8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia

1
State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
2
Department of Pharmacology, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
3
Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, Jiangsu, China
4
Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(11), 2828; https://doi.org/10.3390/molecules23112828
Submission received: 29 September 2018
/
Revised: 28 October 2018
/
Accepted: 29 October 2018
/
Published: 31 October 2018
Abstract
:The inflammatory response mediated by microglia plays a critical role in the progression of ischemic stroke. Phosphoinositide 3-kinase gamma (PI3Kγ) has been implicated in multiple inflammatory and autoimmune diseases, making it a promising target for therapeutic intervention. The aim of this study was to evaluate the efficacy of 8e, a hydrogen sulfide (H2S) releasing derivative of 3-n-butylphthalide (NBP), on brain damage and PI3Kγ signaling following cerebral ischemia injury. 8e significantly reduced sensorimotor deficits, focal infarction, brain edema and neural apoptosis at 72 h after transient middle cerebral artery occlusion (tMCAO). The NOX2 isoform of the NADPH oxidase family is considered a major enzymatic source of superoxide. We found that the release of superoxide, together with the expression of NOX2 subunits p47phox, p-p47phox, and the upstream PI3Kγ/AKT signaling were all down-regulated by 8e, both in the penumbral region of the rat brain and in the primary cultured microglia subjected to oxygen-glucose deprivation (OGD). With the use of siRNA and pharmacological inhibitors, we further demonstrated that 8e regulates the formation of superoxide in activated microglia through the PI3Kγ/AKT/NOX2 signaling pathway and subsequently prevents neuronal death in neighboring neurons. Our experimental data indicate that 8e is a potential candidate for the treatment of ischemic stroke and PI3Kγ-mediated neuroinflammation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
As one of the major causes of death and serious long-term disability, stroke produces immense health and economic burdens globally. In 2013, 6.5 million stroke deaths were reported worldwide, 67% of which were caused by ischemic stroke [1]. Intravenous thrombolysis with tissue-type plasminogen activator (tPA) is the only approved treatment for acute ischemic stroke, but the benefit is strongly time-dependent. Although the therapeutic window of tPA safely extends to three to four and a half hours after the onset of stroke symptoms [2,3], it is not sufficient for patients to receive timely thrombolytic therapy.
A tremendous amount of work has been done on developing neuroprotective agents, which appeared to be quite credible on animal models. However, none of these agents have been demonstrated to be effective where humans are concerned. This may be partly due to overlooking the fact that all cell types in the brain, not only neurons, respond to ischemic brain injury [4]. Homeostatic signaling among cell types maintains normal brain function, while disordered signaling contributes to the progression of injury after stroke [5]. Emerging evidence from recent studies now suggests that therapeutic approaches should target multiple cell types to protect their structural and functional integrity as well as interactions, especially in the salvageable penumbral region [5,6]. Intricate mechanisms are triggered within the penumbra following the onset of cerebral ischemia/reperfusion, and prominent inflammatory response is believed to play a critical role in exacerbating post-ischemic injury, especially neural damage [7,8]. As the major resident immune cells in the brain, microglia change from surveying to activated states when the neuronal death expands in the ischemic core and spreads to the penumbra, which affects the fate of microglia and their neighboring neurons [9,10]. Despite a poor reputation, the activation of microglia is now considered to be a protective response; however, one which the impaired central nervous system (CNS) can barely tolerate and which is therefore destructive [11,12]. From this perspective, we should search for novel treatments which can appropriately regulate the activation of microglia, to save vulnerable neurons from ischemic injury [11].
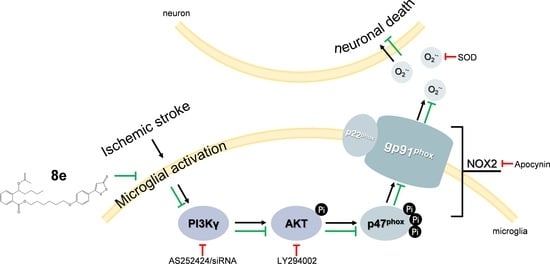
Activated microglia generate excessive amounts of reactive oxygen species (ROS), and ROS in turn promote microglial proliferation in a feed-forward manner [13,14]. Stimulation of NADPH oxidase activity is the predominant mechanism involved in ROS production following cerebral ischemia [15]. Although nearly all cell types in the brain express one or more NADPH oxidase (NOX) isoforms, microglia express the highest levels of NOX, especially NOX2 [16]. NOX2 is composed of membrane subunits (p22phox and gp91phox) and cytosolic subunits (p47phox, p67phox, and p40phox). Phosphorylation of p47phox induces its association with p22phox and translocation to cell membranes, which in turn promotes interaction with p67phox and the small GTPase Rac to form an active enzyme complex [17]. It has been suggested that AKT triggers the phosphorylation of p47phox in leukocytes and endothelial cells [18,19]. The mechanism of p47phox phosphorylation in microglial cells during the process of stroke is not yet fully understood.
Phosphoinositide 3-kinases (PI3Ks) regulate several key functions of immune cells ranging from development, proliferation to migration and cytokine production [20]. A substantial amount of data indicate that the Class IB isoform-PI3Kγ is correlated with many inflammatory and autoimmune diseases, making it a promising target for therapeutic intervention. Numerous efforts have focused on the compelling effects of phosphoinositide 3-kinase gamma (PI3Kγ) in systemic inflammation [21,22,23], but less attention is paid to its contribution to CNS inflammatory response in spite of the high abundance in the brain [24]. It has been shown that genetic and pharmacological inhibition of PI3Kγ prevented Alzheimer’s disease and surgical brain injury [25,26]. However, there is still debate about the role of PI3Kγ during stroke because although genetic deletion of PI3Kγ reduced cerebral ischemic injury in some studies [27,28], it worsened the brain damage in others [29]. Moreover, the cellular location of PI3Kγ in the CNS is also controversial [27,30]. Therefore, further investigation of the role of PI3Kγ signaling in neuroinflammation is urgently needed.
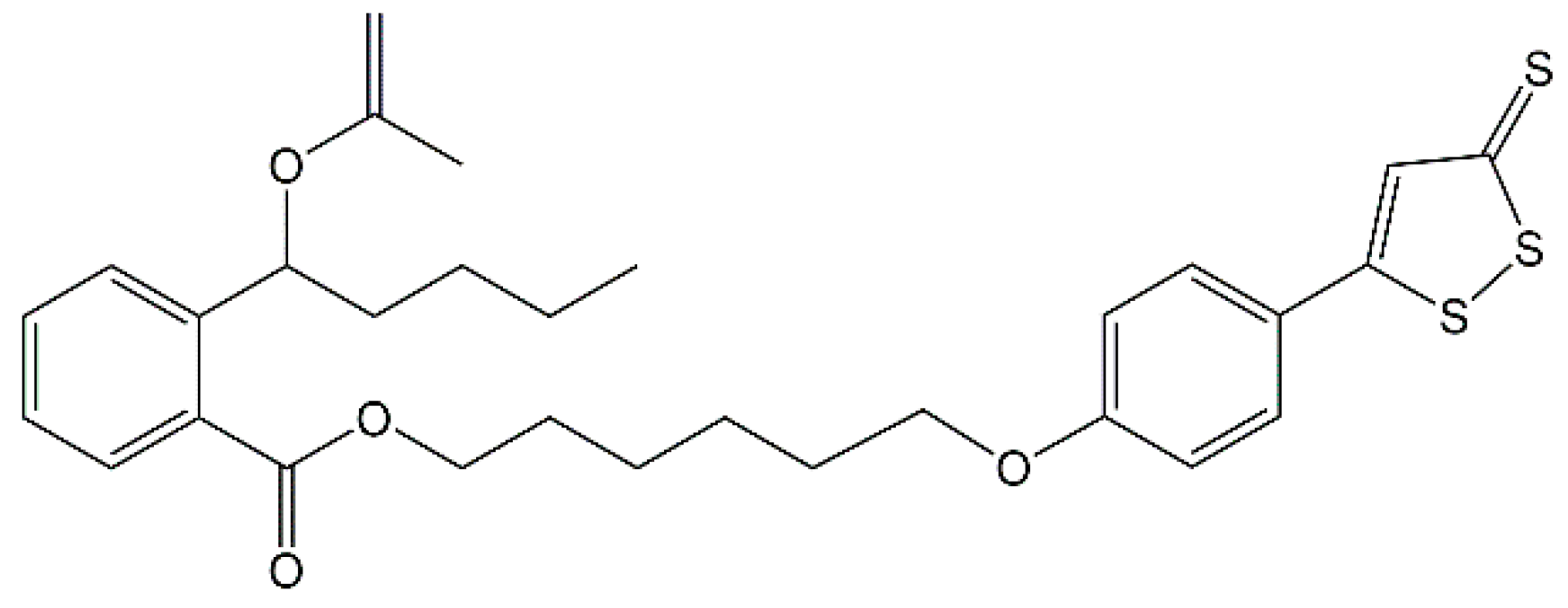
Hydrogen sulfide (H2S) is well known as the third gasotransmitter beside nitric oxide (NO) and carbon monoxide (CO) [31]. Many researchers have found that exogenous H2S could attenuate ischemic injury in various organs and tissues, including the heart, liver and kidney [32,33,34]. The protective effects of H2S-releasing compounds, especially the ones with H2S slow-releasing donors, in cerebral ischemia is also discussed [35], but the underlying mechanism has not been thoroughly investigated. Our previous work has demonstrated that 8e (Figure 1), an NBP-derivative combined with H2S donor ADT-OH (5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione), exhibiting antiplatelet and antithrombotic activities, was a potential agent for the treatment of ischemic stroke [36]. Recent reports by one group showed that ADT-OH suppressed inflammatory response in microglial cells [37] and ADT conferred neuroprotection through regulating NOX4-derived ROS [38]. At the same time, several reports also showed that H2S donor exerts neuroprotection through anti-inflammatory effects in microglia [39,40,41] and anti-oxidative effects via NADPH oxidases [42,43,44]. We hypothesize that the essential mechanisms of H2S and 8e during stroke are involved with microglial activation following oxidative stress.
In the present study, we assessed the therapeutic effects of 8e by studying the sensorimotor dysfunction and brain injury in a rat model of focal ischemic stroke induced by transient middle cerebral artery occlusion (tMCAO). Further investigation into the involvement of NOX2-derived superoxide and PI3Kγ signaling in the protective mechanism of 8e was conducted. To replicate the condition of microglia and the interaction of neuron-microglia after stroke, primary microglia cultures were exposed to oxygen–glucose deprivation (OGD). 8e down-regulated the PI3Kγ/AKT/NOX2 pathway in microglial cells and subsequently attenuated neuronal death in vitro. Our results indicate that 8e is a promising candidate for the treatment of ischemic stroke.
