Ultrasound-Assisted Metal-Mediated Method for the Formation of Tetrahydro-3,3′-Disubstituted Biscoumarins


Department of Organic Chemistry and Pharmacognosy, Faculty of Chemistry and Pharmacy, Sofia University “St. Kliment Ohridski”, 1 J. Bouchier Buld., 1164 Sofia, Bulgaria
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(11), 2810; https://doi.org/10.3390/molecules23112810
Submission received: 27 September 2018
/
Revised: 24 October 2018
/
Accepted: 26 October 2018
/
Published: 29 October 2018
(This article belongs to the Section Organic Chemistry)
Abstract
:A new method for faster and simple preparation of 3,3′,4,4′-tetrahydro-3,3′-disubstituted-4,4′-biscoumarins with participation of an organozinc reagent is reported. The reaction is promoted by ultrasound irradiation and it offers a simple experimental setup and excellent reproducibility of the results. Moreover, homodimers were isolated in yields of 45–92%. The dimerization conditions are applicable to coumarins with electron-withdrawing groups at third position.

1. Introduction
Benzopyrans and their analogs are a large class of compounds characterized by great diversity of pharmacological properties. The main members of this class of compounds are coumarins, 3,4-dihydrocoumarins, flavonoids, and biscoumarins isolated from different medical plants. These substances are synthesized by the plants themselves and have great biochemical significance for the proper functioning of individual plant parts.
Biscoumarins are a relatively unexplored class of compounds, isolated and characterized from different plant species [1]. It has been found that biscoumarins exhibit properties similar to those of coumarins and act as anticoagulants, antioxidants, antitumor, and antifungal agents. Essential and not well studied is their role as inhibitors of various enzymes—urease, anti-HIV-1 protease and integrase, DNA polymerase, and protein kinase [2,3,4,5,6].
A large number of biscoumarins and bis-3,4-dihydrocoumarins, regarding the type, lengths, and locations of the linkage, have been isolated and characterized. There are different dimerized coumarin systems of which 3,3′-, 4,4′-, 3,8′-, 3,6′-, 8,8′-, and 8,6′-biscoumarins are well known [7].
In reference to our ongoing work [8] for the possibility for conjugated addition of organometallic compounds to 3-substituted coumarins under ultrasound irradiation, we became interested in the synthesis of biscoumarin structures where two coumarin fragments of the same or different type are linked through C4-C4′ bond. The sonication method provides an unusual mechanism to generate high-energy chemistry due to the extraordinary temperatures and pressure generated by the cavitation bubble collapse [9,10,11,12], thus enhancing reactions involving radicals and radical ions. Therefore, the present paper reports the application of ultrasound irradiation in the formation of tetrahydroisomers of different 3,3′-disubstituted biscoumarins.
2. Results and Discussion
2.1. Synthesis of 3,3′,4,4′-Tetrahydro-3,3′-disubstituted-4,4′-biscoumarins
Different formation methods of biscoumarin structures are described in the literature. One of the commonly used methods for the formation of dimeric systems is by electrochemical reactions. The reduction of coumarins is accomplished by using different metal electrodes in the presence or absence of variety of catalysts (e.g., amines, alkaloids, etc.) [13,14,15,16,17,18,19,20]. The described electrochemical reactions are applicable to both unsubstituted coumarins and coumarins substituted by electron donor groups in 3-rd or 4-th position in the lactone ring. The formation of the dimeric systems is explained by the formation of radicals and radical anions.
Studies on the photochemically initiated reaction of coumarins and their 3,4-dihydro derivatives have reported radical formation where the dimer was identified as the major product. The substrates used were substituted coumarins with an acetyl and carboxylic group in 3-rd position [21,22,23].
The synthetic procedures for the formation of 4,4′-biscoumarins include the usage of the Wittig reaction where the two fragments can be formed simultaneously or one after another [24]. The formed dimeric structure resembles the structure of 4,4′-biisofraxidin that has been isolated from plants [25]. Later 4,4′-biisofraxidin was synthesized [26] in a multistep procedure with an overall yield of 7%. A Ni-catalyzed cross-coupling was applied in the last step of the study. A few years later the same authors modified the conditions for the Ullmann type cross-coupling reaction between coumarin fragments using a variety of leaving groups in 4-th position [27].
While trying to perform a 1,4-conjugate addition reaction to 3-diethylphosphonocoumarin 1a using zinc and halogen anhydride under ultrasound conditions a dimeric dihydroisomer 3a (Scheme 1) was isolated instead of the expected product 2. Due to the structure of compound 3a we became interested in obtaining tetrahydroisomers which could be synthesized in one step from 3-substituted coumarins under sonication. To design a synthetic strategy, we require high selectivity and quantitative yields. Firstly, we find the suitable conditions which ensure the formation of the dihydrocoumarin dimer as the only product. Secondly, we focus on reactions going smoothly with satisfactory yields. 3-Diethylphosphonocoumarin 1a was chosen as a model compound due to the fact that the first formation of the dimer 3a was observed when using it.
Grignard reagents are strongly basic and nucleophilic as it is known from the literature. Previous studies in our research group report [8,28,29] the 1,2- and 1,4-conjugate addition of organometallic compounds to 3-substituted coumarins. Even when the substituent is a phenyl group and the double bound of the coumarin system is not activated—1,4- and 1,2-addition adducts are observed.
Another intriguing fact is that Grignard reagents could be radical initiators in reactions with different compounds [30]. One of the first examples of producing coumarin radicals using tert-butylmagnesium chloride was the study of Gustafsson and coworkers [31] which presented the formation of bis-3,4-dihydrocoumarin from ethyl 3-coumarincarboxylate. Because of the complex reaction mixture and the low yield (1–10%) of tetrahydrobiscoumarin, its pure isolation was unsuccessful. The authors assumed that the observed results from the reaction could only be explained by radical formation in the transition state.
Regarding the utilized conditions and the known behavior of the coumarin substrates, the most probable path of the studied reaction could be assumed as a single-electron-transfer. Thus, the nature of the organometallic reagent was studied by comparing the applied metal—zinc or magnesium. The results showed equal conversion time in both cases but the yield of the desired product was only 14% for the organomagnesium and 70% for the organozinc reagent (see Table 1). The observed results have impact on the study of the organomagnesium reagents behavior during reactions with coumarins. Most probably the reduced amount of the homodimerization product is due to the small fraction of radicals leaving the magnesium surface. This conclusion is based on the surface nature of the Grignard reagent formation [30]. Therefore, organomagnesium reagents are better nucleophiles [8,28,29] than being suitable for initiation of coumarin radicals.
The organozinc compounds are relatively less reactive then RMgX, however the reactivity could be improved when chelating ligands are used. In the literature there are examples that present the formation of radicals by the assistance of the zinc compounds [32]. Based on the surface nature of the formation of organozinc compounds we suppose that when zinc is used the amount of the radicals that can escape the lattice is larger, resulting in the formation of the homodimerized product. Another advantage in the usage of RZnX for this type of reaction is the enhanced Lewis acidity of the zinc atom in RZnX, resulting in a stabilized radical transition state. This stabilization can be due to formation of a chelate complex with a donor molecule [33,34].
In our previous work [8] the formation of 1,4-addition product of organozinc reagent of ethylbromoacetate to the coumarin system under ultrasound irradiation conditions was observed (Scheme 2). Moreover, additional products or dimers were not observed in that study. Therefore, in the studied reaction conditions the organozinc reagent of chloroacetic anhydride is of a significant importance for the homodimerization of coumarins.
The reaction is sensitive to a number of effects and its outcome cannot be estimated easily. Common solvents for the organometallic reactions are diethyl ether and tetrahydrofuran (Et2O, THF). In the studied reaction THF was the solvent that accelerated the reaction rate with conversion time for compound 1a of 2 h. However, we obtained a complex mixture rendering the separation of individual compounds impossible. The reaction did not occur in Et2O as a solvent (Table 2). A possible assumption for the observed results is the different solvation of the radicals formed in the transition state. It can be implied that THF is the most suitable solvent for single-electron-transfer reactions. Combining the two solvents, we managed to optimize the reaction rate. When the solvent ratio is THF: Et2O = 3.5:5 the reaction goes fluently for 7 h and the desired product is easily isolated with a yield of 70% using zinc as a metalorganic precursor.
The dimerization process for the tetrahydrobiscoumarin 3a was optimized by reflux or ultrasound irradiation. In all the studied cases sonication has an advantage over thermal initiation, hence, we based our investigations are based on applying that technique. The reaction was carried out using different ratios of coumarin 1a, metal, and chloroacetic anhydride. The reagent ratios and the reaction conditions are presented as Method A, B, and C in Table 1. The crucial distinction between the described methods is the amount of the applied zinc powder with respect to the anhydride.
A full conversion of 1a was observed in 420 min when a slight excess of zinc was used (Method A). The addition of a double amount of zinc powder to the anhydride resulted in shorter reaction time and in increased yield of 3a (Method B). To test the reaction rate, the metal to anhydride ratio was kept the same but the amount of reagents used was reduced (Method C) in aim to diminish the quantities. However, we did not observe a full conversion of the coumarin 1a even though the reaction was carried for 24 h. Thus, we conclude that the preferred condition for homodimerization of 3-dialkylphosphonocoumarins 1a–b is Method B.
