In-Silico Prediction and Modeling of the Quorum Sensing LuxS Protein and Inhibition of AI-2 Biosynthesis in Aeromonas hydrophila


Abstract
:1. Introduction
2. Results
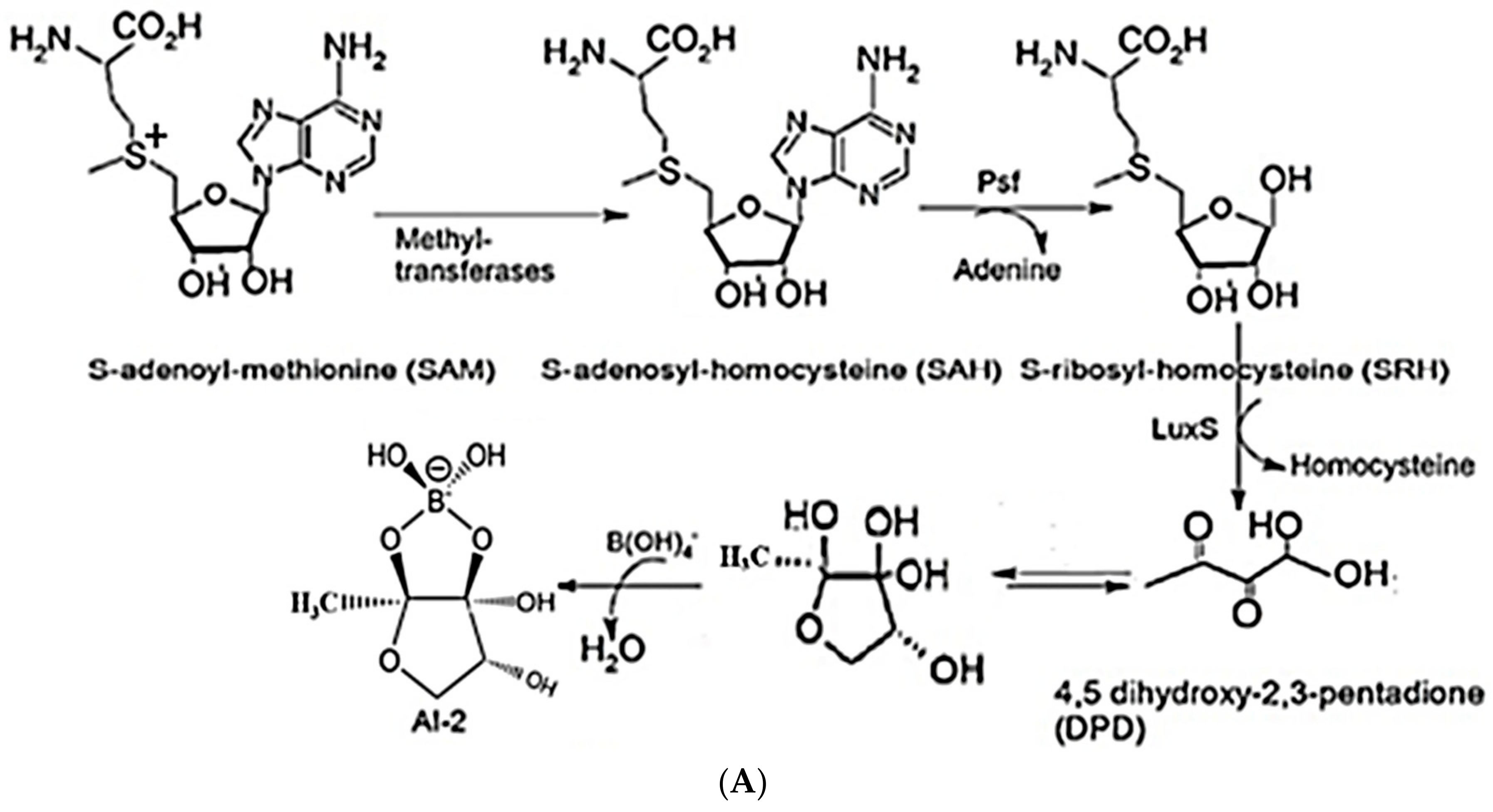
2.1. Synthesis of AI-2 Using a LuxS Ssystem
2.2. LuxS Sequence Analysis
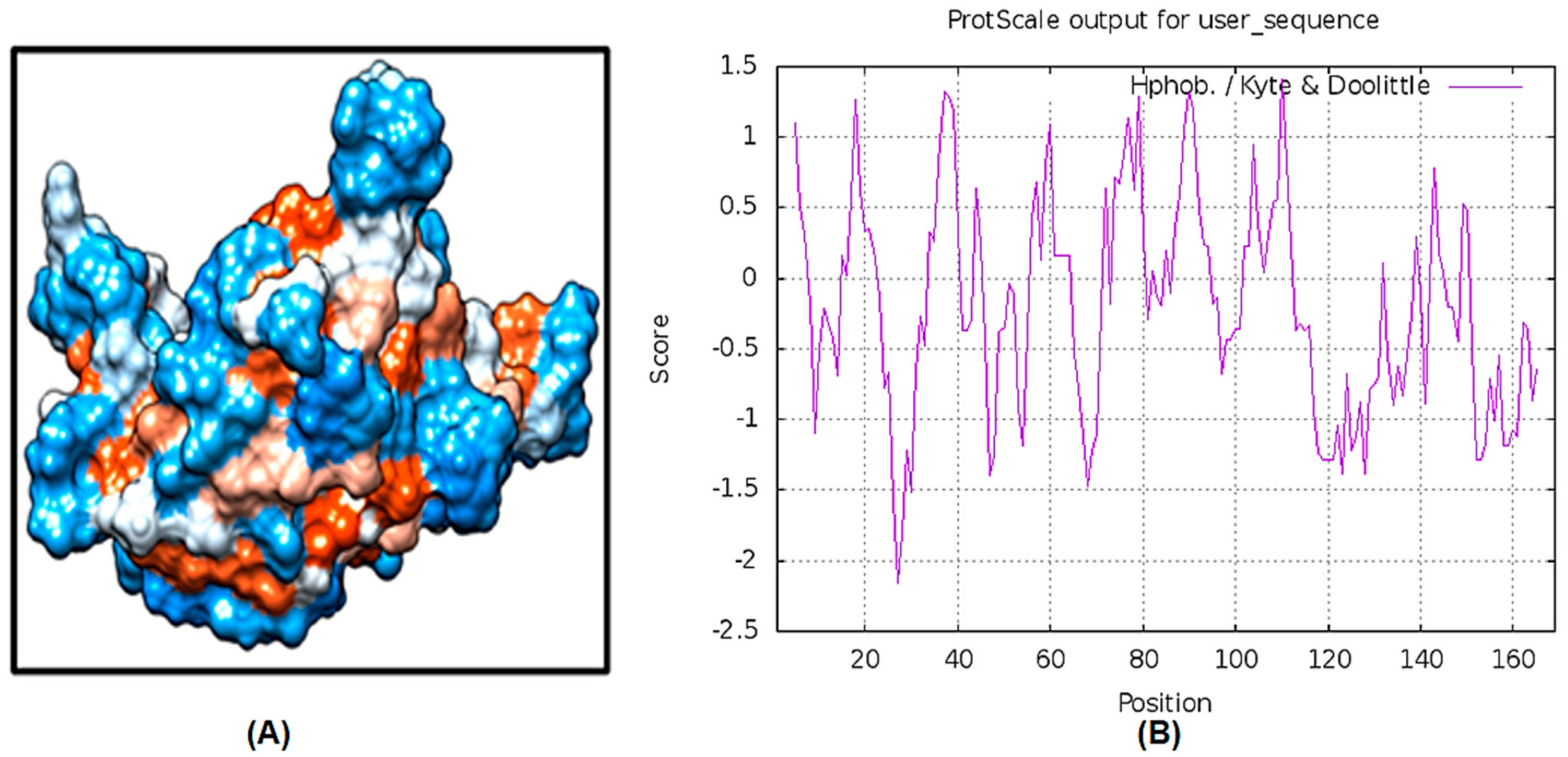
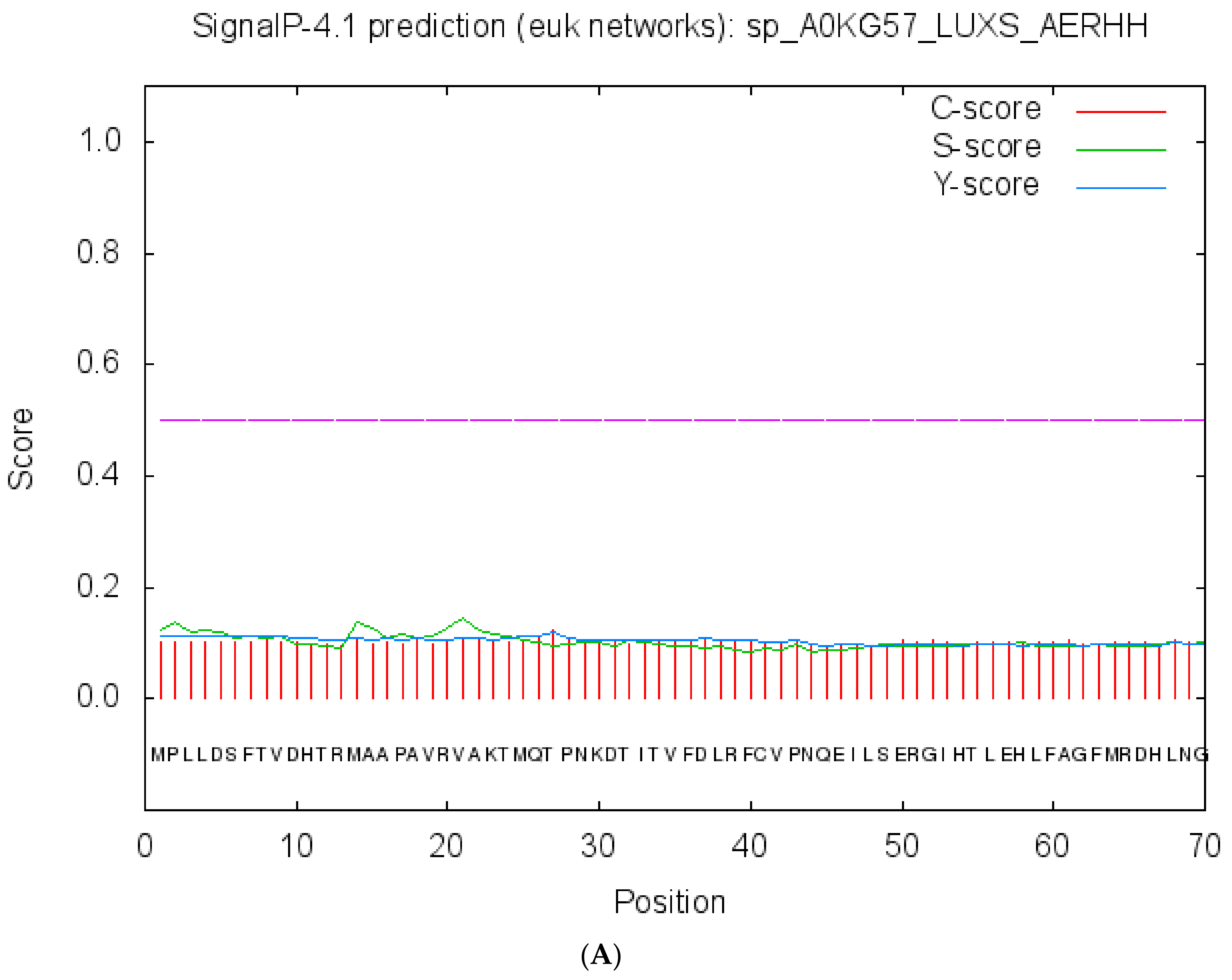
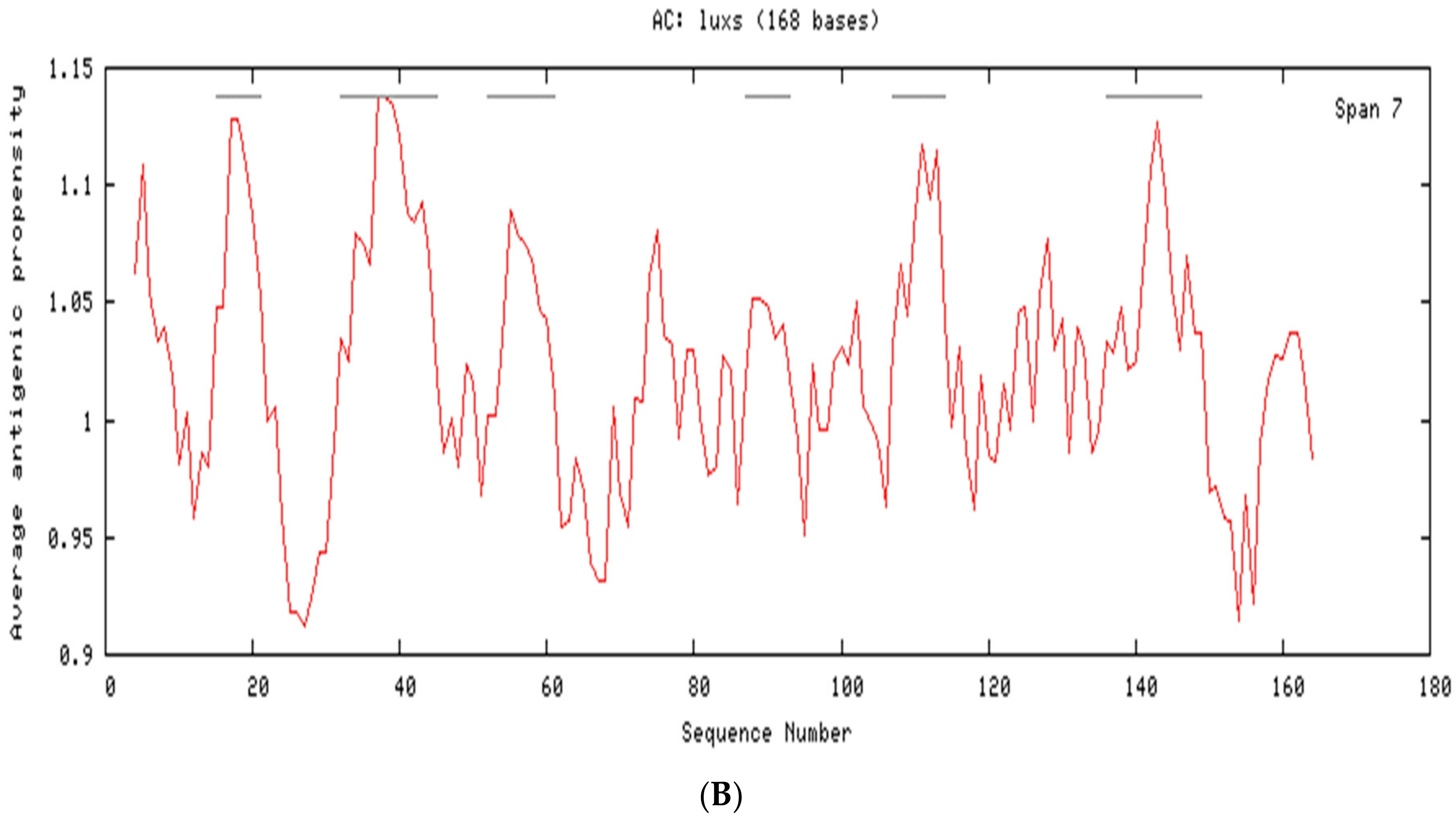
2.3. Cellular Localization and Antigenic Site Prediction
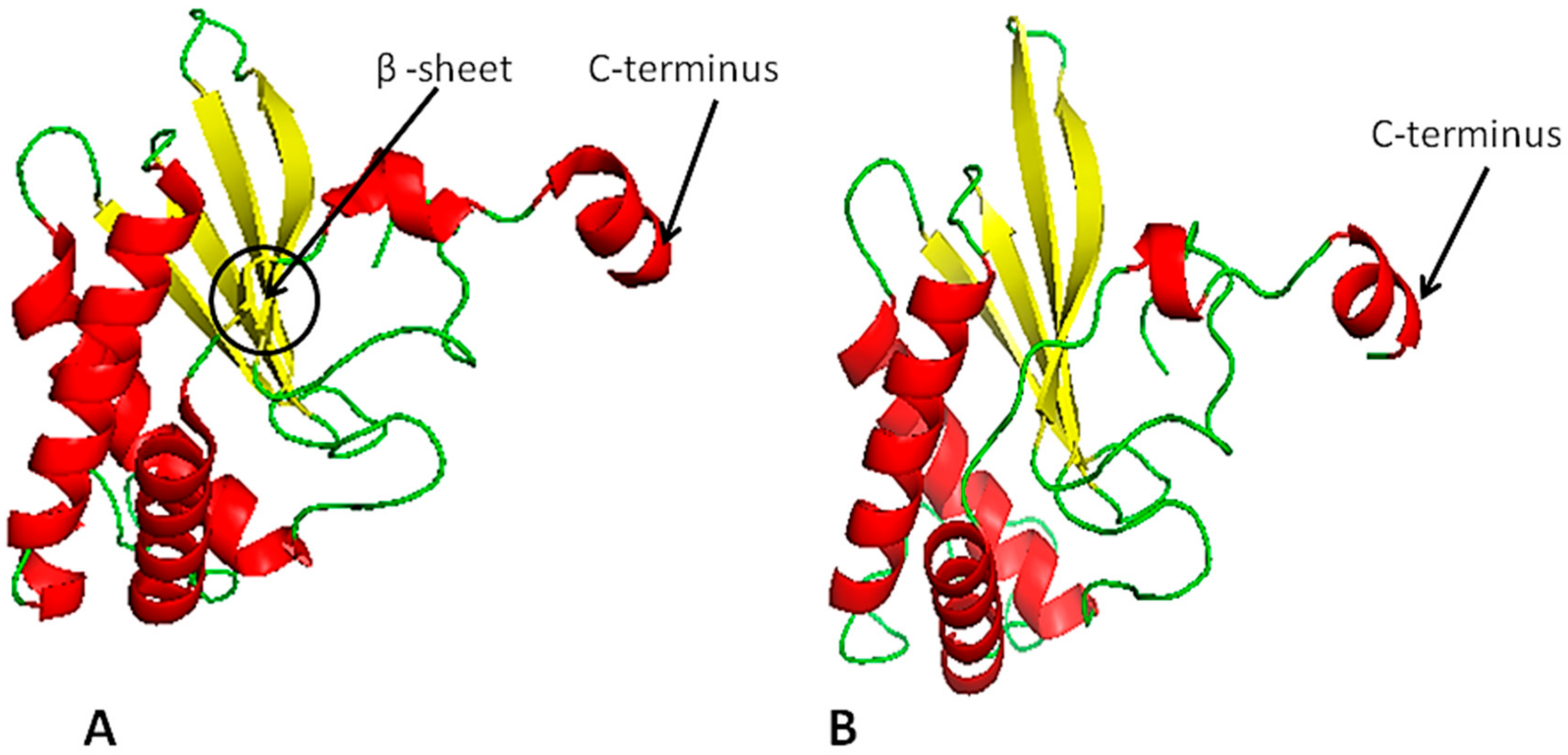
2.4. LuxS Structural Analysis
2.5. Functional Annotation of LuxS
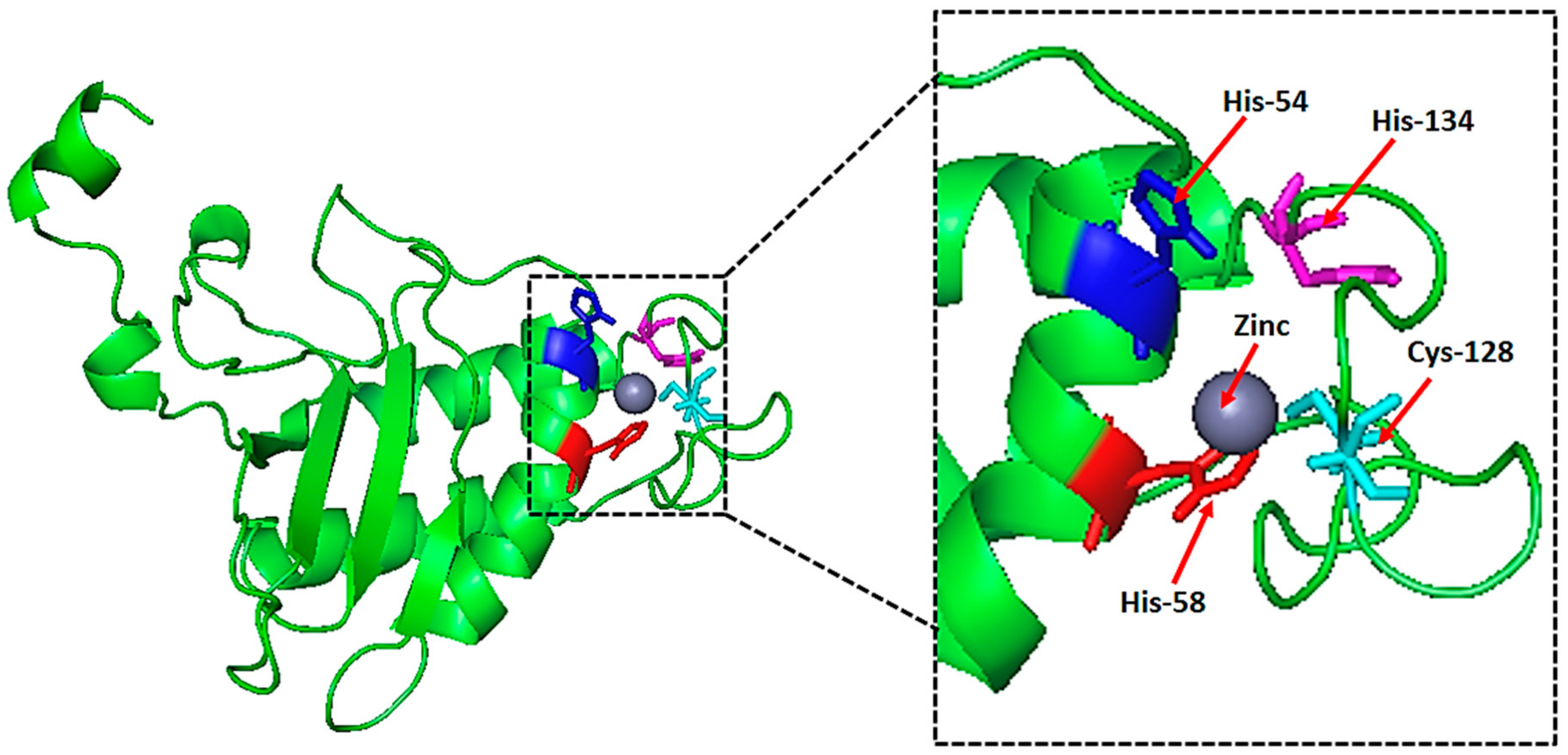
2.6. Natural Ligand, Its Binding Sites and Analysis of Topological Features
2.7. Virtual Screening and Toxicity Studies
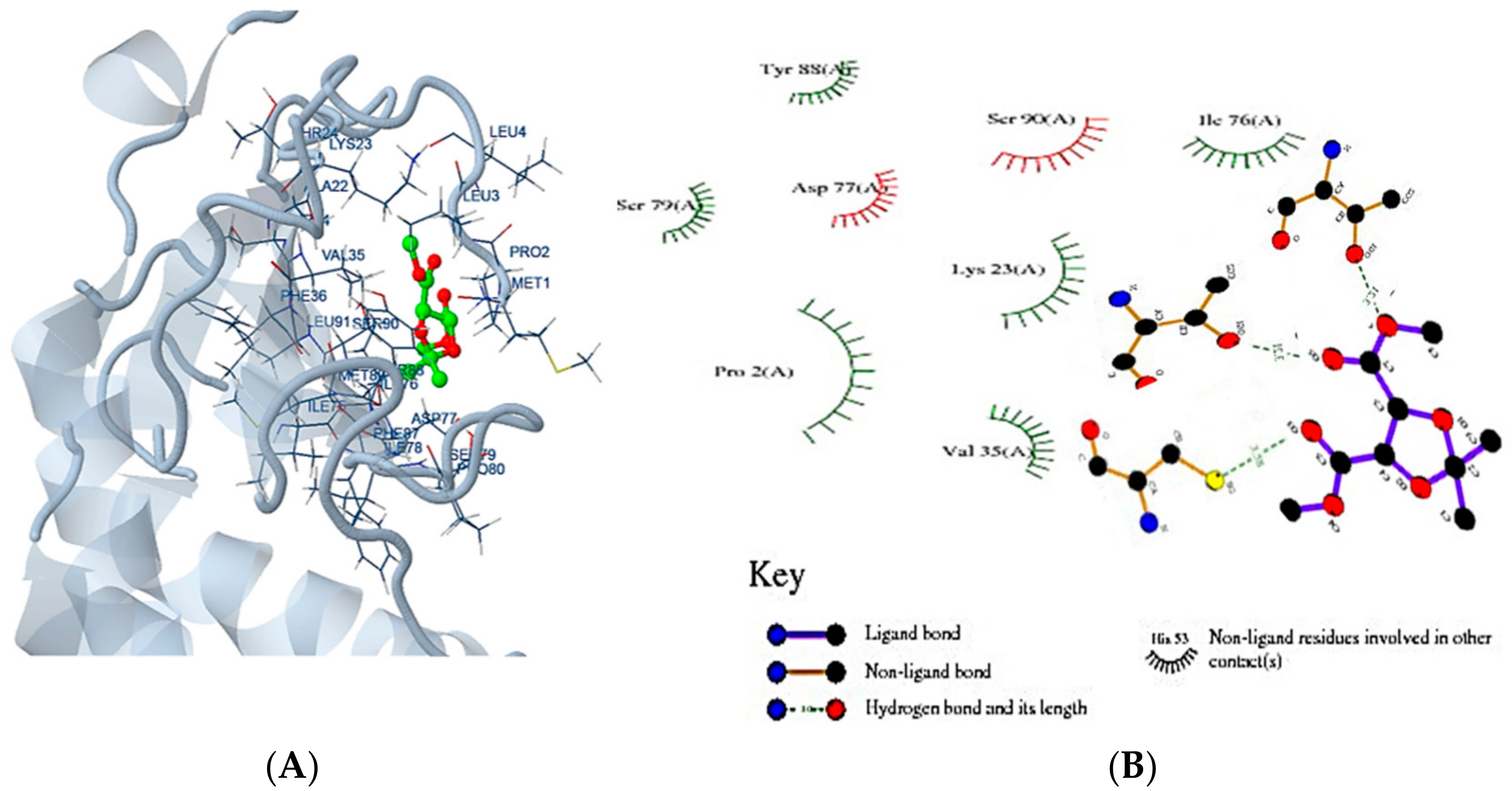
2.8. Molecular Docking
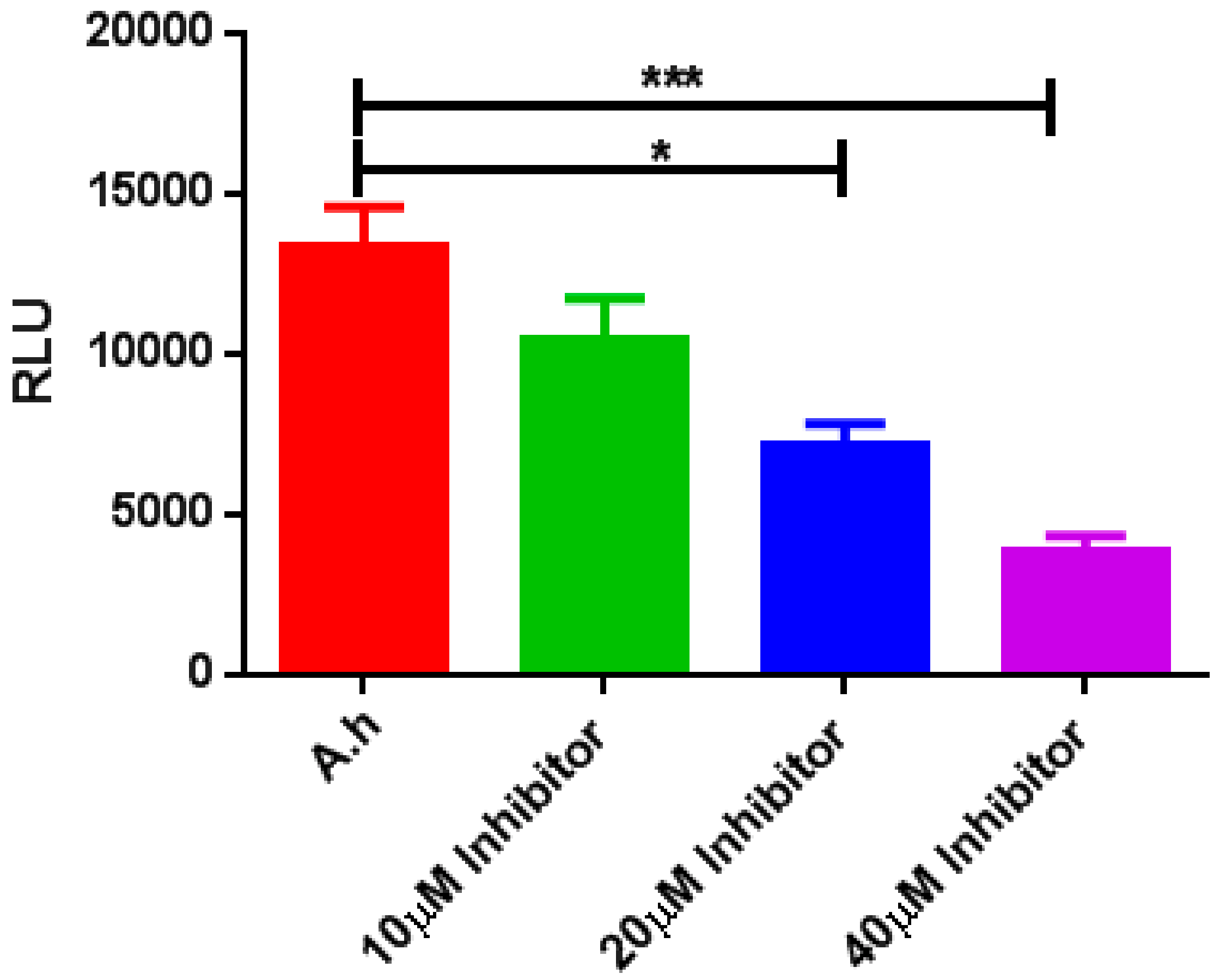
2.9. LuxS AI-2 Biosynthesis Inhibition Using a (−)-Dimethyl 2,3-O-isopropylidene-l-tartrate Inhibitory Compound
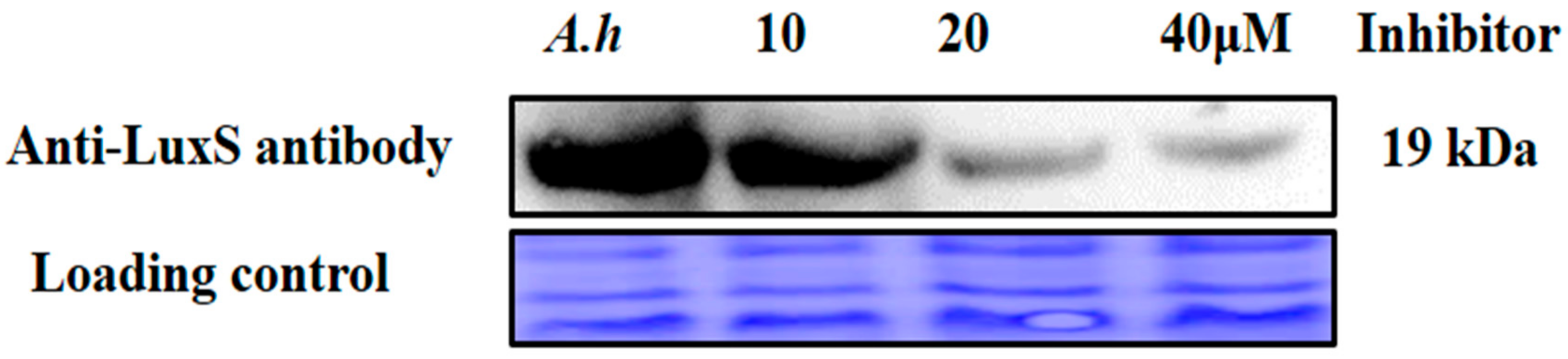
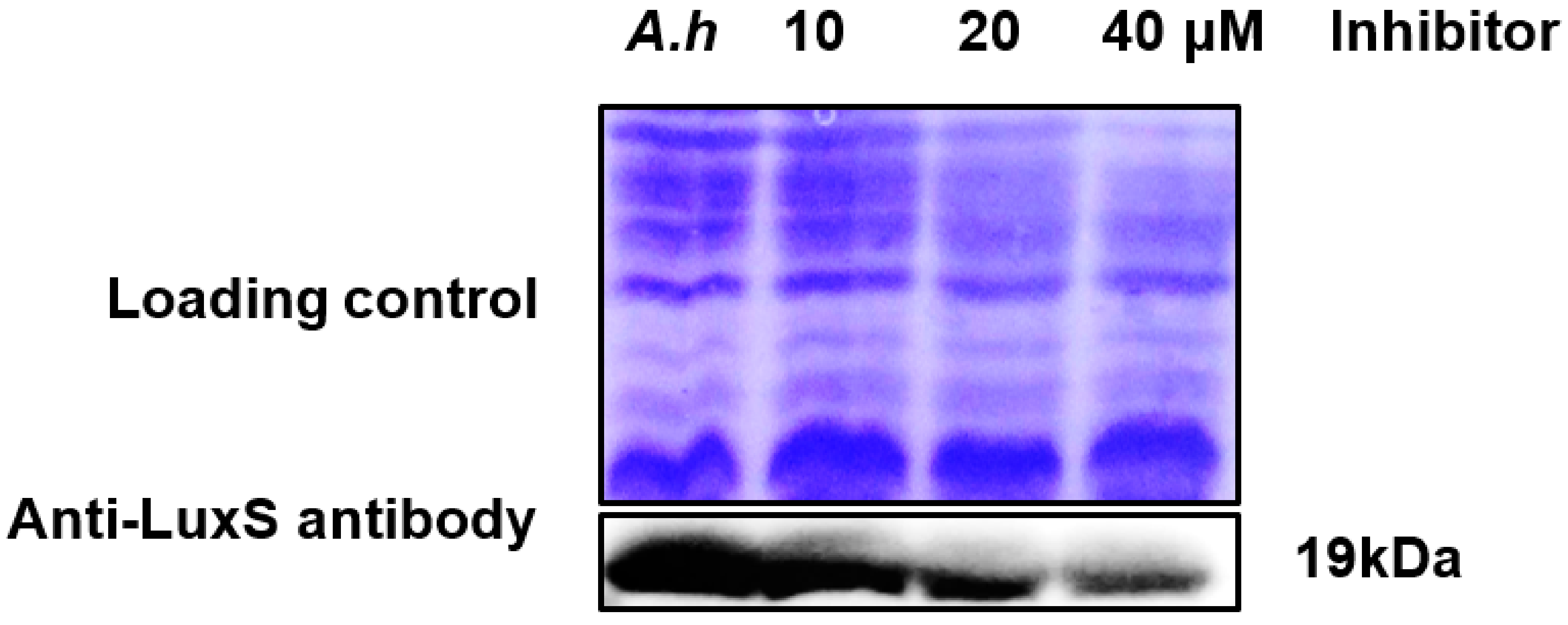
2.10. Validation of AI-2 Inhibition via Analysis of Protein Expression Levels
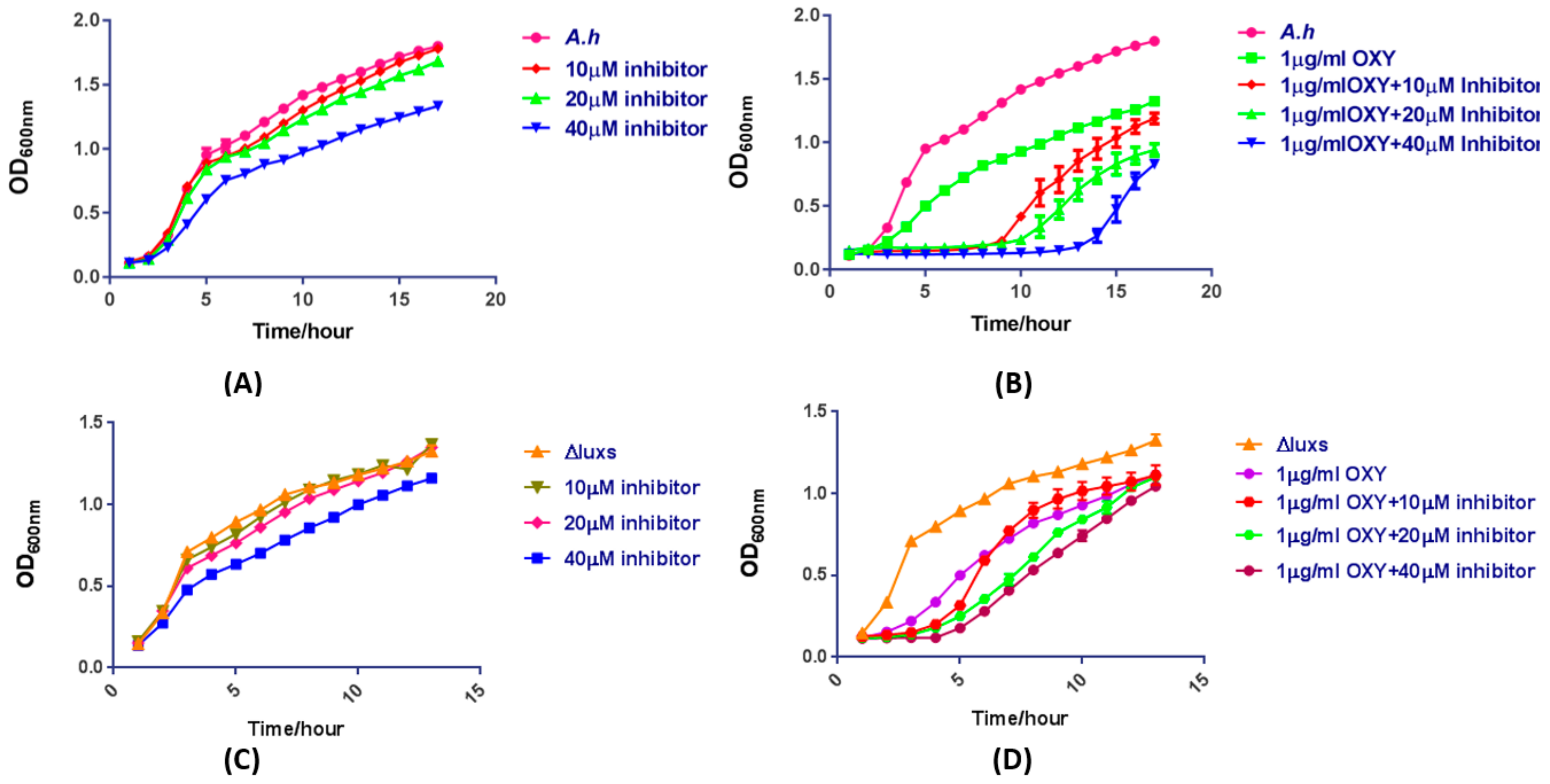
2.11. The Mixture of OXY and AI-2 Inhibitor Is a Potentially Synergistic Strategy for Bacteriostasis
3. Discussion
4. Methodology
4.1. Bioinformatics Analyses
4.1.1. Retrieval of Protein Sequences and Analysis
4.1.2. Assessing Physicochemical Properties
4.1.3. Structural Modeling
4.1.4. Structure Validation and Refinement
4.1.5. Active Site, Ligand, and Ligand Binding Sites Evaluation
4.1.6. High Throughput Virtual Screening and Toxicity Analysis
4.1.7. Ligand Preparation and Molecular Docking of the Receptors to Ligands
4.2. In Vitro Methods
4.2.1. Bacterial Strains and Growth Conditions
4.2.2. AI-2 Inhibition Bioluminescence Assay
4.2.3. Western Blotting
4.2.4. Antimicrobial Survival after Cocktail Therapy
4.2.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, W.; Ali, F.; Cai, Q.; Yao, Z.; Sun, L.; Lin, W.; Lin, X. Quantitative proteomic analysis reveals that chemotaxis is involved in chlortetracycline resistance of Aeromonas hydrophila. J. Proteom. 2018, 172, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-H.; Hsu, R.W.-W.; Huang, T.-J.; Hsu, W.-H.; Huang, K.-C.; Li, Y.-Y.; Peng, K.-T. Necrotizing soft-tissue infections and sepsis caused by Vibrio vulnificus compared with those caused by Aeromonas species. J. Bone Jt. Surg. Am. 2007, 89, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Chompoonuch, S.; Wangsomboonsiri, W.; Wongprasit, P.; Sungkanuparph, S.; Phakdeekitcharoen, B. Aeromonas hydrophila sepsis with septic embolism and rhabdomyolysis in a chronic iron overload haemodialysis patient