2. Results
2.1. 8e Enhanced Sensorimotor Function after Cerebral I/R in Rats
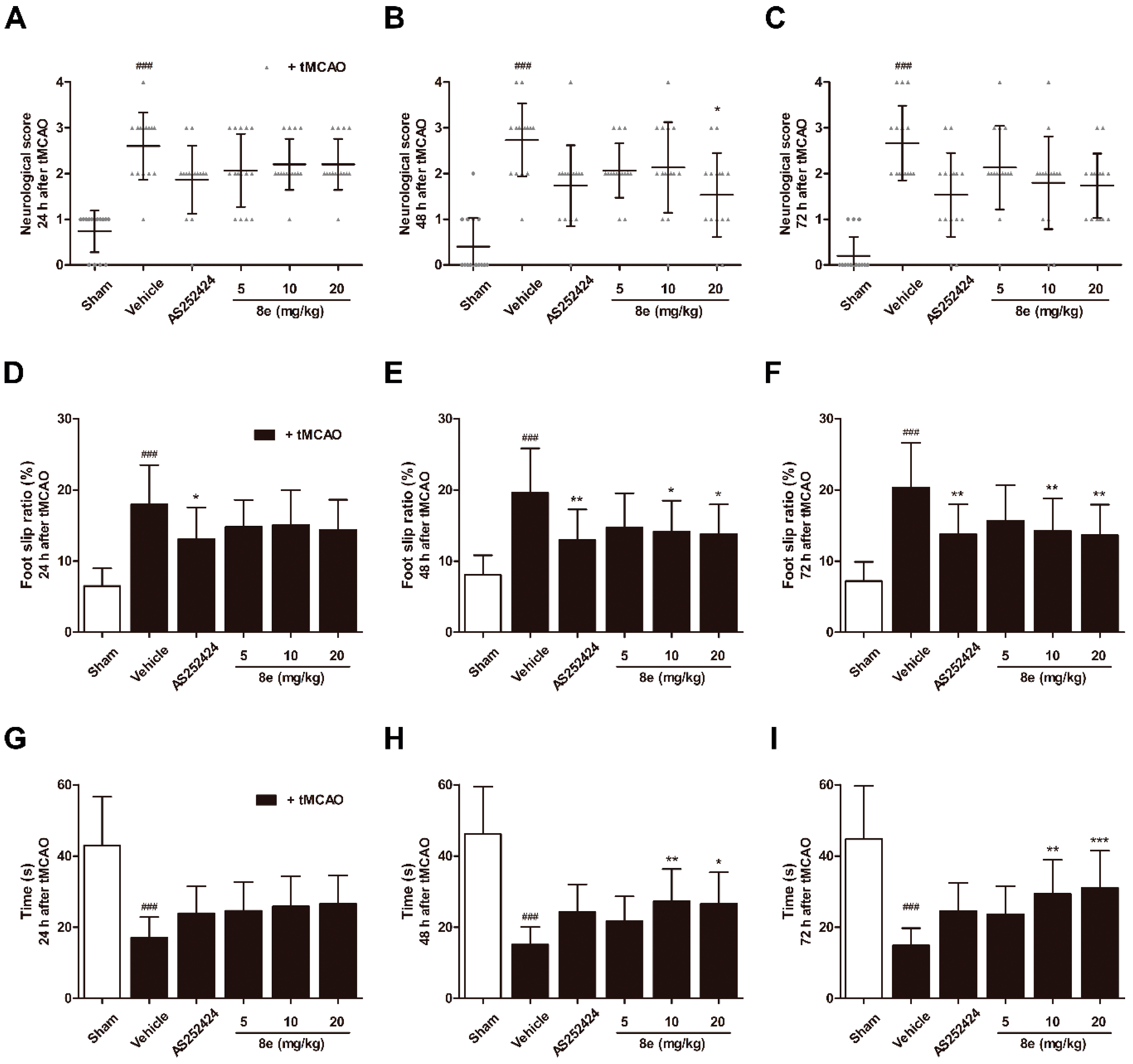
Middle cerebral artery occlusion (MCAO) in rats induces lesions in the cortical and striatal regions which are involved in the processing of sensorimotor information. Impairment of behavior may reflect changes in brain function. A series of behavioral assessments was conducted to determine the neurological and sensorimotor deficits at 24 h, 48 h and 72 h following transient middle cerebral artery occlusion (tMCAO). In the Longa test, which offers a graded scale of body movement post ischemic injury, a repeated measures ANOVA showed a significant group effect (F = 41.88, p < 0.0001), but no time or interaction effect (F = 2.823, p = 0.061; F = 0.810, p = 0.620). Rats in the tMCAO + vehicle group showed severe neurological impairment at every time point tested (p < 0.001, respectively; Figure 2A–C). Trends of functional improvement were observed in the 8e treatment groups, while significant differences were only found between 20 mg/kg 8e and tMCAO + vehicle group at 48 h after tMCAO (p < 0.05).
A significant group effect (F = 33.36, p < 0.0001) in the beam walking test, which reflects motor coordination, was observed without time or interaction effect (F = 0.282, p = 0.755; F = 0.374, p = 0.957). Post hoc analyses showed that beam walking ability was remarkably impaired in vehicle-treated animals for three days after tMCAO (p < 0.001, respectively; Figure 2D–F). The foot slip ratios were notably decreased in rats treated with 10 mg/kg and 20 mg/kg 8e at 48 h (p < 0.05, respectively) and 72 h after tMCAO (p < 0.01, respectively). AS252424, a PI3Kγ pharmacological inhibitor, also reduced foot slip ratios and showed statistical differences in the beam walking test.
The prehensile traction test primarily measures impaired forelimb placement and muscle strength by the length of time spent on the rope. A significant group effect (F = 49.25, p < 0.0001) but no time or interaction effect (F = 0.509, p = 0.602; F = 0.473, p = 0.907) was found. At 24 h, 48 h and 72 h after tMCAO, the vehicle-treated rats spent shorter lengths of time on the rope (p < 0.001, respectively; Figure 2G–I). 8e at a dose of 20 mg/kg profoundly increased muscle strength at 48 h and 72 h after tMCAO (p < 0.05, p < 0.001, respectively). Similar results were obtained after 8e treatment of 10 mg/kg at the same two time points (p < 0.01, respectively). These findings suggested that 8e could improve behavioral outcomes after tMCAO through three days of treatment.
2.2. 8e Exerted Protective Effect against Cerebral I/R Injury, Histological Damage and Neural Apoptosis in Rats
Brain infarct volume after tMCAO was tested by TTC staining to quantify morphological changes (Figure 3A,B). The infarctions declined in rats treated with 8e at 10 mg/kg (p < 0.05) and 20 mg/kg (p < 0.01) compared with the tMCAO + vehicle group. At 72 h after reperfusion, water contents were increased in the ipsilateral hemispheres of the vehicle group (p < 0.05, Figure 3C). All 8e treatment decreased brain edema, but statistical significance was found only in the 20 mg/kg group (p < 0.05).
As shown in Figure 3D, tMCAO surgery induced severe histological damage in rats treated with a vehicle based on hematoxylin and eosin (H&E) staining. The presence of all three doses of 8e notably reduced the numbers of pyknotic nuclei and vacuolization. TUNEL staining was performed to investigate whether 8e could affect neural apoptosis following I/R (Figure 3E). The results showed that the numbers of TUNEL positive nuclei, i.e., the apoptotic nuclei, in the 8e-treated groups were much fewer than those in the tMCAO + vehicle group (10 mg/kg, p < 0.05; 20 mg/kg, p < 0.01). Similarly, PI3Kγ-selective inhibitor AS252424 also exhibited marked protection against I/R injury, proving that inhibition of PI3Kγ is a potential approach to treat cerebral ischemia.
2.3. 8e Attenuated Superoxide Production Following Cerebral I/R in Rats
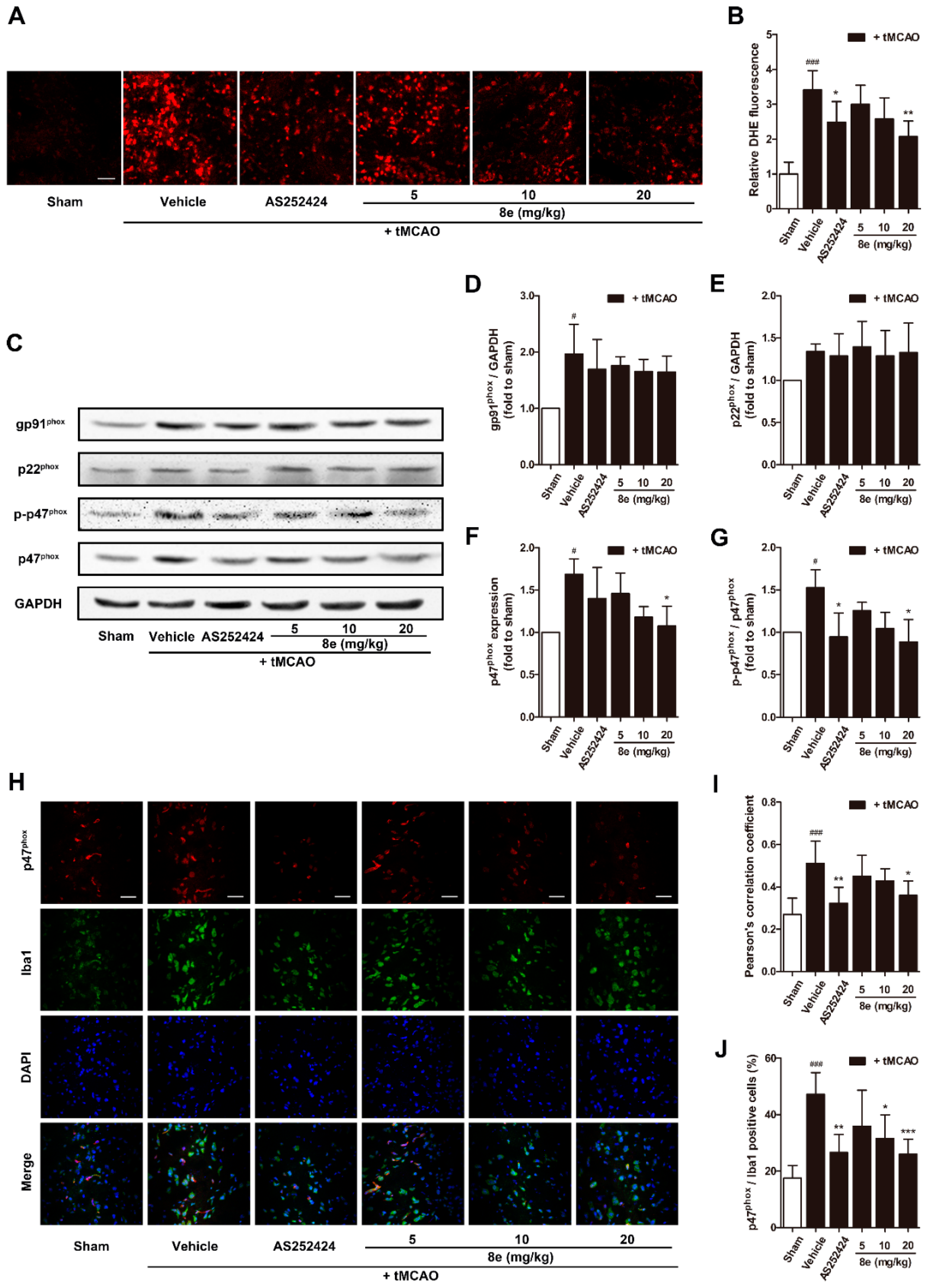
ROS are critical mediators in the pathology of acute ischemic stroke. Superoxide is one of the most important ROS which can directly cause oxidative damage to nearby neural cells or can interact with other molecules to generate “secondary” radicals [45]. The effect of 8e on superoxide production was evaluated with the DHE staining technique which produces red fluorescence when oxidized by superoxide and intercalates into DNA. At 72 h after the tMCAO, although limited fluorescent signal was detected in sham-operated rats, significantly higher fluorescence was found in the penumbral region of vehicle-treated ones (Figure 4A,B). Treatment with 8e markedly attenuated the production of superoxide (20 mg/kg, p < 0.01) as did AS252424 (p < 0.05).
