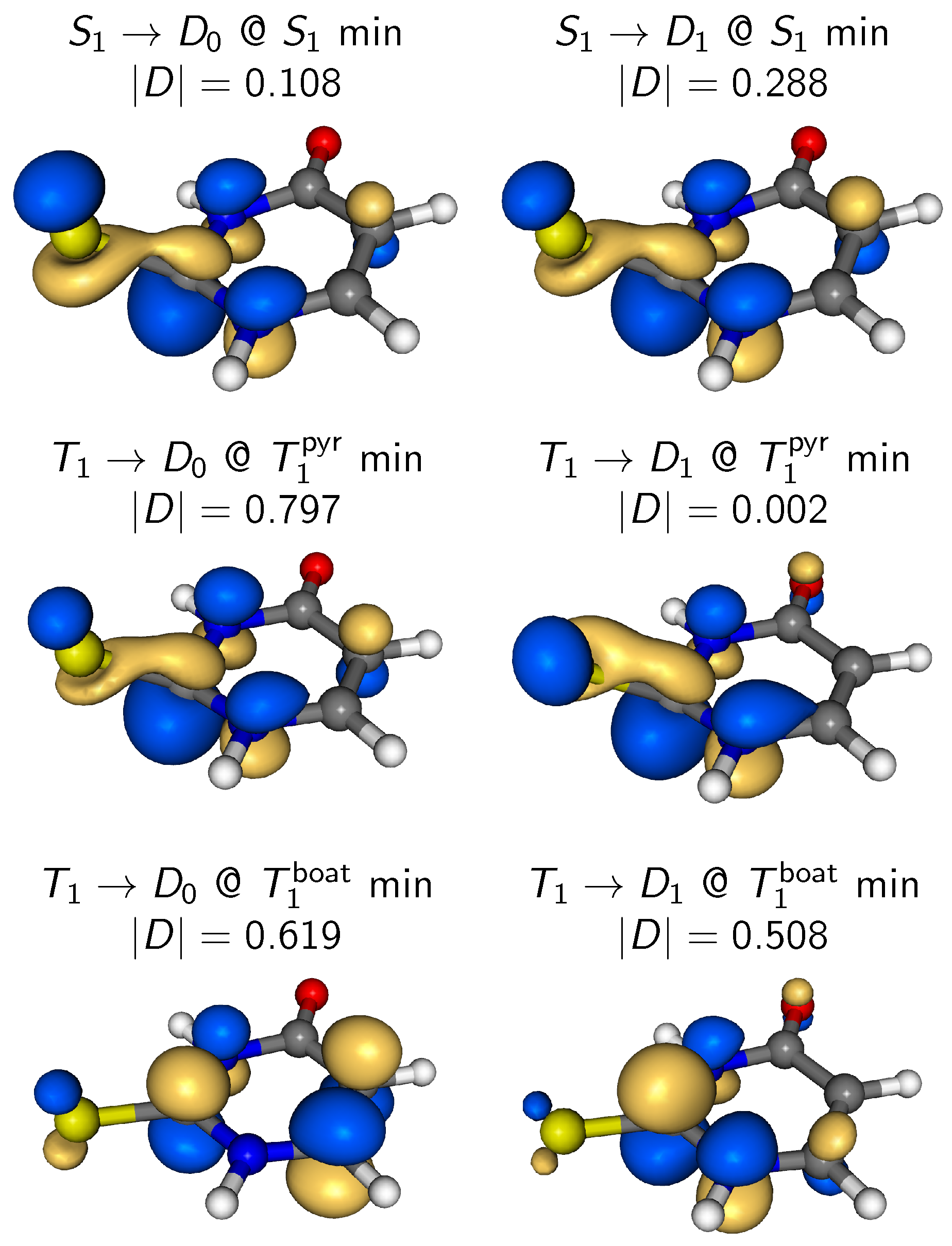
4.3. Simulated TRPE Spectra
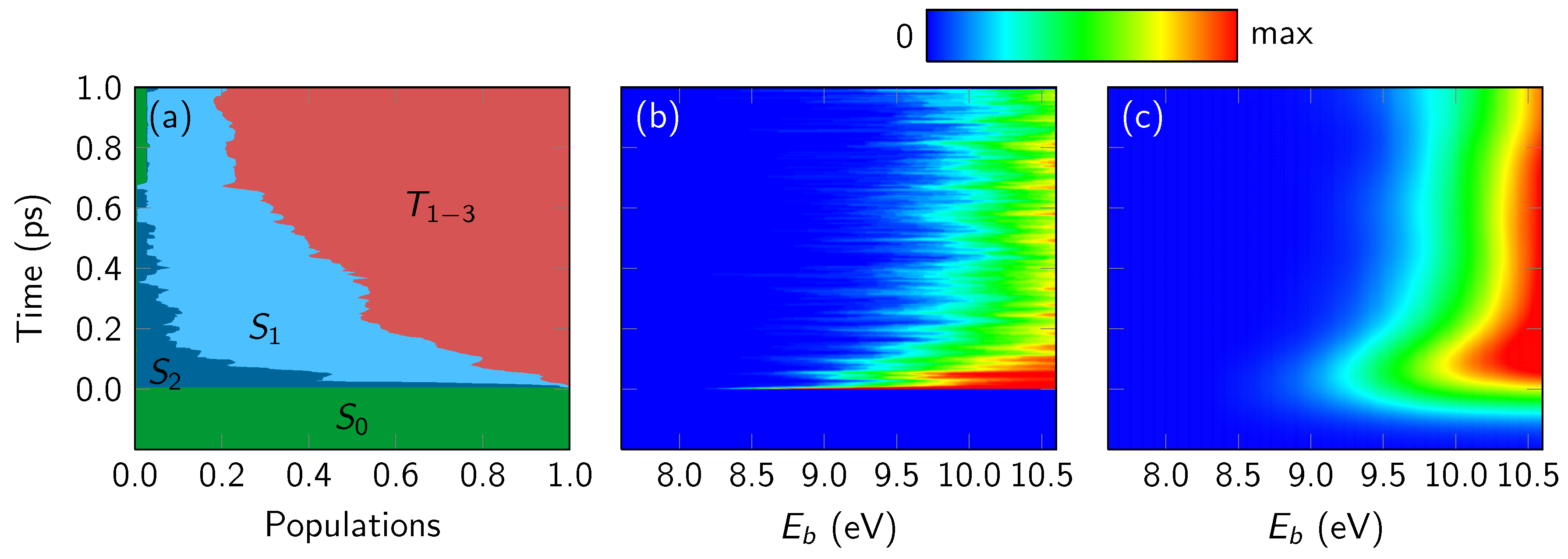
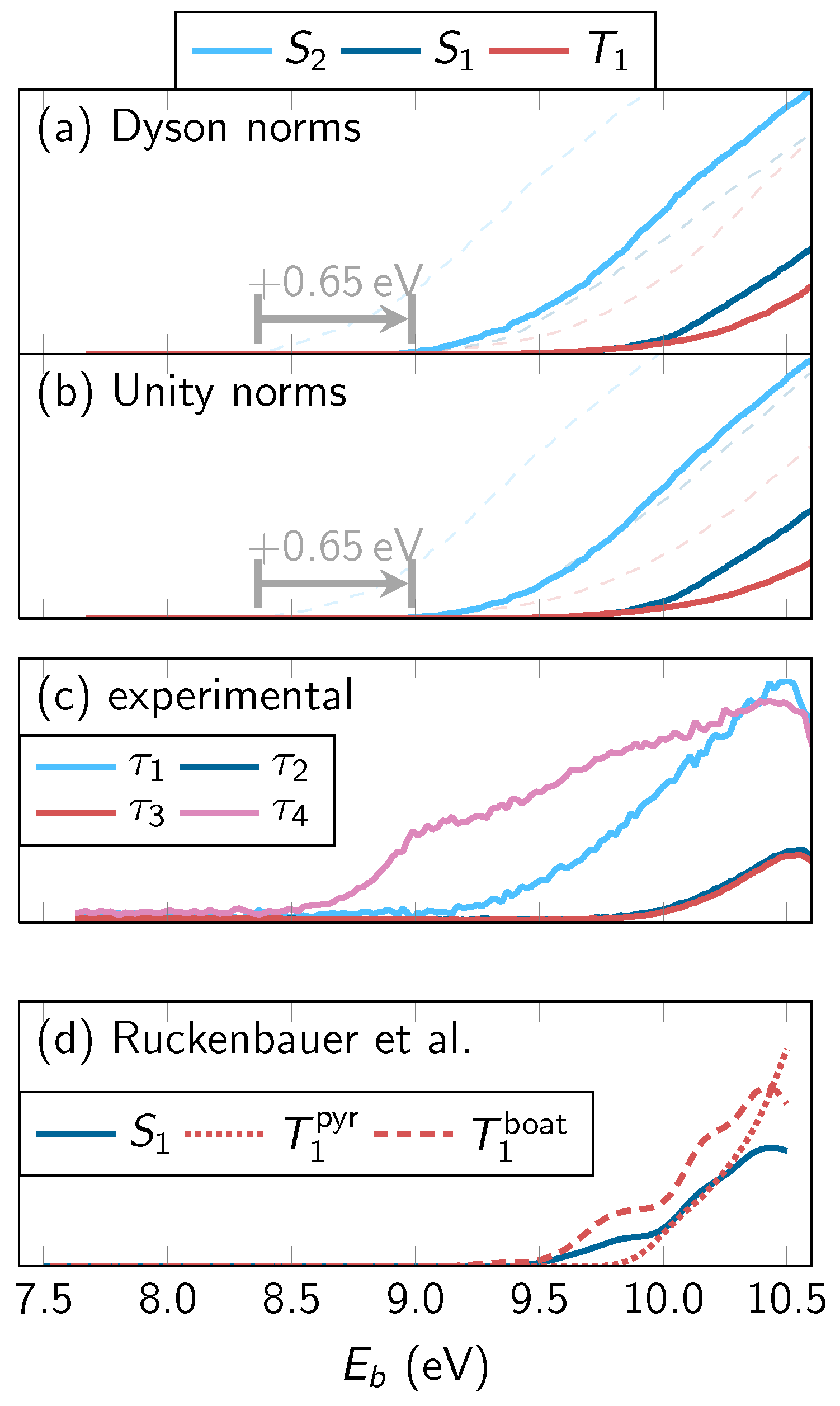
4.3.1. Overall Spectrum
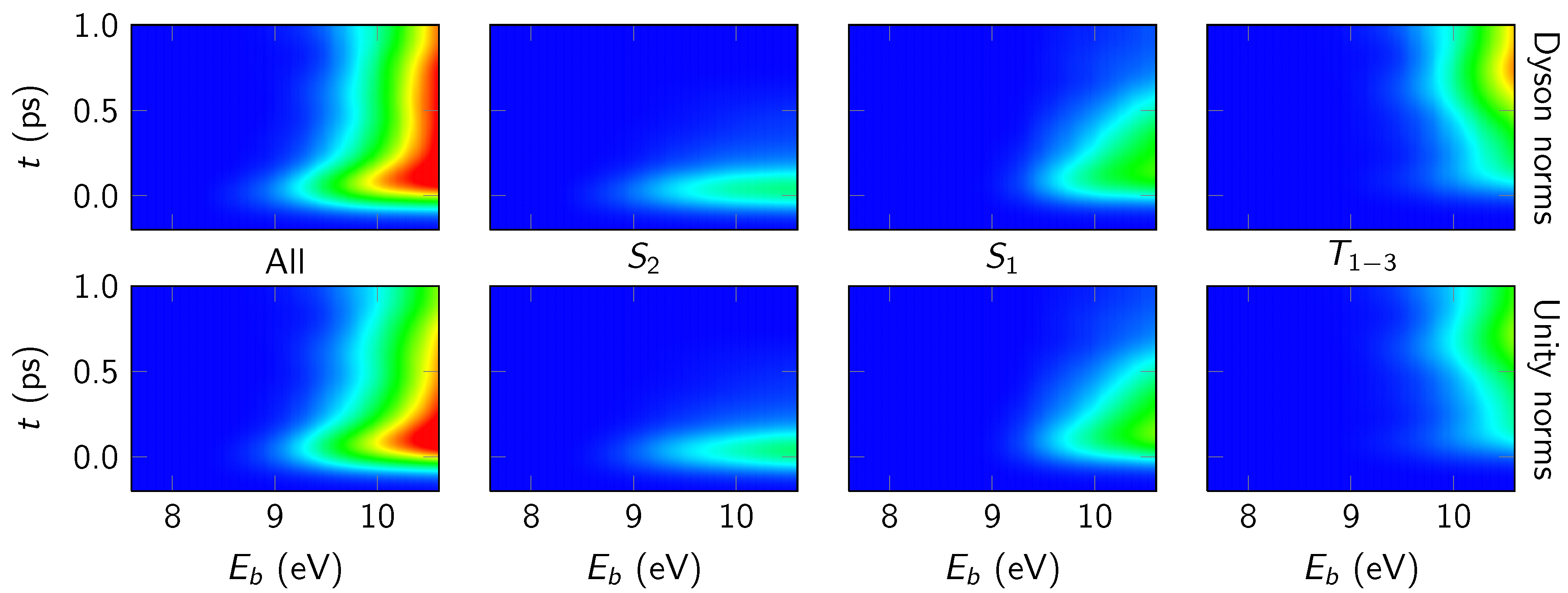
4.3.2. State-Wise Decomposition
4.3.3. Time-Averaged Spectra
4.3.4. Energy-Integrated Yields
4.4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast Excited-State Dynamics in Nucleic Acids. Chem. Rev. 2004, 104, 1977–2020. [Google Scholar] [CrossRef] [PubMed]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernández, C.E.; Kohler, B. DNA Excited-State Dynamics: From Single Bases to the Double Helix. Ann. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Kleinermanns, K.; Nachtigallová, D.; de Vries, M.S. Excited State Dynamics of DNA Bases. Int. Rev. Phys. Chem. 2013, 32, 308–342. [Google Scholar] [CrossRef]
- Barbatti, M.; Borin, A.C.; Ullrich, S. (Eds.) Photoinduced Phenomena in Nucleic Acids I; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2015; Volume 355. [Google Scholar]
- Barbatti, M.; Borin, A.C.; Ullrich, S. (Eds.) Photoinduced Phenomena in Nucleic Acids II; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2015; Volume 356. [Google Scholar]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Carell, T.; Brandmayr, C.; Hienzsch, A.; Müller, M.; Pearson, D.; Reiter, V.; Thoma, I.; Thumbs, P.; Wagner, M. Structure and Function of Noncanonical Nucleobases. Angew. Chem. Int. Ed. 2012, 51, 7110–7131. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Martínez-Fernández, L.; Crespo-Hernández, C.E. Photochemistry of Nucleic Acid Bases and Their Thio- and Aza-Analogues in Solution. In Photoinduced Phenomena in Nucleic Acids I; Topics in Current Chemistry; Barbatti, M., Borin, A.C., Ullrich, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 355, pp. 245–327. [Google Scholar]
- Matsika, S. Modified Nucleobases. In Photoinduced Phenomena in Nucleic Acids I; Topics in Current Chemistry; Barbatti, M., Borin, A.C., Ullrich, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 355, pp. 209–243. [Google Scholar]