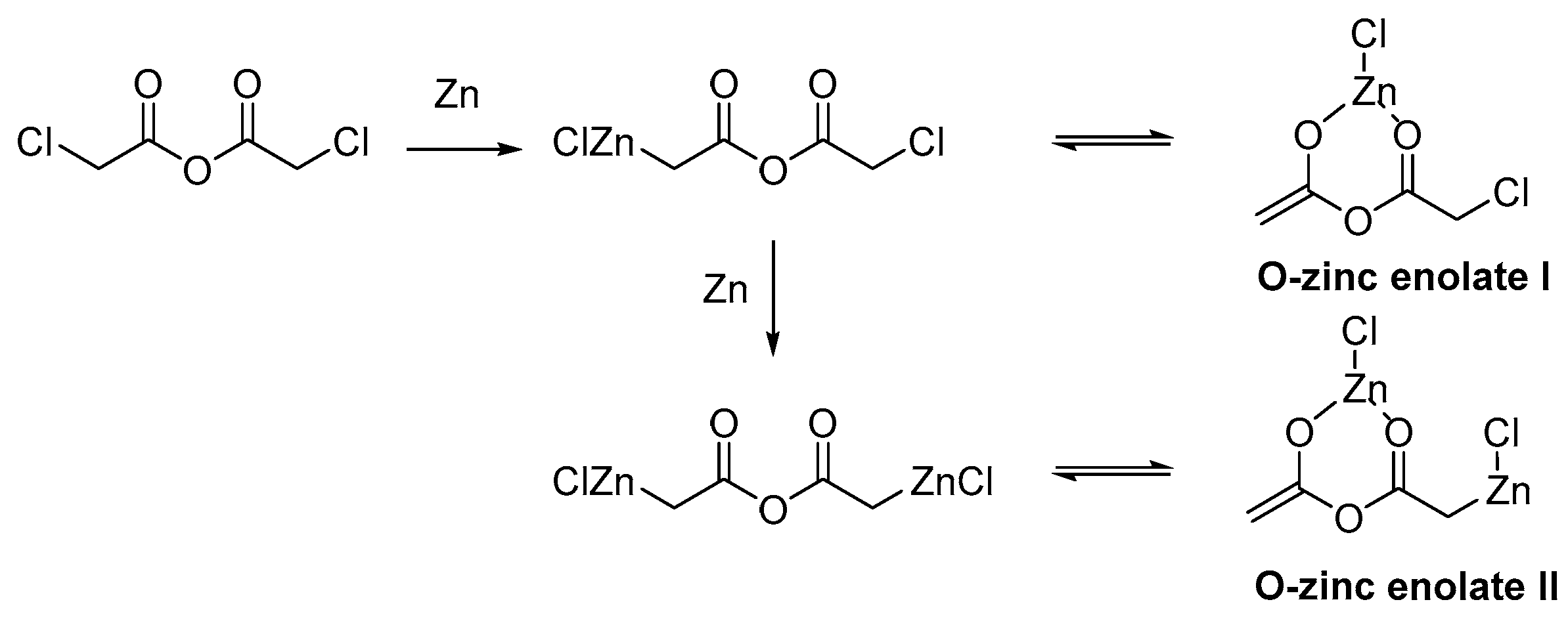
To explain our results from the reaction of 1a, we consider two possible structures for the organozinc reagent. The small amount of zinc suggested a formation of monozinc compound which could be presented as C-zinc and O-zinc enolate given in Scheme 3. Therefore, the long reaction time might be due to the stabilization of the reagent by O-zinc enolate I complex that could react slowly in the implied conditions. When enough metal powder is presented in the reaction mixture there might be a possibility for forming a di-C-zinc enolate with chloroacetic anhydride. The dizinc reagent could as well be stabilized by an O-zinc enolate II, thus, leaving one end of the organozinc compound free to react with the coumarin system (Scheme 3).
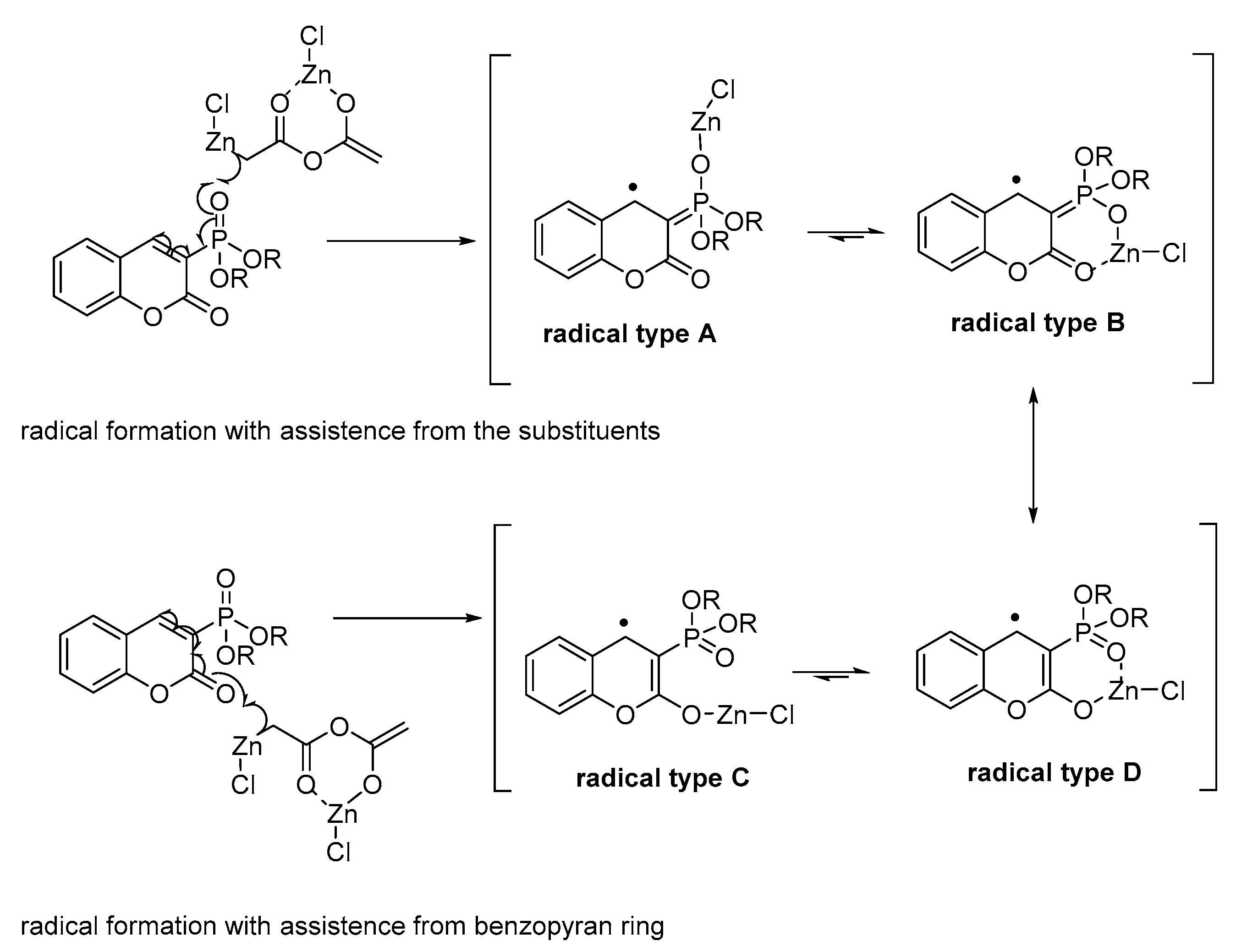
These results suggest that the most probable reaction path for the coumarin 1a includes radical initiation. The examples for radical formation in the presence of organometallic reagents have shown [31,35,36] a coordination of the organometallic reagent to the substrate with a single electron transfer. In case of substituted coumarin compounds there are two possible types of coordination. The first one includes the assistance of the carbonyl group from the benzopyran ring and the other—the substituents in third position. We assume that the initial step might be the formation of the coumarin “radical type A” or “radical type C” (Scheme 4). Both the adjacency of suitable groups with lone pairs and the presence of a metal, indicate a possibility for chelation in the radical with structural stabilization. Therefore, we suppose that, the two resonance structures—“radical type B” and “radical type D”—are more stable and are predominantly presented in the transition state.
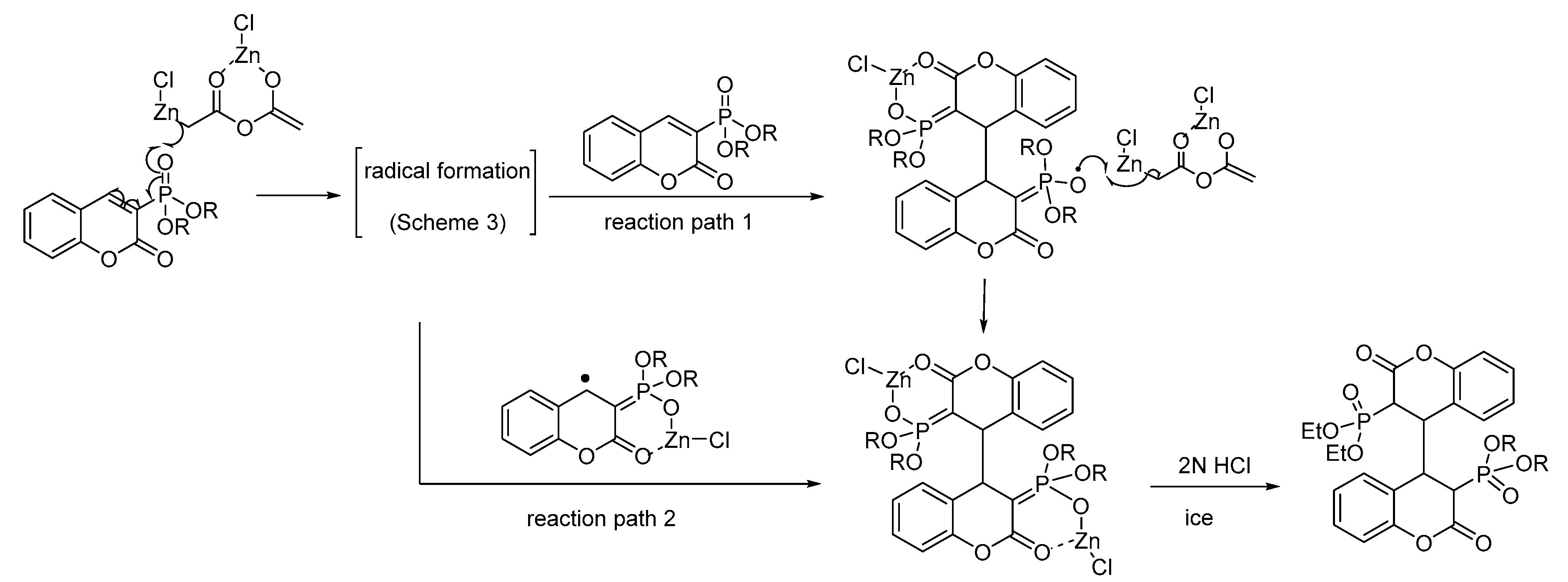
Further analysis of the performed reaction indicates that once the radical generation takes place, there are two possible paths for the formation of bischromanes 3 from 3-diethylphosphonocoumarin 1a, as illustrated in Scheme 5. The first path includes a radical Michael-type addition to a coumarin system, while the second path might involve a coupling of the initiated radicals [37].