We next determined the protein expression of NOX2 subunits which are regarded as the major source of superoxide in brain ischemia (Figure 4C–G). In this experiment, NOX2 was activated after tMCAO, and the levels of its subunits were significantly increased (p < 0.05, respectively) except p22phox. 8e did not affect the elevation of gp91phox although it effectively attenuated the expression of p47phox and phosphorylation of p47phox at Ser328 (20 mg/kg, p < 0.05, respectively). Meanwhile, AS252424 altered the levels of the p-p47phox subunit with statistical significance (p < 0.05).
As mentioned above, abundant expression of NOX2 was found in microglia [16]. We further explored whether 8e could inhibit the activation of NOX2 in this cell type during tMCAO with double immunostaining of p47phox and Iba1 antibodies. Results in Figure 4H–J showed that the Pearson’s correlation coefficient of the vehicle-treated group was sharply raised at 72 h after tMCAO (Rr = 0.512, p < 0.001). Administration of 8e and AS252424 decreased the value of the Pearson’s correlation coefficient (20 mg/kg, Rr = 0.360, p < 0.05; AS252424, Rr = 0.323, p < 0.01). The ratios of p47phox/Iba1 positive cells were profoundly reduced after 8e treatment at doses of 10 mg/kg and 20 mg/kg (p < 0.05, p < 0.001), while AS252424 had a similar effect (p < 0.01). These data indicated that 8e prevents tMCAO-induced elevation of p47phox level in microglial cells. We therefore presumed that the effects of 8e on superoxide production may occur partly through inhibition of p47phox and p-p47phox in microglia after cerebral I/R in rats. Suppression of PI3Kγ by AS252424 could also decrease ROS generation and p47phox activation, indicating potential implication of PI3Kγ signaling in the regulation of superoxide formation.
2.4. 8e Inhibited PI3Kγ Signaling in Microglia after Cerebral I/R in Rats
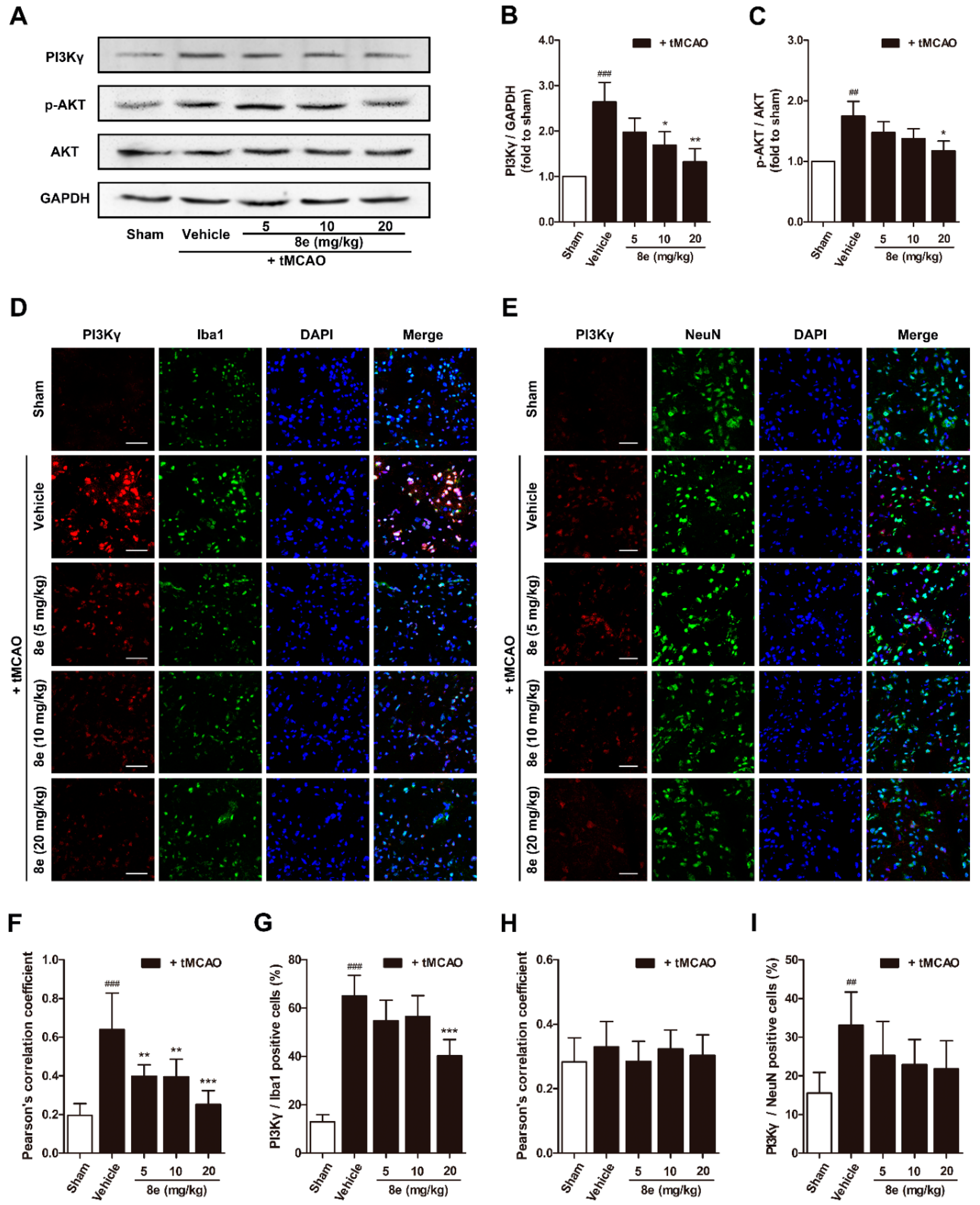
Phosphorylation of AKT is a ubiquitous response to activation of Class I PI3K and has been suggested to trigger p47phox-mediated ROS formation in leukocytes and endothelial cells [46]. We hypothesized that 8e may affect p47phox through the PI3Kγ/AKT signaling pathway in activated microglial cells after stroke. According to our data, PI3Kγ expression in the penumbral region is notably increased at 72 h following tMCAO, and this elevation was effectively eliminated by 8e (10 mg/kg, p < 0.05; 20 mg/kg, p < 0.01). The protein level of p-AKT was also reduced with the presence of 8e (20 mg/kg, p < 0.05; Figure 5A–C). These results revealed that 8e could inhibit the PI3Kγ/AKT pathway in the context of stroke.
Considering the controversial location of PI3Kγ in the CNS, we further assayed the cellular distribution of PI3Kγ in the penumbral region of the ischemic brain with a double immunostaining technique (Figure 5D,E). PI3Kγ signals rose at 72 h after MCAO and were highly co-localized with Iba1 but not with NeuN, the marker for the soma of neurons. The Pearson’s correlation coefficient of the vehicle-treated group in PI3Kγ/Iba1 immunostaining exhibited a dramatic increase at 72 h after tMCAO (Rr = 0.638, p < 0.001), while this value in PI3Kγ/NeuN immunostaining was barely changed (Rr = 0.330, p > 0.05), and those of the 8e-treated groups were reduced (5 mg/kg, Rr = 0.398, p < 0.01; 10 mg/kg, Rr = 0.395, p < 0.01; 20 mg/kg, Rr = 0.251, p < 0.001). The ratio of PI3Kγ/Iba1 positive cells was remarkably reduced when given 20 mg/kg of 8e (p < 0.001). Meanwhile, the ratios of PI3Kγ/NeuN-positive cells were not observably decreased in the presence of 8e. Our data showed that PI3Kγ was highly expressed in the microglia of the ischemic brain, and 8e could prevent cerebral I/R injury via modulating the PI3Kγ/AKT signaling pathway in microglia.