- Carbon, J.; David, H.; Studier, M.H. Thiobases in Escherchia coli Transfer RNA: 2-Thiocytosine and 5-Methylaminomethyl-2-thiouracil. Science 1968, 161, 1146–1147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Blain, J.C.; Zielinska, D.; Gryaznov, S.M.; Szostak, J.W. Fast and Accurate Nonenzymatic Copying of an RNA-like Synthetic Genetic Polymer. Proc. Natl. Acad. Sci. USA 2013, 110, 17732–17737. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W.; Halverstadt, I.F.; Miller, W.H.; Roblin, R.O., Jr. Studies in Chemotherapy. X. Antithyroid Compounds. Synthesis of 5- and 6- Substituted 2-Thiouracils from β-Oxoesters and Thiourea. J. Am. Chem. Soc. 1945, 67, 2197–2200. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S. Antithyroid Drugs. N. Engl. J. Med. 2005, 352, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Cuffari, C.; Li, D.; Mahoney, J.; Barnes, Y.; Bayless, T. Peripheral Blood Mononuclear Cell DNA 6-Thioguanine Metabolite Levels Correlate with Decreased Interferon-γ Production in Patients with Crohn’s Disease on AZA Therapy. Dig. Dis. Sci. 2004, 49, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Elgemeie, G.H. Thioguanine, Mercaptopurine: Their Analogs and Nucleosides as Antimetabolites. Curr. Pharm. Des. 2003, 9, 2627–2642. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzidis, E.; Shavorskaya, O.; Xu, Y.Z.; Trigiante, G. Topical 4-thiothymidine is a viable photosensitiser for the photodynamic therapy of skin malignancies. J. Dermatol. Treat. 2013, 24, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, S.W.; Heer, R.; Taylor, G.A.; Newell, D.R.; O’Toole, K.; Robinson, M.; Xu, Y.Z.; Karran, P.; Boddy, A.V. Thiothymidine Combined with UVA as a Potential Novel Therapy for Bladder Cancer. Br. J. Cancer 2011, 104, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Trigiante, G.; Xu, Y.Z. 4-thiothymidine and its analogues as UVA-activated photosensitizers. In Photodynamic Therapy: Fundamentals, Applications and Health Outcomes; Hugo, A.G., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2015; pp. 193–206. [Google Scholar]