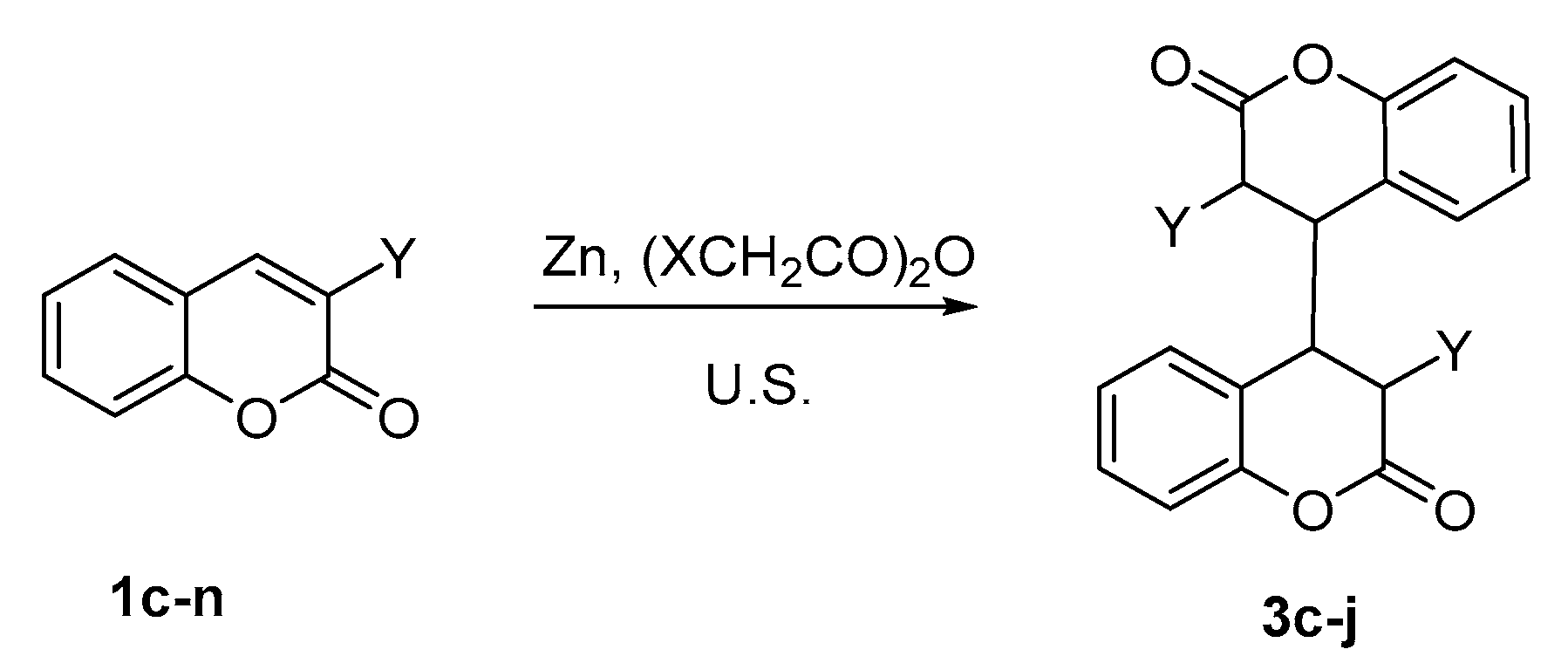
The optimized reaction conditions from Method B were applied to a series of 3-substituted coumarins with different electron-withdrawing and electron-donating groups 1c–n (Scheme 6) and tetrahydroisomers of 4,4′-biscoumarins 3c–j were isolated with yields from 45 to 92% (see Table 3). Identically to the esters of 3-phosphonocoumarins 1a–b, the esters of coumarin-3-carboxylic acid 1c–e reacted smoothly and in shorter reaction times. The reaction with coumarins 1f–i having a carbonyl group in third position was accomplished with the preparation of the all tetrahydrobiscoumarins 3f–i. As shown in Table 3, we observe faster dimerization compared to coumarins carrying ester or phosphonoic group. However, the hindrance effect of these groups is crucial to radical initiation and reaction time. When the conditions of Method A were used for the dimerization reaction, a partial conversion of the substrate or complex mixtures for some of the coumarins were observed.
Interestingly, in cases of coumarins 1l–n no interaction was observed under ultrasound irradiation although 2-oxochromane 1l participates in electrochemical reactions showing excellent yield for tetrahydrocoumarin. Analyzing the results, we assume that if the radical formation occurs with the assistance of the carbonyl group of the pyran ring we should observe the formation of 3l–n because the formation of radical type C (Scheme 7) is not affected by the substituents in 3-rd position. Though, the dimeric product might be with lower yield due to the less stability of the formed radical. The findings during the investigations on compounds 1l–n incontestably illustrate the impossibility of coumarins with electron-donating groups to participate in the described homodimerization conditions. Thus, the assistance of the electron-withdrawing substituent is crucial for the initiation step and further stabilization of the radical as a chelated complex.
The reactions with coumarins 1j and 1k were carried out applying Method B resulting in full conversion. However, only bisoxochromane of 3-nitrocoumarin 3j was characterized after purification with column chromatography. In the case of coumarin 1k, out of many products detected by TLC in the reaction mixture, not one corresponds to a biscoumarin structure. Applying Method A none of the dimeric structures were isolated due to the absence of predominant product.
Regarding the mechanism of coumarin dimerization, another approach was utilized to highlight the behavior of initiated radicals in the reaction mixture. Three approaches for a mixed reaction were chosen comparing coumarin systems with different time for activation or radical formation and their capability to participate in the studied reaction conditions. The reactions were monitored by TLC and 1H NMR. Firstly, coumarins 1a and 1m were mixed in equimolar ratio under the conditions of Method B (Table 4, entry 1) and after three h only the homodimer 3a was isolated. The result was unexpected but it revised our expectation for Michael type addition to the coumarin conjugated system and directed the options towards the radical coupling. The next experiment (Table 4, entry 2) illustrated the properties of two reactive coumarins—1a and 1c—and, for a second time, we observed a dimerization of radicals from the same type. In this entry for a short time (40 min), a conversion of 1c was monitored and the biscoumarin 3c was detected. At the end (Table 4, entry 3), a reaction between 1f and 1i was carried out as an example of two systems giving dimers in a comparable conversion time in the applied conditions. The yields and the reaction time of coumarins 1f and 1i made them the most suitable candidates for testing our predictions. The full conversion of the starting compounds and the registration of the products 3f and 3i had supported the idea of a process that favors the radical homocoupling at the moment of initiation on the metal surface.
2.2. Spectroscopic Data Interpretation of Homodimers 3
The structure of the dimeric compound 3 was mainly analyzed by 1D and 2D NMR spectroscopy. The data for the 3,3′,4,4′-tetrahydro-3,3′-disubstituted-4,4′-biscoumarins present a symmetric structure for all of the compounds. The knowledge on this type of compounds have also demonstrated similar 4,4′-bis-2-oxochromane structures of the dimeric coumarin systems [14,16,19].
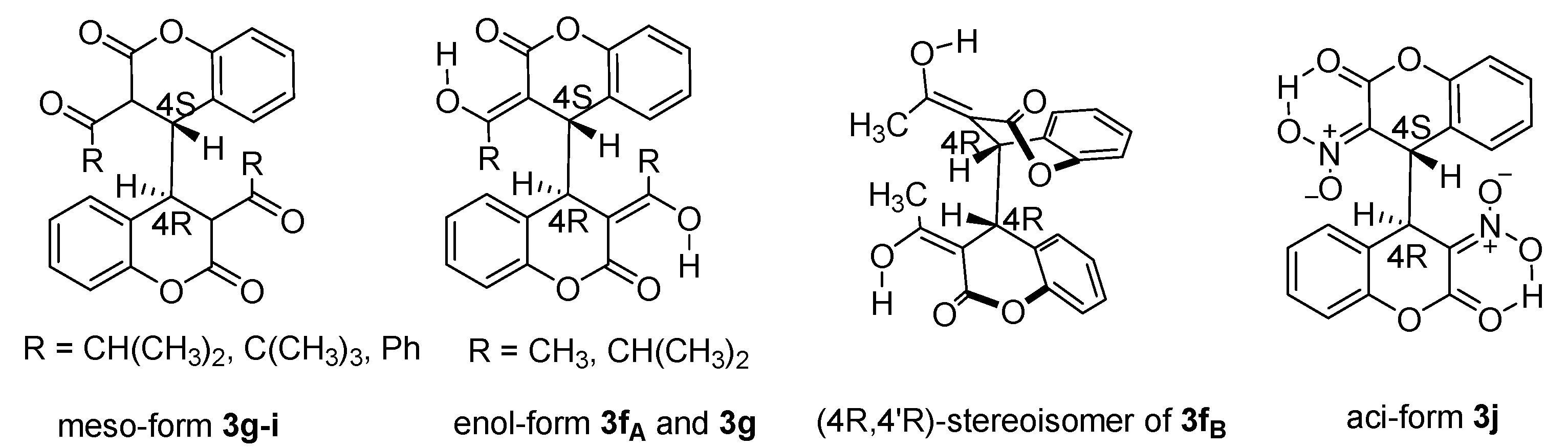
Keeping in mind the presence of four stereocenters in the structure of 3 it is normal to expect several stereoisomers. However, the tetrahydrobiscoumarins 3a–e produced from esters of coumarin-3-carboxylic acid and dialkyl phosphonoesters appear as a single isomer. The signals of H-3 and H-4 protons in 1H NMR of 3c–e are singlets without measurable J-constants. This means the position of the two nuclei determine a very small coupling between them or a constant with value less than 0.9 Hz. The location of the protons could mean 90 degrees between H-3 and H-4. Furthermore, it points to an antiperiplanar disposition of the bulky substituents around the C3-C4 bond. The same conformation was found in the products of conjugate addition of organometallic reagents to 3-substituted coumarins under ultrasound irradiation in our previous study [8]. Protons H-3 and H-4 appeared as doublets in compounds 3a–b with high phosphorus–proton coupling constant. The two characteristic protons were assigned from the HMBC spectra for all tetrahydrobiscoumarins 3.
The structure proposed by NMR spectra was confirmed with a single crystal X-ray structure of homodimer 3a (CCDC1858604, Figure 1a, see also Supporting Materials). Based on the gathered results, we assume that the only isomer to be expected in cases 3a–e is the (3R,4S,3′S,4′R)-stereoisomer or the meso-form where the elements of symmetry minimized the numbers of isomers. Thus, the stereoselectivity of the homodimerization reaction is determined by the steric hindrance of the substituent in position 3 in the pyran ring. Additional evidence for the absence of another diastereomer is the singlet in the 31P NMR spectra of 3a and 3b according to external referent standard H3PO4.
Data from the NOESY spectra for the all meso-dimers demonstrate the correlation between protons H-5/H-5′ from one of the benzene ring with H-3′/H-3 and protons from the substituent in position 3. Another close correlation observed is between H-5 and H-4 from one of the 2-oxo-2H-chromane fragments. These findings supported a stereoisomer in meso-form having formally presented s-trans alignment of the benzopyran fragments around C4-C4′ bond (Figure 2).