2.5. 8e Down-Regulated PI3Kγ Signaling in OGD-Treated Primary Microglial Cells In Vitro
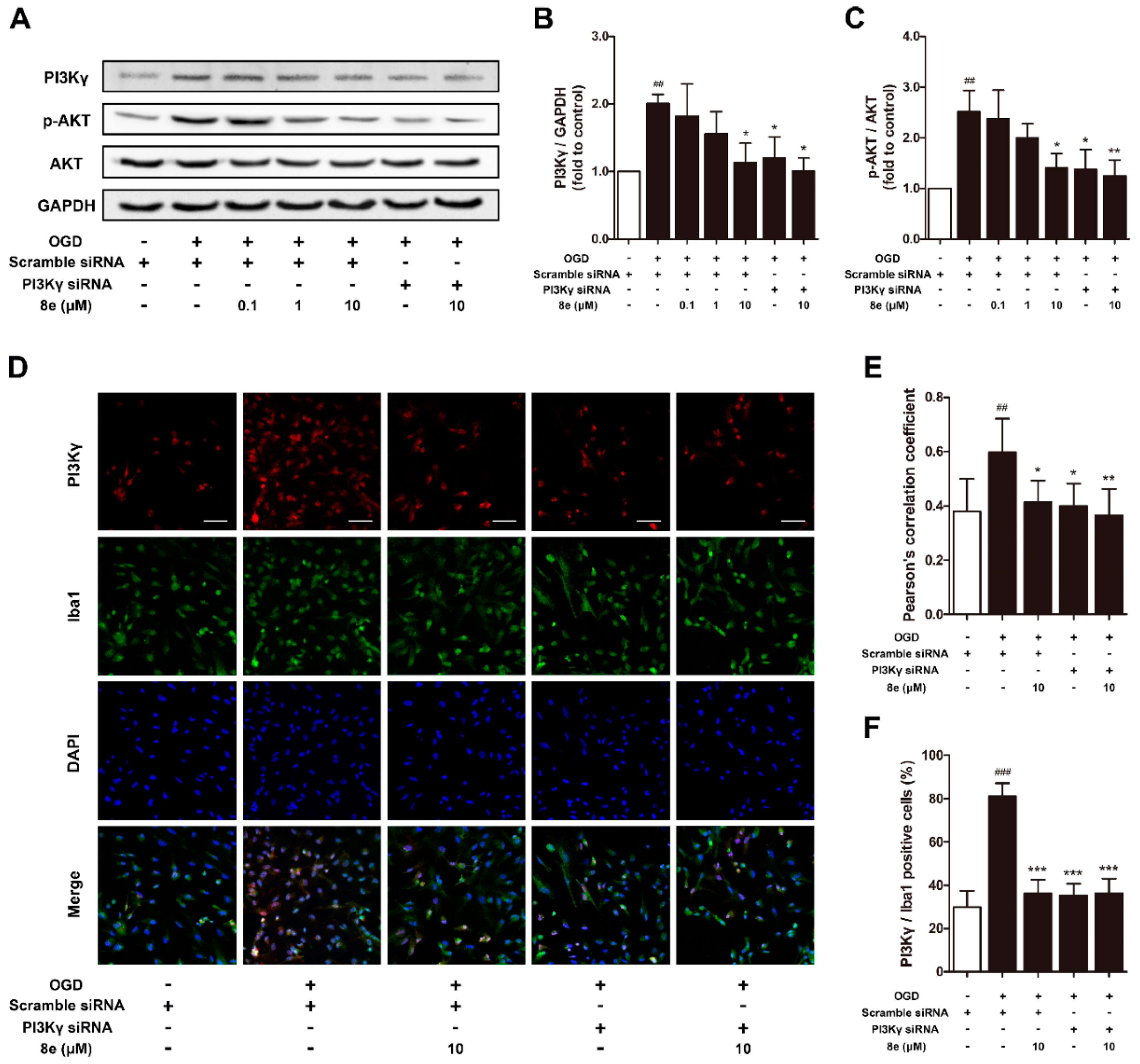
We further conducted in vitro experiments to confirm whether 8e could affect the PI3Kγ/AKT signaling pathway in primary cultured microglial cells. As expected, PI3Kγ siRNA down-regulated the levels of PI3Kγ and p-AKT in microglia subjected to OGD (p < 0.05, respectively; Figure 6A–C). 10 μM 8e significantly decreased the expression of PI3Kγ and p-AKT (p < 0.05, respectively) and so did its combination with PI3Kγ siRNA (PI3Kγ, p < 0.05; p-AKT, p < 0.01).
In addition, for double immunostaining of PI3Kγ and Iba1 antibodies, the value of Pearson’s correlation coefficient (Rr = 0.400, p < 0.05) and the ratio of PI3Kγ/Iba1 positive cells (p < 0.001) were much lessened in primary microglia by the blocking of PI3Kγ with siRNA (Figure 6D–F). 10 μM 8e and its combination with PI3Kγ siRNA exhibited similar inhibition on the fluorescent signal of PI3Kγ: Pearson’s correlation coefficient (8e, Rr = 0.413, p < 0.05; 8e + PI3Kγ siRNA, Rr = 0.365, p < 0.01) and the ratios of PI3Kγ/Iba1 positive cells (p < 0.001, respectively) were considerably reduced. However, no significant difference was found when comparing these two groups (p > 0.05, respectively). These findings were consistent with our in vivo work showing that 8e could regulate PI3Kγ signaling in microglial cells.
2.6. 8e Modulated NOX2 through PI3Kγ/AKT Signaling in Microglia Subjected to OGD In Vitro
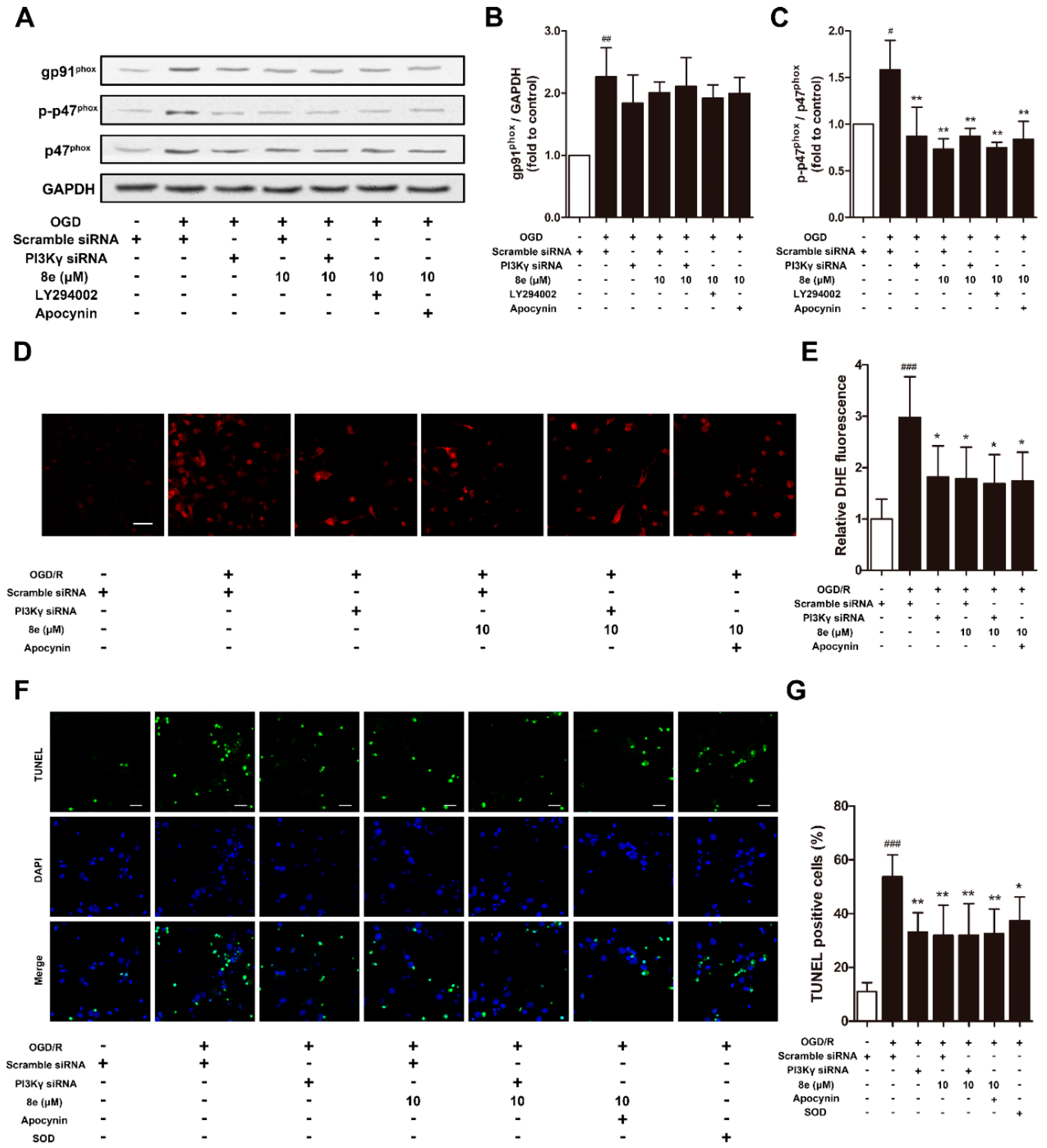
As shown in Figure 7A–C, protein expressions of NOX2 subunits after OGD were monitored to confirm whether 8e regulates NOX2 through PI3Kγ signaling in primary microglia. Cells were treated with LY294002 and apocynin as PI3K/AKT and NOX2 inhibitors, respectively. The level of gp91phox was affected by neither 10 μM 8e nor its combination with PI3Kγ siRNA, LY294002, or even apocynin. These results indicated that 8e and apocynin do not modulate NOX2 via the gp91phox subunit. On the other hand, the elevation of p-p47phox caused by OGD was eliminated by 8e (p < 0.01). Combinations of 8e with neither PI3Kγ siRNA, LY294002 nor apocynin further decrease the expression of p-p47phox when compared with 8e (p > 0.05, respectively). All these data demonstrated that 8e modulates the expression of p47phox and its phosphorylation primarily through blocking of the PI3Kγ/AKT pathway in OGD-treated microglia.
2.7. 8e Protected Neurons against Apoptosis by Down-Regulating Superoxide Production in OGD-Treated Primary Microglia In Vitro
Activated microglia can induce and amplify cell damage to neighboring neurons by the excess production of cytotoxic factors such as superoxide, nitric oxide (NO) and tumor necrosis factor-α (TNFα) [7]. In this part, we investigated whether 8e exerts neuroprotection through inhibition of superoxide formation in microglia subjected to OGD, with DHE staining. Administration of 8e profoundly ameliorated the production of DHE fluorescence (p < 0.05, Figure 7D,E), while its combination with neither PI3Kγ siRNA nor apocynin further enhanced this effect in primary cultured microglia after OGD (p > 0.05, respectively).
Next, we found that neuronal apoptosis of TUNEL staining ascended sharply following co-culture of naïve neurons with OGD-treated microglia (p < 0.001, Figure 7F,G). Single administration of 8e and PI3Kγ siRNA remarkably reduced the ratios of TUNEL positive cells (p < 0.01, respectively), while its combination with neither PI3Kγ siRNA nor apocynin decreased the number further (p > 0.05, respectively). We noticed that treatment with superoxide dismutase-1 (SOD) in culture media also attenuated neuronal apoptosis, but with less potency (p < 0.05). This finding partly confirmed that the superoxide generated in activated microglia enters extracellular space and spreads to nearby neurons [47]. Taken together, these results suggested that 8e could reduce the release of superoxide by microglia, and in turn protect neighboring neurons against apoptosis.
3. Discussion
As a gasotransmitter, H2S can rapidly travel across cell membranes without specific transporters, and hence has an impact on diverse physiological and pathological progresses [48]. A growing number of studies have shown that H2S exerts cytoprotective activity primarily through anti-oxidative, anti-inflammatory, anti-apoptotic and pro-angiogenesis actions [37,49,50,51]. However, H2S gas is not an ideal resource due to difficulties in concentration control and the risk of excessive toxicity [52]. Inorganic sulfide salts such as sodium sulfide (Na2S) and sodium hydrogen sulfide (NaHS) also release H2S rapidly and spontaneously. Recently, a series of slow-releasing H2S donors and hybrid drugs have been developed as new therapeutic approaches. They generate H2S with lower levels of release rates and longer treatment intervals, in contrast to inorganic sulfide salts. A study by Whiteman et al. indicated that the effects of H2S on inflammation depend not only on H2S concentration but also on the rate of H2S generation [53]. That may be one explanation for the seemingly controversial role of H2S in inflammation-related diseases, such as stroke [38,54]. ADT and its main metabolite ADT-OH have been used extensively as a slow-releasing donor of H2S [55]. Combinations of ADT-OH with L-DOPA and aspirin have shown protective properties in neurodegenerative processes [56,57].
