Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
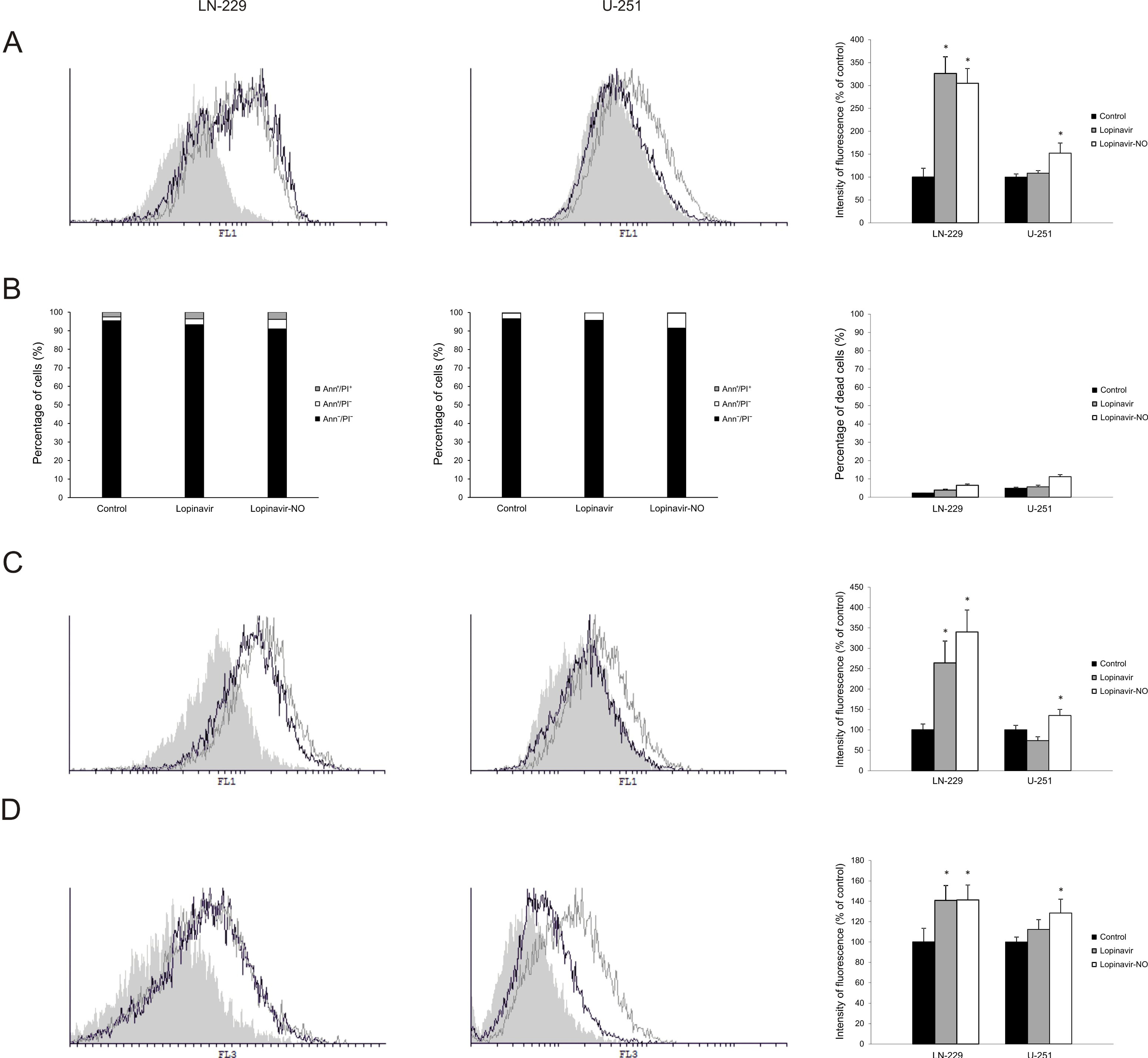
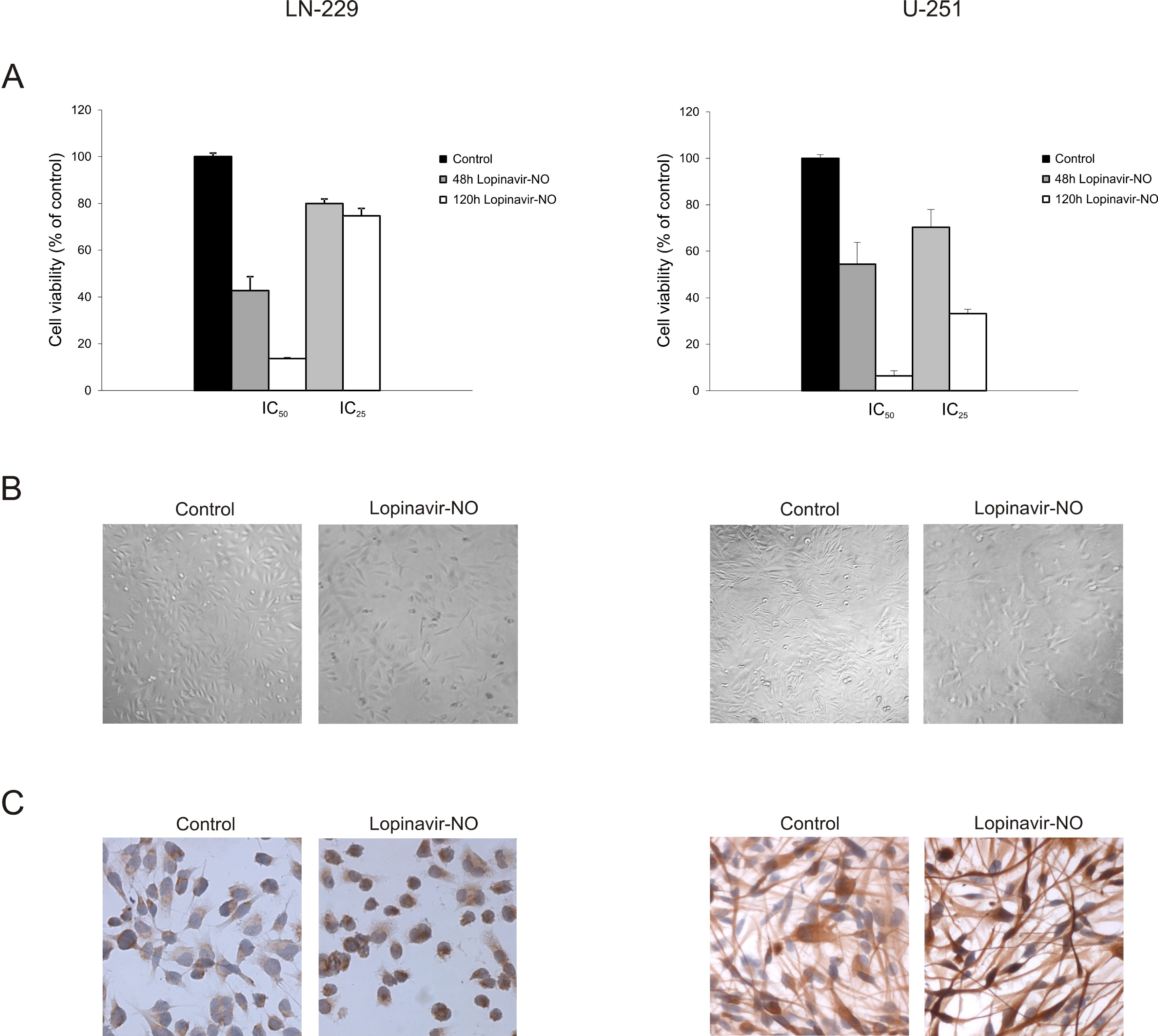
2.1. Lopi-NO Has Stronger Anticancer Action Than Lopi
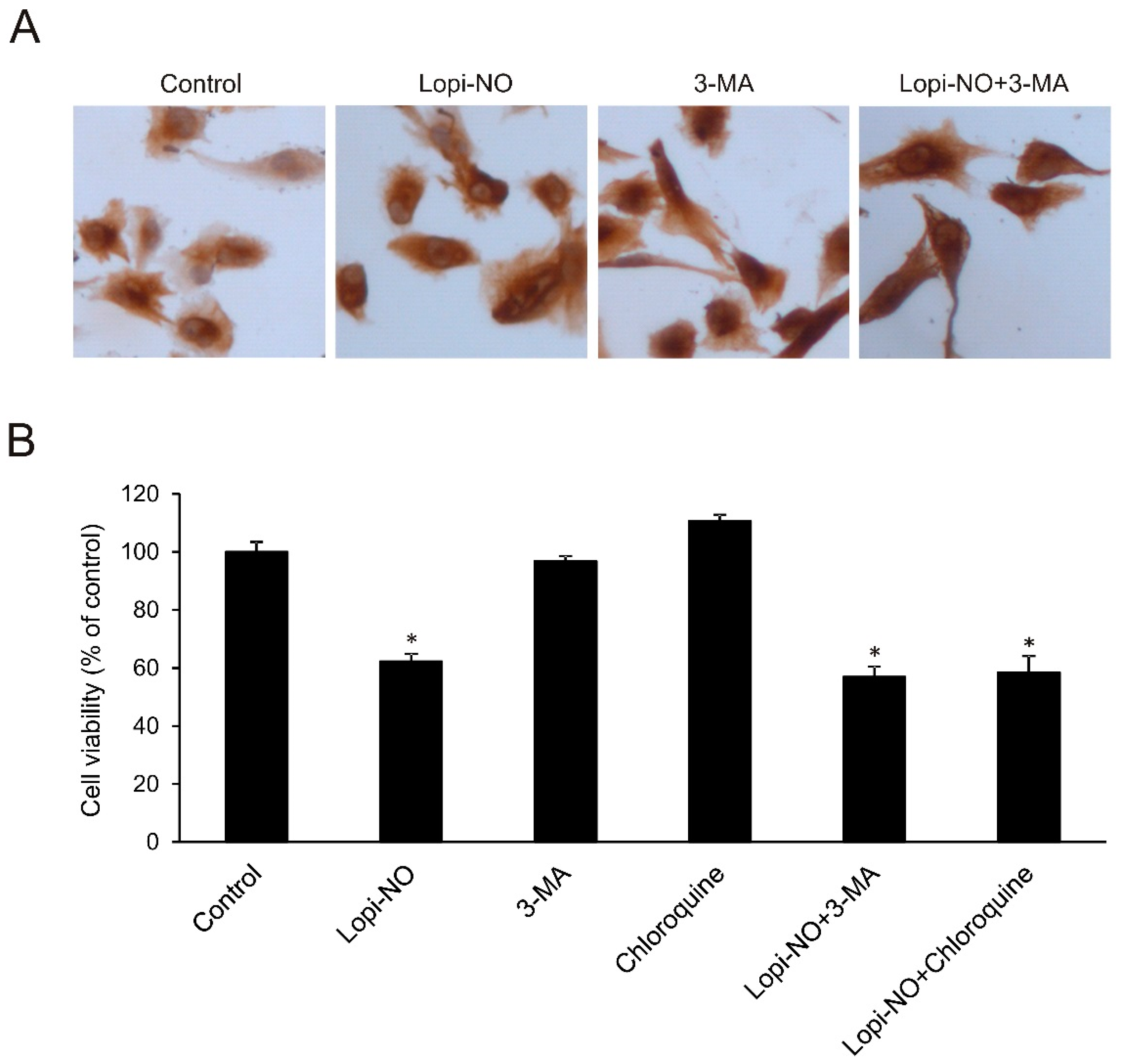
2.2. Lopi-NO Inhibited Tumor Cell Proliferation and Induced Strong Autophagy
2.3. Lopi-NO Triggered Permanent Change in GBM Cell Phenotype
2.4. Autophagy Was Irrelevant for U-251 Differentiation
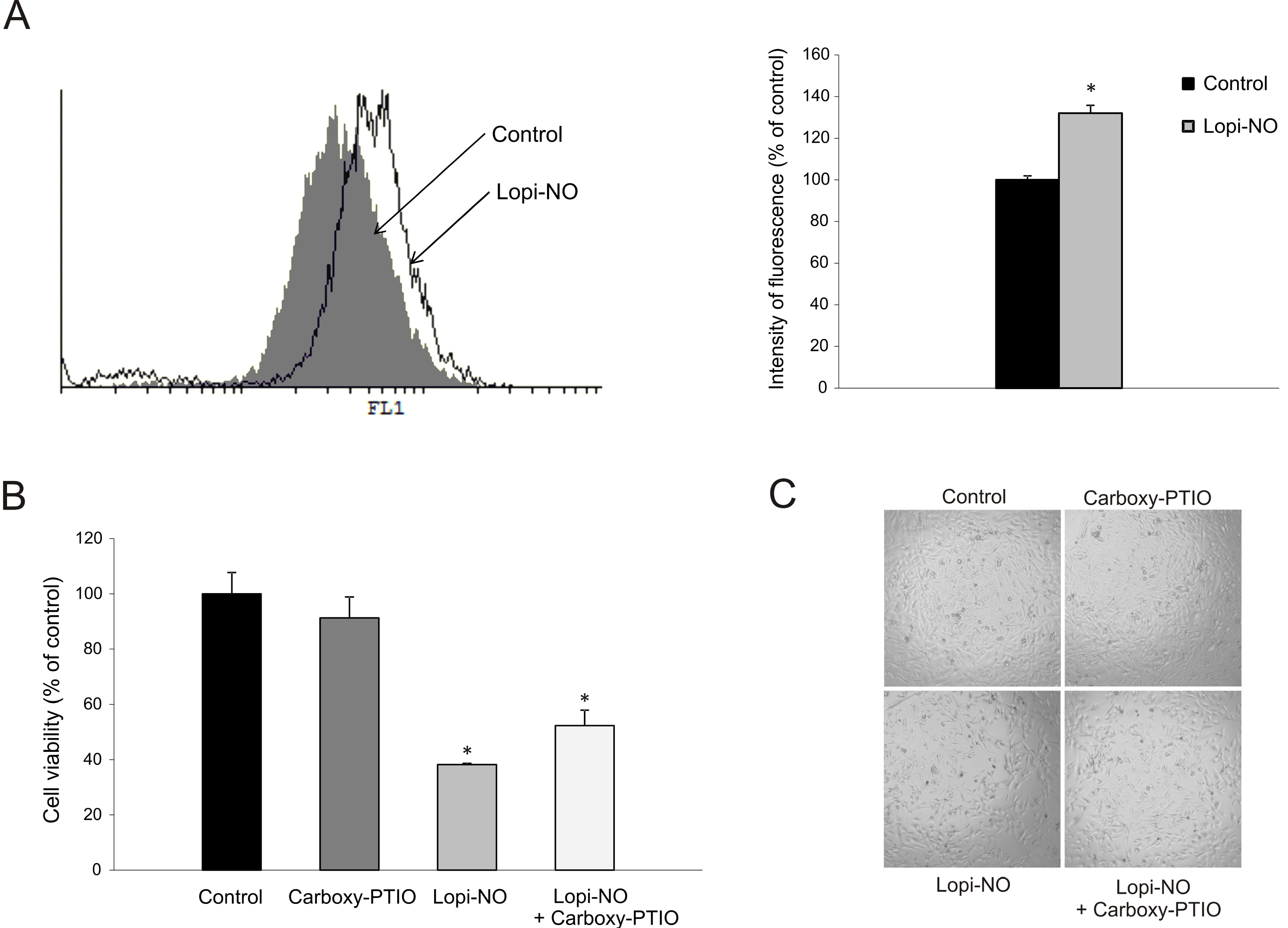
2.5. Lopi-NO Promoted Oxidative/Nitrosative Stress
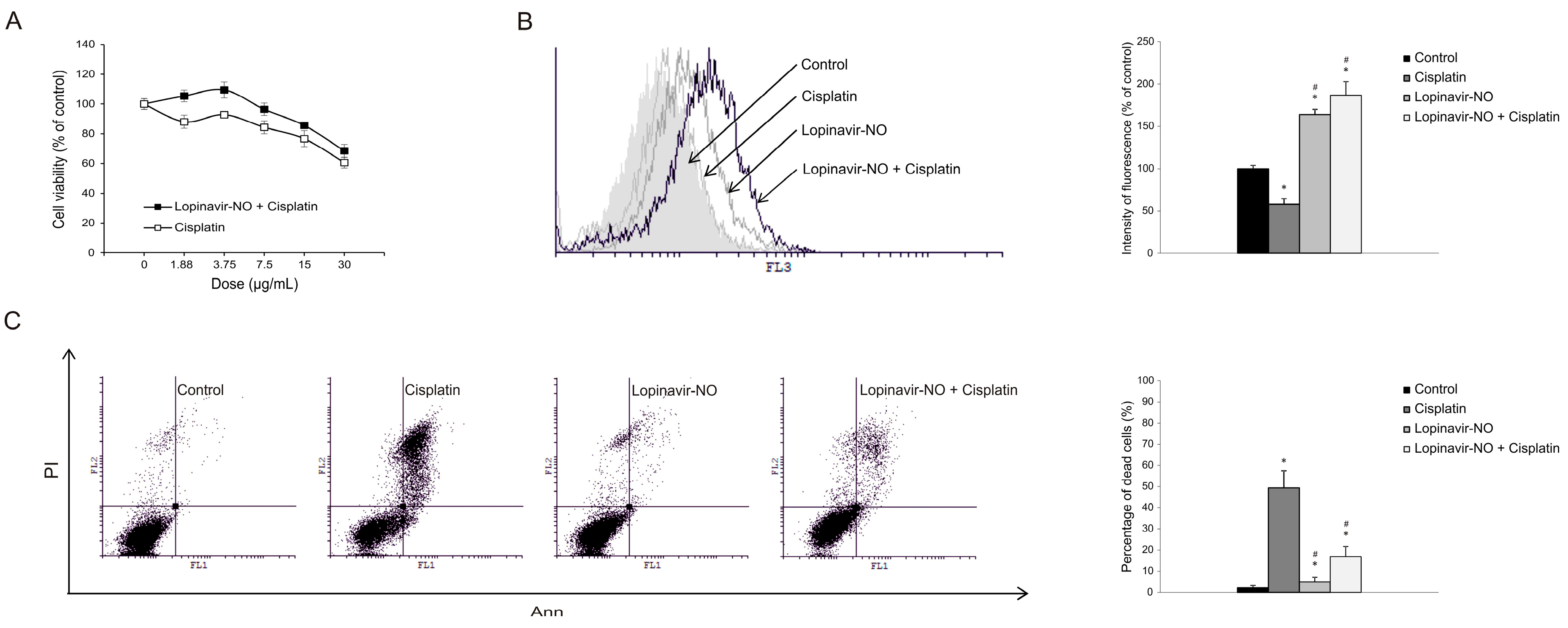
2.6. Lopi-NO Antagonized Cisplatin Activity in Cotreatment
3. Discussion
4. Materials and Methods
4.1. Reagents and Cells
4.2. Determination of Cell Viability by MTT and CV Assays
4.3. AnnV-/PI and Apostat Staining
4.4. CFSE Staining
4.5. Acridine Orange Staining
4.6. Measurement of ROS and RNS
4.7. Immunocytochemistry
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wirsching, H.-G.; Galanis, E.; Weller, M. Glioblastoma. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2016; Volume 134, pp. 381–397. ISBN 9780128029978. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; pp. 143–153. ISBN 978-0-9944381-2-6. [Google Scholar] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, C.; Bruyns, C.; Marchant, A.; Abramowicz, D.; Vandenabeele, P.; Delvaux, A.; Fiers, W.; Goldman, M.; Velu, T. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J. Exp. Med. 1993, 177, 547–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, F.; Mancuso, G.; Cusumano, V.; Di Marco, R.; Zaccone, P.; Bendtzen, K.; Teti, G. Prevention of endotoxin-induced lethality in neonatal mice by interleukin-13. Eur. J. Immunol. 