Difficulties in the stereoselectivity of the reaction arose when ketones were implied due to the small steric hindrance of the substituent and the adjacency of an alpha-proton to the C=O group. While analyzing the NMR spectra for the products of coumarins 1f–i we found two isomers with the tetrahydrobiscoumarin structure as well as enol-forms of the stereoisomers in cases of 3-acetyl- and 3-isopropylcoumarins. The tendency for keto-enol tautomerism of the products of 3-acetylcoumarin is well-known to us [38,39] and also noted in many examples in the literature even for bis-2-oxochromanes [21]. The proton spectrum of 3f has shown two diastereoisomers presented in their enol-forms (Figure 2) in a ratio of 1:0.65. Most probably the appearance of two isomers is defined by the volume and the configuration of the pyran ring in 3f might be close to planar. Definitely the most stable enol-form makes the structure flat; moreover, it might be the driving force for flattening the radical participating in the homodimerization step. This process determines a possibility for coupling from both sides of the intermediate which produces a meso-form (4S,4′R) 3fA and the pair of (4S,4′S)- and (4R,4′R)-stereoisomers 3fB presented in Figure 2, the last are not distinguishable in NMR spectra.
The homodimerization of coumarin 1g is presented by resonances for different stereoisomers in the proton NMR spectrum of the crude reaction mixture. The keto-forms have similar chemical shifts for H-3 and H-4 protons as in the other biscoumarins 3 and we could assign them as meso-forms. The signals for the enol-forms are several with doublets for the H-4 and H-4′ protons where the observed coupling constants of 9.3 Hz is associated with an angle close to 0 degrees between them. We distinguished the keto- and enol-forms by carrying out thin-layer chromatography of the reaction mixture using n-hexane and ethyl acetate as mobile phase; however, the separation by column chromatography did not lead to isolation of individual stereoisomers. Only the meso-isomer 3g (Figure 2) was precipitated from the reaction mixture and its proton and carbon chemical shifts are listed in the Experimental part. From the same proton spectrum, the signals for the enol-form of 3g are assigned and are also presented.
The tetrahydrobiscoumarin of 1h is characterized as a mixture of two stereoisomers, in approximately equal ratio 1:1.1, on the basis of observed signals in the proton NMR spectrum of the crude reaction mixture. The two compounds 3hA and 3hB can be crystalized with minor presence of the other isomer using the appropriate solvents (see Section 4.).
The dimer from 3-benzoylcoumarin 1i is presented by one meso-isomer 3i which is differentiated from all the other products with low solubility in various organic solvents. A notable observation from the proton spectrum is the higher frequency for the H-3 and H-4 protons, compared to the values in the other homodimers 3, resulted from the magnetic anisotropy of the carbonyl group.
The reaction with coumarin 1j was carried out applying Method B and the desired product 3j was isolated after column chromatography as aci-enol form (Figure 2). Such tautomers of nitro compounds are not frequently detected by spectroscopy or reported in the literature. Previously, we reported [40] the 1,4-addition of nitromethane to 3-nitrocoumarin that undergoes nitro- to aci-nitro-form in solution. Here the disposition of the substituents around the C3-C4 bond have similar configuration and it is not surprising to assign an enol-structure. Moreover, chelation between aci-form of nitro group and the C=O from the lactone ring could additionally stabilize this configuration. The proton spectrum of 3j in CDCl3 represents a broad signal at 4.24 ppm for the enol and disappeared in deuterated methanol which proves the participation of the proton in chemical exchange process. Another interesting fact is the anisotropic effect of the nitro group on H-4, shifting it to a higher frequency of 6.71 ppm. If we compare the data from 13C NMR of 3j and the other enol forms from the current investigation, 3f and 3g, the chemical shifts of C-3 and C-4 nuclei changed from around 92 and 46 ppm to higher frequency 131 and 110.9 ppm, respectively. Moreover, the data from 1H-15N HMBC show a signal at 50.474 ppm for the nitrogen nuclei as a long range correlation with the H4 which is another fact supporting the proposed structure.
3. Conclusions
The current investigations present a new method for faster and simple preparation of 3,3′,4,4′-tetrahydro-3,3′-disubstituted-4,4′-biscoumarins when an organozinc reagent is used. The ultrasound promoted reaction offers a simple experimental setup and reproducibility of the results. The homodimers are isolated with the highest yields reported in the literature about such compounds. Unlike previous synthetic procedures, our dimerization conditions are applicable for coumarins with electron-withdrawing groups in third position. We hypothesize that the formed radical, stabilized by the substituent and assisted by the organozinc compound, plays a major role on the dimerization mechanism. The structures of tetrahydrocoumarins are characterized by IR, NMR, MS, and X-ray data and the main product was assigned to be the meso-stereoisomer.
4. Experimental Section
4.1. Materials
Melting points were determined with a Kofler hot-stage apparatus (Reichert Technologies, New York, NY, USA) and are used without correction. The IR spectra were recorded with a Specord IR 71, IR 75 spectrophotometer (Carl Zeiss, 73447 Oberkochen, Germany). 1Н-, 13С-, and 31P-NMR spectra were recorded on a Bruker Avance III 500 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) (at 500 MHz for 1H, 125.7 MHz for 13C and 202.4 MHz for 31P, respectively). Chemical shifts are given in ppm downfield from tetramethylsilane as internal standard with CDCl3, DMSO, and CD3OD as solvents. 31P-NMR spectra were recorded with 85% H3PO4 as an external standard. Analyses were carried out on Q Exactive® mass analyzer equipped with TurboFlow® LC system and IonMax II® electrospray ionization module (ThermoScientific Co., Waltham, MA, USA). Data acquisition and processing were carried out using the XCalibur® 4.2 software package (ThetmoScientific Co., Waltham, MA, USA). Chromatographic conditions: Column: Syncronis C18, 1.7 μm (100 × 2.1 mm) (ThetmoScientific Co., Waltham, MA, USA). Mobile phase: A = 0.1 % formic acid in water and B = 0.1% formic acid in acetonitrile with flow rate: 300 μL/min. Mass spectrometric conditions: Full-scan spectra over the m/z range 120 to 1200 were acquired in positive ion mode at resolution settings of 140,000 (m/z = 200). All MS parameters were optimized for sensitivity to the target analyzes using the instrument control software program. Operating parameters: spray voltage 4.0 kV, sheath gas flow rate 32, auxiliary gas flow rate 10, spare gas flow rate 3, capillary temperature 280 °C, probe heater temperature 320 °C, and S-lens RF level 50. Data acquisition and processing were carried out with Xcalibur 4.2® software package (ThetmoScientific Co., Waltham, MA, USA). Ultrasonic irradiation was performed in an ultrasonic cleaner with a frequency of 20 kHz and power 250 W. Reactions were monitored by TLC on silica gel 60 F254. Column chromatography was carried out on silica gel (Merck 0.043–0.063 mm) (Merck, Kenilworth, NJ, USA) using as eluent n-hexane/EtOAc mixture with increasing polarity. Elemental analyses of C, H, and N were carried out in the Laboratory of Elemental Analysis at the Department of Organic Chemistry and Pharmacognosy, University of Sofia, Bulgaria. The X-ray analysis was performed on Bruker Apex-II CCD diffractometer at the Laboratory of Molecular Spectroscopy of Structural Analysis, University of Sofia, Bulgaria. The crystal was kept at 300.15 K during data collection. Using Olex2 [41], the structure was solved with the SIR2004 [42] structure solution program using Direct Methods and refined with the ShelXL [43] refinement package using “Least Squares” minimization.
All chemical reagents were purchased from Merck and Sigma Aldrich (Taufkirchen, Germany). The starting 3-substitueted 2-oxo-2H-1-benzopyrans 1 were prepared according to a procedures reported by us [44].
4.2. General Methods
Method A: A mixture of 1 (0.001 mol), Zn (0.183 g, 0.0028 mol), (ClCH2CO)2O (0.410 g, 0.0024 mol) in Et2O/THF (10 mL/7 mL), and a catalytic amount of I2 was sonicated until the coumarin 1 was consumed (TLC-monitoring). The reaction mixture was poured onto a 2 N solution of hydrochloric acid and ice, extracted with chloroform (3 × 20 mL), and the organic extracts were washed several times with saturated solution of NaHCO3 and then dried with anhydrous sodium sulfate. After the evaporation of the solvent, to the residue were added 3 mL Et2O and 1 mL acetone and the resulting mixture was left in a fridge overnight. Compound 3 was obtained as a solid. After the filtration of the crystals, the solvent of the mother liquor was evaporated and the residue was purified by column chromatography using n-hexane/EtOAc as an eluent system.
Method B: Performed according to Method A, however the used metallic Zn powder was 0.366 g (0.0056 mol).
Method C: A mixture of 1a (0.282 g, 0.001 mol), Zn (0.222 g, 0.0034 mol), (ClCH2CO)2O (0.256 g, 0.0015 mol) in Et2O/THF (10 mL/7 mL), and a catalytic amount of I2 was sonicated for 24 h.
Tetraethyl (2,2′-dioxo-[4,4′-bichroman]-3,3′-diyl)bis(phosphonate), 3a—Method B. The product was isolated from Et2O/acetone: 0.251 g, 89% white crystals, m.p. = 225–228 °C. IR (nujol): ν = 1775, 1060, 1035, cm−1. 1H NMR (500 MHz, DMSO) δ = 7.45 (dd, J = 7.0, 1.0 Hz, 2H, H-5/H-5′), 7.27 (td, J = 7.4, 1.1 Hz, 2H, H-7/H-7′), 7.02 (td, J = 6.5, 0.9 Hz, 2H, H-8/H-8′), 6.76 (dd, J = 7.9, 0.9 Hz, 2H, H-6/H-6′), 4.53 (d, J = 28.3, 2H, H-3/H-3′), 4.01–4.14 (m, 4H, POCH2CH3), 3.95 (d, J = 15, 2H, H-4/H-4′), 3.62–3.71 (m, 2H, POCH2CH3), 3.35–3.43 (m, 2H, POCH2CH3), 1.21 (t, J = 7.0 Hz, 6H, POCH2CH3), 0.72 (t, J = 7.1 Hz, 6H, POCH2CH3); 13C NMR (125.7 MHz, DMSO) δ = 163.2 (d, J = 6.3 Hz, C-2/C-2′), 151.9 (s, C-8a/C-8a′), 130.9 (s, C-7/C-7′), 130.2 (s, C-5/C-5′), 124.6 (s, C-8/C-8′), 119.2 (s, C-4a/C-4a′), 116.3 (s. C-6/C-6′), 63.2 (d, J = 6.3 Hz, POCH2CH3), 62.6 (d, J = 7.2 Hz, POCH2CH3), 43.3 (d, J = 122.1 Hz, C-3/C-3′), 41.7 (d, J = 3.5 Hz, C-4/C-4′), 16.5 (d, J = 6.5 Hz, POCH2CH3), 16.0 (d, J = 6.4 Hz, POCH2CH3); 31P NMR (202.4 MHz, DMSO): δ = 20.75 (s). HRMS (FTMS+p ESI) m/z calculated for C26H32O10P2 [M]+ 567.1549 found 567.1528.