3-n-butylphthalide (NBP) was approved by the China Food and Drug Administration (CFDA) in 2002 as a new drug for ischemic stroke treatment. Our group investigated a series of NBP derivatives combined with ADT-OH and reported that oral treatment with 8e provides superior antiplatelet and antithrombotic activities than NBP [36]. In this study, we verified the therapeutic effect of 8e in a rat model of transient focal cerebral ischemia. After a three-day intravenous treatment post tMCAO, 8e improved sensorimotor outcomes in three different behavioral assessments. The Longa test is one of the most frequently used neurological exams in rodent stroke studies. Although improvement trends were observed in all doses of 8e-treated groups, statistical significance was only found at 20 mg/kg following 48 h of reperfusion. This may be partly due to the fact that the Longa test focuses only on motor deficits. A recent study recommended the use of both Garcia and Longa tests to capture a broader range of neurological deficits [58]. The beam walking and prehensile traction tests are also widely applied in models of brain damage [59]. 8e markedly reduced the foot slip ratios and increased the length of time spent on the rope at 48 h and 72 h after tMCAO, indicating a protective capability of 8e in the sub-acute phase of ischemic stroke (Figure 2D–I). Meanwhile, we found that infarction volume and brain edema were notably decreased in the presence of 8e, which provided solid evidence for its effectiveness. The results of histological examination and TUNEL staining in the penumbral region further confirmed these findings (Figure 3).
After stroke, inflammatory response is initiated in ischemic penumbra which is a metabolically active, but neurophysiologically silent, region surrounding the infarct core [60]. This response is characterized by rapid activation of microglia, production of proinflammatory mediators, and infiltration of various types of inflammatory cells including neutrophils, monocytes/macrophages, T cells and other cells [61]. Resident microglial cells are among the first cells to respond to brain injury. They are activated within minutes of ischemia onset and peak at 48–72 h [62]. Considering that activated microglia are morphologically and functionally indistinguishable from blood-derived monocytes/macrophages in the brain, Schilling et al. designed a series of experiments and confirmed that blood-derived macrophages are recruited at a later time and become abundant on days 3–7 [63,64,65]. Resident microglial activation precedes and predominates over blood-derived macrophage infiltration during the first three or four days of cerebral ischemia. Invasion of neutrophils occurs slightly later at 48 h. However, the existence of neutrophils is largely masked after day three by the large-scale accumulation of activated microglia and macrophages in the penumbra region [66].
The experimental observations that PI3Kγ is an important mediator in the signaling cascade of inflammatory response, have attracted worldwide attention to this kinase. But the role of PI3Kγ in the context of brain injury, has not been extensively studied. The level of PI3Kγ was significantly increased at one day and, up to seven days, after surgical brain injury in rats [26]. Similar up-regulation was found by Jin et al. in a mouse model of tMCAO within 24 h [27]. Here we reported for the first time that the protein expression of PI3Kγ is elevated in the penumbral region of rats at 72 h following cerebral ischemia (Figure 5A,B). We also detected the cellular location of PI3Kγ by a double immunostaining technique. PI3Kγ was colocalized with Iba1, a cell marker for microglia, while limited fluorescence signals of PI3Kγ were expressed in NeuN-positive neurons (Figure 5D–I). These data indicated that microglia may be the major cellular source of PI3Kγ in the penumbral region at day three after ischemic stroke. Our findings are consistent with the time course of inflammatory cell recruitment motioned above, suggesting PI3Kγ’s involvement in microglial activation after tMCAO. Genetic ablation and pharmacological inhibition of PI3Kγ confer enhanced protection against Alzheimer’s disease, surgical brain injury and cerebral ischemia [25,26,28]. On the other hand, the selective PI3Kγ inhibitor AS252424 at a dose of 10 mg/kg significantly attenuated surgical brain injury-induced brain edema and neurological deficits at 24 and 72 h [26]. In our research, administration of AS252424, a PI3Kγ-selective inhibitor, attenuated sensorimotor deficits, infarction size and neural apoptosis following tMCAO (Figure 2 and Figure 3), proving that inhibition of PI3Kγ is a potential therapeutic strategy for neuroinflammation-mediated disorders. At the same time, 8e effectively down-regulated the level of PI3Kγ and improved I/R injury in vivo and in vitro (Figure 5 and Figure 6). The combination of 8e with PI3Kγ siRNA did not further reduce the protein expression in primary cultured microglia, indicating that 8e exerts protective effect primarily through inhibition of PI3Kγ signaling. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) negatively regulates the PI3K pathway and AKT activation. Recent reports showed that treatment with 25–100 μM NaHS reduced PTEN expression, whereas 800–1000 μM NaHS increased the level of PTEN protein in hepatocellular carcinoma cells [67]; however, the relative roles of H2S donor in PTEN regulation in innate immune cells are still mostly unexplored.
Oxidative stress is implicated in acute brain injury and chronic neurodegenerative disorders [68]. Because of its high metabolic rate and relatively reduced capacity for cellular regeneration, the brain is extremely vulnerable to oxidative damage [69]. Unfortunately, therapies based on free radical scavenger or antioxidant principles have generally failed in clinical trials, despite the promising efficacy in preclinical studies. More and more researchers have shifted their focus onto ROS-generating pathways in various pathological situations. NADPH oxidases are considered the major source of ROS production, among which NOX2 is one of the most extensively studied isoforms during ischemic injury [70,71]. A strong increase of NOX2 was reported in resident microglial cells and recruited neutrophils after stroke onset [72]. A previous study has detected the robust production of superoxide at 4 h and its decrease within 24 h after reperfusion in WT mice, which were attributed to the enhanced neutrophil infiltration into the ischemic area [28]. They also found that the protein expression of NOX2 continuously increased until 72 h after tMCAO in WT mice. In the present study, elevation in the expression of NOX2 subunits and superoxide were observed at 72 h following cerebral ischemia in rats. This ROS generation and NOX2 activation at later time-points appears to be the consequence of microglial response to brain injury, as the infiltration of neutrophils has not reached its peak and their existence is masked in the sub-acute phase of stroke. Inhibition of PI3Kγ with AS252424 showed significant reductions in superoxide formation, the protein level of p-p47phox and the fluorescence signal of p47phox in microglia at 72 h after tMCAO (Figure 4). Similar results were found with the use of PI3Kγ siRNA in primary cultured microglia following OGD. 8e also exhibited equivalent effects on regulation of phosphorylated p47phox and NOX2-derived superoxide in vivo and in vitro (Figure 7A–E). Furthermore, combination of 8e with neither PI3Kγ siRNA nor apocynin enhanced this effect in microglial cells. On the other hand, AKT is an established downstream of Class I PI3K, and it has been suggested that it is involved in the activation of p47phox in leukocytes and endothelial cells [18,19]. Our experimental data showed that, both 8e and blocking PI3Kγ eliminated the increase of p-AKT following I/R injury. The combination of 8e with LY294002, a PI3K/AKT inhibitor, did not further decrease the expression of p-p47phox when compared with 8e. Taken together, these results demonstrate that 8e modulates p47phox and its phosphorylation through PI3Kγ/AKT signaling. With the use of a co-culture model, we also observed that superoxide generation from activated microglia enters extracellular space and spreads to nearby neurons. Administration of 8e and PI3Kγ siRNA to OGD-treated microglia markedly attenuated neuronal apoptosis in naïve neurons, suggesting that 8e protects neighboring neurons from apoptosis via control of superoxide generation in microglia (Figure 7F,G).
4. Materials and Methods
4.1. Chemicals
8e (purity >98%) was synthesized by Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University. PI3Kγ-selective inhibitor AS252424, PI3K/AKT inhibitor LY294002, NOX2 inhibitor apocynin and superoxide dismutase-1 (SOD) were purchased from Aladdin Biotechnology, China. 0.9% NaCl solution containing 5% ethanol and 5% polyethylene glycol 400 (v/v, both from Sinopharm Chemical Reagent, Shanghai, China) was utilized as the vehicle.
4.2. Animals
All animal experimental protocols were approved by Ethics Committee of China Pharmaceutical University and performed in compliance with the Guideline on Administration of Laboratory Animals (China). Efforts were made to minimize the number of animals used and their suffering. Adult male Sprague-Dawley rats (250–280 g) were provided by the Comparative Medicine Centre of Yangzhou University (China) and housed under standard laboratory conditions of controlled temperature, humidity and a 12-hour light/dark cycle. The animals were allowed free access to food and water during one week of adaptation and fasted overnight before surgery.
4.3. tMCAO Surgery and Drug Treatment
Transient middle cerebral artery occlusion was accomplished as previously described [73] to mimic human cerebral ischemia/reperfusion (I/R) injury. Briefly, after achieving general anesthesia by intraperitoneal injection of chloral hydrate (300 mg/kg, Sinopharm Chemical Reagent, Shanghai, China), the regional cerebral blood flow (rCBF) was monitored by Iaser Doppler flowmetry (LDF 100C, BIOPAC Systems, Goleta, California, USA). A 4-0 nylon monofilament suture with a rounded tip (Sunbio Biotechnology, Beijing, China) was inserted into the right internal carotid artery (ICA) through the external carotid artery (ECA) stump and gently advanced to block the origin of the middle cerebral artery (MCA). Restoration of MCA blood flow was accomplished by withdrawing the suture 60 min later. The sham-operated rats received the same surgery without suture insertion. Rats with unsuccessful occlusion (reduction of rCBF <75%) and unsuccessful reperfusion (recovery of rCBF <70%) were excluded from the study [74]. The core body temperature was maintained at 37 ± 0.5 °C throughout the procedure.
Before surgery, rats were randomly divided into six groups, including sham, tMCAO + vehicle, tMCAO + 10 mg/kg AS252424, tMCAO + 5 mg/kg 8e, tMCAO + 10 mg/kg 8e, and tMCAO + 20 mg/kg 8e groups. Among the 120 rats that underwent the tMCAO surgery, 4 rats were excluded for unsuccessful occlusion and 5 rats were excluded for unsuccessful reperfusion; 7 rats died during the surgery and 9 after the surgery. AS252424 and other drugs were administered intraperitoneally or intravenously at the beginning of reperfusion and repeated every 24 h for 3 days. The doses of 8e and AS252424 were calculated based on the clinical use of NBP and sodium chloride injection and previous report [26].
4.4. Behavioral Assessment
4.4.1. Longa Test
This neurological assessment is a 5-point scale: 0, no neurological deficit; 1, failure to extend the left forepaw fully; 2, circling to the left; 3, falling to the left; and 4, no spontaneous walking and being depressed [75]. At 24 h, 48 h and 72 h after tMCAO, the performance of rats was evaluated by a blinded investigator.
4.4.2. Beam Walking
Fine motor coordination was assessed with the beam walking task. For 2 days before surgery, the rats were trained to traverse an elevated narrow beam (2.5 cm in width, 1.2 m in length and 0.8 m in height) to enter a darkened goal box at the opposite end of beam. The test was conducted at 24 h, 48 h and 72 h after tMCAO and the performance of the rats was videotaped and analyzed by calculating the foot slip ratios of the impaired limbs (number of foot faults/number of total steps) by a blinded investigator. A fault was defined as any foot slip off the top surface of the beam or any limb use on the side of the beam [76].