treated with deferoxamine. NDT Plus 2009, 2, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, M.K.P.; Nakandakare, I.B.; Terhune, J.S.; Wood, T.; Ranzani-Paiva, M.J.T. Dietary supplementation with Bacillus subtilis, Saccharomyces cerevisiae and Aspergillus oryzae enhance immunity and disease resistance against Aeromonas hydrophila and Streptococcus iniae infection in juvenile tilapia Oreochromis niloticus. Fish Shellfish Immunol. 2015, 43, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.G.; Miller, M.B.; Bassler, B.L. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: A new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. USA 1999, 96, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Bassler, B.L. LuxS quorum sensing: More than just a numbers game. Curr. Opin. Microbiol. 2003, 6, 191–197. [Google Scholar] [CrossRef]
- Winzer, K.; Hardie K, W.P. LuxS and autoinducer-2: Their contribution to quorum. Adv. Appl. Microbiol. 2003, 53, 291–396. [Google Scholar] [PubMed]
- Schauder, S.; Shokat, K.; Surette, M.G.; Bassler, B.L. The LuxS family of bacterial autoinducers: Biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 2001, 41, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, V.; Torres, A.G.; Jarvis, B.; Nataro, J.P.; Kaper, J.B. Bacteria-host communication: The language of hormones. Proc. Natl. Acad. Sci. USA 2003, 100, 8951–8956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winzer, K.; Hardie, K.R.; Williams, P. Bacterial cell-to-cell communication: Sorry, can’t talk now—Gone to lunch! Curr. Opin. Microbiol. 2002, 5, 216–222. [Google Scholar] [CrossRef]
- De Kievit, T.R.; Iglewski, B.H. Bacterial quorum sensing in pathogenic relationships. Infect. Immun. 2000, 68, 4839–4849. [Google Scholar] [CrossRef] [PubMed]
- Plummer, P.J. LuxS and quorum-sensing in Campylobacter. Front. Cell. Infect. Microbiol. 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Schauder, S.; Bassler, B.L. The languages of bacteria. Genes Dev. 2001, 15, 1468–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, L.; Goraya, M.U.; Arafat, Y.; Ajmal, M.; Chen, J.-L.; Yu, D. Molecular mechanism of quorum-sensing in Enterococcus faecalis: Its role in virulence and therapeutic approaches. Int. J. Mol. Sci. 2017, 18, 960. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schauder, S.; Potier, N.; Van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.S.; Thompson, J.A.; Xavier, K.B. AI-2-mediated signalling in bacteria. FEMS Microbiol. Ecol. 2013, 37, 156–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.; Shang, W.; Yang, Z.; Sun, Z.; Li, Y.; Guo, J.; Wang, X.; Zou, D.; Wang, S.; Lei, H. LuxS-dependent AI-2 regulates versatile functions in Enterococcus faecalis V583. J. Proteome Res. 2012, 11, 4465–4475. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.J.; Swift, S.; Kirke, D.F.; Keevil, C.W.; Dodd, C.E.; Williams, P. The regulation of biofilm development by quorum sensing in Aeromonas hydrophila. Environ. Microbiol. 2002, 4, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Jahid, I.K.; Mizan, M.F.R.; Ha, A.J.; Ha, S.-D. Effect of salinity and incubation time of planktonic cells on biofilm formation, motility, exoprotease production, and quorum sensing of Aeromonas hydrophila. Food Microbiol. 2015, 49, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Defoirdt, T.; Bossier, P.; Sorgeloos, P.; Verstraete, W. The impact of mutations in the quorum sensing systems of Aeromonas hydrophila, Vibrio anguillarum and Vibrio harveyi on their virulence towards gnotobiotically cultured Artemia franciscana. Environ. Microbiol. 2005, 7, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Zhu, J. Mechanism of action of S-ribosylhomocysteinase (LuxS). Curr. Opin. Chem. Biol. 2004, 8, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.M.; Pasha, S.N.; Sowdhamini, R. Genome-wide survey and phylogeny of S-Ribosylhomocysteinase (LuxS) enzyme in bacterial genomes. BMC Genom. 2016, 17, 742. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, R.; Joseph, S.W.; Chopra, A.K.; Sha, J.; Shaw, J.; Graf, J.; Haft, D.; Wu, M.; Ren, Q.; Rosovitz, M. Genome sequence of Aeromonas hydrophila ATCC 7966T: Jack of all trades. J. Bacteriol. 2006, 188, 8272–8282. [Google Scholar] [CrossRef] [PubMed]
- Ruzheinikov, S.; Das, S.; Sedelnikova, S.; Hartley, A.; Foster, S.; Horsburgh, M.; Cox, A.; McCleod, C.; Mekhalfia, A.; Blackburn, G.; et al. The 1.2 Å structure of a novel quorum-sensing protein, Bacillus subtilis LuxS. J. Mol. Biol. 2001, 313, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, M.T.; Ludwig, M.L. Crystal structure of the quorum-sensing protein LuxS reveals a catalytic metal site. Proc. Natl. Acad. Sci. USA 2001, 98, 11169–11174. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.A.; Furlong, E.B.; Laubert, B.; Eroshkina, G.A.; Batiyenko, Y.; Adams, J.M.; Bergseid, M.G.; Marsh, C.D.; Peat, T.S.; Sanderson, W.E.; et al. A structural genomics approach to the study of quorum sensing: Crystal structures of three LuxS orthologs. Structure 2001, 9, 527–537. [Google Scholar] [CrossRef]