1997, 27, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- McCartney-Francis, N.; Jin, W.; Wahl, S.M. Aberrant Toll receptor expression and endotoxin hypersensitivity in mice lacking a functional TGF-beta 1 signaling pathway. J. Immunol. 2004, 172, 3814–3821. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Presti, M.; Mazzon, E.; Basile, M.S.; Petralia, M.C.; Bramanti, A.; Colletti, G.; Bramanti, P.; Nicoletti, F.; Fagone, P. Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol. Lett. 2018, 16, 2881–2886. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Hu, M.; Iyer, V.; Yu, J. Blocking the PD-1/PD-L1 pathway in glioma: A potential new treatment strategy. J. Hematol. Oncol. 2017, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS 2015, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic-Ivanic, D.; Mijatovic, S.; Miljkovic, D.; Harhaji-Trajkovic, L.; Timotijevic, G.; Mojic, M.; Dabideen, D.; Cheng, K.F.; McCubrey, J.A.; Mangano, K.; et al. The antitumor properties of a nontoxic, nitric oxide-modified version of saquinavir are independent of Akt. Mol. Cancer Ther. 2009, 8, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic-Ivanic, D.; Fagone, P.; McCubrey, J.; Bendtzen, K.; Mijatovic, S.; Nicoletti, F. HIV-protease inhibitors for the treatment of cancer: Repositioning HIV protease inhibitors while developing more potent NO-hybridized derivatives? Int. J. Cancer 2017, 140, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Sgadari, C.; Monini, P.; Barillari, G.; Ensoli, B. Use of HIV protease inhibitors to block Kaposi’s sarcoma and tumour growth. Lancet Oncol. 2003, 4, 537–547. [Google Scholar] [CrossRef]