4.4.3. Prehensile Traction
A 0.6 m long and 4 mm diameter wire was placed horizontally 0.4 m above a foam pad [59]. At 24 h, 48 h and 72 h after tMCAO, the animals were released with their forepaws placed onto the rope to evaluate muscle strength and equilibrium. The time to falling (maximum 60 s) was noted.
4.5. Infarct Volume and Brain Edema Measurement
At 72 h after tMCAO, rats were deeply anesthetized with a lethal dose of chloral hydrate (350 mg/kg) and the brains were frozen at −20 °C for 20 min. The frozen brains were cut into 2-mm-thick coronal sections and stained with 2% of 2,3,5-Triphenyltetrazolium chloride (TTC, Sigma-Aldrich, St. Louis, MO, USA) for 20 min at 37 °C. The brain slices were then fixed in 4% formaldehyde overnight at room temperature and photographed with a digital camera. The area of infarction in each slice was measured using Image J (NIH, Bethesda, MD, USA), and infarct volume was obtained from the product of average thickness (2 mm) and the sum of infarction areas in all slices examined. Infarct volume (%) = total infarct volume/total brain volume × 100%.
Brain edema was evaluated by measuring the brain water content as previously described [77]. Brains were rapidly removed at 72 h post reperfusion and both hemispheres were weighed before and after drying at 100 °C for 24 h. The brain water content (%) = (1—dry weight/wet weight) × 100%.
4.6. Primary Microglia Culture
Microglial culture was prepared as previously described with modification [74]. The brain of postnatal (P0-2) Sprague-Dawley (SD) rats was separated and the meninges were carefully removed. The cerebral cortex was cut into small fragments and digested with trypsin (0.25%, Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature. The cell suspension was filtered through 100-μm and 40-μm cell strainers (BD Falcon, Bedford, MA, USA). After centrifugation for 5 min at 100 g, the pallet was resuspended in Dulbecco’s modified Eagle’s medium/Ham′s nutrient mixture F-12 (DMEM/F12) containing 10% fetal bovine serum (FBS) and seeded into poly-lysine (0.1 mg/mL, Sigma-Aldrich, St. Louis, MO, USA) pre-coated 75-cm2 flasks in a humidified incubator (95% O2, 5% CO2, 37 °C). Culture media were changed every 2 days. Two weeks later, the isolation of microglia was accomplished by gentle shaking of the culture flask. The collected cells were seeded into 24-well plates with coverslips, or the upper inserts of Transwell chambers (0.4 μm diameter pores, Corning, NY, USA).
4.7. siRNA Transfection and OGD Treatment
PI3Kγ siRNA and negative control (40 nM, all from GenePharma, Shanghai, China) were transfected into primary cultured microglia using Lipofectamine 2000 (Thermo Fisher Scientific, San Jose, CA, USA) 24 h post cell seeding. PI3Kγ siRNA consisted of an RNA duplex containing a sense strand 5′-GCA UUG AUA UCC CUG UCU UTT-3′ and an antisense strand 5’-AAG ACA GGG AUA UCA AUG CTT-3′.
48 h after transfection, microglia were subjected to OGD by replacing the culture media with glucose free DMEM containing 2% FBS and transferring to an OGD incubator chamber (95% N2, 5% CO2, 37 °C). After 1 h of OGD, the plates were removed from the chamber and the culture media were replaced with DMEM/F12 and 2% FBS to achieve reoxygenation. Microglial cells were harvested for further experimentation 24 h post reoxygenation. Drug treatments including LY294002 (10 μM), apocynin (1 mM) and SOD (200 U/mL) were applied to the cell culture 2 h before and during OGD/reoxygenation.
4.8. Primary Neuron Culture and Neuron-Microglia Co-Culture
Primary neuron cultures were prepared using embryos (E16-18) of SD rats. The cortex of the brainwas dissected and digested with 800 U DNase I and 2 mg/mL papain (both from Biosharp, Hefei, Anhui, China) at 37 °C for 30 min [78]. The cell suspension was filtered through 40-μm cell strainers (BD Falcon, Bedford, MA, USA). After centrifugation for 5 min at 100 g, the cells were resuspended in DMEM/F12 containing 10% FBS and seeded into poly-l-lysine pre-coated lower wells of Transwell chambers with coverslips. The culture media were replaced by Neurobasal medium supplemented with 2% B27 (both from Gibco, Carlsbad, CA, USA) after 4 h of incubation. Half of the media were replaced with fresh Neurobasal/B27 every 4 days.
After 7–10 days of incubation, the lower wells containing naïve neurons were allowed to communicate with microglia subjected to 1 h of OGD in the upper inserts of the Transwell chambers for 24 h (95% N2, 5% CO2, 37 °C). The culture media were all replaced with Neurobasal/B27.
4.9. Histological Observation and Apoptosis Analysis
Rats were anesthetized with 350 mg/kg of chloral hydrate and intracardially perfused with iced normal saline followed by 4% paraformaldehyde (PFA, Sinopharm Chemical Reagent, Shanghai, China) at 72 h after tMCAO. The brains were post-fixed in PFA overnight and embedded in paraffin. Microglia-bearing coverslips were removed from the 24-well plates 24 h after OGD, then fixed with 4% PFA for 30 min and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA). For histological observation, 5-μm brain slices at the level of bregma +0.7 to −4.3 mm were counterstained with hematoxylin and eosin. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining were conducted on both brain slices and coverslips according to manufacturer’s instructions (KeyGEN Biotech, Nanjing, China). Tissue sections of the penumbral regions were observed under a microscope with 200× magnification (CKX41, Olympus, Tokyo, Japan). Cell TUNEL staining was captured with a confocal microscope (LSM700, Zeiss, Oberkochen, Germany). The numbers of TUNEL-positive cells were analyzed by a blinded investigator with Image J.
4.10. Oxidative Stress Evaluation
72 h after tMCAO, the animals were anesthetized with 350 mg/kg of chloral hydrate and perfused with iced normal saline through the heart. 5-μm frozen brain slices at the level of bregma +0.7 to −4.3 mm were cut on a CM1950 cryostat (Leica, Wetzlar, Germany) and further fixed in acetone for 10 min at 4 °C. Microglia-bearing coverslips were prepared as mentioned above. The slides and coverslips were incubated with 2.5 μM dihydroethidium (DHE, Beyotime Biotechnology, Shanghai, China) at 37 °C for 30 min in a humidified chamber which was shielded from light [79]. DHE produces red fluorescence when oxidized to ethidium by superoxide anion and intercalates into DNA. Fluorescent images were acquired using a confocal microscope. The mean fluorescence intensity for each region (integrated density/area) of the penumbral regions was blind-analyzed by Image J and normalized to the level of the sham or control group.
4.11. Immunofluorescent Staining
Frozen brain slices and microglia-bearing coverslips mentioned above were immunostained as previously described [80]. Briefly, after incubation with primary antibodies of p47phox (1:200, Sigma-Aldrich, St. Louis, MO, USA), PI3Kγ (1:500, Novus, Littleton, CO, USA), Iba1 (1:500, Millipore, Billerica, MA, USA) and NeuN (1:500, Millipore, Billerica, MA, USA), antibody binding was visualized with Alexa Fluor 488-labeled goat anti-mouse IgG and Cy3-labeled goat anti-rabbit IgG (1:200, both from Beyotime Biotechnology, Shanghai, China). Cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Images of the penumbral regions were obtained using a confocal microscope. The Pearson’s correlation coefficient and the numbers of antibody-positive cells were analyzed by a blinded investigator using Image J.
4.12. Western Blotting
Total protein was prepared from the penumbral regions at the level of bregma +0.7 to −4.3 mm 72 h after tMCAO, and primary cultured cells 24 h after OGD as reported previously [81]. Samples were homogenized in iced radio immunoprecipitation assay (RIPA) buffer supplemented with protease and phosphatase inhibitor cocktail (all from Beyotime Biotechnology, Shanghai, China). Following centrifugation at 13,000 g for 20 min at 4 °C, the supernatant was collected. Equal amounts of protein (20 μg) were loaded onto 8–10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% non-fat milk in Tris-Buffered Saline with Tween 20 (0.1%, v/v, TBST) for 1 h at room temperature, and incubated overnight at 4 °C with primary antibodies against PI3Kγ, gp91phox, p22phox, p47phox (all from Santa Cruz Biotechnology, Dallas, TX, USA), phosphorylation-p47phox at Ser328 (Abcam, Cambridge, MA, USA), AKT, phosphorylation-AKT at Ser473 and GAPDH (all from Cell Signaling Technology, Danvers, MA, USA). Then the membranes were hybridized with the appropriate horseradish peroxidase (HRP) conjugated secondary antibodies (1:5000, Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature. Protein expression was detected by the enhanced chemiluminescence (ECL) method on an imaging system (ChmiScope 2850; Clinx Science Instruments, Shanghai, China) and normalized to internal reference.
4.13. Statistical Analysis
Data are presented as the mean ± SD. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). All behavioral data were analyzed using repeated measures ANOVA. Group differences were ascertained using Dunn or Tukey post hoc comparisons where appropriate. As the normality test with the Kolmogorov-Smirnov test was passed, the rest of the data were analyzed by one-way ANOVA with Tukey’s post hoc test. p < 0.05 was considered statistically significant.
5. Conclusions
Our study demonstrated that 8e attenuates sensorimotor deficits, focal infarction and neural apoptosis in cerebral ischemia. 8e may exert protective effects partly thorough regulation of superoxide production in activated microglia via the PI3Kγ/AKT/NOX2 signaling pathway. These findings about 8e widen our knowledge of H2S-releasing compounds and provide a novel direction for stroke research.
Author Contributions
L.W., Y.Z. and H.J. conceived and designed the experiments; X.W. synthesized the compound 8e; L.W., X.W. and T.L. performed the experiments; T.L. analyzed the data; L.W. and H.J. wrote the paper.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81872867, No. 81503150, No. 21502071), the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Policy Directive Program of Jiangsu Province (BY2015072-03).
