New Layered Polythiophene-Silica Composite Through the Self-Assembly and Polymerization of Thiophene-Based Silylated Molecular Precursors




Abstract
:
{kind=link}
{kind=link}
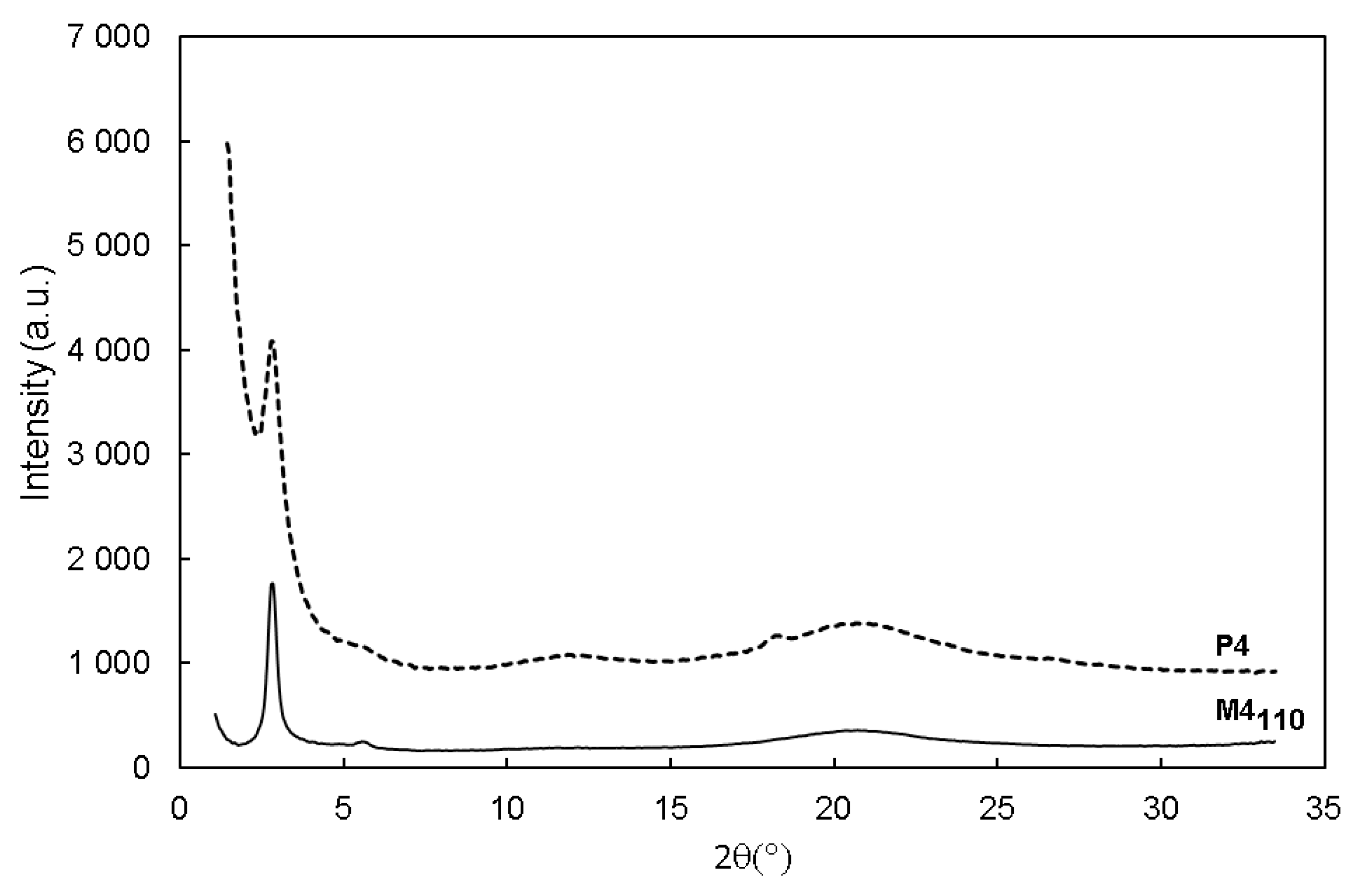{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis of the Silylated-Thiophene Precursor
2.2. Synthesis of the Lamellar Thiophene-Silica Hybrid Material
2.3. The preparation of Lamellar Polythiophene-Silica Hybrid Material
3. Experimental Method
3.1. Instrumentation and Methods
3.2. Synthesis of 3-(8′-Azidooctyl)thiophene (2)
3.3. Synthesis of 3-(8′-Aminooctyl)thiophene (3)
3.4. Synthesis of the Thiophene Silylated Precursor (4)
3.5. Synthesis of the Lamellar Thiophene-silica Hybrid Materials
3.6. In Situ Polymerization of Thiophene Groups
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, J.-S.; Cheng, S.-W.; Cheng, Y.-J.; Hsu, C.-S. Donor-acceptor conjugated polymers based on multifused ladder-type arenes for organic solar cells. Chem. Soc. Rev. 2015, 44, 1113–1154. [Google Scholar] [CrossRef] [PubMed]
- Richard, H.; Anna, K.; Peter, M.-B.; Fabian, P.; Mukundan, T. π-conjugated donor polymers: Structure formation and morphology in solution, bulk and photovoltaic blends. Adv. Energy Mater. 2017, 7, 1700314. [Google Scholar] [CrossRef]
- Kang, H.; Lee, W.; Oh, J.; Kim, T.; Lee, C.; Kim, B.J. From fullerene–polymer to all-polymer solar cells: The importance of molecular packing, orientation, and morphology control. Acc. Chem. Res. 2016, 49, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-H.; Chang, H.-C.; Liu, C.-L.; Chen, W.-C. Polymeric charge storage electrets for non-volatile organic field effect transistor memory devices. Polym. Chem. 2015, 6, 341–352. [Google Scholar] [CrossRef]
- Henning, S. Organic Field-Effect Transistors: The path beyond amorphous silicon. Adv. Mater. 2014, 26, 1319–1335. [Google Scholar] [CrossRef]
- Facchetti, A. Semiconductors for organic transistors. Mater. Today 2007, 10, 28–37. [Google Scholar] [CrossRef]
- Lei, Y.; Cheuk-Lam, H.; Hongbin, W.; Yong, C.; Wai-Yeung, W. White polymer light-emitting devices for solid-state lighting: Materials, devices, and recent progress. Adv. Mater. 2014, 26, 2459–2473. [Google Scholar] [CrossRef]
- Dai, L.; Winkler, B.; Dong, L.; Tong, L.; Mau, A.W.H. Conjugated Polymers for Light-Emitting Applications. Adv. Mater. 2001, 13, 915–925. [Google Scholar] [CrossRef]
- Xie, J.; Zhao, C.; Lin, Z.-Q.; Gu, P.-Y.; Zhang, Q. Nanostructured Conjugated Polymers for Energy-Related Applications beyond Solar Cells. Chem. Asian J. 2016, 11, 1489–1511. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gu, P.; Zhang, Q. Nanostructured Conjugated Polymers: Toward High-Performance Organic Electrodes for Rechargeable Batteries. ACS Energy Lett. 2017, 2, 1985–1996. [Google Scholar] [CrossRef]
- Wang, J.; Lv, F.; Liu, L.; Ma, Y.; Wang, S. Strategies to design conjugated polymer based materials for biological sensing and imaging. Coord. Chem. Rev. 2018, 354, 135–154. [Google Scholar] [CrossRef]