- Jiang, Z.; Pore, N.; Cerniglia, G.J.; Mick, R.; Georgescu, M.-M.; Bernhard, E.J.; Hahn, S.M.; Gupta, A.K.; Maity, A. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007, 67, 4467–4473. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Kardosh, A.; Wang, W.; Xiong, W.; Schönthal, A.H.; Chen, T.C. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007, 67, 10920–10928. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Halatsch, M.-E. Matrix metalloproteinase-2 and -9 in glioblastoma: A trio of old drugs-captopril, disulfiram and nelfinavir-are inhibitors with potential as adjunctive treatments in glioblastoma. Arch. Med. Res. 2012, 43, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Basanta, M.; Fang, P.; Maity, A.; Hahn, S.M.; Lustig, R.A.; Dorsey, J.F. A phase I study of nelfinavir concurrent with temozolomide and radiotherapy in patients with glioblastoma multiforme. J. Neurooncol. 2014, 116, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Laurent, N.; de Boüard, S.; Guillamo, J.-S.; Christov, C.; Zini, R.; Jouault, H.; Andre, P.; Lotteau, V.; Peschanski, M. Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol. Cancer Ther. 2004, 3, 129–136. [Google Scholar] [PubMed]
- Kast, R.E.; Ramiro, S.; Lladó, S.; Toro, S.; Coveñas, R.; Muñoz, M. Antitumor action of temozolomide, ritonavir and aprepitant against human glioma cells. J. Neurooncol. 2016, 126, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, P.; Phillips, J.; Conget, I.; Cooke, A.; Nicoletti, F. IL-18 binding protein fusion construct delays the development of diabetes in adoptive transfer and cyclophosphamide-induced diabetes in NOD mouse. Clin. Immunol. 2005, 115, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. The role of interleukin-18 in glioblastoma pathology implies therapeutic potential of two old drugs-disulfiram and ritonavir. Chin. J. Cancer 2015, 34, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Patton, C.; Stevens, G.; Tekautz, T.; Angelov, L.; Vogelbaum, M.A.; Weil, R.J.; Chao, S.; Elson, P.; Suh, J.H.; et al. Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas. J. Neurooncol. 2011, 102, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Darini, C.Y.; Martin, P.; Azoulay, S.; Drici, M.-D.; Hofman, P.; Obba, S.; Dani, C.; Ladoux, A. Targeting cancer stem cells expressing an embryonic signature with anti-proteases to decrease their tumor potential. Cell Death Dis. 2013, 4, e706. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; O’Connell, M.; Pilcher, W. Lopinavir inhibits meningioma cell proliferation by Akt independent mechanism. J. Neurooncol. 2011, 101, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Kariya, R.; Taura, M.; Suzu, S.; Kai, H.; Katano, H.; Okada, S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-κB pathway. Cancer Lett. 2014, 342, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Song, J.M.; Upadhyaya, P.; Kassie, F. Nitric oxide-donating aspirin (NO-Aspirin) suppresses lung tumorigenesis in vitro and in vivo and these effects are associated with modulation of the EGFR signaling pathway. Carcinogenesis 2018, 39, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Canducci, F.; Ceresola, E.R.; Saita, D.; Al-Abed, Y.; Garotta, G.; Clementi, M.; Nicoletti, F. The new and less toxic protease inhibitor saquinavir—NO maintains anti-HIV-1 properties in vitro indistinguishable from those of the parental compound saquinavir. Antivir. Res. 2011, 91, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, S.; Maksimovic-Ivanic, D.; Mojic, M.; Timotijevic, G.; Miljkovic, D.; Mangano, K.; Donia, M.; Di Cataldo, A.; Al-Abed, Y.; Cheng, K.F.; et al. Cytotoxic and immune-sensitizing properties of nitric oxide-modified Saquinavir in iNOS-positive human melanoma cells. J. Cell. Physiol. 