- Mai, S.; Pollum, M.; Martínez-Fernández, L.; Dunn, N.; Marquetand, P.; Corral, I.; Crespo-Hernández, C.E.; González, L. The Origin of Efficient Triplet State Population in Sulfur-Substituted Nucleobases. Nat. Commun. 2016, 7, 13077. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, H.; Kobayashi, T.; Suzuki, T.; Ichimura, T. Excited-State Dynamics of 6-Aza-2-thiothymine and 2-Thiothymine: Highly Efficient Intersystem Crossing and Singlet Oxygen Photosensitization. J. Phys. Chem. B 2010, 114, 8782–8789. [Google Scholar] [CrossRef] [PubMed]
- Vendrell-Criado, V.; Saez, J.A.; Lhiaubet-Vallet, V.; Cuquerella, M.C.; Miranda, M.A. Photophysical Properties of 5-Substituted 2-Thiopyrimidines. Photochem. Photobiol. Sci. 2013, 12, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Taras-Goślińska, K.; Burdziński, G.; Wenska, G. Relaxation of the T1 Excited State of 2-Thiothymine, its Riboside and Deoxyriboside-Enhanced Nonradiative Decay Rate Induced by Sugar Substituent. J. Photochem. Photobiol. A 2014, 275, 89–95. [Google Scholar] [CrossRef]
- Pollum, M.; Crespo-Hernández, C.E. Communication: The Dark Singlet State as a Doorway State in the Ultrafast and Efficient Intersystem Crossing Dynamics in 2-Thiothymine and 2-Thiouracil. J. Chem. Phys. 2014, 140, 071101. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. 2,4-Dithiothymine as a Potent UVA Chemotherapeutic Agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the Photoreactivity of Uracil Derivatives by Doubling Thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Koyama, D.; Milner, M.J.; Orr-Ewing, A.J. Evidence for a Double Well in the First Triplet Excited State of 2-Thiouracil. J. Phys. Chem. B 2017, 121, 9274–9280. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sánchez-Rodríguez, J.A.; Pollum, M.; Crespo-Hernández, C.E.; Mai, S.; Marquetand, P.; González, L.; Ullrich, S. Internal Conversion And Intersystem Crossing Pathways In UV Excited, Isolated Uracils And Their Implications In Prebiotic Chemistry. Phys. Chem. Chem. Phys. 2016, 18, 20168–20176. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, J.A.; Mohamadzade, A.; Mai, S.; Ashwood, B.; Pollum, M.; Marquetand, P.; González, L.; Crespo-Hernández, C.E.; Ullrich, S. 2-Thiouracil Intersystem Crossing Photodynamics Studied by Wavelength-Dependent Photoelectron And Transient Absorption Spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 19756–19766. [Google Scholar] [CrossRef] [PubMed]
