Genome-Wide Investigation of Genes Regulated by ERα in Breast Cancer Cells

 ,
,
Abstract
:1. Introduction
2. Results
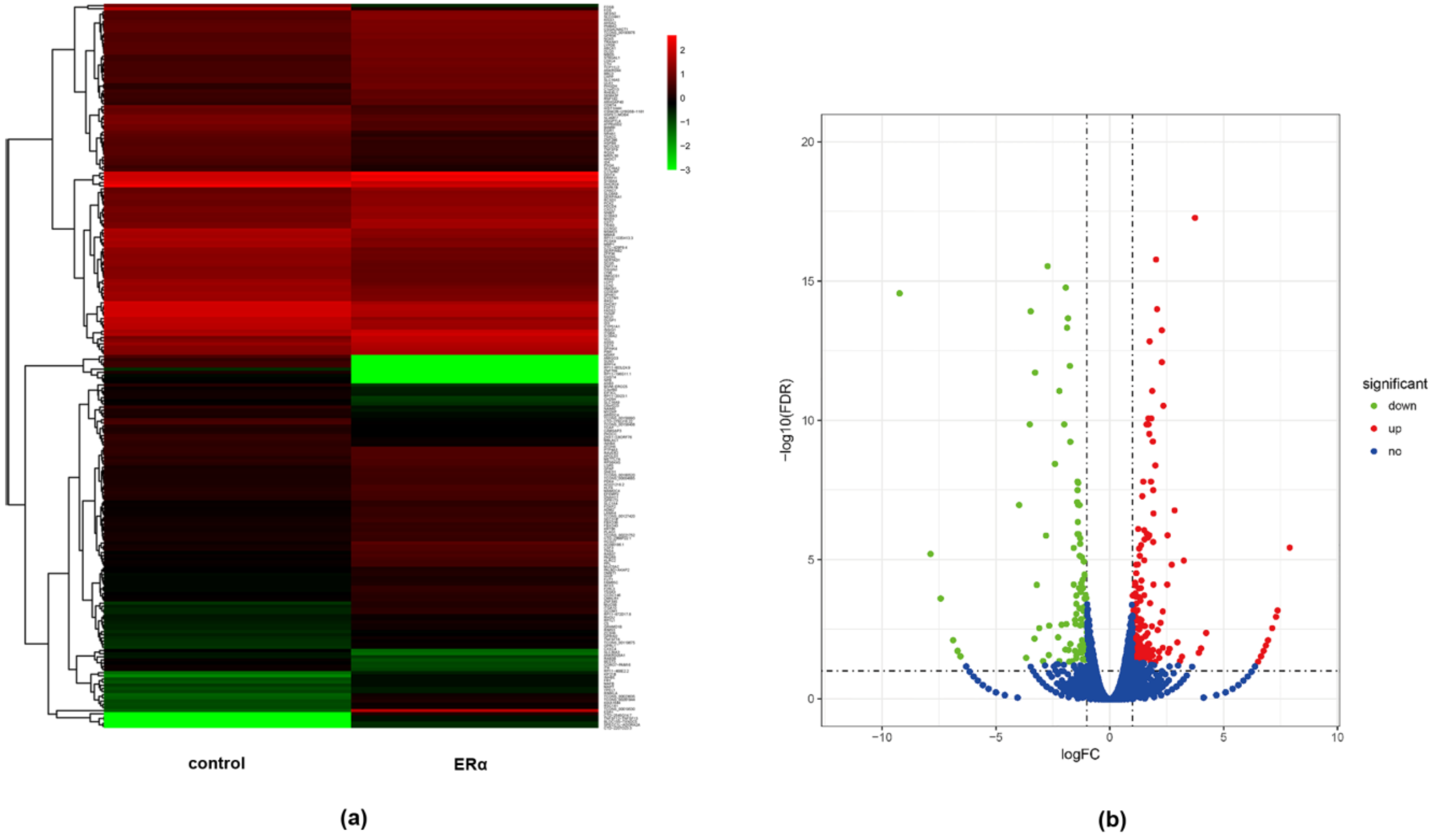
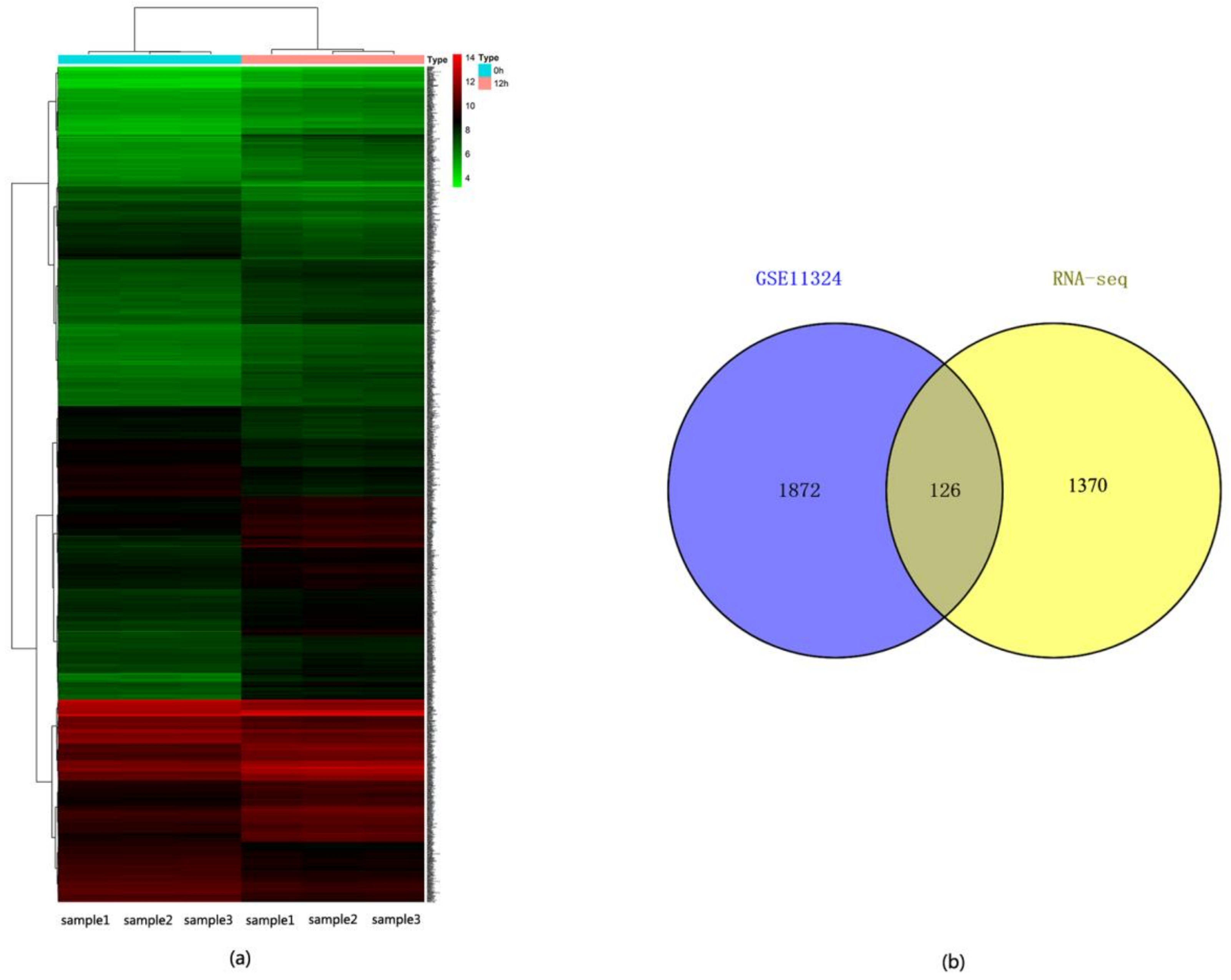
2.1. Identification of DEGs between ERα Transgenic MDA-MB-231 and Wild Type MDA-MB-231 Cells
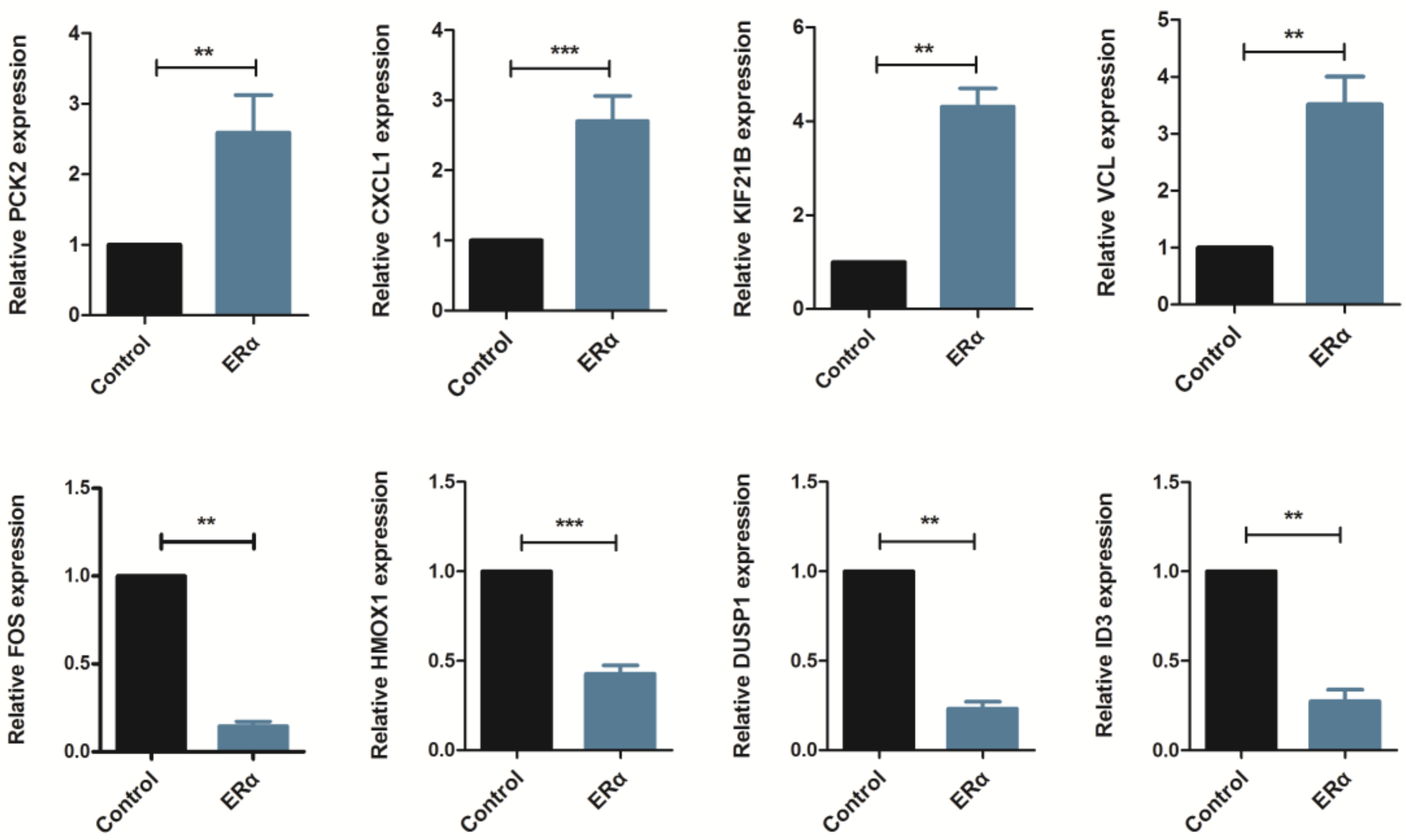
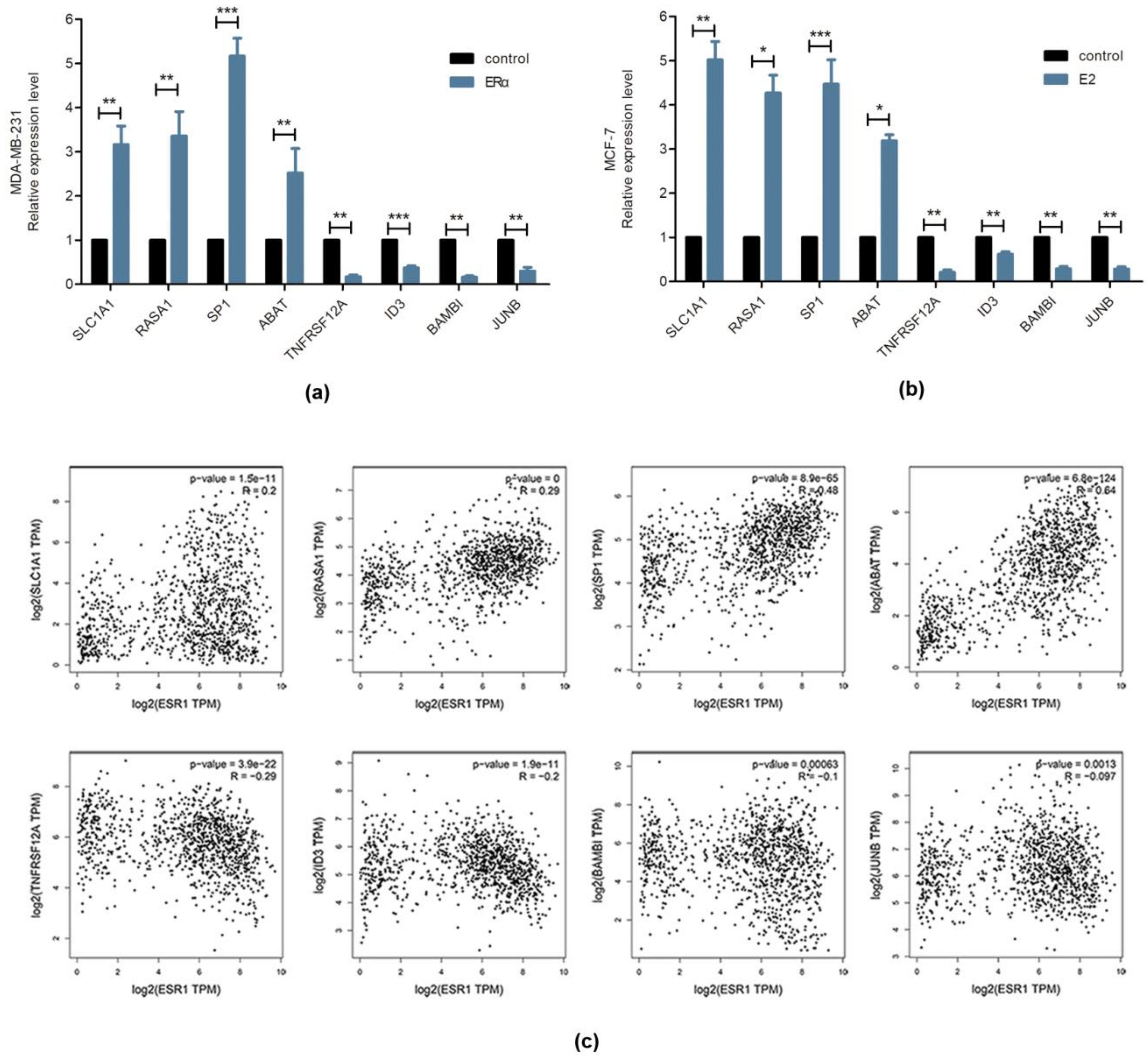
2.2. Validation of Gene Expression Data by Real-Time PCR
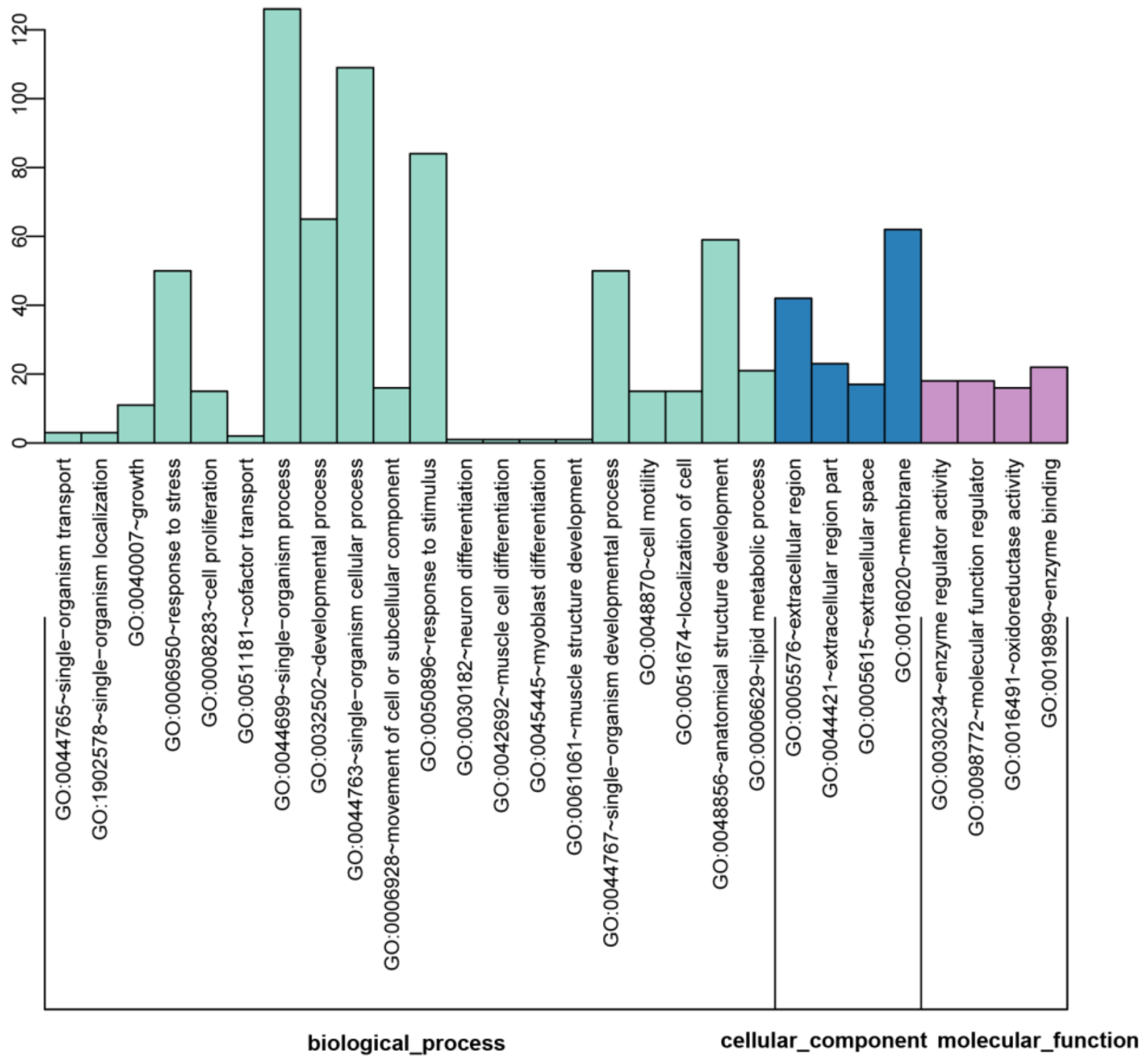
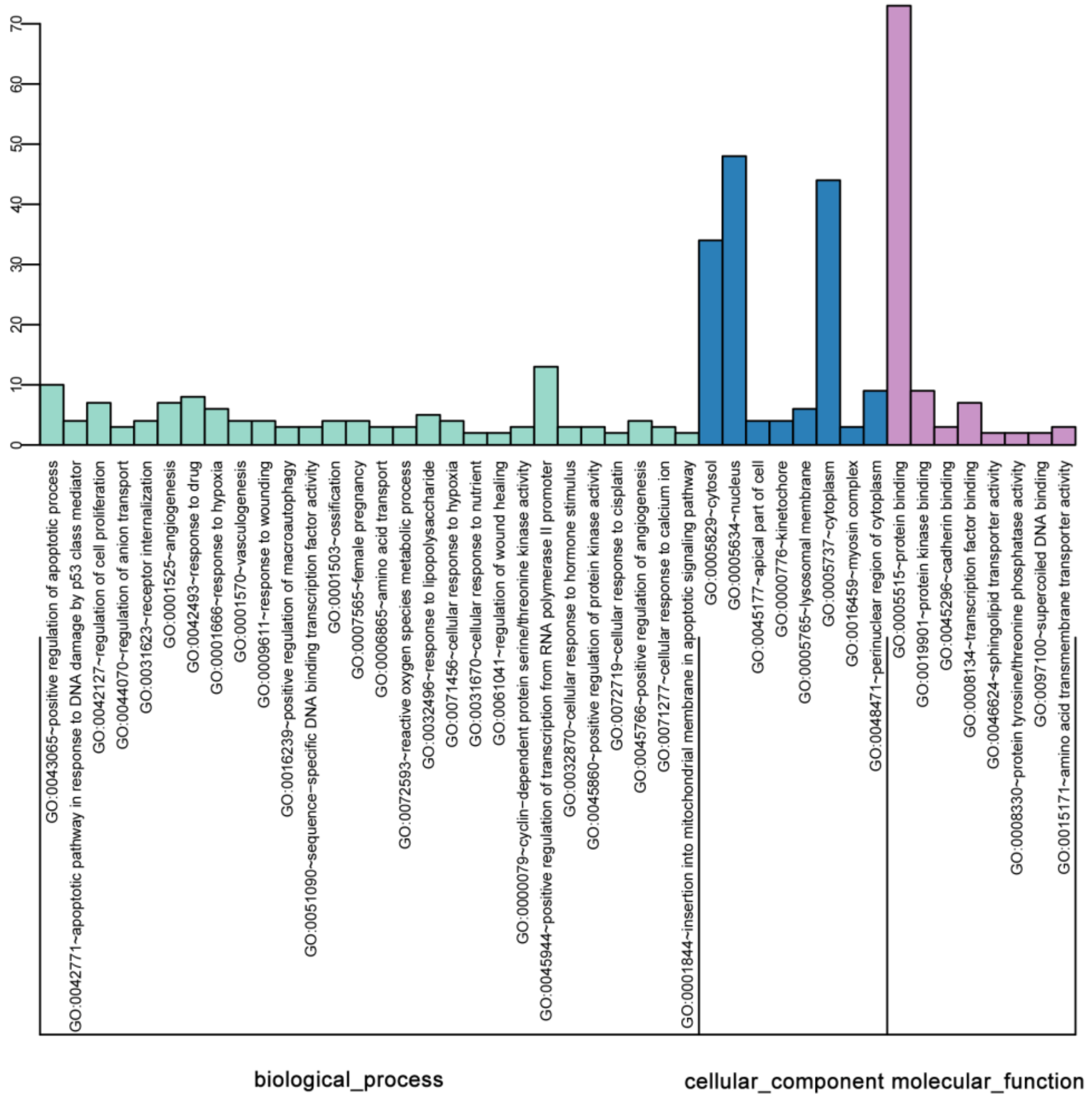
2.3. GO Enrichment of DEGs
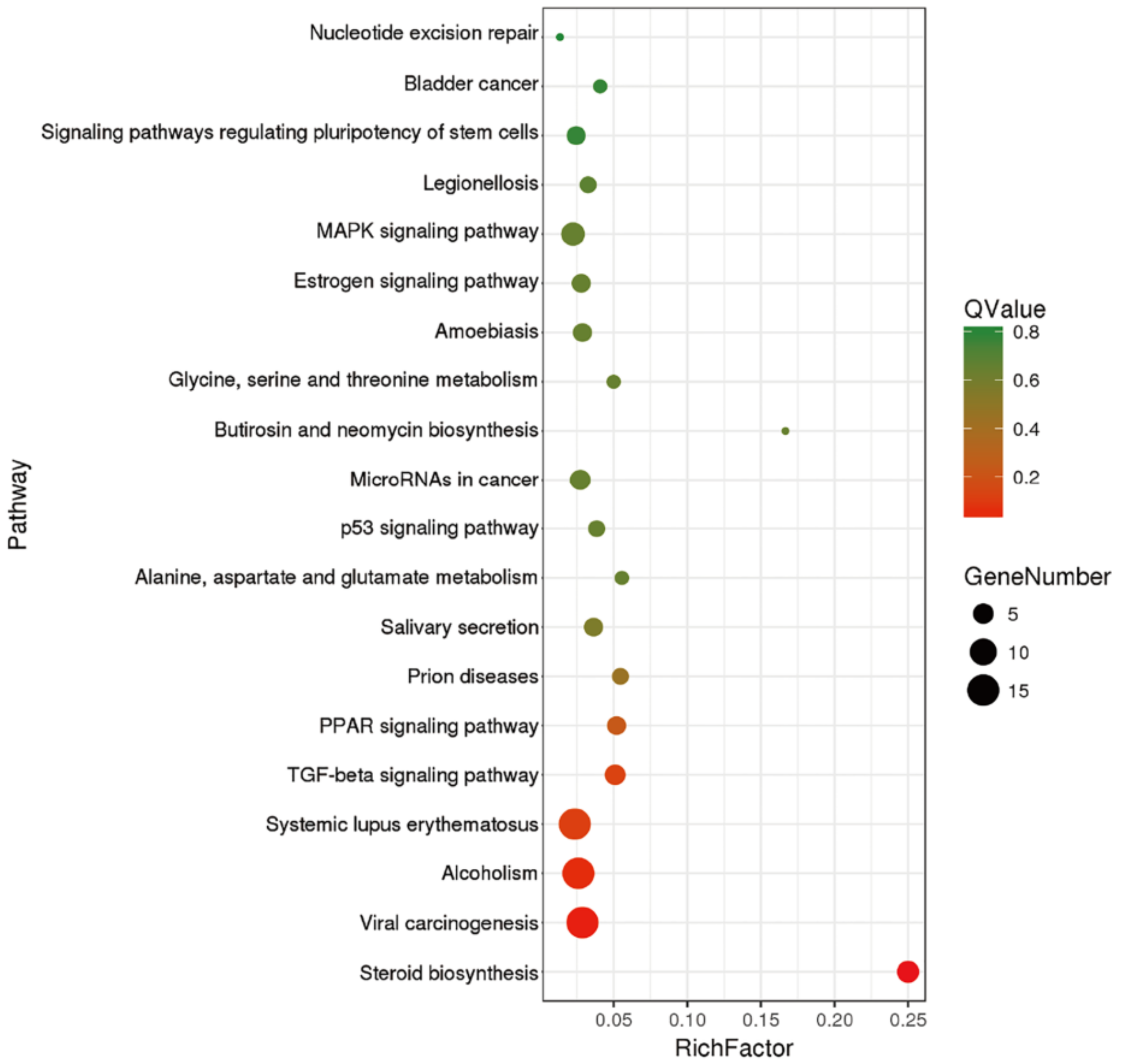
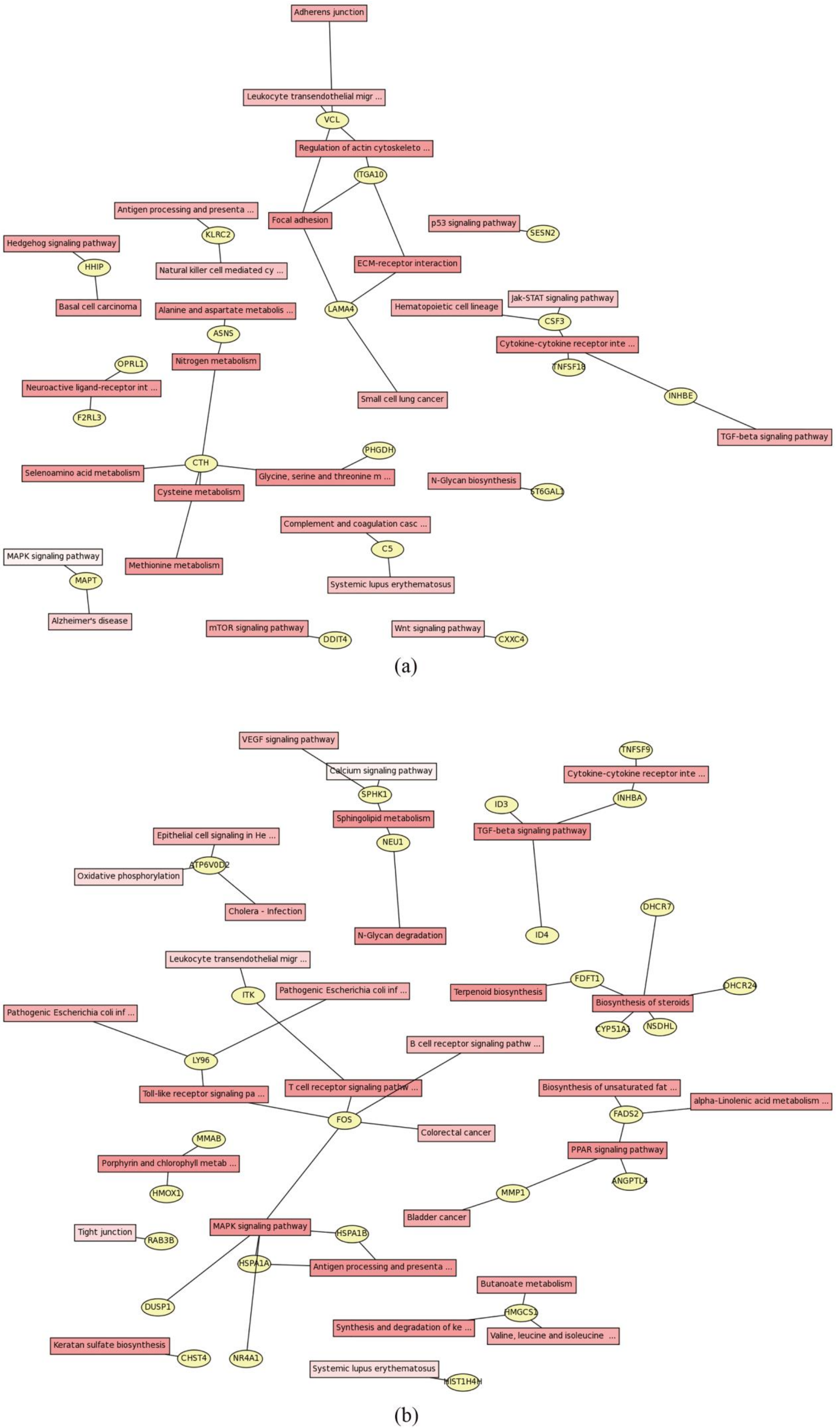
2.4. Pathway Analysis of DEGs
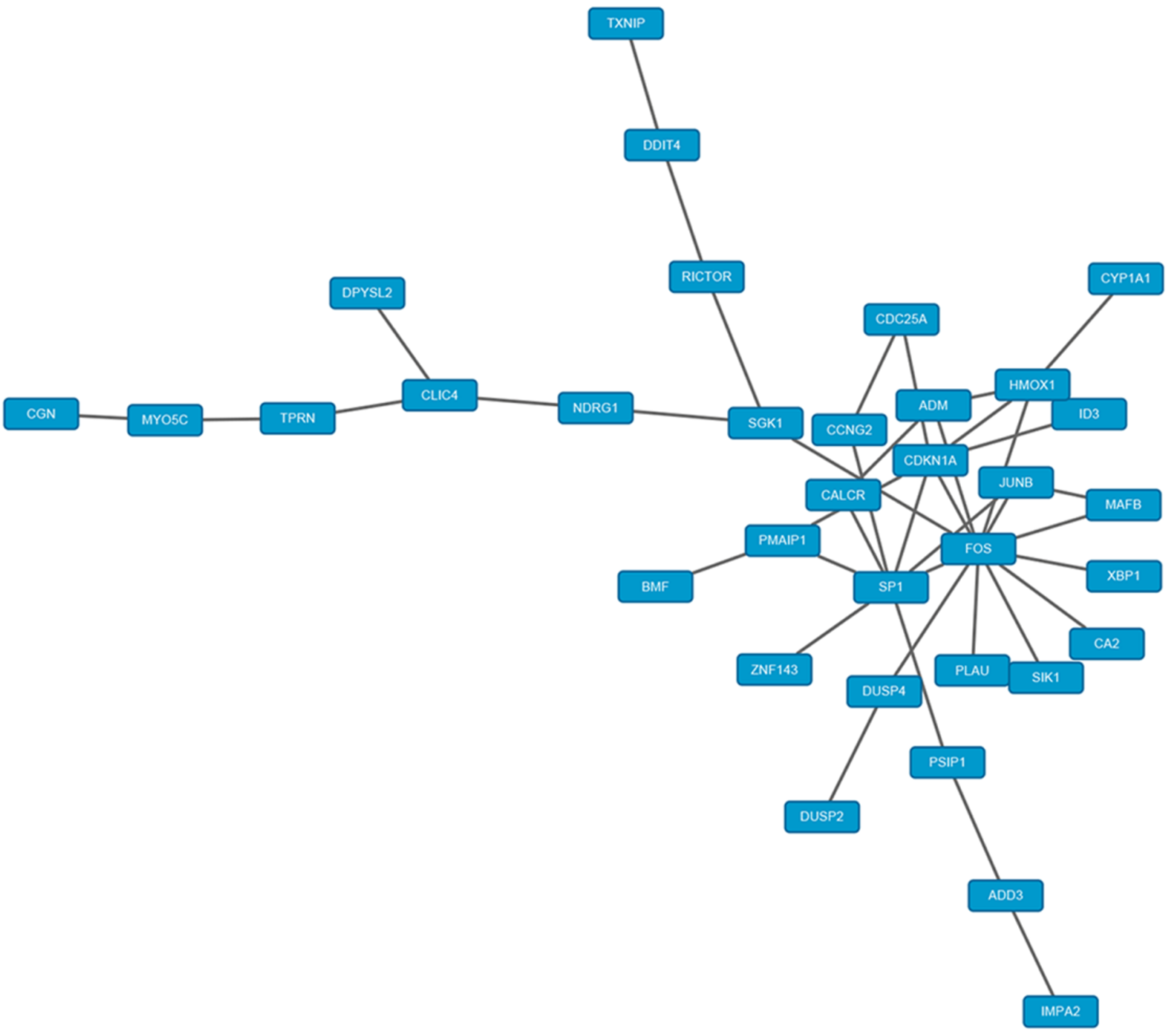
2.5. PPI Network Analysis
2.6. Comparison of the DEGs Identified in ERα Transgenic MDA-MB-231 versus E2 Stimulated MCF-7 Cells
2.7. Bioinformatics Analysis of Common Genes Detected in ERα Transgenic 231 versus E2 Stimulated MCF-7 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construction of ERα Transgenic MDA-MB-231Cell Line
4.3. Western-Blotting
4.4. RNA-Seq Analysis
4.5. Identification of Differentially Expressed Genes (DEGs)
4.6. Validation by RT-PCR
4.7. GO Enrichment and KEGG Pathway Analysis
4.8. Construction of Protein-Protein Interaction (PPI) Networks
4.9. Microarray Data Analysis
4.10. Validation Overlapped Genes by RT-PCT and The Cancer Genome Atlas (TCGA) Analysis
4.11. Verification Genes with ERα Binding Sites
4.12. Identification of DEGs between ER Positive and Negative Breast Cancer Patients
4.13. Kaplan–Meier Plotter Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of naturally occurring esr1 mutations in breast cancer cell lines modelling endocrine resistance. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Green, K.A.; Carroll, J.S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Key, T.; Appleby, P.; Barnes, I.; Reeves, G. Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002, 94, 606–616. [Google Scholar] [PubMed]