Tetramethyl (2,2′-dioxo-[4,4′-bichroman]-3,3′-diyl)bis(phosphonate), 3b—Method B. The product was isolated from Et2O/acetone: 0.122 g, 61%, white crystals, m.p. = 265–266 °C. IR (CHCl3): ν = 1760, 1050, 1035 cm−1. 1H NMR (500 MHz, DMSO) δ = 7.45 (td, J = 6.5, 1.5 Hz, 2H, H-5/H-5′), 7.23 (td, J = 6.5, 1.0 Hz, 2H, H-7/H-7′), 7.12 (dd, J = 8.2, 0.8 Hz, 2H, H-8/H-8′), 6.76 (dd, J = 7.6, 1.4 Hz, 2H, H-6/H-6′), 3.57 (d, J = 11.3, 3H, POCH3), 3.55–3.57 (m, 2H, H-3/H-3′), 3.52 (d, J = 26.5, 2H, H-4/H-4′); 13C NMR (125.7 MHz, DMSO) δ = 163.1 (d, J = 3.3 Hz, C-2/C-2′), 150.9 (s, C-8a/C-8a′), 130.3 (s, C-7/C-7′), 129.1 (s, C-5/C-5′), 125.1 (s, C-8/C-8′), 119.3 (s, C-4a/C-4a′), 116.8 (s, C-6/C-6′), 53.4 (d, J = 6.9 Hz, POCH3), 53.3 (d, J = 6.9 Hz, POCH3), 42.9 (dd, J = 134.5, 1.3 Hz, C-3/C-3′), 41.7 (dd, J = 19, 3.8 Hz, C-4/C-4′), 31P NMR (202.4 MHz, DMSO): δ = 20.19 (s). Anal. Calcd for C22H24O10P2: C,51.55; H,5.12. Found: C, 51.75; H, 5.28. HRMS (FTMS-p ESI) m/z calculated for C22H24O10P2 [M]− 509.0767 found 509.0756.
Diethyl 2,2′-dioxo-[4,4′-bichroman]-3,3′-dicarboxylate, 3c—Method B. The product was isolated from Et2O/acetone: 0.159 g, 73%, white crystals, m.p. = 212–214 °C. IR (nujol): ν = 1795, 1710, 1460 cm−1. 1H NMR (500 MHz, CDCl3) δ = 7.41 (td, J = 7.5, 1.8 Hz, 2H, H-7/H-7′), 7.26 (td, J = 7.4, 1.7 Hz, 2H, H-5/H-5′), 7.23 (dq, J = 14.8, 7.4, 1.1 Hz, 2H, H-8/H-8′), 7.19 (dd, J = 8.1, 0.8 Hz, 2H, H-6/H-6′), 4.02–3.91 (m, 6H, COOCH2CH3), 3.78 (s, 2H, H-3/H-3′), 3.29 (s, 2H, H-4/H-4‘), 0.94 (t, 6H, COOCH2CH3); 13C NMR (125.7 MHz, CDCl3) δ = 165.9 (s, COOCH2CH3), 163.9 (s, C-2/C-2′), 150.8 (s, C-8a/C-8a’), 130.4 (s, C-7/C-7′), 130.1 (s, C-5/C-5′), 125.4 (s, C-8/C-8′), 120.6 (s, C-4a/C-4a’), 117.9 (s, C-6/C-6′), 62.5 (s, COOCH2CH3), 49.9 (s, C-4/C-4′), 42.3 (s, C-3/C-3′), 13.7 (s, COOCH2CH3). Anal. Calcd for C24H22O8: C,65.74; H,5.06. Found: C, 65.70; H, 5.03. HRMS (FTMS+p ESI) m/z calculated for C24H22O8 [M + H + NH3]+ 456.1658 found 456.1649.
Dimethyl 2,2′-dioxo-[4,4′-bichroman]-3,3′-dicarboxylate, 3d—Method B. The product was isolated from Et2O/acetone: 0.194g, 73%, white crystals, m.p. = 225–226 °C. IR (nujol): ν = 1795, 1725, 1455 cm−1. 1H NMR (500 MHz, CDCl3) δ = 7.41 (ddd, J = 8.1, 7.0, 2.0 Hz, 2H, H-7/H-7′), 7.26 (td, J = 5.5, 2.0 Hz, 2H, H-5/H-5′), 7.24 (dq, J = 14.5, 6.4, 1.0 Hz, 2H, H-8/H-8′), 7.19 (dd, J = 8.1, 0.9 Hz, 2H, H-6/H-6′), 3.82 (s, 2H, H-3/H-3′), 3.52 (s, 6H, COOCH3), 3.29 (s, 2H, H-4/H-4′); 13C NMR (125.7 MHz, CDCl3) δ = 166.3 (s, COOCH3), 163.6 (s, C-2/C-2′), 150.6 (s, C-8a/C-8a′), 130.4 (s, C-7/C-7′), 130.1 (s, C-5/C-5′), 125.5 (s, C-8/C-8′), 120.4 (s, C-4a/C-4a′), 117.9 (s, C-6/C-6′), 49.7 (s, C-4/C-4′), 42.0 (s, C-3/C-3′), 53.3 (s, COOCH3). HRMS (FTMS-p ESI) m/z calculated for C22H18O8 [M]− 409.0924 found 409.0918.
Diphenyl 2,2′-dioxo-[4,4′-bichroman]-3,3′-dicarboxylate, 3e—Method B. The product was isolated from Et2O/acetone: 0.176 g, 66%, white crystals, m.p. = 180–183 °C. IR (nujol): ν = 1775, 1745, 1460 cm−1. 1H NMR (500 MHz, CDCl3) δ = 7.49 (td, J = 7.5, 1.6 Hz, 2H, aromatic), 7.39 (dd, J = 7.4, 1.5 Hz, 2H, H-5/H-5′), 7.33 (td, J = 6.3, 1.0 Hz, 2H, H-4′′ and aromatic), 7.29 (dd, J = 8.0, 0.8 Hz, 2H, H-4′′ and aromatic), 7.22–7.25 (m, 4H, COOPh), 7.15–7.18 (m, 2H, aromatic), 6.60 (dd, J = 8.2, 1.0 Hz, 4H, COOPh), 4.07 (s, 2H, H-3/H-3′), 3.49 (s, 2H, H-4/H-4′); 13C NMR (125.7 MHz, CDCl3) δ = 164.8 (s, COOPh), 163.3 (s, C-2/C-2′), 150.9 (s, C-8a/C-8a′), 149.6 (s, C-1′′ from COOPh), 130.8 (s, C-7/C-7′), 130.2 (s, C-5/C-5′), 129.5 (s, C-2′′ and C-6′′ from COOPh), 126.6 (s, C-4′′ from COOPh), 125.8 (s, C-8/C-8′), 120.7 (s, C-3′′ and C-5′′ from COOPh), 120.4 (s, C-4a/C-4a′), 118.2 (s, C-6/C-6′), 50.0 (s, C-4/C-4′), 42.6 (s, C-3/C-3′). HRMS (FTMS-p ESI) m/z calculated for C32H22O8 [M]− 533.1236 found 533.1223.
3,3′-Diacetyl-[4,4′-bichroman]-2,2′-dione, 3f—Method B. The product was isolated as two isomers 3fA and 3fB from Et2O/acetone: 0.173 g, 92%, white crystals, m.p. = 196–198 °C. IR (nujol): ν = 1595, 1645, 1450 cm−1.
3fA (major isomer): 1H NMR (500 MHz, CDCl3) δ = 12.95 (d, J = 0.6 Hz, 2H, enol form =C(CH3)OH), 7.34 (td, J = 8.0, 1.5 Hz, 2H, H-7/H-7′), 7.13 (td, J = 7.4, 1.1 Hz, 2H, H-8/H-8′), 7.08 (dd, J = 8.4, 1.0 Hz, 2H, H-6/H-6′), 6.86 (td, J = 8.2, 1.5 Hz, 2H, H-5/H-5′), 3.49 (s, 2H, H-4/H-4′), 1.41 (s, 6H, CH3); 13C NMR (125.7 MHz, CDCl3) δ = 178.3 (s, enol form =C(CH3)OH), 169.7 (s, C-2/C-2′), 150.9 (s, C-8a/C-8a′), 129.4 (s, C-7/C-7′), 129.1 (s, C-5/C-5′), 124.6 (s, C-8/C-8′), 122.9 (s, C-4a/C-4a′), 116.8 (s, C-6/C-6′), 93.9 (s, C-3/C-3′), 45.1 (s, C-4/C-4′), 17.6 (s, CH3). HRMS (FTMS+p ESI) m/z calculated for C22H18O6 [M + H + NH3]+ 396.1447 found 396.1522.
3fB (minor isomer): 1H NMR (500 MHz, CDCl3) δ = 13.19 (d, J = 0.5 Hz, 2H, enol form =C(CH3)OH), 7.29 (td, J = 8.0, 1.5 Hz, 2H, H-7/H-7′), 7.05 (d, J = 6.4 Hz, 2H, H-8/H-8′), 7.03 (dd, J = 8.1, 0.9 Hz, 2H, H-6/H-6′), 6.86 (td, J = 8.2, 1.5 Hz, 2H, H-5/H-5′), 3.73 (s, 2H, H-4/H-4′), 1.69 (s, 6H, CH3); 13C NMR (125.7 MHz, CDCl3) δ = 177.5 (s, enol form =C(CH3)OH), 168.9 (s, C-2/C-2′), 151.0 (s, C-8a/C-8a′), 129.2 (s, C-7/C-7′), 128.9 (s, C-5/C-5′), 124.7 (s, C-8/C-8′), 122.6 (s, C-4a/C-4a′), 116.9 (s, C-6/C-6′), 93.0 (s, C-3/C-3′), 46.4 (s, C-4/C-4′), 18.1 (s, CH3). HRMS (FTMS+p ESI) m/z calculated for C22H18O6 [M + H + NH3]+ 396.1447 found 396.152.
3,3′-Diisobutyryl-[4,4′-bichroman]-2,2′-dione, 3g—Method B. The product was isolated in meso- and enol-forms from Et2O/acetone: 0.186 g, 86%, white crystals, m.p. = 256−257 °C.
3g meso-form–isolated from Et2O/acetone 0.036 g: IR (nujol): ν = 1750, 1700, 1450 cm−1 1H NMR (500 MHz, CDCl3) δ = 7.39 (ddd, J = 8.1, 6.9, 2.0 Hz, 2H, aromatic), 7.24–7.27 (m, 4H, aromatic), 7.16 (d, J = 8.0 Hz, 2H, aromatic), 3.94 (s, 2H, H-3/H-3′), 3.26 (s, 2H, H-4/H-4′), 2.62–2.7 (sept, 2H, CH(CH3)2), 0.91 (d, J = 6.9 Hz, 6H, CH3), 0.78 (d, J = 6.9 Hz, 6H, CH3); 13C NMR (125.7 MHz, CDCl3) δ = 204.6 (s, C=0), 165.1 (s, C-2/C-2′), 150.4 (s, C-8a/C-8a′), 130.3 (s, C-7/C-7′), 130.0 (s, C-5/C-5′), 125.5 (s, C-8/C-8′), 120.9 (s, C-4a/C-4a′), 117.8 (s, C-6/C-6′), 55.8 (s, C-3/C-3′), 45.1 (s, C-4/C-4′), 38.6 (s, CH(CH3)2), 18.1 (s, CH3), 17.9 (s, CH3). HRMS (FTMS+p ESI) m/z calculated for C26H26O6 [M + H + NH3]+ 452.2073 found 452.2159.
