Brush Polymer of Donor-Accepter Dyads via Adduct Formation between Lewis Base Polymer Donor and All Carbon Lewis Acid Acceptor

Abstract
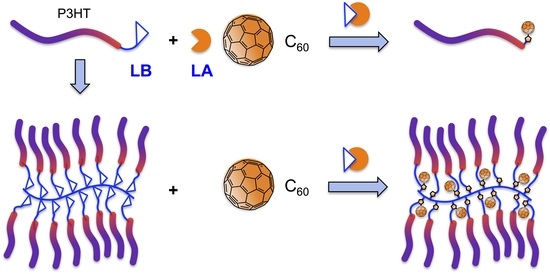
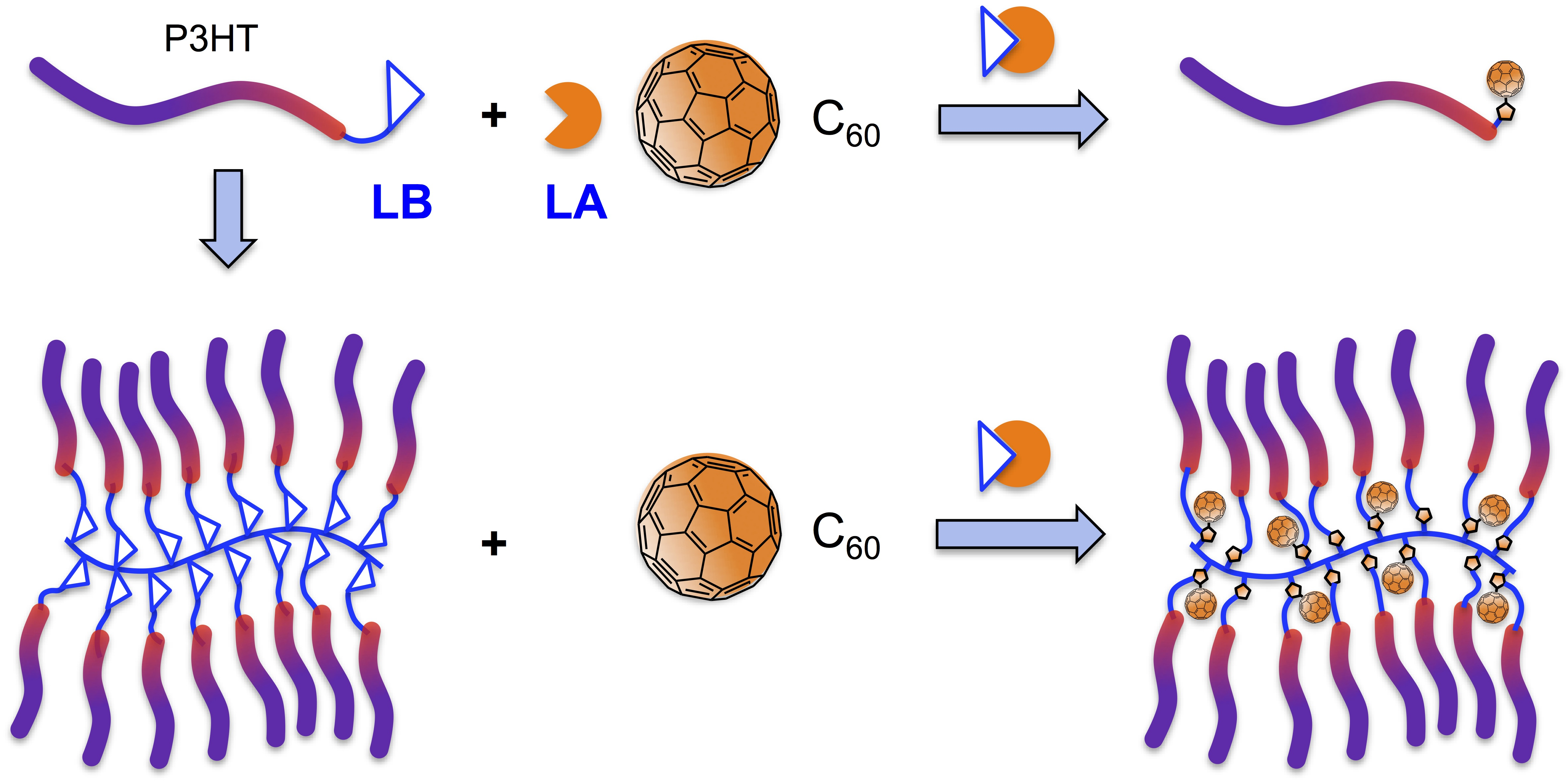