- Liang, J.; Li, K.; Liu, B. Visual sensing with conjugated polyelectrolytes. Chem. Sci. 2013, 4, 1377–1394. [Google Scholar] [CrossRef]
- Park, J.; Lee, C.; Jung, J.; Kang, H.; Kim, K.-H.; Ma, B.; Kim, B.J. Facile photo-crosslinking of azide-containing hole-transporting polymers for highly efficient, solution-processed, multilayer organic light emitting devices. Adv. Funct. Mater. 2014, 24, 7588–7596. [Google Scholar] [CrossRef]
- Trattnig, R.; Pevzner, L.; Jäger, M.; Schlesinger, R.; Nardi, M.V.; Ligorio, G.; Christodoulou, C.; Koch, N.; Baumgarten, M.; Müllen, K.; et al. Bright blue solution processed triple-layer polymer light-emitting diodes realized by thermal layer stabilization and orthogonal solvents. Adv. Funct. Mater. 2013, 23, 4897–4905. [Google Scholar] [CrossRef]
- Maturová, K.; van Bavel Svetlana, S.; Wienk Martijn, M.; Janssen, R.A.J.; Kemerink, M. Description of the morphology dependent charge transport and performance of polymer: Fullerene bulk heterojunction solar cells. Adv. Funct. Mater. 2010, 21, 261–269. [Google Scholar] [CrossRef]
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 2005, 4, 80–84. [Google Scholar] [CrossRef]
- Ruderer, M.A.; Muller-Buschbaum, P. Morphology of polymer-based bulk heterojunction films for organic photovoltaics. Soft Matter 2011, 7, 5482–5493. [Google Scholar] [CrossRef]
- Kim, Y.; Cook, S.; Tuladhar, S.M.; Choulis, S.A.; Nelson, J.; Durrant, J.R.; Bradley, D.D.C.; Giles, M.; McCulloch, I.; Ha, C.-S.; et al. A strong regioregularity effect in self-organizing conjugated polymer films and high-efficiency polythiophene:fullerene solar cells. Nat. Mater. 2006, 5, 63–69. [Google Scholar] [CrossRef]
- Sirringhaus, H.; Brown, P.J.; Friend, R.H.; Nielsen, M.M.; Bechgaard, K.; Langeveld-Voss, B.M.W.; Spiering, A.J.H.; Janssen, R.A.J.; Meijer, E.W.; Herwig, P.; et al. Two-dimensional charge transport in self-organized, high-mobility conjugated polymers. Nature 1999, 401, 685–688. [Google Scholar] [CrossRef]
- Beljonne, D.; Pourtois, G.; Silva, C.; Hennebicq, E.; Herz, L.M.; Friend, R.H.; Scholes, G.D.; Setayesh, S.; Müllen, K.; Brédas, J.L. Interchain vs. intrachain energy transfer in acceptor-capped conjugated polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 10982–10987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivaton, A.; Tournebize, A.; Gaume, J.; Bussière, P.-O.; Gardette, J.-L.; Therias, S. Photostability of organic materials used in polymer solar cells. Polym. Int. 2013, 63, 1335–1345. [Google Scholar] [CrossRef]
- Manceau, M.; Rivaton, A.; Gardette, J.-L.; Guillerez, S.; Lemaître, N. The mechanism of photo- and thermooxidation of poly(3-hexylthiophene) (P3HT) reconsidered. Polym. Degrad. Stab. 2009, 94, 898–907. [Google Scholar] [CrossRef]
- Yang, B.; Xiao, M.; Zhao, C.; Zhang, S.; Jiang, A.; Wang, J. Alignment control of polythiophene chains with mesostructured silica nanofibers having different pore orientations. Small 2012, 8, 2021–2026. [Google Scholar] [CrossRef]