Acknowledgments
The authors would like to express their gratitude to the core facility of State Key Laboratory of Natural Medicines, China Pharmaceutical University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update A Report from the American Heart Association. Circulation 2017, 135, E146–E603. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Davalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.A. Reperfusion Therapies for Acute Ischemic Stroke Current Pharmacological and Mechanical Approaches. Stroke 2011, 42, S16–S19. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H. Experimental models, neurovascular mechanisms and translational issues in stroke research. Br. J. Pharmacol. 2008, 153, S396–S405. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med. 2008, 14, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. JPPP 2013, 5, 73–90. [Google Scholar]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Butovsky, O.; Bruck, W.; Hanisch, U.K. Microglial phenotype: Is the commitment reversible? Trends Neurosci. 2006, 29, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Owens, T.; Boddeke, E. What is microglia neurotoxicity (Not)? Glia 2014, 62, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Browning, E.A.; Hong, N.K.; DeBolt, K.; Sorokina, E.M.; Liu, W.D.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart C 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okkenhaug, K. Signaling by the Phosphoinositide 3-Kinase Family in Immune Cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camps, M.; Ruckle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Francon, B.; et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.F.; Bartolome, A.; Hernandez, C.; Flores, J.M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; Ruckle, T.; Schwarz, M.K.; Rodriguez, S.; et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005, 11, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Katare, R.; Meloni, M.; Damilano, F.; Hirsch, E.; Emanueli, C.; Madeddu, P. Involvement of Phosphoinositide 3-Kinase gamma in Angiogenesis and Healing of Experimental Myocardial Infarction in Mice. Circ. Res. 2010, 106, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Ruckle, T.; Schwarz, M.K.; Rommel, C. PI3K gamma inhibition: Towards an ‘aspirin of the 21st century’? Nat. Rev. Drug Discov. 2006, 5, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Passos, G.F.; Figueiredo, C.P.; Prediger, R.D.S.; Silva, K.A.B.S.; Siqueira, J.M.; Duarte, F.S.; Leal, P.C.; Medeiros, R.; Calixto, J.B. Involvement of phosphoinositide 3-kinase gamma in the neuro-inflammatory response and cognitive impairments induced by beta-amyloid 1–40 peptide in mice. Brain Behav. Immun. 2010, 24, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sherchan, P.; Wang, Y.; Reis, C.; Applegate, R.L., 2nd; Tang, J.; Zhang, J.H. Phosphoinositide 3-Kinase Gamma Contributes to Neuroinflammation in a Rat Model of Surgical Brain Injury. J. Neurosci. 2015, 35, 10390–10401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Yu, S.; Song, Z.; Quillin, J.W.; Deasis, D.P.; Penninger, J.M.; Nanda, A.; Granger, D.N.; Li, G. Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem. Biophys. Res. Commun. 2010, 399, 458–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Song, Z.F.; Yu, S.Y.; Piazza, A.; Nanda, A.; Penninger, J.M.; Granger, D.N.; Li, G.H. Phosphatidylinositol-3-Kinase Gamma Plays a Central Role in Blood-Brain Barrier Dysfunction in Acute Experimental Stroke. Stroke 2011, 42, 2033–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.; Frahm, C.; Schneble, N.; Muller, J.P.; Brodhun, M.; Franco, I.; Witte, O.W.; Hirsch, E.; Wetzker, R.; Bauer, R. Phosphoinositide 3-Kinase gamma Restrains Neurotoxic Effects of Microglia After Focal Brain Ischemia. Mol. Neurobiol. 2016, 53, 5468–5479. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Lee, H.R.; Sim, S.E.; Baek, J.; Yu, N.K.; Choi, J.H.; Ko, H.G.; Lee, Y.S.; Park, S.W.; Kwak, C.; et al. PI3K gamma is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat. Neurosci. 2011, 14, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen Sulfide and Cell Signaling. Annu. Rev. Pharmacol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yao, X.; Zhang, Y.; Li, W.; Kang, K.; Sun, L.; Sun, X. The protective role of hydrogen sulfide in myocardial ischemia-reperfusion-induced injury in diabetic rats. Int. J. Cardiol. 2011, 152, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Calvert, J.W.; Duranski, M.R.; Ramachandran, A.; Lefer, D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.P.; Hosgood, S.A.; Patel, M.; Rose, R.; Read, K.; Nicholson, M.L. Effects of hydrogen sulphide in an experimental model of renal ischaemia-reperfusion injury. Br. J. Surg. 2012, 99, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Kosugi, S.; Tokuda, K.; Khatri, A.; Nguyen, R.; Atochin, D.N.; Kida, K.; Van Leyen, K.; Arai, K.; Ichinose, F. A novel hydrogen sulfide-releasing N-methyl-D-aspartate receptor antagonist prevents ischemic neuronal death. J. Biol. Chem. 2012, 287, 32124–32135. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Sheng, X.; Huang, Z.; Li, T.; Zhang, M.; Xu, J.; Ji, H.; Yin, J.; Zhang, Y. Design, synthesis and biological evaluation of hydrogen sulfide releasing derivatives of 3-n-butylphthalide as potential antiplatelet and antithrombotic agents. Org. Biomol. Chem. 2014, 12, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cao, Y.; Ao, G.; Hu, L.; Liu, H.; Wu, J.; Wang, X.; Jin, M.; Zheng, S.; Zhen, X.; et al. CaMKKbeta-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid. Redox Signal. 2014, 21, 1741–1758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Jia, J.; Ao, G.Z.; Hu, L.F.; Liu, H.; Xiao, Y.Q.; Du, H.P.; Alkayed, N.J.; Liu, C.F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; McGeer, E.; Kodela, R.; Kashfi, K.; McGeer, P.L. NOSH-aspirin (NBS-1120), a novel nitric oxide and hydrogen sulfide releasing hybrid, attenuates neuroinflammation induced by microglial and astrocytic activation: A new candidate for treatment of neurodegenerative disorders. Glia 2013, 61, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yuan, L.; Liu, D.X.; Wang, J.M.; Wang, S.L.; Zhang, Q.R.; Gong, Y.F.; Liu, H.D.; Hao, A.J.; Wang, Z. Hydrogen sulfide attenuates hypoxia-induced neurotoxicity through inhibiting microglial activation. Pharmacol. Res. 2014, 84, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Sodium thiosulfate attenuates glial-mediated neuroinflammation in degenerative neurological diseases. J. Neuroinflamm. 2016, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.M.; Chen, G.W.; Liu, Y.; Xiao, X.Z. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Lu, M.; Tiong, C.X.; Dawe, G.S.; Hu, G.; Bian, J.S. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 2010, 9, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Lee, D.Y.; Mariappan, M.M.; Feliers, D.; Ghosh-Choudhury, G.; Abboud, H.E.; Gorin, Y.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J. Biol. Chem. 2017, 292, 5665–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Minnella, A.M.; Shen, Y.; El-Benna, J.; Swanson, R.A. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Z.; Shi, M.M.; Xie, L.; Wu, Z.Y.; Li, G.; Hua, F.; Bian, J.S. Sulfhydration of p66Shc at Cysteine59 Mediates the Antioxidant Effect of Hydrogen Sulfide. Antioxid. Redox Signal. 2014, 21, 2531–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.L.; Huang, X.W.; Wang, Y.G.; Cao, Y.X.; Zhang, C.C.; Zhu, Y.C. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3 beta-dependent opening of mPTP. Am. J. Physiol. Heart C 2010, 298, H1310–H1319. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Biggs, T.D.; Xian, M. Hydrogen sulfide (H2S) releasing agents: Chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.H.; Parkinson, D.B.; Moore, P.K. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, K.; Chen, C.P.L.H.; Halliwell, B.; Moore, P.K.; Wong, P.T.H. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 2006, 37, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Olson, K.R. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. 2013, 85, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Tazzari, V.; Giustarini, D.; Rossi, R.; Sparatore, A.; Del Soldato, P.; McGeer, E.; McGeer, P.L. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: Potential for treating Parkinson disease. J. Biol. Chem. 2010, 285, 17318–17328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Sparatore, A.; Del Soldato, P.; Bian, J.S. ACS84, a novel hydrogen sulfide-releasing compound, protects against amyloid beta-induced cell cytotoxicity. Neurochem. Int. 2011, 58, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bachour, S.P.; Hevesi, M.; Bachour, O.; Sweis, B.M.; Mahmoudi, J.; Brekke, J.A.; Divani, A.A. Comparisons between Garcia, Modo, and Longa rodent stroke scales: Optimizing resource allocation in rat models of focal middle cerebral artery occlusion. J. Neurol. Sci. 2016, 364, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zausinger, S.; Hungerhuber, E.; Baethmann, A.; Reulen, H.J.; Schmid-Elsaesser, R. Neurological impairment in rats after transient middle cerebral artery occlusion: A comparative study under various treatment paradigms. Brain Res. 2000, 863, 94–105. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Koerner, I.P.; Moller, T. Microglia in ischemic brain injury. Future Neurol. 2010, 5, 227–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Yang, G.J.; Li, G.H. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukocyte Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Strecker, J.K.; Schabitz, W.R.; Ringelstein, E.B.; Kiefer, R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience 2009, 161, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Besselmann, M.; Leonhard, C.; Mueller, M.; Ringelstein, E.B.; Kiefer, R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: A study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2003, 183, 25–33. [Google Scholar] [CrossRef]
- Schilling, M.; Besselmann, M.; Muller, M.; Strecker, J.K.; Ringelstein, E.B.; Kiefer, R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2005, 196, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Weston, R.M.; Jones, N.M.; Jarrott, B.; Callaway, J.K. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: Histochemical and myeloperoxidase correlation with temporal changes in brain injury. J. Cereb. Blood Flow Metab. 2007, 27, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.D.; Li, M.L.; Tian, W.K.; Wang, S.W.; Cui, L.Z.; Li, H.; Wang, H.J.; Ji, A.L.; Li, Y.Z. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci. Rep. 2017, 7, 5134. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Kahles, T.; Brandes, R.P. Which NADPH Oxidase Isoform Is Relevant for Ischemic Stroke? The Case for Nox 2. Antioxid. Redox Signal. 2013, 18, 1400–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, F.; Teixeira, P.C.; Braunersreuther, V.; Mach, F.; Vuilleumier, N.; Montecucco, F. Pathophysiology and Treatments of Oxidative Injury in Ischemic Stroke: Focus on the Phagocytic NADPH Oxidase 2. Antioxid. Redox Signal. 2015, 23, 460–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.P.; Cairns, B.; Rae, J.; Errett-Baroncini, C.; Hongo, J.A.S.; Erickson, R.W.; Curnutte, J.T. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 2001, 21, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Wang, L.N.; Li, T.T.; Huang, Z.J.; Lai, Y.S.; Ji, H.; Wan, X.L.; Xu, J.Y.; Tian, J.D.; Zhang, Y.H. Novel Hybrids of Optically Active Ring-Opened 3-n-Butylphthalide Derivative and Isosorbide as Potential Anti-Ischemic Stroke Agents. J. Med. Chem. 2013, 56, 3078–3089. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Wang, J.; Fang, Y.Q.; Liu, Y.; Chen, T.; Sun, H.; Zhou, X.F.; Liao, H. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav. Immun. 2016, 56, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Biernaskie, J.; Chernenko, G.; Corbett, D. Efficacy of rehabilitative experience declines with time after focal ischemic brain injury. J. Neurosci. 2004, 24, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Huang, P.L.; Panahian, N.; Fishman, M.C.; Moskowitz, M.A. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J. Cereb. Blood Flow Metab. 1996, 16, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Schlichter, L.C. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008, 28, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, S.J.; Song, E.C.; Kim, E.H.; Park, D.K.; Sinn, D.I.; Kim, J.M.; Kim, M.; et al. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral Hemorrhage. J. Pharmacol. Exp. Ther. 2007, 322, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, T.; Yu, H.; Shen, H.; Xia, W. Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood-brain barrier disruption. J. Neuroinflamm. 2014, 11, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Xiang, P.; Li, T.; Lan, L.; Xu, X.; Lu, G.; Ji, H.; Zhang, Y.; Li, Y. NOSH-NBP, a Novel Nitric Oxide and Hydrogen Sulfide- Releasing Hybrid, Attenuates Ischemic Stroke-Induced Neuroinflammatory Injury by Modulating Microglia Polarization. Front. Cell. Neurosci. 2017, 11, 154. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound 8e is not available from the authors. |
Figure 1.