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
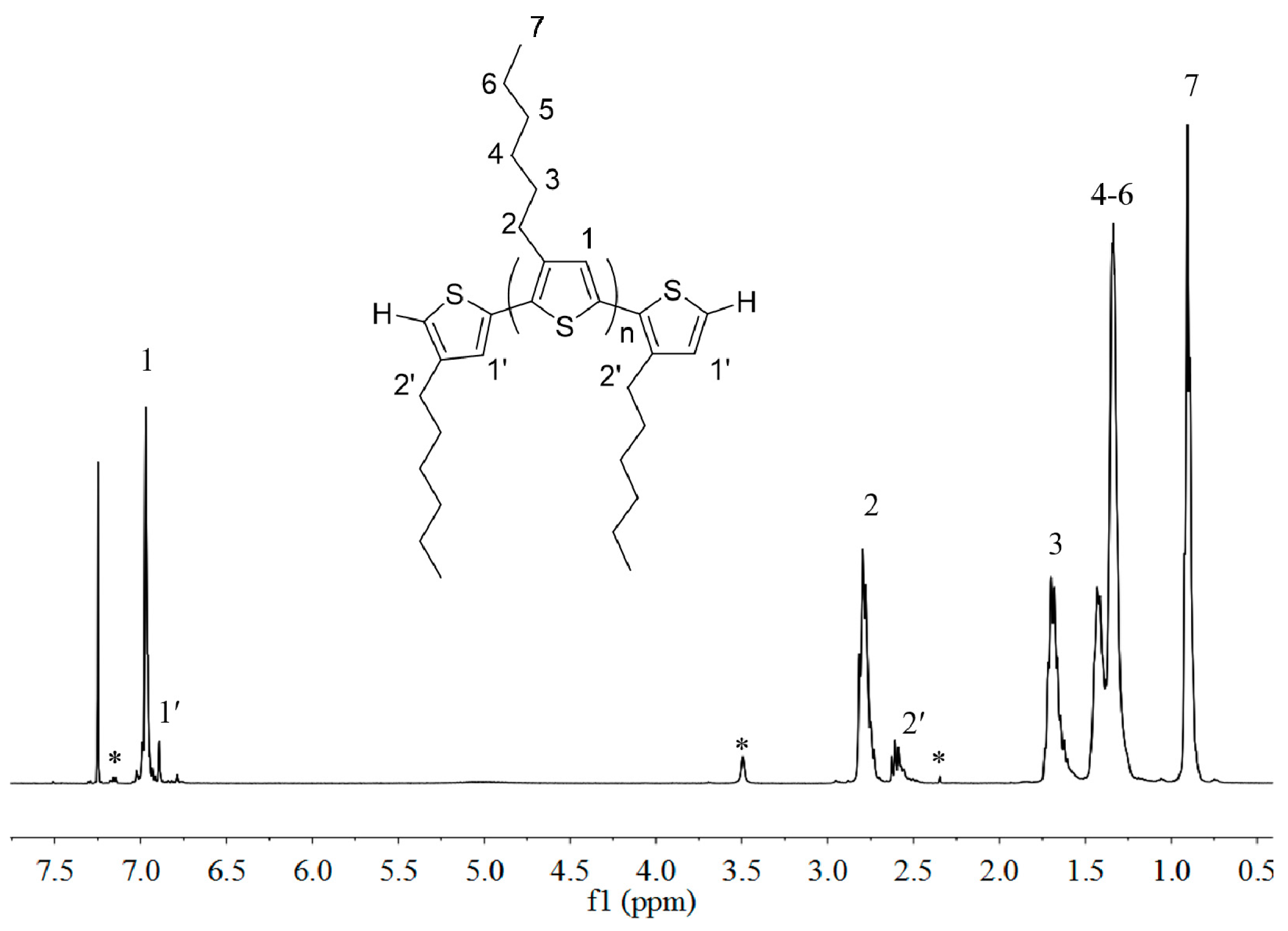