2011, 226, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Maksimovic-Ivanic, D.; Mijatovic, S.; Mojic, M.; Miljkovic, D.; Timotijevic, G.; Fagone, P.; Caponnetto, S.; Al-Abed, Y.; McCubrey, J.; et al. In vitro and in vivo anticancer action of Saquinavir-NO, a novel nitric oxide-derivative of the protease inhibitor saquinavir, on hormone resistant prostate cancer cells. Cell Cycle 2011, 10, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momčilović, M.; Mangano, K.; Jevtić, B.; Mammana, S.; Stošić-Grujičić, S.; Nicoletti, F.; Miljković, D. Saquinavir-NO inhibits IL-6 production in macrophages. Basic Clin. Pharmacol. Toxicol. 2014, 115, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Petković, F.; Blaževski, J.; Momčilović, M.; Timotijević, G.; Zocca, M.-B.; Mijatović, S.; Maksimović-Ivanić, D.; Mangano, K.; Fagone, P.; Stošić-Grujičić, S.; et al. NSaquinavir-NO inhibits S6 kinase activity, impairs secretion of the encephalytogenic cytokines interleukin-17 and interferon-gamma and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 259, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic-Ivanic, D.; Mojic, M.; Bulatovic, M.; Radojkovic, M.; Kuzmanovic, M.; Ristic, S.; Stosic-Grujicic, S.; Miljkovic, D.; Cavalli, E.; Libra, M.; et al. The NO-modified HIV protease inhibitor as a valuable drug for hematological malignancies: Role of p70S6K. Leuk. Res. 2015, 39, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Cavalli, E.; Mammana, S.; Lombardo, G.A.G.; Pennisi, V.; Zocca, M.-B.; He, M.; Al-Abed, Y.; et al. Effects of NO-Hybridization on the Immunomodulatory Properties of the HIV Protease Inhibitors Lopinavir and Ritonavir. Basic Clin. Pharmacol. Toxicol. 