- Li, H.; Zhao, H.; Zhu, L.; Hong, L.; Zhang, H.; Lin, F.; Xu, C.; Li, S.; Zhang, Z. Crystallization and preliminary X-ray analysis of S-ribosylhomocysteinase from Streptococcus mutans. Acta Crystallogr. F 2012, 68, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Daniel, R.; Wagner-Döbler, I.; Zeng, A.-P. Is autoinducer-2 a universal signal for interspecies communication: A comparative genomic and phylogenetic analysis of the synthesis and signal transduction pathways. BMC Evol. Biol. 2004, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezzonico, F.; Duffy, B. Lack of genomic evidence of AI-2 receptors suggests a non-quorum sensing role for luxS in most bacteria. BMC Microbiol. 2008, 8, 154. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Meng, F.; Ma, X.; Song, X.; Liu, X.; Gao, W.; Dang, Y.; Meng, Y.; Cao, M.; Song, C. Regulation of bacteria population behaviors by AI-2 “consumer cells” and “supplier cells”. BMC Microbiol. 2017, 17, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L.C.; Federle, M.J. Peptide pheromone signaling in Streptococcus and Enterococcus. FEMS Microbiol. Rev. 2014, 38, 473–492. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, S.C.; Varszegi, C.; van Boxel, N.; Habel, L.W.; Metzger, K.; Daniels, R.; Marchal, K.; De Vos, D.; Vanderleyden, J. Chemical synthesis of (S)-4, 5-dihydroxy-2, 3-pentanedione, a bacterial signal molecule precursor, and validation of its activity in Salmonella typhimurium. J. Biol. Chem. 2005, 280, 19563–19568. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Zhu, J.; Hu, X.; Pei, D.; Bell, C.E. Crystal Structure of S-Ribosylhomocysteinase (LuxS) in Complex with a Catalytic 2-Ketone Intermediate. Biochemistry 2005, 44, 3745–3753. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Bowie, J.U.; Luthy, R. Method to Identify Protein Sequences that Fold into a Known Three-Dimensional Structure. Google Patents US5436850A, 25 July 1995. [Google Scholar]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Liithy, R.; Bowie, J.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Wang, Z.; Sun, L.; Li, W.; Shi, Y.; Lin, L.; Lin, W.; Lin, X. Quantitative proteomic analysis of cell envelope preparations under iron starvation stress in Aeromonas hydrophila. BMC Microbiol. 2016, 16, 161. [Google Scholar] [CrossRef] [PubMed]
- Brackman, G.; Celen, S.; Baruah, K.; Bossier, P.; Van Calenbergh, S.; Nelis, H.J.; Coenye, T. AI-2 quorum-sensing inhibitors affect the starvation response and reduce virulence in several Vibrio species, most likely by interfering with LuxPQ. Microbiology 2009, 155, 4114–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.; Zhou, S.; Zhu, W.; Zhuang, X. Quorum quenching bacteria Bacillus sp. QSI-1 protect zebrafish (Danio rerio) from Aeromonas hydrophila infection. Sci. Rep. 2014, 4, 5446. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, E.V.; Khajanchi, B.K.; Popov, V.L.; Wen, J.; Chopra, A.K. Impact of QseBC system in c-di-GMP-dependent quorum sensing regulatory network in a clinical isolate SSU of Aeromonas hydrophila. Microb. Pathog. 