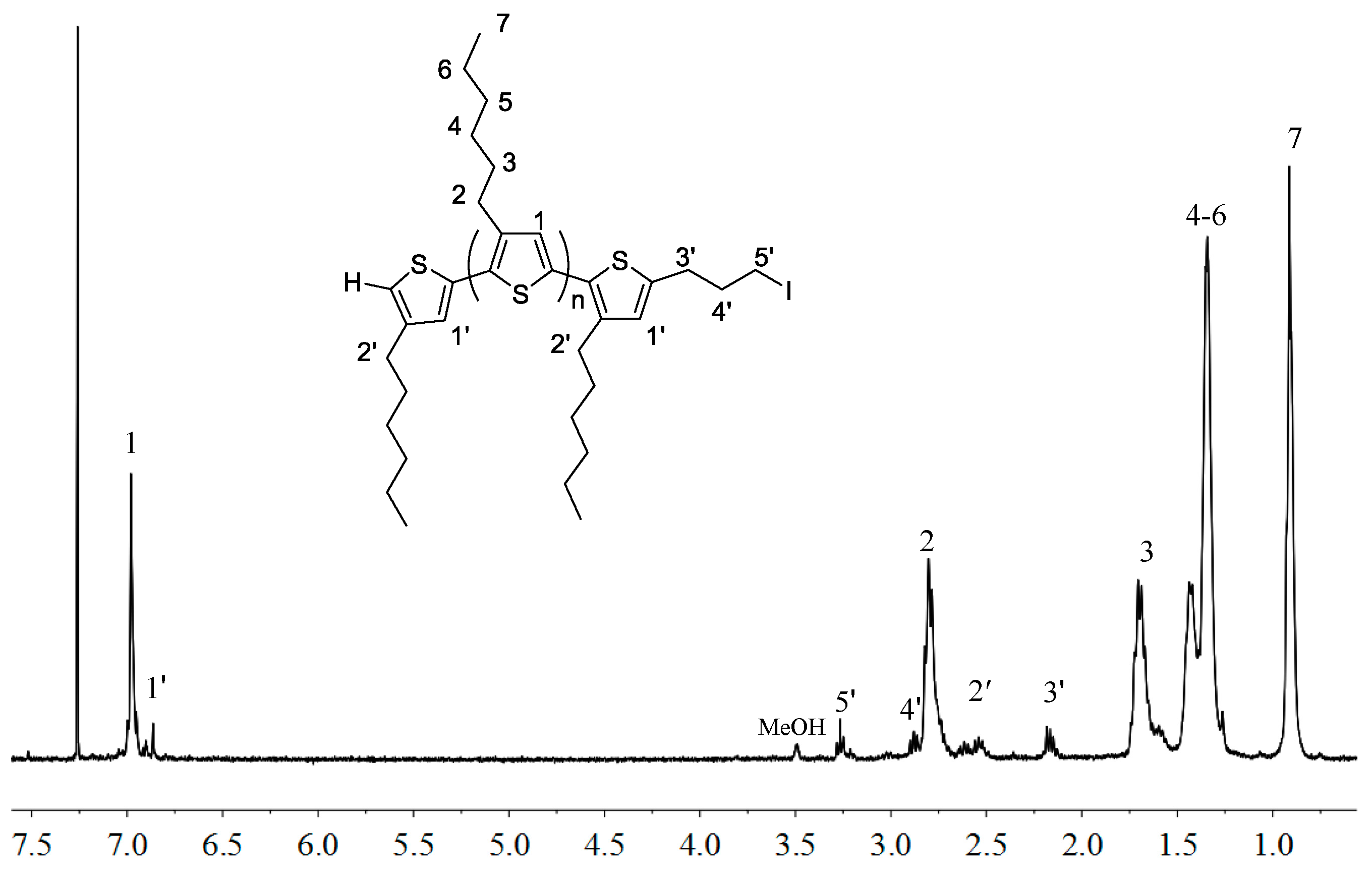
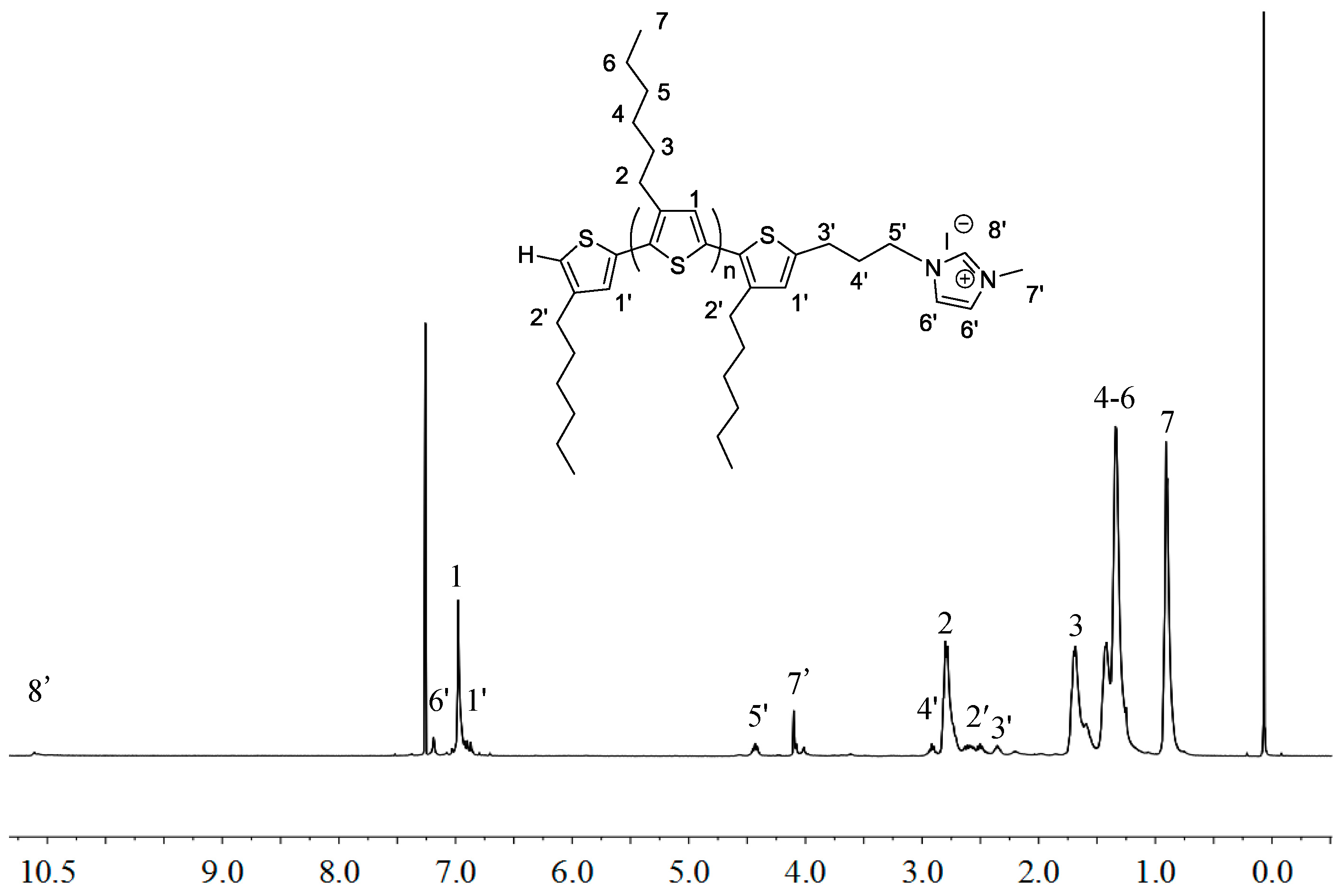