3g enol form: 1H NMR (500 MHz, CDCl3) δ = 13.05 (d, 1H, J = 1.5 Hz, =C(CH)OH), 7.31 (td, J = 8.1, 1.5 Hz, 2H, aromatic), 7.19–7.22 (m, 2H, aromatic), 7.11–7.13 (m, 2H, aromatic), 7.02 (dd, J = 8.2, 0.9, 2H, aromatic), 3.66 (d, 1H, J = 9.3 Hz, H-4), 3.26 (d, 1H, J = 9.3 Hz, H-4′), 2.56–2.62 (sept, 2H, CH(CH3)2), 0.97 (d, J = 6.9 Hz, CH3), 0.88 (d, J = 6.9 Hz, CH3), 0.77 (d, J = 6.9 Hz, CH3), 0.45 (d, J = 6.9 Hz, CH3); 13C NMR (125.7 MHz, CDCl3) δ = 170.1 (s, =C(CH)OH), 165.1 (s, C-2/C-2′), 150.8 (s, C-8a/C-8a′), 129.8 (s, C-7/C-7′), 139.5 (s, C-5/C-5′), 124.9 (s, C-8/C-8′), 119.1 (s, C-4a/C-4a′), 117.1 (s, C-6/C-6′), 92.1 (s, C-3/C-3′), 45.4 (s, C-4/C-4′), 38.7 (s, CH(CH3)2), 18.6 (s, CH3), 18.3 (s, CH3). HRMS (FTMS+p ESI) m/z calculated for C26H26O6 [M + H + NH3]+ 452.2073 found 452.2159.
3,3′-Dipivaloyl-[4,4′-bichroman]-2,2′-dione, 3h—Method B. The product was isolated as two isomers 3hA and 3hB from Et2O/acetone: 0.212 g, 92%, white crystals, m.p. = 308–310 °C. IR (nujol): ν = 1745, 1695, 1450 cm−1.
3hA: 1H NMR (500 MHz, CDCl3) δ = 7.30 (td, J = 8.1, 1.5 Hz, 2H, aromatic), 7.11 (dd, J = 8.1, 0.9 Hz, 2H, aromatic), 6.91 (td, J = 7.5, 1.1 Hz, 2H, aromatic), 6.48 (dd, J = 7.5, 1.4 Hz, 2H, H-5/H-5′), 4.52 (s, 2H, H-3/H-3′), 3.19 (s, 2H, H-4/H-4′), 1.13 (s, 18H, CH3); 13C NMR (125.7 MHz, CDCl3) δ = 206.2 (s, C=O), 164.1 (s, C-2/C-2′), 151.2 (s, C-8a/C-8a′), 129.9 (s, C-7/C-7′), 129.6 (s, C-5/C-5′), 124.6 (s, C-8/C-8′), 119.4 (s, C-4a/C-4a′), 116.8 (s, C-6/C-6′), 50.4 (s, C-3/C-3′), 46.1 (s, C(CH3)3), 44.7 (s, C-4/C-4′), 26.3 (s, CH3). HRMS (FTMS+p ESI) m/z calculated for C28H30O6 [M + H + NH3]+ 480.2382 found 480.2481.
3hB: 1H NMR (500 MHz, CDCl3) δ = 7.46 (dd, J = 7.6, 1.6 Hz, 1H, aromatic), 7.44 (dd, J = 7.6, 1.6 Hz, 1H, aromatic), 7.28 (td, J = 7.4, 1.1 Hz, 2H, aromatic), 7.22 (dd, J = 6.1, 1.5 Hz, 2H, H-5/H-5′), 7.21 (dd, J = 6.3, 1.0 Hz, 2H, aromatic), 4.41 (s, 2H, H-3/H-3′), 2.97 (s, 2H, H-4/H-4′), 0.88 (s, 18H, CH3); 13C NMR (125.7 MHz, CDCl3) δ = 206.4 (s, C=O), 164.6 (s, C-2/C-2′), 151.2 (s, C-8a/C-8a′), 130.4 (s, C-7/C-7′), 130.0 (s, C-5/C-5′), 125.2 (s, C-8/C-8′), 119.7 (s, C-4a/C-4a′), 117.5 (s, C-6/C-6′), 50.8 (s, C-3/C-3′), 45.7 (s, C(CH3)3), 43.3 (s, C-4/C-4′), 25.9 (s, CH3). HRMS (FTMS+p ESI) m/z calculated for C28H30O6 [M + H + NH3]+ 480.2382 found 480.2481.
3,3′-Dibenzoyl-[4,4′-bichroman]-2,2′-dione, 3i—Method B. The product was isolated from Et2O/acetone: 0.209 g, 84%, white crystals, m.p. = 188–192 °C. IR (nujol): ν = 1775, 1675, 1450, 1445 cm−1. 1H NMR (500 MHz, DMSO) δ = 8.07 (d, J = 7.8 Hz, 4H, H-2′′ and H-6′′), 7.83 (t, J = 7.2 Hz, 2H, H-4′′), 7.67 (d, J = 7.2 Hz, 4H, H-3′′ and H-5′′), 7.29 (t, J = 8.1 Hz, 2H, H-7/H-7′), 7.26 (d, J = 7.4 Hz, 2H, H-5/H-5′), 6.98 (t, J = 7.1 Hz, 2H, H-8/H-8′), 6.89 (d, J = 8.1 Hz, 2H, H-6/H-6′), 5.81 (s, 2H, H-3/H-3′), 4.09 (s, 2H, H-4/H-4′); 13C NMR (125.7 MHz, DMSO) δ = 195.2 (s, C=O), 165.2 (s, C-2/C-2′), 151.5 (s, C-8a/C-8a′), 135.3 (s, C-4′′), 133.6 (s, C-1′′), 130.9 (s, C-7/C-7′), 130.5 (s, C-5/C-5′), 129.7 (s, C-2′′ and C-6′′), 129.6 (s, C-3′′ and C-5′′), 124.7 (s, C-8/C-8′), 118.2 (s, C-4a/C-4a′), 116.7 (s, C-6/C-6′), 53.8 (s, C-3/C-3′), 44.02 (s, C-4/C-4′). HRMS (FTMS+p ESI) m/z calculated for C32H22O6 [M + H + NH3]+ 520.1863 found 520.1760.
3,3′-Dinitro-[4,4′-bichroman]-2,2′-dione, 3j—Method B. The product was purified by column chromatography using n-hexane/EtOAc: 0.085 g, 45%, pale yellow crystals, m.p. = 131–133 °C. IR (nujol): ν = 3410, 3320, 1695, 1630, 1590, 1445 cm−1.
1H NMR (500 MHz, CDCl3) δ = 7.31–7.28 (dd, J = 8.1, 1.0 Hz, 2H, aromatic), 7.28–7.25 (m, 4H, aromatic), 7.22–7.19 (m, 2H, aromatic), 6.71 (s, 2H, H-4/H-4′), 4.24 (bs, =N(O)OH); 13C NMR (125.7 MHz, CDCl3) δ = 159.4 (s, C-2/C-2′), 149.1 (s, C-8a/C-8a′), 126.6 (s, C-7/C-7′), 125.1 (s, C-5/C-5′), 124.6 (s, C-8/C-8′), 116.2 (s, C-4a/C-4a′), 116.2 (s, C-6/C-6′), 131.9 (s, C-3/C-3′), 110.9 (s, C-4/C-4′).
1H NMR (500 MHz, CD3OD) δ = 7.26–7.24 (dd, J = 7.1, 1.2 Hz, 2H, aromatic), 7.17–7.14 (m, 1H, aromatic), 7.144–7.141 (m, 3H, aromatic), 7.12–7.09 (m, 2H, aromatic), 6.68 (s, 2H, H-4/H-4′); 13C NMR (125.7 MHz, CD3OD) δ = 159.7 (s, C-2/C-2′), 148.6 (s, C-8a/C-8a′), 132.9 (s, C-3/C-3′), 125.7 (s, C-7/C-7′), 124.7 (s, C-5/C-5′), 124.3 (s, C-8/C-8′), 121.8 (s, C-4a/C-4a′), 115.3 (s, C-6/C-6′), 109.4 (s, C-4/C-4′). 1H-15N-HMBC: 50.474 ppm. Anal. Calcd for C18H12O8N2: C, 56.26; H, 3.15; N, 7.29. Found: C, 56.08; H, 3.07; N, 7.12.
Author Contributions
R.D.N. and N.I.P.-Y. conceived and planned the experiments. A.I.K. carried out the experiments. N.P. performed the NMR experiments and analyzed the spectra data. R.D.N., N.I.P.-Y., and A.I.K. contributed to the interpretation of the observed results. N.I.P.-Y. and A.I.K. took the lead in writing the manuscript, drafted the manuscript and designed the figures. R.D.N. provided helpful review on an early draft of the paper. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Funding
This research was funded by University Scientific Fund (Grant No. 80-10-63/2018).