2012, 53, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, J.F.; Zhang, T.; Wynn, D.P.; Karschner, E.L.; Zhou, Z.S. Synthesis of LuxS inhibitors targeting bacterial cell−cell communication. Org. Lett. 2004, 6, 3043–3046. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, W.; Duan, Q.; Li, F.; Cheng, H. Sodium houttuyfonate affects production of N-acyl homoserine lactone and quorum sensing-regulated genes expression in Pseudomonas aeruginosa. Front. Microbiol. 2014, 5, 635. [Google Scholar] [CrossRef] [PubMed]
- De Nys, R.; Givskov, M.C.; Kumar, N.; Kjelleberg, S.; Steinberg, P. Furanones: Progress in molecular and subcellular biology. Subseries marine molecular biotechnology. In Antifouling Compounds; Springer: Berlin, Germany, 2006. [Google Scholar]
- Defoirdt, T.; Boon, N.; Bossier, P. Can bacteria evolve resistance to quorum sensing disruption? PLoS Pathog. 2010, 6, e1000989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Contreras, R.; Maeda, T.; Wood, T.K. Resistance to quorum quenching compounds. Appl. Environ. Microbiol. 2013, 79, 6840–6846. [Google Scholar] [CrossRef] [PubMed]
- García-Contreras, R.; Peréz-Eretza, B.; Jasso-Chávez, R.; Lira-Silva, E.; Roldán-Sánchez, J.A.; González-Valdez, A.; Soberón-Chávez, G.; Coria-Jiménez, R.; Martínez-Vázquez, M.; Alcaraz, L.D.; et al. High variability in quorum quenching and growth inhibition by furanone C-30 in Pseudomonas aeruginosa clinical isolates from cystic fibrosis patients. Pathog. Dis. 2015, 73, ftv040. [Google Scholar] [CrossRef] [PubMed]
- Quave, C.L.; Lyles, J.T.; Kavanaugh, J.S.; Nelson, K.; Parlet, C.P.; Crosby, H.A.; Heilmann, K.P.; Horswill, A.R. Castanea sativa (European Chestnut) leaf extracts rich in ursene and oleanene derivatives block Staphylococcus aureus virulence and pathogenesis without detectable resistance. PLoS ONE 2015, 10, e0136486. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gamby, S.; Zheng, Y.; Sintim, H.O. Small molecule inhibitors of AI-2 signaling in bacteria: State-of-the-art and future perspectives for anti-quorum sensing agents. Int. J. Mol. Sci. 2013, 14, 17694–17728. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yi, L.; Wang, S.; Fan, H.; Ding, C.; Mao, X.; Lu, C. Crystal structure and identification of two key amino acids involved in AI-2 production and biofilm formation in Streptococcus suis LuxS. PLoS ONE 2015, 10, e0138826. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.; Hodgkinson, J.T.; Bowden, S.D.; Welch, M.; Spring, D.R. Quorum sensing in Gram-negative bacteria: Small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem. Rev. 2010, 111, 28–67. [Google Scholar] [CrossRef] [PubMed]