- Ghafur, O.; Crane, S.W.; Ryszka, M.; Bockova, J.; Rebelo, A.; Saalbach, L.; De Camillis, S.; Greenwood, J.B.; Eden, S.; Townsend, D. Ultraviolet relaxation dynamics in uracil: Time-resolved photoion yield studies using a laser-based thermal desorption source. J. Chem. Phys. 2018, 149, 034301. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez, L.; Fahleson, T.; Norman, P.; Santoro, F.; Coriani, S.; Improta, R. Optical absorption and magnetic circular dichroism spectra of thiouracils: A quantum mechanical study in solution. Photochem. Photobiol. Sci. 2017, 16, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Fang, W.H. State-Specific Heavy-Atom Effect on Intersystem Crossing Processes in 2-Thiothymine: A Potential Photodynamic Therapy Photosensitizer. J. Chem. Phys. 2013, 138, 044315. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, J.P.; Borin, A.C. 2-Thiouracil Deactivation Pathways and Triplet States Population. Comput. Theor. Chem. 2014, 1040–1041, 195–201. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. A Static Picture of the Relaxation and Intersystem Crossing Mechanisms of Photoexcited 2-Thiouracil. J. Phys. Chem. A 2015, 119, 9524–9533. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Barbatti, M. On the decay of the triplet state of thionucleobases. Phys. Chem. Chem. Phys. 2017, 19, 12674–12682. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Marquetand, P.; González, L. Intersystem Crossing Pathways in the Noncanonical Nucleobase 2-Thiouracil: A Time-Dependent Picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Plasser, F.; Pabst, M.; Neese, F.; Köhn, A.; González, L. Surface Hopping Dynamics Including Intersystem Crossing using the Algebraic Diagrammatic Construction Method. J. Chem. Phys. 2017, 147, 184109. [Google Scholar] [CrossRef] [PubMed]
- Hudock, H.R.; Levine, B.G.; Thompson, A.L.; Satzger, H.; Townsend, D.; Gador, N.; Ullrich, S.; Stolow, A.; Martínez, T.J. Ab Initio Molecular Dynamics and Time-Resolved Photoelectron Spectroscopy of Electronically Excited Uracil and Thymine. J. Phys. Chem. A 2007, 111, 8500–8508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrić, R.; Werner, U.; Bonačić-Koutecký, V. Nonadiabatic dynamics and simulation of time resolved photoelectron spectra within time-dependent density functional theory: Ultrafast photoswitching in benzylideneaniline. J. Chem. Phys. 2008, 129, 164118. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.; Kowalewski, M.; Mukamel, S. Probing electronic and vibrational dynamics in molecules by time-resolved photoelectron, Auger-electron, and X-ray photon scattering spectroscopy. Faraday Discuss. 2015, 177, 405–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckenbauer, M.; Mai, S.; Marquetand, P.; González, L. Revealing Deactivation Pathways Hidden in Time-Resolved Photoelectron Spectra. Sci. Rep. 2016, 6, 35522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbelo-González, W.; Crespo-Otero, R.; Barbatti, M. Steady and Time-Resolved Photoelectron Spectra Based on Nuclear Ensembles. J. Chem. Theory Comput. 2016, 12, 5037–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, N.L.; Ullrich, S. Wavelength Dependence of Electronic Relaxation in Isolated Adenine Using UV Femtosecond Time-Resolved Photoelectron Spectroscopy. J. Phys. Chem. A 2010, 114, 11225–11230. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, T.J.; Yu, H.; Biddle, M.S.; Ullrich, S. A wavelength dependent investigation of the indole photophysics via ionization and fragmentation pump-probe spectroscopies. Phys. Chem. Chem. Phys. 2015, 17, 25197–25209. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Evans, N.L.; Chatterley, A.S.; Roberts, G.M.; Stavros, V.G.; Ullrich, S. Tunneling Dynamics of the NH3 () State Observed by Time-Resolved Photoelectron and H Atom Kinetic Energy Spectroscopies. J. Phys. Chem. A 2014, 118, 9438–9444. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Evans, N.L.; Stavros, V.G.; Ullrich, S. Investigation of multiple electronic excited state relaxation pathways following 200 nm photolysis of gas-phase imidazole. Phys. Chem. Chem. Phys. 2012, 14, 6266–6272. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.L.; Yu, H.; Roberts, G.M.; Stavros, V.G.; Ullrich, S. Observation of ultrafast NH3 () state relaxation dynamics using a combination of time-resolved photoelectron spectroscopy and photoproduct detection. Phys. Chem. Chem. Phys. 2012, 14, 10401–10409. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, T.J.; Yu, H.; Ullrich, S. Investigation of electronically excited indole relaxation dynamics via photoionization and fragmentation pump-probe spectroscopy. J. Chem. Phys. 2014, 141, 044314. [Google Scholar] [CrossRef] [PubMed]
- Eland, J. Photoelectron spectra of conjugated hydrocarbons and heteromolecules. Int. J. Mass Spectrom. Ion Phys. 1969, 2, 471–484. [Google Scholar] [CrossRef]
- Pulay, P. A perspective on the CASPT2 method. Int. J. Quantum Chem. 2011, 111, 3273–3279. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.Å.; Roos, B.O.; Serrano-Andrés, L. The Multi-State CASPT2 Method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The Atoms Aluminum Through Argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Reiher, M. Relativistic Douglas-Kroll-Hess Theory. WIREs Comput. Mol. Sci. 2012, 2, 139–149. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin–orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- Schimmelpfennig, B. Atomic Spin-Orbit Mean-Field Integral Program AMFI; Stockholms Universitet: Stockholm, Sweden, 1996. [Google Scholar]
- Ghigo, G.; Roos, B.O.; Malmqvist, P.Å. A Modified Definition of the Zeroth-Order Hamiltonian in Multiconfigurational Perturbation Theory (CASPT2). Chem. Phys. Lett. 2004, 396, 142–149. [Google Scholar] [CrossRef]
- Zobel, J.P.; Nogueira, J.J.; González, L. The IPEA Dilemma in CASPT2. Chem. Sci. 2017, 8, 1482–1499. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, N.; Malmqvist, P.Å. Multiconfiguration Perturbation Theory with Imaginary Level Shift. Chem. Phys. Lett. 1997, 274, 196–204. [Google Scholar] [CrossRef]
- Schinke, R. Photodissociation Dynamics: Spectroscopy and Fragmentation of Small Polyatomic Molecules; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Dahl, J.P.; Springborg, M. The Morse oscillator in position space, momentum space, and phase space. J. Chem. Phys. 1988, 88, 4535–4547. [Google Scholar] [CrossRef] [Green Version]