- Li, C.I.; Malone, K.E.; Weiss, N.S.; Daling, J.R. Tamoxifen therapy for primary breast cancer and risk of contralateral breast cancer. J. Natl. Cancer Inst. 2001, 93, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, Z.; Hao, Q.; Li, W.; Xu, Y.; Zhang, J.; Zhang, W.; Wang, S.; Liu, S.; Li, M.; et al. Loss of eralpha induces amoeboid-like migration of breast cancer cells by downregulating vinculin. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. Rna-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bellin, D.; Ferrarini, A.; Chimento, A.; Kaiser, O.; Levenkova, N.; Bouffard, P.; Delledonne, M. Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC Genomics 2009, 10, 555. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using illumina paired-end sequencing and development of cssr markers in sweet potato (ipomoea batatas). BMC Genomics 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.; Goebel, G.; Hackl, H.; Fiegl, H. Reduced mrna expression levels of nfe2l2 are associated with poor outcome in breast cancer patients. BMC Cancer 2016, 16, 821. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, P.; She, J.; Cao, L.; Li, Y.; Xia, H. Loss of zg16 is regulated by mir-196a and contributes to stemness and progression of colorectal cancer. Oncotarget 2016, 7, 86695–86703. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Vega, V.B.; Thomsen, J.S.; Zhang, T.; Kong, S.L.; Xie, M.; Chiu, K.P.; Lipovich, L.; Barnett, D.H.; Stossi, F.; et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007, 3, e87. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.S.; Garcia-Bassets, I.; Hutt, K.R.; Cheng, C.S.; Jin, M.; Liu, D.; Benner, C.; Wang, D.; Ye, Z.; Bibikova, M.; et al. Sensitive chip-dsl technology reveals an extensive estrogen receptor alpha-binding program on human gene promoters. Proc. Natl. Acad. Sci. USA 2007, 104, 4852–4857. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002, 4, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; Wang, Y.C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Welboren, W.J.; van Driel, M.A.; Janssen-Megens, E.M.; van Heeringen, S.J.; Sweep, F.C.; Span, P.N.; Stunnenberg, H.G. Chip-seq of eralpha and rna polymerase ii defines genes differentially responding to ligands. Embo J. 2009, 28, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Frasor, J.; Komm, B.; Chang, K.C.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen-regulated gene networks in human breast cancer cells: Involvement of e2f1 in the regulation of cell proliferation. Mol. Endocrinol. 2007, 21, 2112–2123. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell Biol. 2010, 30, 3943–3955. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gustafsson, J.A. The different roles of er subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I. Sp1: Emerging roles—Beyond constitutive activation of tata-less housekeeping genes. Biochem. Biophys. Res. Commun. 2008, 372, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Black, A.R.; Black, J.D.; Azizkhan-Clifford, J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell Physiol. 2001, 188, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Beresford, P.J.; Zhang, D.; Lieberman, J. Hmg2 interacts with the nucleosome assembly protein set and is a target of the cytotoxic t-lymphocyte protease granzyme A. Mol. Cell. Biol. 2002, 22, 2810–2820. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Li, J.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. Hmgb2 is associated with malignancy and regulates warburg effect by targeting ldhb and fbp1 in breast cancer. Cell. Commun. Signal. 2018, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- List, H.J.; Lauritsen, K.J.; Reiter, R.; Powers, C.; Wellstein, A.; Riegel, A.T. Ribozyme targeting demonstrates that the nuclear receptor coactivator aib1 is a rate-limiting factor for estrogen-dependent growth of human mcf-7 breast cancer cells. J. Biol. Chem. 2001, 276, 23763–23768. [Google Scholar] [CrossRef] [PubMed]
- Bays, J.L.; DeMali, K.A. Vinculin in cell-cell and cell-matrix adhesions. Cell Mol. Life Sci. 2017, 74, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Papadopoulou, N.; Nasir, O.; Foller, M.; Alevizopoulos, K.; Lang, F.; Stournaras, C. Activation of membrane androgen receptors in colon cancer inhibits the prosurvival signals akt/bad in vitro and in vivo and blocks migration via vinculin/actin signaling. Mol. Med. 2011, 17, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guo, H.; Song, Y.; Zhao, X.; Shi, Y.; Lu, Y.; Hu, S.; Nie, Y.; Fan, D.; Wu, K. Loss of vinculin and membrane-bound beta-catenin promotes metastasis and predicts poor prognosis in colorectal cancer. Mol. Cancer 2014, 13, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Ma, S.; Qi, X.; Tang, X.; Cui, D.; Wang, Z.; Chi, J.; Li, P.; Zhai, B. Knockdown of the differentially expressed gene tnfrsf12a inhibits hepatocellular carcinoma cell proliferation and migration in vitro. Mol. Med. Rep. 2017, 15, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liu, Y.N.; Tillman, H.; Barrett, B.; Hewitt, S.; Ylaya, K.; Fang, L.; Lake, R.; Corey, E.; Morrissey, C.; et al. Ar-regulated tweak-fn14 pathway promotes prostate cancer bone metastasis. Cancer Res. 2014, 74, 4306–4317. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Schafer, R. Meta-analysis of gene expression profiles related to relapse-free survival in 1079 breast cancer patients. Breast Cancer Res. Treat. 2009, 118, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Emt, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.L.; Dou, X.W.; Bai, J.W.; Luo, X.R.; Qiu, S.Q.; Xi, D.D.; Huang, W.H.; Du, C.W.; Man, K.; Zhang, G.J. Eralpha inhibits epithelial-mesenchymal transition by suppressing bmi1 in breast cancer. Oncotarget 2015, 6, 21704–21717. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.M.; Jung, Y.; Lee, J.M.; Kim, W.; Cho, J.K.; Jeong, J.; Kim, S.J. Loss of tbk1 induces epithelial-mesenchymal transition in the breast cancer cells by eralpha downregulation. Cancer Res. 2013, 73, 6679–6689. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xiao, Y.; Wang, W.; Yearsley, K.; Gao, J.X.; Shetuni, B.; Barsky, S.H. Eralpha signaling through slug regulates e-cadherin and emt. Oncogene 2010, 29, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Agrawal, A.; Bhushan, R.; Chavalmane, A.K.; Kalathur, R.K.; Takahashi, T.; Kondaiah, P. Expression profiling of genes regulated by tgf-beta: Differential regulation in normal and tumour cells. BMC Genomics 2007, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Kirkbride, K.C.; How, T.; Blobe, G.C. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2009, 30, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Gervasi, M.; Bianchi-Smiraglia, A.; Cummings, M.; Zheng, Q.; Wang, D.; Liu, S.; Bakin, A.V. Junb contributes to id2 repression and the epithelial-mesenchymal transition in response to transforming growth factor-beta. J. Cell Biol. 2012, 196, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perk, J.; Iavarone, A.; Benezra, R. Id family of helix-loop-helix proteins in cancer. Nat. Rev. Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Strong, N.; Millena, A.C.; Walker, L.; Chaudhary, J.; Khan, S.A. Inhibitor of differentiation 1 (id1) and id3 proteins play different roles in tgfbeta effects on cell proliferation and migration in prostate cancer cells. Prostate 2013, 73, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Perk, J.; Acharyya, S.; de Candia, P.; Mittal, V.; Todorova-Manova, K.; Gerald, W.L.; Brogi, E.; Benezra, R.; Massague, J. Id genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 19506–19511. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.; Teo, W.S.; Mittal, V.; Swarbrick, A. Id proteins regulate diverse aspects of cancer progression and provide novel therapeutic opportunities. Mol. Ther. 2014, 22, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The ras/raf/mek/erk and the pi3k/akt signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 Suppl 2, S17–S27. [Google Scholar] [CrossRef]
- Song, X.; Wei, Z.; Shaikh, Z.A. Requirement of eralpha and basal activities of egfr and src kinase in cd-induced activation of mapk/erk pathway in human breast cancer mcf-7 cells. Toxicol. Appl. Pharmacol. 2015, 287, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R. Cancer: Tumour stem-cell surprises. Nature 2017, 543, 626–627. [Google Scholar] [CrossRef] [PubMed]