2015, 117, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Chedid, K.; Kalkanis, S.N. Glioblastoma cell line-derived spheres in serum-containing medium versus serum-free medium: A comparison of cancer stem cell properties. Int. J. Oncol. 2012, 41, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Mojic, M.; Mijatovic, S.; Maksimovic-Ivanic, D.; Dinic, S.; Grdovic, N.; Miljkovic, D.; Stosic-Grujicic, S.; Tumino, S.; Fagone, P.; Mangano, K.; et al. Saquinavir-NO-targeted S6 protein mediates sensitivity of androgen-dependent prostate cancer cells to TRAIL. Cell Cycle 2012, 11, 1174–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojic, M.; Mijatovic, S.; Maksimovic-Ivanic, D.; Miljkovic, D.; Stosic-Grujicic, S.; Stankovic, M.; Mangano, K.; Travali, S.; Donia, M.; Fagone, P.; et al. Therapeutic potential of nitric oxide-modified drugs in colon cancer cells. Mol. Pharmacol. 2012, 82, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.; Bandyopadhyay, S.; Messing, A.; Brenner, M. Transgenic analysis of GFAP promoter elements. Glia 2013, 61, 1488–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, Y.; Marshall, H.E. S-nitrosylation in the regulation of gene transcription. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakshinamoorthy, S.; Sridharan, S.R.; Li, L.; Ng, P.Y.; Boxer, L.M.; Porter, A.G. Protein/DNA arrays identify nitric oxide-regulated cis-element and trans-factor activities some of which govern neuroblastoma cell viability. Nucleic Acids Res. 2007, 35, 5439–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Fitzwalter, B.E.; Thorburn, A. Autophagy inhibition improves anti-cancer drugs via FOXO3a activation. Oncotarget 2018, 9, 25384–25385. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pasqualis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Rothweiler, F.; Michaelis, M.; Brauer, P.; Otte, J.; Weber, K.; Fehse, B.; Doerr, H.W.; Wiese, M.; Kreuter, J.; Al-Abed, Y.; et al. Anticancer effects of the nitric oxide-modified saquinavir derivative saquinavir-NO against multidrug-resistant cancer cells. Neoplasia 2010, 12, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Harhaji-Trajkovic, L.; Vilimanovich, U.; Kravic-Stevovic, T.; Bumbasirevic, V.; Trajkovic, V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J. Cell. Mol. Med. 2009, 13, 3644–3654. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, L.; Zhou, H.; Wang, W.; Luo, Y.; Yang, H.; Yi, H. Inhibition of autophagy promotes cisplatin-induced apoptotic cell death through Atg5 and Beclin 1 in A549 human lung cancer cells. Mol. Med. Rep. 2018, 17, 6859–6865. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-F.; Lin, Y.-C.; Tsai, T.-F.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Dev. Ther. 2017, 11, 1517–1533. [Google Scholar] [CrossRef] [PubMed]
- Mijatović, S.; Bramanti, A.; Nicoletti, F.; Fagone, P.; Kaluđerović, G.N.; Maksimović-Ivanić, D. Naturally occurring compounds in differentiation based therapy of cancer. Biotechnol. Adv. 2018, 36, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gomez, G.A.; Zhao, Y.; Yang, Y.; Cao, D.; Lu, J.; Yang, H.; Lin, S. ETV2 mediates endothelial transdifferentiation of glioblastoma. Signal Transduct. Target. Ther. 2018, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenkamp, A.M.E.; Boddeke, E.; Balasubramanyian, V.; Wagemakers, M.; et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Romero-López, M.; Benitez, L.I.; Di, K.; Frieboes, H.B.; Hughes, C.C.W.; Bota, D.A.; Lowengrub, J.S. 3D Mathematical Modeling of Glioblastoma Suggests That Transdifferentiated Vascular Endothelial Cells Mediate Resistance to Current Standard-of-Care Therapy. Cancer Res. 2017, 77, 4171–4184. [Google Scholar] [CrossRef] [PubMed]