- Barbatti, M.; Granucci, G.; Persico, M.; Ruckenbauer, M.; Vazdar, M.; Eckert-Maksić, M.; Lischka, H. The on-the-fly surface-hopping program system Newton-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems. J. Photochem. Photobiol. A 2007, 190, 228–240. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. A General Method to Describe Intersystem Crossing Dynamics in Trajectory Surface Hopping. Int. J. Quantum Chem. 2015, 115, 1215–1231. [Google Scholar] [CrossRef]
- Mai, S.; Richter, M.; Ruckenbauer, M.; Oppel, M.; Marquetand, P.; González, L. SHARC: Surface Hopping Including Arbitrary Couplings—Program Package for Non-Adiabatic Dynamics; University of Vienna: Vienna, Austria, 2014. [Google Scholar]
- Granucci, G.; Persico, M.; Toniolo, A. Direct Semiclassical Simulation of Photochemical Processes with Semiempirical Wave Functions. J. Chem. Phys. 2001, 114, 10608–10615. [Google Scholar] [CrossRef]
- Plasser, F.; Granucci, G.; Pittner, J.; Barbatti, M.; Persico, M.; Lischka, H. Surface Hopping Dynamics using a Locally Diabatic Formalism: Charge Transfer in The Ethylene Dimer Cation and Excited State Dynamics in the 2-Pyridone Dimer. J. Chem. Phys. 2012, 137, 22A514. [Google Scholar] [CrossRef] [PubMed]
- Granucci, G.; Persico, M. Critical appraisal of the fewest switches algorithm for surface hopping. J. Chem. Phys. 2007, 126, 134114. [Google Scholar] [CrossRef] [PubMed]
- Plasser, F.; Ruckenbauer, M.; Mai, S.; Oppel, M.; Marquetand, P.; González, L. Efficient and Flexible Computation of Many-Electron Wave Function Overlaps. J. Chem. Theory Comput. 2016, 12, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Gozem, S.; Gunina, A.O.; Ichino, T.; Osborn, D.L.; Stanton, J.F.; Krylov, A.I. Photoelectron Wave Function in Photoionization: Plane Wave or Coulomb Wave? J. Phys. Chem. Lett. 2015, 6, 4532–4540. [Google Scholar] [CrossRef] [PubMed]
- Spanner, M.; Patchkovskii, S.; Zhou, C.; Matsika, S.; Kotur, M.; Weinacht, T.C. Dyson norms in XUV and strong-field ionization of polyatomics: Cytosine and uracil. Phys. Rev. A 2012, 86, 053406. [Google Scholar] [CrossRef]
- Fuji, T.; Suzuki, Y.I.; Horio, T.; Suzuki, T.; Mitrić, R.; Werner, U.; Bonačić-Koutecký, V. Ultrafast photodynamics of furan. J. Chem. Phys. 2010, 133, 234303. [Google Scholar] [CrossRef] [PubMed]