- Kakarala, M.; Wicha, M.S. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J. Clin. Oncol. 2008, 26, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Taylor, J.C.; Black, M.A.; Nicolas, E.; Slifker, M.J.; Azzam, D.J.; Boyd, J. A new role for eralpha: Silencing via DNA methylation of basal, stem cell, and emt genes. Mol. Cancer Res. 2017, 15, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Durchdewald, M.; Angel, P.; Hess, J. The transcription factor fos: A janus-type regulator in health and disease. Histol. Histopathol. 2009, 24, 1451–1461. [Google Scholar] [PubMed]
- Elias, D.; Vever, H.; Laenkholm, A.V.; Gjerstorff, M.F.; Yde, C.W.; Lykkesfeldt, A.E.; Ditzel, H.J. Gene expression profiling identifies fyn as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene 2015, 34, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Anurag, M.; Punturi, N.; Hoog, J.; Bainbridge, M.N. Comprehensive profiling of DNA repair defects in breast cancer identifies a novel class of endocrine therapy resistance drivers. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Roswall, P.; Bocci, M.; Bartoschek, M.; Li, H.; Kristiansen, G.; Jansson, S.; Lehn, S.; Sjolund, J.; Reid, S.; Larsson, C.; et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-cc signaling. Nat. Med. 2018, 24, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the kegg orthology (ko) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The string database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–568. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. Gepia: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Gene ID | RNA-Seq | |
|---|---|---|---|
| Fold Change | Significant | ||
| ESR1 | 2099 | 1053 | Up |
| PCK2 | 5106 | 2.57 | Up |
| CXCL1 | 2919 | 2.29 | Up |
| KIF21B | 23046 | 7 | Up |
| VCL | 7414 | 6.63 | Up |
| FOS | 2353 | 0.02 | Down |
| HMOX1 | 3162 | 0.41 | Down |