- Pistell, P.J.; Gupta, S.; Knight, A.G.; Domingue, M.; Uranga, R.M.; Ingram, D.K.; Kheterpal, I.; Ruiz, C.; Keller, J.N.; Bruce-Keller, A.J. Metabolic and neurologic consequences of chronic lopinavir/ritonavir administration to C57BL/6 mice. Antivir. Res. 2010, 88, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compound Lopinavir-NO is available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Assay | IC50 (µg/mL) | |
|---|---|---|---|
| Lopinavir | Lopinavir-NO | ||
| LN-229 | MTT | 13.80 ± 2.40 | 8.70 ± 1.41 |
| CV | 13.75 ± 2.90 | 7.30 ± 1.70 | |
| U-251 | MTT | 21.50 ± 0.85 | 13.30 ± 1.13 |
| CV | 22.35 ± 0.07 | 12.50 ± 0.00 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basile, M.S.; Mazzon, E.; Krajnovic, T.; Draca, D.; Cavalli, E.; Al-Abed, Y.; Bramanti, P.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells. Molecules 2018, 23, 2463. https://doi.org/10.3390/molecules23102463
Basile MS, Mazzon E, Krajnovic T, Draca D, Cavalli E, Al-Abed Y, Bramanti P, Nicoletti F, Mijatovic S, Maksimovic-Ivanic D. Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells. Molecules. 2018; 23(10):2463. https://doi.org/10.3390/molecules23102463
Chicago/Turabian StyleBasile, Maria Sofia, Emanuela Mazzon, Tamara Krajnovic, Dijana Draca, Eugenio Cavalli, Yousef Al-Abed, Placido Bramanti, Ferdinando Nicoletti, Sanja Mijatovic, and Danijela Maksimovic-Ivanic. 2018. "Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells" Molecules 23, no. 10: 2463. https://doi.org/10.3390/molecules23102463