Acknowledgments
This work was supported by the Horizon 2020 program of the European Commission (project Materials Networking—grant agreement 692146). The investigations were assisted by University Scientific Fund (Grant No. 80-10-63/2018). The authors would like to thank their colleagues from the Laboratory of Molecular Spectroscopy of Structural Analysis, University of Sofia for the single crystal X-ray analysis and Medical University of Sofia, Department of Chemistry and Biochemistry for the MS spectra.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pratap, R.; Ram, V.J. Natural and synthetic chromenes, fused chromenes, and versatility of dihydrobenzo[h]chromenes in organic synthesis. Chem. Rev. 2014, 114, 10476–10526. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple coumarins and analogues in medicinal chemistry: Occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef] [PubMed]
- Kancheva, V.D.; Boranova, P.V.; Nechev, J.T.; Manolov, I.I. Structure-activity relationships of new 4-hydroxy bis-coumarins as radical scavengers and chain-breaking antioxidants. Biochimie 2010, 92, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, B.K.; Sharma, N.K.; Gyanda, K.; Jain, S.K.; Tyagi, Y.K.; Baghel, A.S.; Pandey, M.; Sharma, S.K.; Prasad, A.K.; et al. Specificities of acetoxy derivatives of coumarins, biscoumarins, chromones, flavones, isoflavones and xanthones for acetoxy drug: Protein transacetylase. Eur. J. Med. Chem. 2007, 42, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Dutra, P.K.; Majumder, P.C.; Dutia, N.L. Synthetic approaches towards bicoumarins: Synthesis of euphorbetin and isoeuphorbetin. Tetrahedron 1975, 31, 1167–1170. [Google Scholar] [CrossRef]
- Spencer, R.R.; Witt, S.C.; Lundin, R.E.; Bickoff, E.M. Bicoumol, a new bicoumarinyl, from ladino clover. J. Agric. Food Chem. 1967, 3, 536–538. [Google Scholar] [CrossRef]
- Hussain, H.; Hussain, J.; Al-Harrasi, A.; Krohn, K. The chemistry and biology of bicoumarins. Tetrahedron 2012, 68, 2553–2578. [Google Scholar] [CrossRef]
- Koleva, A.I.; Petkova, N.I.; Nikolova, R.D. Ultrasound-assisted conjugate addition of organometallic reagents to 3-diethylphosphonocoumarin. Synlett 2016, 27, 2676–2680. [Google Scholar] [CrossRef]
- Luche, J.-L. Synthetic Organic Sonochemistry; Springer: New York, NY, USA, 1998; ISBN 978-1-4899-1910-6. [Google Scholar]
- Mason, T.J.; Lorimer, P.J. Applied Sonochemistry: The Uses of Power Ultrasound in Chemistry and Processing, 1st ed.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Mason, T.J.; Peters, D. Practical Sonochemistry: Uses and Applications of Ultrasound, 2nd ed.; Woodhead Publishing: Cambridge, UK, 2002. [Google Scholar]
- Gallego-Juáres, A.J.; Graff, F.K. Power Ultrasonics: Applications of High-Intensity Ultrasound, 1st ed.; Woodhead Publishing: Cambridge, UK, 2014. [Google Scholar]
- Nielsen, M.F.; Batanero, B.; Löhl, T.; Schafer, H.J.; Wurthwein, E.; Frohlich, R. Enantioselective cathodic reduction of 4-methylcoumarin: Dependence of selectivity on reaction conditions and investigation of the mechanism. Chem. Eur. J. 1997, 3, 2011–2024. [Google Scholar] [CrossRef]
- Pasciak, E.M.; Rittichier, J.T.; Chen, C.; Mubarak, M.S.; VanNieuwenhze, M.S.; Peters, D.G.J. Electroreductive dimerization of coumarin and coumarin analogues at carbon cathodes. Org. Chem. 2015, 80, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Tuğral, S.; Berkemb, M.L. Electrochemical behavior of some ethylenedioxycoumarins: Cathodic dimerization. J. Mol. Liq. 2014, 196, 363–369. [Google Scholar] [CrossRef]
- Kise, N. Density functional theory study of electroreductive hydrocoupling of α,β-unsaturated carbonyl compounds. J. Org. Chem. 2006, 71, 9203–9207. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Sakai, M.; Ishino, Y.; Shibata, T.; Maekawa, H.; Nishiguchi, I. Mg-promoted regio- and stereoselective C-acylation of aromatic α,β-unsaturated carbonyl compounds. Org. Lett. 2001, 3, 3439–3442. [Google Scholar] [CrossRef] [PubMed]
- Gourley, R.N.; Grimshaw, J.; Millar, P.G. Electrochemical reactions. Part VIII. Asymmetric induction during the reduction of coumarins modified by the presence of tertiary amines. J. Chem. Soc. (C) 1970, 17, 2318–2323. [Google Scholar] [CrossRef]
- Kise, N.; Iitaka, S.; Iwasaki, K.; Ueda, N. Stereoselective hydrocoupling of cinnamic acid esters by electroreduction: Application to asymmetric synthesis of hydrodimers. J. Org. Chem. 2002, 67, 8305–8315. [Google Scholar] [CrossRef] [PubMed]
- Schoo, N.; Schäfer, H.-J. Electroorganic synthesis, 54. Enantioselective cathodic reduction of 4-substituted coumarins with alkaloids as catalysts, 1. Liebigs Ann. Chem. 1993, 1993, 601–607. [Google Scholar] [CrossRef]
- Pfoertner, K.-H. Photoreaktionen von 3-substituierten cumarinen. Helv. Chim. Acta 1976, 59, 834–840. [Google Scholar] [CrossRef]
- Petkov, I.; Bojilova, A.; Markov, P. Photochemical dehydrogenation of 3-acetyl-3,4-dihydrocoumarin. Monatshefte fur Chemie 1990, 121, 85–87. [Google Scholar] [CrossRef]
- Kawata, H.; Ichikawa, S.; Kumagai, T.; Niizuma, S. A new type of photodimerization reaction for coumarin derivatives. Tetrahedron Lett. 2002, 43, 5161–5163. [Google Scholar] [CrossRef]
- Konde Deshmukh, R.S.; Paradkar, M.V. A facile synthesis of new [4,4′-BI-2H-1-Benzopyran]-2,2′-Diones. Synth. Commun. 1988, 18, 589–596. [Google Scholar] [CrossRef]
- Panichayupakaranant, P.; Noguchi, H.; Ke-Eknamkul, W. A new biscoumarin from Impatiens balsamina root cultures. Planta Med. 1998, 64, 774–775. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.G.; Lin, G.Q. The first total synthesis of 4,4′-biisofraxidin. Chin. J. Chem. 2002, 20, 1263–1267. [Google Scholar] [CrossRef]
- Lei, J.G.; Xu, M.H.; Lin, G.Q. Nickel-catalyzed cross-coupling reactions of 4-mesylcoumarins with aryl halides: Facile synthesis of 4-substituted coumarins. Synlett 2004, 13, 2364–2368. [Google Scholar] [CrossRef]
- Ivanov, C.; Bojilova, A. On the reaction of 3-phenylcoumarin with organomagnesium compounds. Synthesis 1974, 1974, 708–709. [Google Scholar] [CrossRef]
- Bojilova, A.; Ivanov, C. Two new routes to esters of 2-oxochroman-4-acetic acid. Synthesis 1976, 1976, 267–268. [Google Scholar] [CrossRef]
- Silverman, G.S.; Rakita, P.E. Handbook of Grignard Reagents; Marcel Dekker, Inc.: New York, NY, USA, 1996; ISBN 0-8247-9545-8. [Google Scholar]
- Gustafsson, B. Case of radical formation in the reactions between ethyl 3-coumarincarboxylate and Grignard reagents. Finn. Chem. Lett. 1975, 2, 49–50. [Google Scholar]
- Eistert, B.; Klein, L. Das verschiedene verhalten einiger o-chinone und des benzils gegen dimethyl- und diäthylzink; Analogie zum verhalten gegen diazomethan und -äthan. Chem. Ber. 1968, 101, 900–907. [Google Scholar] [CrossRef]
- Rappoport, Z.; Marek, I. The Chemistry of Organozinc Compounds: R-Zn 2 Part Set (Patai’s Chemistry of Functional Groups), 1st ed.; John Wiley & Sons Ltd.: New York, NY, USA, 2006; Print ISBN 9780470093375, Online ISBN 9780470093399. [Google Scholar] [CrossRef]
- Corey, E.J.; Pyne, S.G. Conversion of ketones having δ, ϵ-π-functions to cyclopentanols by zinc-trimethylchlorosilane. Tetrahedron Lett. 1983, 24, 2821–2824. [Google Scholar] [CrossRef]
- Blomberg, C.; Mosher, H. A radical process in a reaction of a Grignard compound. J. Organomet. Chem. 1968, 13, 519–522. [Google Scholar] [CrossRef]
- Blomberg, C.; Grootveld, H.H.; Gerner, T.H.; Bickelhaupt, F. Radical formation during reactions of Grignard reagents with quinones. J. Organomet. Chem. 1970, 24, 549–553. [Google Scholar] [CrossRef]
- Streuff, J.; Gansauer, A. Metal-catalyzed β-functionalization of Michael acceptors through reductive radical addition reactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 14232–14242. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, R.D.; Bojilova, A.G.; Rodios, N.A. A new and efficient method for conjugate addition of trialkylphosphites to 3-acylsubstituted coumarins. Tetrahedron 2004, 60, 10335–10342. [Google Scholar] [CrossRef]
- Simeonov, M.F.; Spassov, S.L.; Bojilova, A.; Ivanov, C. Conformation and tautomeric equilibria of 3-acyl-3,4-dihydrocoumarins: A 1H and 13C NMR study. J. Mol. Struct. 1985, 127, 127–133. [Google Scholar] [CrossRef]
- Ilieva, E.D.; Petkova, N.I.; Nikolova, R.D. A new and efficient method for the synthesis of 3,4-disubstituted pyrrolidine-2,5-diones. Molecules 2012, 17, 4936–4949. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. IL MILIONE: A suite of computer programs for crystal structure solution of proteins. J. Appl. Cryst. 2007, 40, 609–613. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.A.; Terzis, A.; Raptopoulou, C.P. A comparative study of the interaction of salicylaldehydes with phosphonoacetates under Knoevenagel reaction conditions. Synthesis of 1,2-benzoxaphosphorines and their dimers. Tetrahedron 1996, 52, 12597–12612. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds will be available on request from the authors. |
Scheme 1.
Dimeric compound of 1a–b.
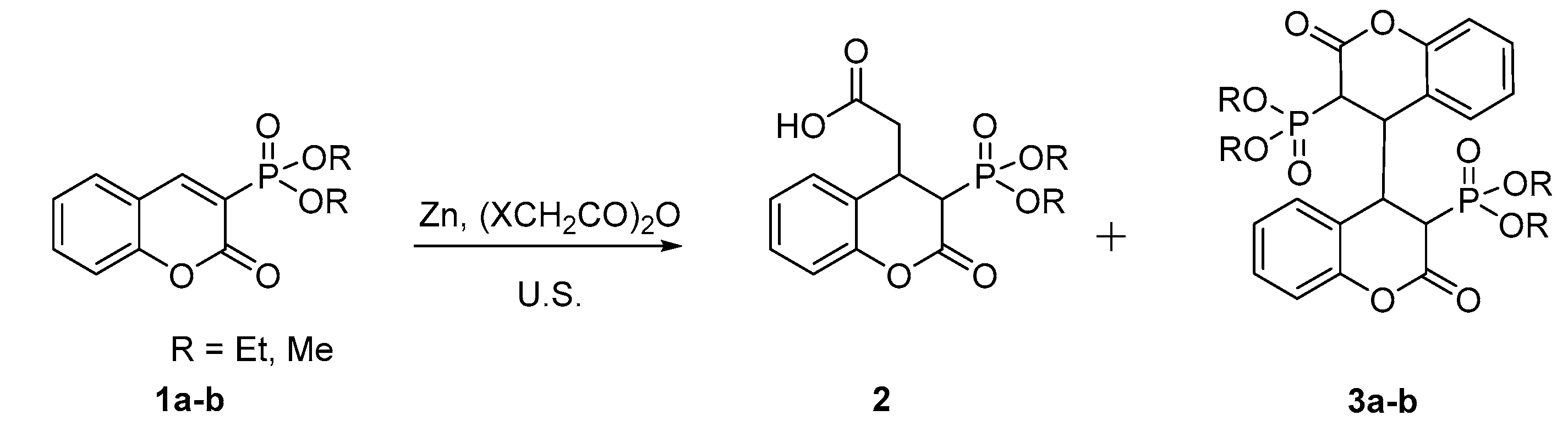
Scheme 2.