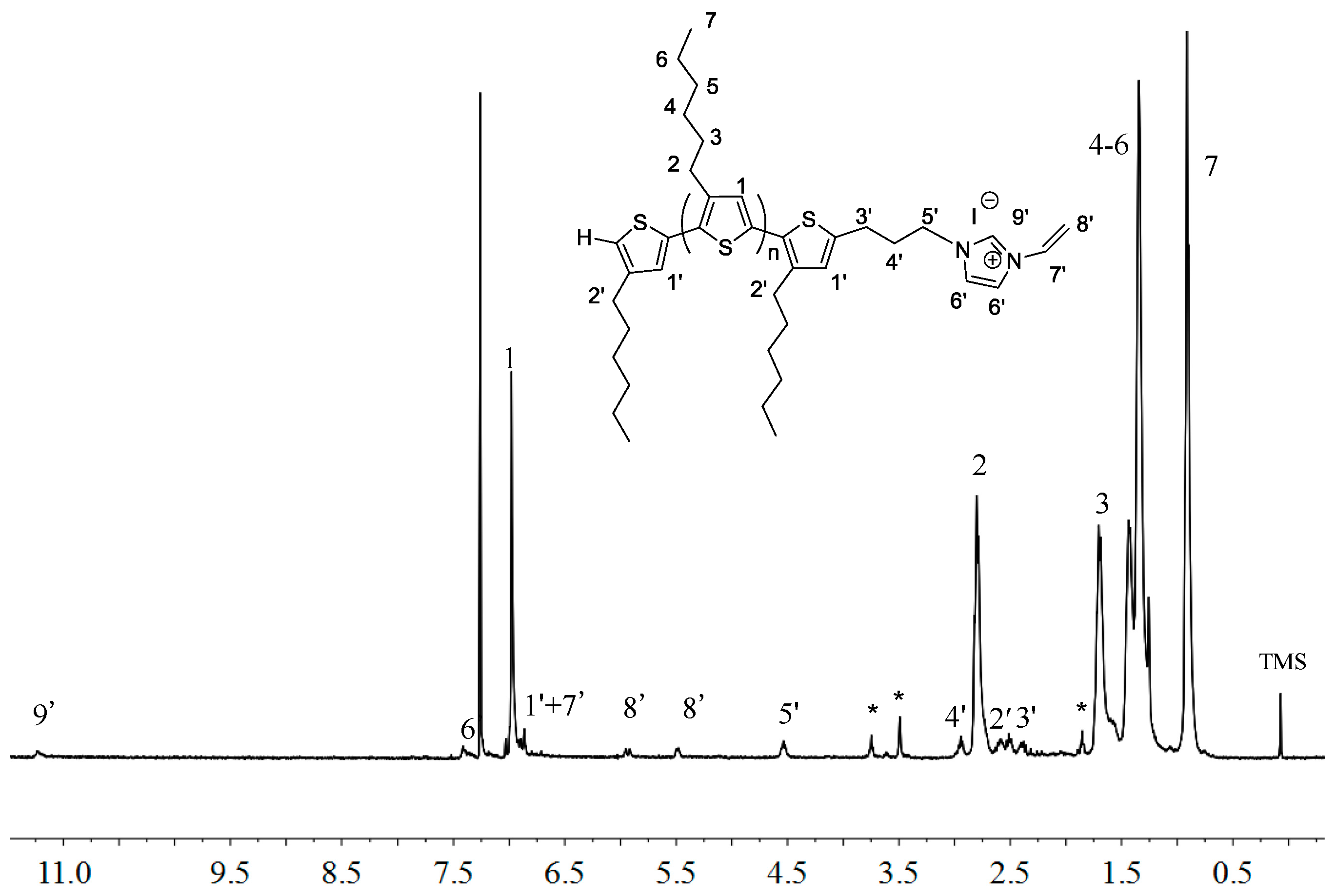