- Syed, N.; Ahmed, A.; Moin, S.T. Understanding LuxS-based quorum sensing and its inhibition–molecular dynamics simulation study. Mol. Simul. 2018, 44, 558–567. [Google Scholar] [CrossRef]
- Lowery, C.A.; Abe, T.; Park, J.; Eubanks, L.M.; Sawada, D.; Kaufmann, G.F.; Janda, K.D. Revisiting AI-2 quorum sensing inhibitors: Direct comparison of alkyl-DPD analogues and a natural product fimbrolide. J. Am. Chem. Soc. 2009, 131, 15584–15585. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, E.V.; Popov, V.L.; Sha, J.; Foltz, S.M.; Erova, T.E.; Agar, S.L.; Horneman, A.J.; Chopra, A.K. Mutation in the S-ribosylhomocysteinase (luxS) gene involved in quorum sensing affects biofilm formation and virulence in a clinical isolate of Aeromonas hydrophila. Microb. Pathog. 2008, 45, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Du, Y.; Liu, P.; Li, X.; Liu, Y. Involvement of LuxS in Aeromonas salmonicida metabolism, virulence and infection in Atlantic salmon (Salmo salar L.). Fish Shellfish Immunol. 2017, 64, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Kumari, S.; Banerjee, R.; Samanta, M.; Das, S. Disruption of the quorum sensing regulated pathogenic traits of the biofilm-forming fish pathogen Aeromonas hydrophila by tannic acid, a potent quorum quencher. Biofouling 2017, 33, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; McLean, R.J. Quorum signal inhibitors and their potential use against fish diseases. J. Aquat. Anim. Health 2016, 28, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, M. Analysis of the antibacterial effect of an Edwardsiella tarda LuxS inhibitor. SpringerPlus 2016, 5, 92. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins 2007, 67, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Arvidson, D.; Shapiro, M.; Youderian, P. Mutant tryptophan aporepressors with altered specificities of corepressor recognition. Genetics 1991, 128, 29–35. [Google Scholar] [PubMed]
- Howard, A.E.; Kollman, P.A. Molecular dynamics studies of a DNA-binding protein: 1. A comparison of the trp repressor and trp aporepressor aqueous simulations. Protein Sci. 1992, 1, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.T.; Wang, Q.; Harshey, R.M. Cell density and mobility protect swarming bacteria against antibiotics. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Zhao, L.; Sun, B. LuxS/AI-2 system is involved in antibiotic susceptibility and autolysis in Staphylococcus aureus NCTC 8325. Int. J. Antimicrob. Agents 2013, 41, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Su, Y.-b.; Li, H.; Han, Y.; Guo, C.; Tian, Y.-m.; Peng, X.-x. Exogenous alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell Metab. 2015, 21, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.-M.; Zhang, K.; Shen, D.-S.; Wang, M.-Z.; Shentu, J.-L.; Li, N. Control of the pollution of antibiotic resistance genes in soils by quorum sensing inhibition. Environ. Sci. Pollut. Res. 2017, 24, 5259–5267. [Google Scholar] [CrossRef] [PubMed]