- Evans, R.C.; Marr, P.C. Chain confinement promotes β-phase formation in polyfluorene-based photoluminescent ionogels. Chem. Commun. 2012, 48, 3742–3744. [Google Scholar] [CrossRef] [PubMed]
- Cheminet, N.; Jarrosson, T.; Lere-Porte, J.-P.; Serein-Spirau, F.; Cury, L.; Moreau, J.; Viau, L.; Vioux, A. One pot synthesis of fluorescent π-conjugated materials: Immobilization of phenylene-ethynylene polyelectrolytes in silica confined ionogels. J. Mater. Chem. 2011, 21, 13588–13593. [Google Scholar] [CrossRef]
- Ayumi, M.; Yuta, M.; Takahiro, U.; Takahito, I.; Takayuki, Y.; Akira, K.; Masataka, K. Incorporation of fluorene-based emitting polymers into silica. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 5322–5328. [Google Scholar] [CrossRef]
- Evans, R.C.; Macedo, A.G.; Pradhan, S.; Scherf, U.; Carlos, L.D.; Burrows, H.D. Fluorene based conjugated polyelectrolyte/silica nanocomposites: Charge-mediated phase aggregation at the organic-inorganic interface. Adv. Mater. 2010, 22, 3032–3037. [Google Scholar] [CrossRef] [PubMed]
- Aharon, E.; Kalina, M.; Frey, G.L. Inhibition of energy transfer between conjugated polymer chains in host/guest nanocomposites generates white photo- and electroluminescence. J. Am. Chem. Soc. 2006, 128, 15968–15969. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.L.; Yamada, Y.; Schneider, C.; Yano, K.; Wolf, M.O. Enhanced optical properties and opaline self-assembly of ppv encapsulated in mesoporous silica spheres. Adv. Funct. Mater. 2009, 19, 3737–3745. [Google Scholar] [CrossRef]
- Kubo, M.; Takimoto, C.; Minami, Y.; Uno, T.; Itoh, T.; Shoyama, M. Incorporation of π-conjugated polymer into silica: preparation of poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]/silica and poly(3-hexylthiophene)/silica composites. Macromolecules 2005, 38, 7314–7320. [Google Scholar] [CrossRef]
- Rothberg, L.J.; Yan, M.; Papadimitrakopoulos, F.; Galvin, M.E.; Kwock, E.W.; Miller, T.M. Photophysics of phenylenevinylene polymers. Synth. Met. 1996, 80, 41–58. [Google Scholar] [CrossRef]
- Jakubiak, R.; Collison, C.J.; Wan, W.C.; Rothberg, L.J.; Hsieh, B.R. Aggregation quenching of luminescence in electroluminescent conjugated polymers. J. Phys. Chem. A 1999, 103, 2394–2398. [Google Scholar] [CrossRef]
- Evans, R.C. Harnessing self-assembly strategies for the rational design of conjugated polymer based materials. J. Mater. Chem. C 2013, 1, 4190–4200. [Google Scholar] [CrossRef]
- Tamaki, R.; Samura, K.; Chujo, Y. Synthesis of polystyrene and silica gel polymer hybrids via π-π interactions. Chem. Commun. 1998, 1131–1132. [Google Scholar] [CrossRef]
- Ogoshi, T.; Chujo, Y. Synthesis of amorphous and nanostructured cationic polyacetylene/silica hybrids by using ionic interactions. Macromolecules 2005, 38, 9110–9116. [Google Scholar] [CrossRef]