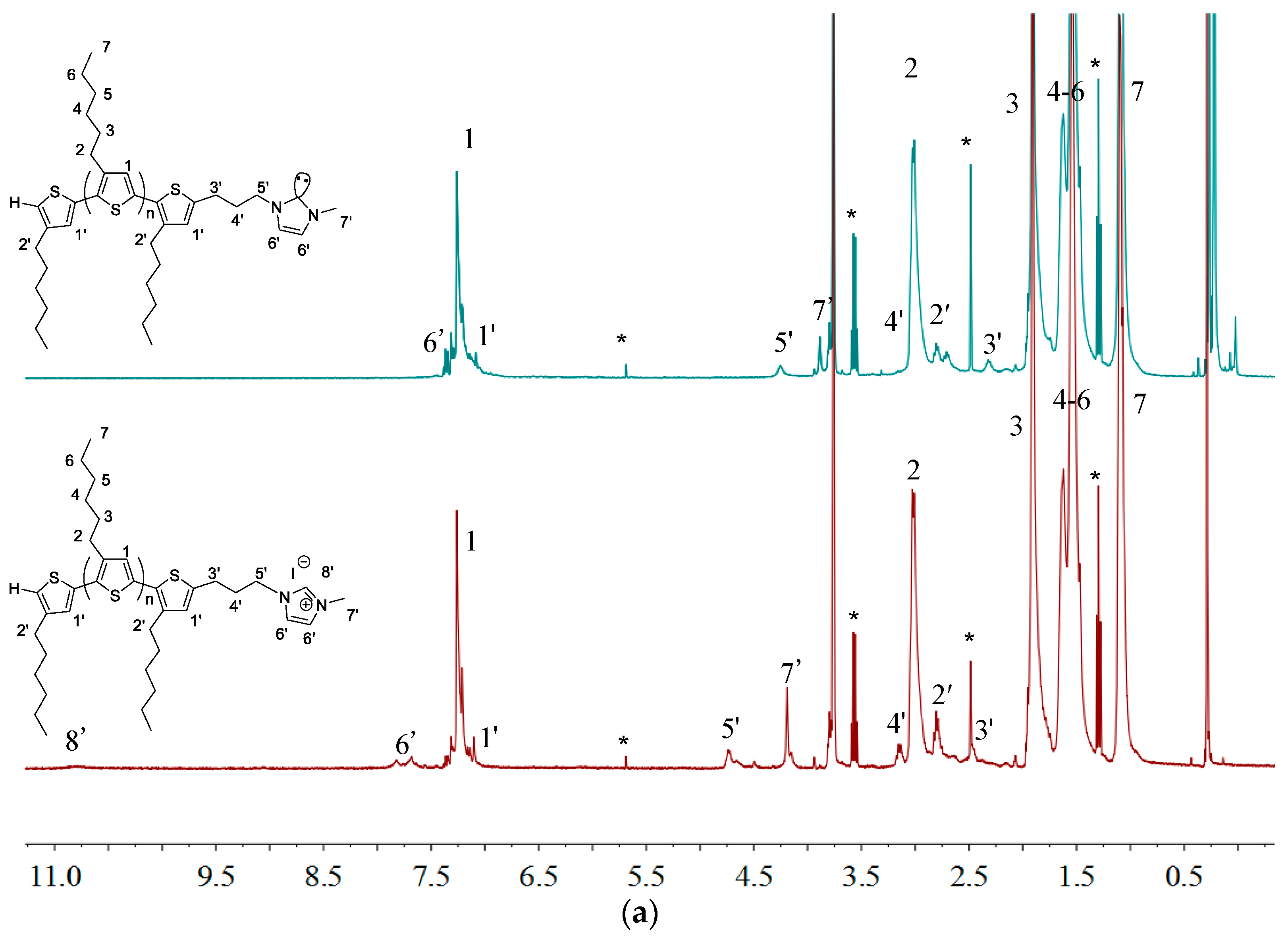
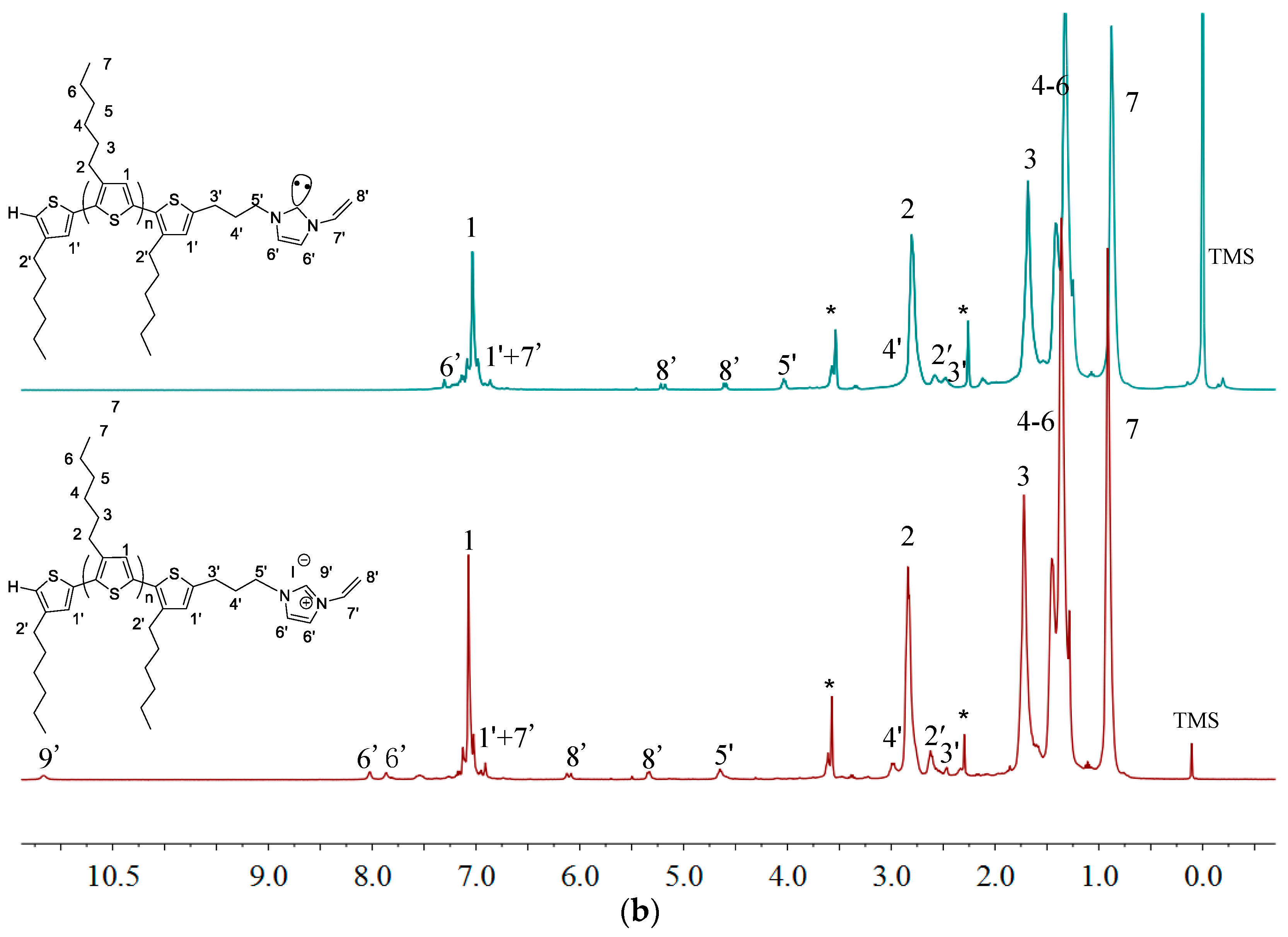