Chemical structure of 8e.

Figure 2.
8e enhanced sensorimotor function after cerebral ischemia/reperfusion (I/R) in rats. (A–C) Neurological scores in Longa test; (D–F) foot slip ratios in beam walking test; (G–I) time spent on the rope in prehensile traction test at 24 h, 48 h and 72 h after transient middle cerebral artery occlusion (tMCAO). Data are presented as mean ± SD (n = 15). ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. tMCAO + vehicle group.
Figure 2.
8e enhanced sensorimotor function after cerebral ischemia/reperfusion (I/R) in rats. (A–C) Neurological scores in Longa test; (D–F) foot slip ratios in beam walking test; (G–I) time spent on the rope in prehensile traction test at 24 h, 48 h and 72 h after transient middle cerebral artery occlusion (tMCAO). Data are presented as mean ± SD (n = 15). ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. tMCAO + vehicle group.

Figure 3.
8e protected against cerebral I/R injury, histological damage and neural apoptosis at 72 h after tMCAO in rats. (A) Representative images of TTC staining on rat brain sections; (B) quantitative analysis of infarct volumes in TTC staining (n = 6); (C) brain water contents in ipsilateral hemispheres (n = 6); (D) representative images of H&E staining; (E) TUNEL staining at 200 × magnification. Scale bar = 100 μm; (F) quantitative analysis of the percentage ratios of TUNEL positive cells (n = 3). Data are presented as mean ± SD. # p < 0.05, ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01 vs. tMCAO + vehicle group.
Figure 3.
8e protected against cerebral I/R injury, histological damage and neural apoptosis at 72 h after tMCAO in rats. (A) Representative images of TTC staining on rat brain sections; (B) quantitative analysis of infarct volumes in TTC staining (n = 6); (C) brain water contents in ipsilateral hemispheres (n = 6); (D) representative images of H&E staining; (E) TUNEL staining at 200 × magnification. Scale bar = 100 μm; (F) quantitative analysis of the percentage ratios of TUNEL positive cells (n = 3). Data are presented as mean ± SD. # p < 0.05, ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01 vs. tMCAO + vehicle group.

Figure 4.
8e attenuated superoxide production and the expression of p47phox in microglia at 72 h after tMCAO in rats. (A) Representative images of DHE fluorescence in penumbral region. Scale bar = 50 μm; (B) semiquantitative analysis of superoxide production in DHE fluorescence; (C) representative bands of Western blot assay for gp91phox, p22phox, p47phox and p-p47phox (Ser328) in penumbral region; (D–G) semiquantitative analysis of gp91phox, p22phox, p47phox and p-p47phox protein levels; (H) representative confocal fluorescence images of penumbral region labeled with p47phox (red), Iba1 (green) antibodies and DAPI (blue). Scale bar = 50 μm; (I) Pearson’s correlation coefficients between p47phox (red) and Iba1 (green) fluorescence; (J) quantitative analysis of the percentage ratios of p47phox/Iba1 positive cells to Iba1 positive cells. Data are presented as mean ± SD (n = 3). # p < 0.05, ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. tMCAO + vehicle group.
Figure 4.
8e attenuated superoxide production and the expression of p47phox in microglia at 72 h after tMCAO in rats. (A) Representative images of DHE fluorescence in penumbral region. Scale bar = 50 μm; (B) semiquantitative analysis of superoxide production in DHE fluorescence; (C) representative bands of Western blot assay for gp91phox, p22phox, p47phox and p-p47phox (Ser328) in penumbral region; (D–G) semiquantitative analysis of gp91phox, p22phox, p47phox and p-p47phox protein levels; (H) representative confocal fluorescence images of penumbral region labeled with p47phox (red), Iba1 (green) antibodies and DAPI (blue). Scale bar = 50 μm; (I) Pearson’s correlation coefficients between p47phox (red) and Iba1 (green) fluorescence; (J) quantitative analysis of the percentage ratios of p47phox/Iba1 positive cells to Iba1 positive cells. Data are presented as mean ± SD (n = 3). # p < 0.05, ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. tMCAO + vehicle group.

Figure 5.
8e inhibited phosphoinositide 3-kinase gamma (PI3Kγ) signaling in microglia at 72 h after tMCAO in rats. (A) Representative bands of Western blot for PI3Kγ, AKT and p-AKT in penumbral region; (B,C) semiquantitative analysis of PI3Kγ and p-AKT protein levels; (D) representative confocal fluorescence images of penumbral region labeled with PI3Kγ (red), Iba1 (green) antibodies and DAPI (blue). Scale bar = 50 μm; (E) representative confocal fluorescence images of penumbral region labeled with PI3Kγ (red), NeuN (green) antibodies and DAPI (blue). Scale bar = 50 μm; (F) Pearson’s correlation coefficients between PI3Kγ (red) and Iba1 (green) fluorescence; (G) quantitative analysis of the percentage ratios of PI3Kγ/Iba1 positive cells to Iba1 positive cells; (H) Pearson’s correlation coefficients between PI3Kγ (red) and NeuN (green) fluorescence; (I) quantitative analysis of the percentage ratios of PI3Kγ/NeuN positive cells to NeuN positive cells. Data are presented as mean ± SD (n = 3). ## p < 0.01, ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. tMCAO + vehicle group.
Figure 5.
8e inhibited phosphoinositide 3-kinase gamma (PI3Kγ) signaling in microglia at 72 h after tMCAO in rats. (A) Representative bands of Western blot for PI3Kγ, AKT and p-AKT in penumbral region; (B,C) semiquantitative analysis of PI3Kγ and p-AKT protein levels; (D) representative confocal fluorescence images of penumbral region labeled with PI3Kγ (red), Iba1 (green) antibodies and DAPI (blue). Scale bar = 50 μm; (E) representative confocal fluorescence images of penumbral region labeled with PI3Kγ (red), NeuN (green) antibodies and DAPI (blue). Scale bar = 50 μm; (F) Pearson’s correlation coefficients between PI3Kγ (red) and Iba1 (green) fluorescence; (G) quantitative analysis of the percentage ratios of PI3Kγ/Iba1 positive cells to Iba1 positive cells; (H) Pearson’s correlation coefficients between PI3Kγ (red) and NeuN (green) fluorescence; (I) quantitative analysis of the percentage ratios of PI3Kγ/NeuN positive cells to NeuN positive cells. Data are presented as mean ± SD (n = 3). ## p < 0.01, ### p < 0.001 vs. sham group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. tMCAO + vehicle group.

Figure 6.
8e downregulated PI3Kγ signaling in primary microglial cells at 24 h after oxygen–glucose deprivation (OGD). (A) Representative bands of Western blot for PI3Kγ, AKT and p-AKT in primary microglia; (B,C) semiquantitative analysis of PI3Kγ and p-AKT protein levels; (D) representative confocal fluorescence images of primary microglia labeled with PI3Kγ (red), Iba1 (green) antibodies and DAPI (blue). Scale bar = 50 μm; (E) Pearson’s correlation coefficients between PI3Kγ (red) and Iba1 (green) fluorescence; (F) quantitative analysis of the percentage ratios of PI3Kγ/Iba1 positive cells to Iba1 positive cells. Data are presented as mean ± SD (n = 3). ## p < 0.01, ### p < 0.001 vs. control group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. OGD group.
Figure 6.
8e downregulated PI3Kγ signaling in primary microglial cells at 24 h after oxygen–glucose deprivation (OGD). (A) Representative bands of Western blot for PI3Kγ, AKT and p-AKT in primary microglia; (B,C) semiquantitative analysis of PI3Kγ and p-AKT protein levels; (D) representative confocal fluorescence images of primary microglia labeled with PI3Kγ (red), Iba1 (green) antibodies and DAPI (blue). Scale bar = 50 μm; (E) Pearson’s correlation coefficients between PI3Kγ (red) and Iba1 (green) fluorescence; (F) quantitative analysis of the percentage ratios of PI3Kγ/Iba1 positive cells to Iba1 positive cells. Data are presented as mean ± SD (n = 3). ## p < 0.01, ### p < 0.001 vs. control group. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. OGD group.

Figure 7.
8e modulated NOX2 through PI3Kγ/AKT signaling in primary microglial cells and protected neurons from apoptosis at 24 h after OGD. (A) Representative images of Western blot for gp91phox, p47phox and p-p47phox expressions in primary microglia; (B,C) semiquantitative analysis of gp91phox and p-p47phox protein levels; (D) representative DHE fluorescence images of primary microglia at 24 h after OGD. Scale bar = 50 μm; (E) semiquantitative analysis of superoxide production in DHE fluorescence images; (F) representative TUNEL images of primary neurons counterstained with DAPI (blue). Scale bar = 20 μm; (G) quantitative analysis of the percentage ratios of TUNEL positive cells. Data are presented as mean ± SD (n = 3). # p < 0.05, ## p < 0.01, ### p < 0.001 vs. control group. * p < 0.05, ** p < 0.01, vs. OGD group.
Figure 7.
8e modulated NOX2 through PI3Kγ/AKT signaling in primary microglial cells and protected neurons from apoptosis at 24 h after OGD. (A) Representative images of Western blot for gp91phox, p47phox and p-p47phox expressions in primary microglia; (B,C) semiquantitative analysis of gp91phox and p-p47phox protein levels; (D) representative DHE fluorescence images of primary microglia at 24 h after OGD. Scale bar = 50 μm; (E) semiquantitative analysis of superoxide production in DHE fluorescence images; (F) representative TUNEL images of primary neurons counterstained with DAPI (blue). Scale bar = 20 μm; (G) quantitative analysis of the percentage ratios of TUNEL positive cells. Data are presented as mean ± SD (n = 3). # p < 0.05, ## p < 0.01, ### p < 0.001 vs. control group. * p < 0.05, ** p < 0.01, vs. OGD group.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, L.; Wang, X.; Li, T.; Zhang, Y.; Ji, H. 8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia. Molecules 2018, 23, 2828. https://doi.org/10.3390/molecules23112828
AMA Style
Wang L, Wang X, Li T, Zhang Y, Ji H. 8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia. Molecules. 2018; 23(11):2828. https://doi.org/10.3390/molecules23112828
Chicago/Turabian StyleWang, Linna, Xiaoli Wang, Tingting Li, Yihua Zhang, and Hui Ji. 2018. "8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia" Molecules 23, no. 11: 2828. https://doi.org/10.3390/molecules23112828