- Gamby, S.; Roy, V.; Guo, M.; Smith, J.A.; Wang, J.; Stewart, J.E.; Wang, X.; Bentley, W.E.; Sintim, H.O. Altering the communication networks of multispecies microbial systems using a diverse toolbox of AI-2 analogues. ACS Chem. Biol. 2012, 7, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Solis, F.J.; Wets, R.J.-B. Minimization by random search techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Gorenc, G.; Lukas, F.; Avgustin, G. Examination of ai-2 quorum sensing system in Prevotella bryantii and Prevotella ruminicola-like strains by using bioluminiscence assay. Acta Chim. Slov. 2007, 90, 107–113. [Google Scholar]
- Vilchez, R.; Lemme, A.; Thiel, V.; Schulz, S.; Sztajer, H.; Wagner-Döbler, I. Analysing traces of autoinducer-2 requires standardization of the Vibrio harveyi bioassay. Anal. Bioanal. Chem. 2007, 387, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yao, Z.; Sun, L.; Hu, W.; Cao, J.; Lin, W.; Lin, X. Proteomics analysis reveals a potential antibiotic cocktail therapy strategy for Aeromonas hydrophila infection in biofilm. J. Proteome Res. 2016, 15, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Li, W.; Lin, Y.; Wu, Q.; Yu, F.; Lin, W.; Lin, X. Proteomic analysis reveals that metabolic flows affect the susceptibility of Aeromonas hydrophila to antibiotics. Sci. Rep. 2016, 6, 39413. [Google Scholar] [CrossRef] [PubMed]
- Sasikala, D.; Jeyakanthan, J.; Srinivasan, P. Structure-based virtual screening and biological evaluation of LuxT inhibitors for targeting quorum sensing through an in vitro biofilm formation. J. Mol. Struct. 2017, 1127, 322–336. [Google Scholar] [CrossRef]
Sample Availability: Not available. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Start Position | Sequence | End Position |
|---|---|---|---|
| 1 | 15 | AAPAVRV | 21 |
| 2 | 32 | TITVFDLRFCVPNQ | 45 |
| 3 | 52 | GIHTLEHLFA | 61 |
| 4 | 87 | FYMSLIG | 93 |
| 5 | 107 | AMSDVLTV | 114 |
| 6 | 136 | LEEAHAIARHVLER | 149 |
| Molecules | Chemical Structure | Mutageic | Tumorigenic | Irritant | Reproductive Effects |
|---|---|---|---|---|---|
| N-(4-Methyl-2-pyridinyl)-2-(1-naphthyl)acetamide |  | Non | Non | Non | No |
| 3-(1,2-Diazabicyclo[2.2.2]oct-2-yl)-1-phenylpropyl acetate |  | Non | Non | Medium risk of irritation | Mediumrisk of Reproductive effect |