- Clement, S.; Tizit, A.; Desbief, S.; Mehdi, A.; De Winter, J.; Gerbaux, P.; Lazzaroni, R.; Boury, B. Synthesis and characterisation of π-conjugated polymer/silica hybrids containing regioregular ionic polythiophenes. J. Mater. Chem. 2011, 21, 2733–2739. [Google Scholar] [CrossRef]
- Willis-Fox, N.; Kraft, M.; Arlt, J.; Scherf, U.; Evans, R.C. Tunable white-light emission from conjugated polymer-di-ureasil materials. Adv. Funct. Mater. 2016, 26, 532–542. [Google Scholar] [CrossRef]
- Willis-Fox, N.; Marques, A.-T.; Arlt, J.; Scherf, U.; Carlos, L.D.; Burrows, H.D.; Evans, R.C. Synergistic photoluminescence enhancement in conjugated polymer-di-ureasil organic-inorganic composites. Chem. Sci. 2015, 6, 7227–7237. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, J.M.; Foster, A.B.; McCairn, M.C.; Willcock, H.; O’Reilly, R.K.; Turner, M.L. Hybrid inorganic-organic composite nanoparticles from crosslinkable polyfluorenes. J. Mater. Chem. C 2013, 1, 3297–3304. [Google Scholar] [CrossRef]
- Meazzini, I.; Behrendt, J.M.; Turner, M.L.; Evans, R.C. Targeted β-phase formation in poly(fluorene)–ureasil grafted organic-inorganic hybrids. Macromolecules 2017, 50, 4235–4243. [Google Scholar] [CrossRef]
- Wu, C.-G.; Bein, T. Conducting polyaniline filaments in a mesoporous channel host. Science 1994, 264, 1757–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, T.L.; Che, S.P.Y.; Yamada, Y.; Yano, K.; Wolf, M.O. Influence of surface morphology on the colloidal and electronic behavior of conjugated polymer-silica microspheres. Langmuir 2008, 24, 9809–9815. [Google Scholar] [CrossRef] [PubMed]
- Lin, V.S.Y.; Radu, D.R.; Han, M.-K.; Deng, W.; Kuroki, S.; Shanks, B.H.; Pruski, M. Oxidative polymerization of 1,4-diethynylbenzene into highly conjugated poly(phenylene butadiynylene) within the channels of surface-functionalized mesoporous silica and alumina materials. J. Am. Chem. Soc. 2002, 124, 9040–9041. [Google Scholar] [CrossRef] [PubMed]
- Cardin, D.J.; Constantine, S.P.; Gilbert, A.; Lay, A.K.; Alvaro, M.; Galletero, M.S.; Garcia, H.; Marquez, F. Polymerization of alkynes in the channels of mesoporous materials containing Ni and Zn cations: Almost complete filling of the voids. J. Am. Chem. Soc. 2001, 123, 3141–3142. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, Y.; Lu, M.; Huang, J.; Haddad, R.; Xomeritakis, G.; Liu, N.; Malanoski, A.P.; Sturmayr, D.; Fan, H.; et al. Functional nanocomposites prepared by self-assembly and polymerization of diacetylene surfactants and silicic acid. J. Am. Chem. Soc. 2003, 125, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kou, X.; Ni, W.; Sun, Z.; Li, L.; Wang, J. Fluorescent mesostructured polythiophene-silica composite particles synthesized by in situ polymerization of structure-directing monomers. Chem. Mater. 2007, 19, 6222–6229. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; Sellinger, A.; Lu, M.; Huang, J.; Fan, H.; Haddad, R.; Lopez, G.; Burns, A.R.; Sasaki, D.Y.; et al. Self-assembly of mesoscopically ordered chromatic polydiacetylene/silica nanocomposites. Nature 2001, 410, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Takuzo, A.; Keisuke, T. Photoluminescent silicate microsticks containing aligned nanodomains of conjugated polymers by sol-gel-based in situ polymerization. Angew. Chem. Int. Ed. 2001, 40, 3803–3806. [Google Scholar] [CrossRef]