- Ruckenbauer, M.; Mai, S.; Marquetand, P.; González, L. Photoelectron spectra of 2-thiouracil, 4-thiouracil, and 2,4-dithiouracil. J. Chem. Phys. 2016, 144, 074303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Fernández, L.; González, L.; Corral, I. An Ab Initio Mechanism for Efficient Population of Triplet States in Cytotoxic Sulfur Substituted DNA Bases: The Case of 6-Thioguanine. Chem. Commun. 2012, 48, 2134–2136. [Google Scholar] [CrossRef] [PubMed]
- Arslancan, S.; Martínez-Fernández, L.; Corral, I. Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules 2017, 22, 998. [Google Scholar] [CrossRef]
- Gobbo, J.P.; Borin, A.C. On The Population of Triplet Excited States of 6-Aza-2-Thiothymine. J. Phys. Chem. A 2013, 117, 5589–5596. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Pump (nm) | Probe (nm) | (fs) | (fs) | (ps) | (fs) | Remark |
|---|---|---|---|---|---|---|---|
| —experimental— | |||||||
| Two-photon TRPES | 293 | 2 × 330 | <50 | 775 | 203 | [27,28] | |
| Two-photon TRPES | 260 | 2 × 330 | 67 | 285 | 85.6 | [28] | |
| One-photon TRPES | 293 | 194 | 83 | 750 | 203 | −80 | [present work] |
| One-photon TRPES | 260 | 194 | <50 | 246 | 85.6 | −80 | [present work] |
| —simulated— | |||||||
| SHARC populations | 295–317 | none | ∼60 | ∼400 | [35] | ||
| Simulated TRPES | 295–317 | 194 | ∼45 | ∼500 | [present work] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, S.; Mohamadzade, A.; Marquetand, P.; González, L.; Ullrich, S. Simulated and Experimental Time-Resolved Photoelectron Spectra of the Intersystem Crossing Dynamics in 2-Thiouracil. Molecules 2018, 23, 2836. https://doi.org/10.3390/molecules23112836
Mai S, Mohamadzade A, Marquetand P, González L, Ullrich S. Simulated and Experimental Time-Resolved Photoelectron Spectra of the Intersystem Crossing Dynamics in 2-Thiouracil. Molecules. 2018; 23(11):2836. https://doi.org/10.3390/molecules23112836
Chicago/Turabian StyleMai, Sebastian, Abed Mohamadzade, Philipp Marquetand, Leticia González, and Susanne Ullrich. 2018. "Simulated and Experimental Time-Resolved Photoelectron Spectra of the Intersystem Crossing Dynamics in 2-Thiouracil" Molecules 23, no. 11: 2836. https://doi.org/10.3390/molecules23112836