| N-(3,5-Dimethylphenyl)-2-{4-hydroxy-2-[(1-methylethylidene)hydrazono]-2,5-dihydro-1,3-thiazol-5-yl}acetamide |  | Non | Non | Non | No |
| (−)-Dimethyl (−)-2,3-O-isopropylidene-l-tartrate |  | Non | Non | Non | No |
| 1-(3-Methylphenyl)-2,5-dioxo-3-pyrrolidinyl N’-phenylimidothiocarbamate |  | Non | Non | Non | No |
| Methyl (4R,5R)-5-Bromomethyl-2,2-dimethyl [1,3]dioxolane4-carboxylate |  | Non | Non | Non | No |
| tert-Butyl(2S)-2-tert-Butoxycarbonylamino-6-[3-(4-meth oxyphenyl)oxaziridin-2-yl] hexanoate |  | Non | Non | Non | No |
| Name of Drug | Chemical Structure | cLogP | Solubility | Mol.Wt | TPSA | Drug Likeliness | Drug Score |
|---|---|---|---|---|---|---|---|
| N-(4-methyl-2-pyridinyl)-2-(1-naphthyl)acetamide |  | −0.54 | −4.04 | 276 | 89.42 | 2.61 | 0.90 |
| N-(3,5-dimethylphenyl)-2-{4-hydroxy-2-[(1-methylethylidene)hydrazono]-2,5-dihydro-1,3-thiazol-5-yl}acetamide |  | −1.27 | −0.42 | 332 | 93.06 | −5.79 | 0.67 |
| (−)-dimethyl(−)-2,3-O-isopropylidene-l-tartrate |  | −1.68 | −0.14 | 219 | 113.9 | 1.16 | 0.97 |
| 1-(3-methylphenyl)-2,5-dioxo-3-pyrrolidinyl N’-phenylimidothiocarbamate |  | 0.45 | −1.28 | 339 | 93.78 | 1.06 | 0.89 |
| Methyl (4R,5R)-5-Bromomethyl-2,2-dimethyl [1,3]dioxolane4-carboxylate |  | −3.02 | −0.41 | 245 | 119.5 | −10.75 | 0.39 |
| tert-Butyl(2S)-2-tert-Butoxycarbonylamino-6-[3-(4-meth oxyphenyl)oxaziridin-2-yl] hexanoate |  | −2.17 | −0.58 | 211 | 105.4 | 2.52 | 0.87 |
| Protein PDB: ID and Species Name | Energy Binding Affinity | Total Intermolecular Energy |
|---|---|---|
| LuxS (predicted model) A. hydrophila | −3.06 (Kcal/mol) | −4.26 kcal/mol |
| LuxS (5e68) Salmonella typhi | −2.50 (Kcal/mol) | −3.63 kcal/mol |
| LuxS (4XCH) Streptococcus suis | −1.76 (Kcal/mol) | −2.90 kcal/mol |
| LuxS (1VJE) Deinococcus radiodurans | −2.55 (Kcal/mol) | −3.69 kcal/mol |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, F.; Yao, Z.; Li, W.; Sun, L.; Lin, W.; Lin, X. In-Silico Prediction and Modeling of the Quorum Sensing LuxS Protein and Inhibition of AI-2 Biosynthesis in Aeromonas hydrophila. Molecules 2018, 23, 2627. https://doi.org/10.3390/molecules23102627
Ali F, Yao Z, Li W, Sun L, Lin W, Lin X. In-Silico Prediction and Modeling of the Quorum Sensing LuxS Protein and Inhibition of AI-2 Biosynthesis in Aeromonas hydrophila. Molecules. 2018; 23(10):2627. https://doi.org/10.3390/molecules23102627
Chicago/Turabian StyleAli, Farman, Zujie Yao, Wanxin Li, Lina Sun, Wenxiong Lin, and Xiangmin Lin. 2018. "In-Silico Prediction and Modeling of the Quorum Sensing LuxS Protein and Inhibition of AI-2 Biosynthesis in Aeromonas hydrophila" Molecules 23, no. 10: 2627. https://doi.org/10.3390/molecules23102627