- Guangtao, L.; Sheshanath, B.; Tianyu, W.; Yang, Z.; Hesun, Z.; Jürgen-Hinrich, F. Gram-scale synthesis of submicrometer-long polythiophene wires in mesoporous silica matrices. Angew. Chem. Int. Ed. 2003, 42, 3818–3821. [Google Scholar] [CrossRef]
- Peng, H.; Tang, J.; Yang, L.; Pang, J.; Ashbaugh, H.S.; Brinker, C.J.; Yang, Z.; Lu, Y. Responsive periodic mesoporous polydiacetylene/silica nanocomposites. J. Am. Chem. Soc. 2006, 128, 5304–5305. [Google Scholar] [CrossRef] [PubMed]
- Roncali, J. Conjugated poly(thiophenes): Synthesis, functionalization, and applications. Chem. Rev. 1992, 92, 711–738. [Google Scholar] [CrossRef]
- Mouawia, R.; Mehdi, A.; Reyé, C.; Corriu, R.J.P. From simple molecules to highly functionalised lamellar materials. J. Mater. Chem. 2008, 18, 2028–2035. [Google Scholar] [CrossRef]
- Moreau, J.J.E.; Vellutini, L.; Wong Chi Man, M.; Bied, C.; Dieudonné, P.; Bantignies, J.-L.; Sauvajol, J.-L. Lamellar bridged silsesquioxanes: Self-assembly through a combination of hydrogen bonding and hydrophobic interactions. Chem. Eur. J. 2005, 11, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- van Esch, J.H.; Schoonbeek, F.; de Loos, M.; Kooijman, H.; Spek, A.L.; Kellogg, R.M.; Feringa, B.L. Cyclic bis-urea compounds as gelators for organic solvents. Chem. Eur. J. 1999, 5, 937–950. [Google Scholar] [CrossRef]
- Besson, E.; Mehdi, A.; Reyé, C.; Gaveau, P.; Corriu, R.J.P. Self-assembly of layered organosilicas based on weak intermolecular interactions. Dalton Trans. 2010, 39, 7534–7539. [Google Scholar] [CrossRef] [PubMed]
- Besson, E.; Mehdi, A.; Van der Lee, A.; Chollet, H.; Reyé, C.; Guilard, R.; Corriu, R.J.P. Selective lanthanides sequestration based on a self-assembled organosilica. Chem. Eur. J. 2010, 16, 10226–10233. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Honda, K.; Azumi, R. Model chemistry calculations of thiophene dimer interactions: Origin of π-stacking. J. Am. Chem. Soc. 2002, 124, 12200–12209. [Google Scholar] [CrossRef] [PubMed]
- Alauzun, J.; Mehdi, A.; Reyé, C.; Corriu, R.J.P. Hydrophilic conditions: A new way for self-assembly of hybrid silica containing long alkylene chains. J. Mater. Chem. 2005, 15, 841–843. [Google Scholar] [CrossRef]
- Niemi, V.M.; Knuuttila, P.; Österholm, J.E.; Korvola, J. Polymerization of 3-alkylthiophenes with FeCl3. Polymer 1992, 33, 1559–1562. [Google Scholar] [CrossRef]
- Qiao, X.; Wang, X.; Mo, Z. The effects of different alkyl substitution on the structures and properties of poly(3-alkylthiophenes). Synth. Met. 2001, 118, 89–95. [Google Scholar] [CrossRef]
- Łużny, W.; Trznadel, M.; Proń, A. X-ray diffraction study of regioregular poly(3-alkylthiophenes). Synth. Met. 1996, 81, 71–74. [Google Scholar] [CrossRef]