| DUSP1 | 1843 | 0.3 | Down |
| ID3 | 3399 | 0.45 | Down |
| Genes | RNA-Seq | Microarray Data | Significance | ||
|---|---|---|---|---|---|
| Fold Change | p-Value | Fold Change | p-Value | ||
| SLC1A1 | 1.503 | 0.0406 | 1.878 | 2.18 × 10−6 | up |
| RASA1 | 1.71 | 0.000281 | 1.879 | 2.89 × 10−6 | up |
| SP1 | 1.603 | 0.005176 | 1.801 | 6.38 × 10−6 | up |
| ABAT | 2.545 | 0.005576 | 1.857 | 4.42 × 10−8 | up |
| TNFRSF12A | 0.67 | 0.0029 | 0.65 | 1.84 × 10−5 | down |
| ID3 | 0.446 | 3.05 × 10−08 | 0.5 | 8.36 × 10−8 | down |
| BAMBI | 0.42 | 7.05 × 10−07 | 0.491 | 1.65 × 10−8 | down |
| JUNB | 0.65 | 0.003262 | 0.55 | 5.20 × 10−7 | down |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Li, X.; Zhang, W.; Gao, Y.; Zhang, K.; Hao, Q.; Li, W.; Wang, Z.; Li, M.; Zhang, W.; et al. Genome-Wide Investigation of Genes Regulated by ERα in Breast Cancer Cells. Molecules 2018, 23, 2543. https://doi.org/10.3390/molecules23102543
Wang S, Li X, Zhang W, Gao Y, Zhang K, Hao Q, Li W, Wang Z, Li M, Zhang W, et al. Genome-Wide Investigation of Genes Regulated by ERα in Breast Cancer Cells. Molecules. 2018; 23(10):2543. https://doi.org/10.3390/molecules23102543
Chicago/Turabian StyleWang, Shuning, Xiaoju Li, Wangqian Zhang, Yuan Gao, Kuo Zhang, Qiang Hao, Weina Li, Zhaowei Wang, Meng Li, Wei Zhang, and et al. 2018. "Genome-Wide Investigation of Genes Regulated by ERα in Breast Cancer Cells" Molecules 23, no. 10: 2543. https://doi.org/10.3390/molecules23102543
APA StyleWang, S., Li, X., Zhang, W., Gao, Y., Zhang, K., Hao, Q., Li, W., Wang, Z., Li, M., Zhang, W., Zhang, Y., & Zhang, C. (2018). Genome-Wide Investigation of Genes Regulated by ERα in Breast Cancer Cells. Molecules, 23(10), 2543. https://doi.org/10.3390/molecules23102543