Reaction of 1a with the Reformatsky reagent [8].
Scheme 2.
Reaction of 1a with the Reformatsky reagent [8].

Scheme 3.
Structures of the possible organozinc enolates.
Scheme 4.
Possibilities for radical formation during the reaction.
Scheme 5.
Probable mechanism for the formation of 3,3′,4,4′-tetrahydro-3,3′-disubstituted-4,4′-biscoumarins using 3-dialkylphosphocoumarins 1a–b as model compounds.
Scheme 5.
Probable mechanism for the formation of 3,3′,4,4′-tetrahydro-3,3′-disubstituted-4,4′-biscoumarins using 3-dialkylphosphocoumarins 1a–b as model compounds.
Scheme 6.
Homodimerization of coumarins 1c–n under ultrasound irradiation.
Scheme 7.
Structures of a possible radicals type C for compounds 1l–n.

Figure 1.
Structure of compound 3a: (a) ORTEP representation with thermal ellipsoids 40%—Crystal Data for C26H32O10P2 (M = 566.45 g/mol): monoclinic, space group P21/n (no. 14), a = 12.3959(15) Å, b = 9.3824(10) Å, c = 12.4119(16) Å, β = 101.126(5)°, V = 1416.4(3) Å3, Z = 2, T = 300.15 K, μ(MoKα) = 0.207 mm−1, Dcalc = 1.328 g/cm3, 9883 reflections measured (5.17° ≤ 2Θ ≤ 52.788°), 2888 unique (Rint = 0.0528, Rsigma = 0.0587) which were used in all calculations. The final R1 was 0.0507 (I > 2σ(I)) and wR2 was 0.1269 (all data). (b) Meso-form of compound 3a.
Figure 1.
Structure of compound 3a: (a) ORTEP representation with thermal ellipsoids 40%—Crystal Data for C26H32O10P2 (M = 566.45 g/mol): monoclinic, space group P21/n (no. 14), a = 12.3959(15) Å, b = 9.3824(10) Å, c = 12.4119(16) Å, β = 101.126(5)°, V = 1416.4(3) Å3, Z = 2, T = 300.15 K, μ(MoKα) = 0.207 mm−1, Dcalc = 1.328 g/cm3, 9883 reflections measured (5.17° ≤ 2Θ ≤ 52.788°), 2888 unique (Rint = 0.0528, Rsigma = 0.0587) which were used in all calculations. The final R1 was 0.0507 (I > 2σ(I)) and wR2 was 0.1269 (all data). (b) Meso-form of compound 3a.
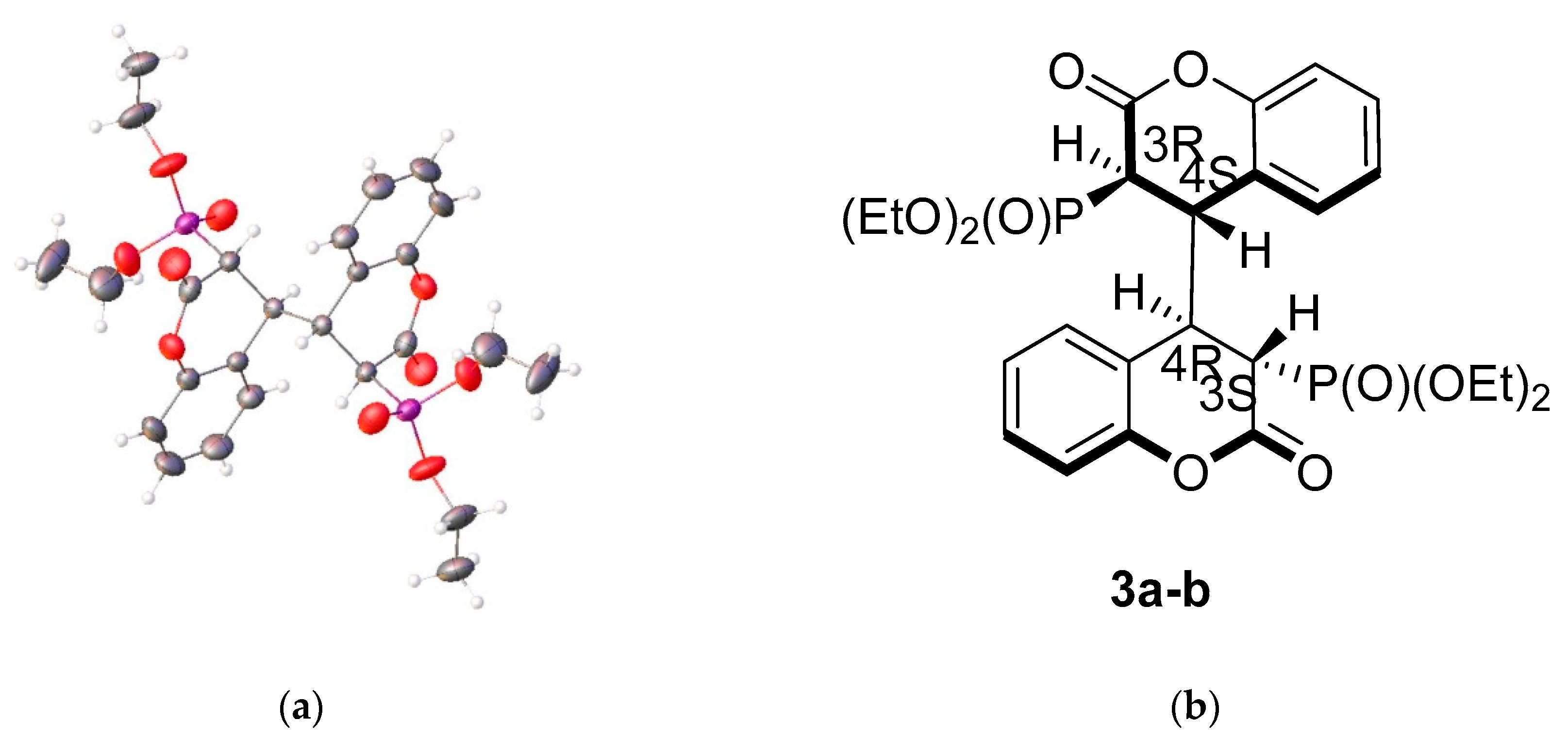
Figure 2.
Stereoisomers of tetrahydrobiscoumarins 3g–j.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Homodimerization of 3-dialkylphosphonocoumarins 1a–b under ultrasound irradiation.
| Product | Substituent | Metal | Reaction Conditions–1: Metal: (ClCH2CO)2O | |||||
|---|---|---|---|---|---|---|---|---|
| Method A 1:2.8:2.4 | Method B 1:5.6:2.4 | Method C 1:3.4:1.5 | ||||||
| Time [min] | Yield [%] | Time [min] | Yield [%] | Time [min] | Yield [%] | |||
| 3a | P(O)(OEt)2 | Mg | 420 | 14 | - | - | - | - |
| 3a | P(O)(OEt)2 | Zn | 420 | 70 | 180 | 89 | 2040 | 30 a |
| 3b | P(O)(OMe)2 | Zn | 420 | 25 a | 200 | 61 | - | - |
a There was not full conversion of 1a.
Table 2.
Solvents used for the homodimerization of 3-dialkylphosphonocoumarins 1a–b (Method A).
| Solvent | Reaction Time [min] | Yield |
|---|---|---|
| THF | 120 | Complex mixture |
| Et2O | 420 | N/A |
| THF:Et2O (3.5:5) | 420 | 70% |
Table 3.
Homodimerization of 3-substituted coumarins 1c–n under ultrasound reaction conditions.
| Coumarin | Product | Y | Method A | Method B | ||
|---|---|---|---|---|---|---|
| Reaction time [min] | Yield [%] | Reaction time [min] | Yield [%] | |||
| 1c | 3c | COOEt | 300 | 60 | 30 | 73 |
| 1d | 3d | COOMe | 240 | 51 | 30 | 73 |
| 1e | 3e | COOPh | 120 | 55 | 40 | 67 |
| 1f | 3f | COMe | 180 | Complex mixture | 10 | 92 a |
| 1g | 3g | COiPr | 1020 | Partial conversion | 15 | 86 a |
| 1h | 3h | COtBu | 390 | 71 | 180 | 92 a |
| 1i | 3i | COPh | 270 | 52 | 10 | 84 |
| 1j | 3j | NO2 | 840 | Complex mixture | 90 | 45 |
| 1k | 3k | CN | 840 | Complex mixture | 270 | Complex mixture |
| 1l | 3l | H | 1020 | N/A | 120 | N/A |
| 1m | 3m | Ph | 1020 | N/A | 120 | N/A |
| 1n | 3n | NHCOMe | 720 | N/A | - | - |
a Two or more isomers see Section 2.2. Spectroscopic data interpretation of compounds 3.
Table 4.
Mixed reactions on the coumarin dimerization.
| Entry | Mixed Coumarin Reaction (Method B, Equimolar Ratio for the Coumarins) | Reaction Time [min] | Products |
|---|---|---|---|
| 1 | 1a:1m | 180 | 3a:1m |
| 2 | 1a:1c | 40 | 1a:3c |
| 3 | 1f:1i | 10 | 3f:3i |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Koleva, A.I.; Petkova-Yankova, N.I.; Nikolova, R.D. Ultrasound-Assisted Metal-Mediated Method for the Formation of Tetrahydro-3,3′-Disubstituted Biscoumarins. Molecules 2018, 23, 2810. https://doi.org/10.3390/molecules23112810
AMA Style
Koleva AI, Petkova-Yankova NI, Nikolova RD. Ultrasound-Assisted Metal-Mediated Method for the Formation of Tetrahydro-3,3′-Disubstituted Biscoumarins. Molecules. 2018; 23(11):2810. https://doi.org/10.3390/molecules23112810
Chicago/Turabian StyleKoleva, Ana I., Nevena I. Petkova-Yankova, and Rositca D. Nikolova. 2018. "Ultrasound-Assisted Metal-Mediated Method for the Formation of Tetrahydro-3,3′-Disubstituted Biscoumarins" Molecules 23, no. 11: 2810. https://doi.org/10.3390/molecules23112810