- Hundt, N.; Hoang, Q.; Nguyen, H.; Sista, P.; Hao, J.; Servello, J.; Palaniappan, K.; Alemseghed, M.; Biewer, M.C.; Stefan, M.C. Synthesis and Characterization of a Block Copolymer Containing Regioregular Poly(3-hexylthiophene) and Poly(γ-benzyl-l-glutamate). Macromol. Rapid Commun. 2011, 32, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, M.P.; Sista, P.; Hao, J.; Hundt, N.; Biewer, M.C.; Stefan, M.C. Electronic properties-morphology correlation of a rod-rod semiconducting liquid crystalline block copolymer containing poly(3-hexylthiophene). Langmuir 2012, 28, 12762–12770. [Google Scholar] [CrossRef] [PubMed]
- Peter, B. End-capped oligothiophenes-new model compounds for polythiophenes. Adv. Mater. 1992, 4, 102–107. [Google Scholar] [CrossRef]
- Gierschner, J.; Cornil, J.; Egelhaaf, H.-J. Optical bandgaps of π-conjugated organic materials at the polymer limit: Experiment and theory. Adv. Mater. 2007, 19, 173–191. [Google Scholar] [CrossRef]
- Chen, S.-A.; Ni, J.-M. Facile Structure/properties of conjugated conductive polymers. 1. Neutral poly(3-alkythiophene)s. Macromolecules 1992, 25, 6081–6089. [Google Scholar] [CrossRef]
- Ho, K.-S.; Bartus, J.; Levon, K.; Mao, J.; Zheng, W.-Y.; Laakso, J.; Taka, T. Layered structure with side chain crystallinity in undoped poly(3-alkyl thiophenes). Synth. Met. 1993, 55, 384–387. [Google Scholar] [CrossRef]
- Peter, B.; Frank, W.; Stephan, H. Facile synthesis of 3-(ω-haloalkyl)thiophenes as key building blocks for functionalized thiophenes and polythiophenes. Angew. Chem. Int. Ed. Engl. 1990, 29, 419–420. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds described in this manuscript are not available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacca, M.-J.; Laurencin, D.; Richeter, S.; Clément, S.; Mehdi, A. New Layered Polythiophene-Silica Composite Through the Self-Assembly and Polymerization of Thiophene-Based Silylated Molecular Precursors. Molecules 2018, 23, 2510. https://doi.org/10.3390/molecules23102510
Zacca M-J, Laurencin D, Richeter S, Clément S, Mehdi A. New Layered Polythiophene-Silica Composite Through the Self-Assembly and Polymerization of Thiophene-Based Silylated Molecular Precursors. Molecules. 2018; 23(10):2510. https://doi.org/10.3390/molecules23102510
Chicago/Turabian StyleZacca, Marie-José, Danielle Laurencin, Sébastien Richeter, Sébastien Clément, and Ahmad Mehdi. 2018. "New Layered Polythiophene-Silica Composite Through the Self-Assembly and Polymerization of Thiophene-Based Silylated Molecular Precursors" Molecules 23, no. 10: 2510. https://doi.org/10.3390/molecules23102510
APA StyleZacca, M.-J., Laurencin, D., Richeter, S., Clément, S., & Mehdi, A. (2018). New Layered Polythiophene-Silica Composite Through the Self-Assembly and Polymerization of Thiophene-Based Silylated Molecular Precursors. Molecules, 23(10), 2510. https://doi.org/10.3390/molecules23102510