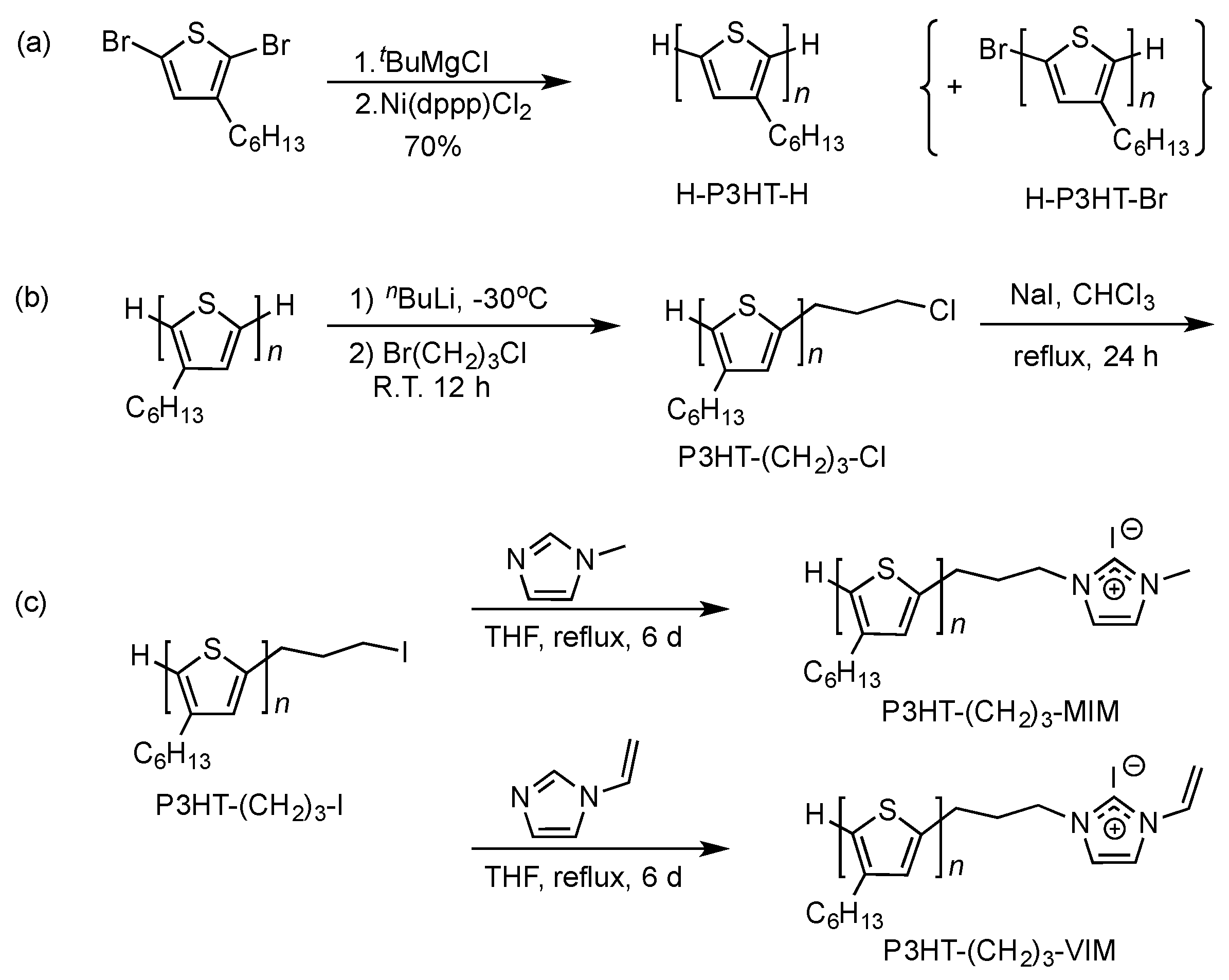
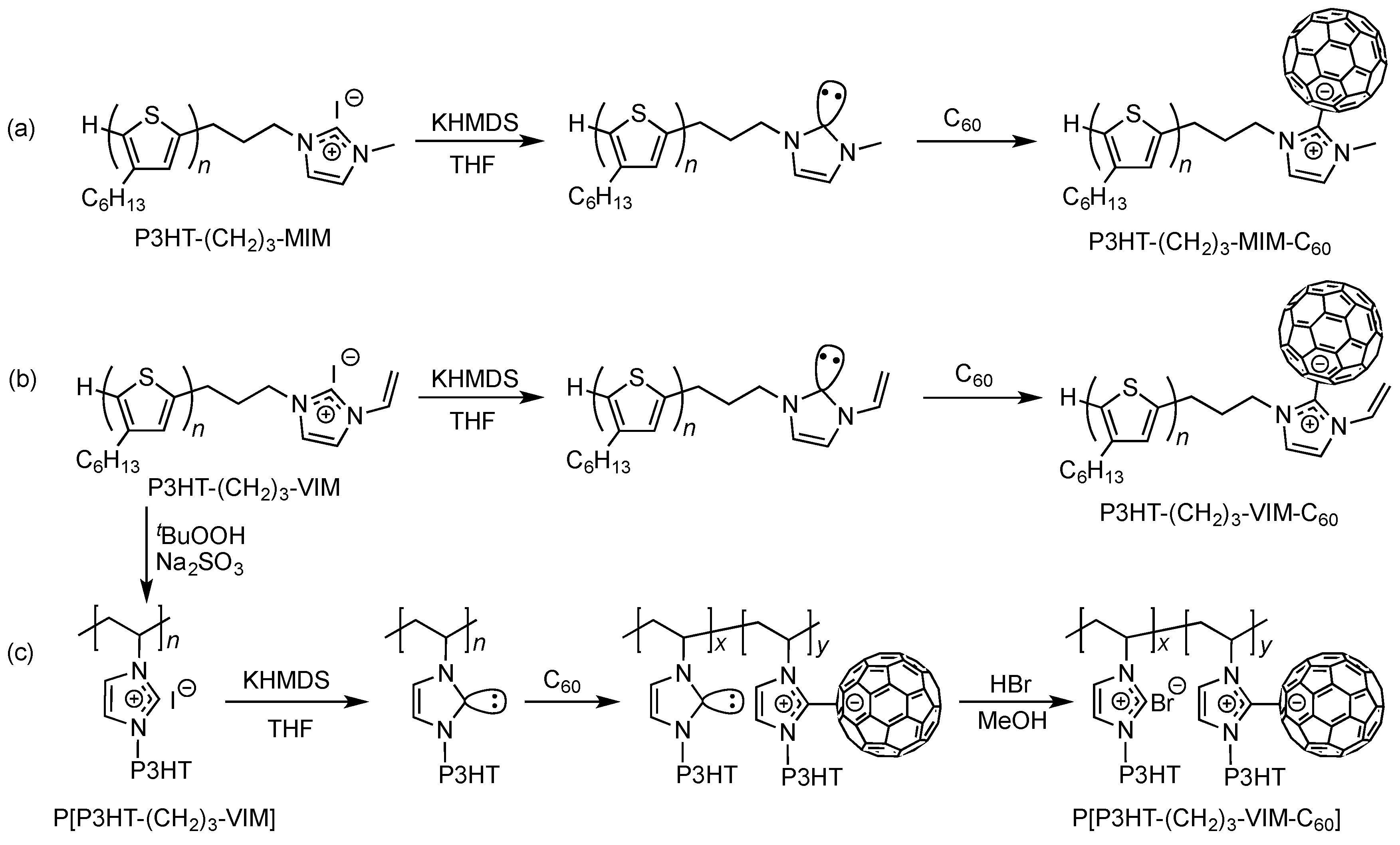
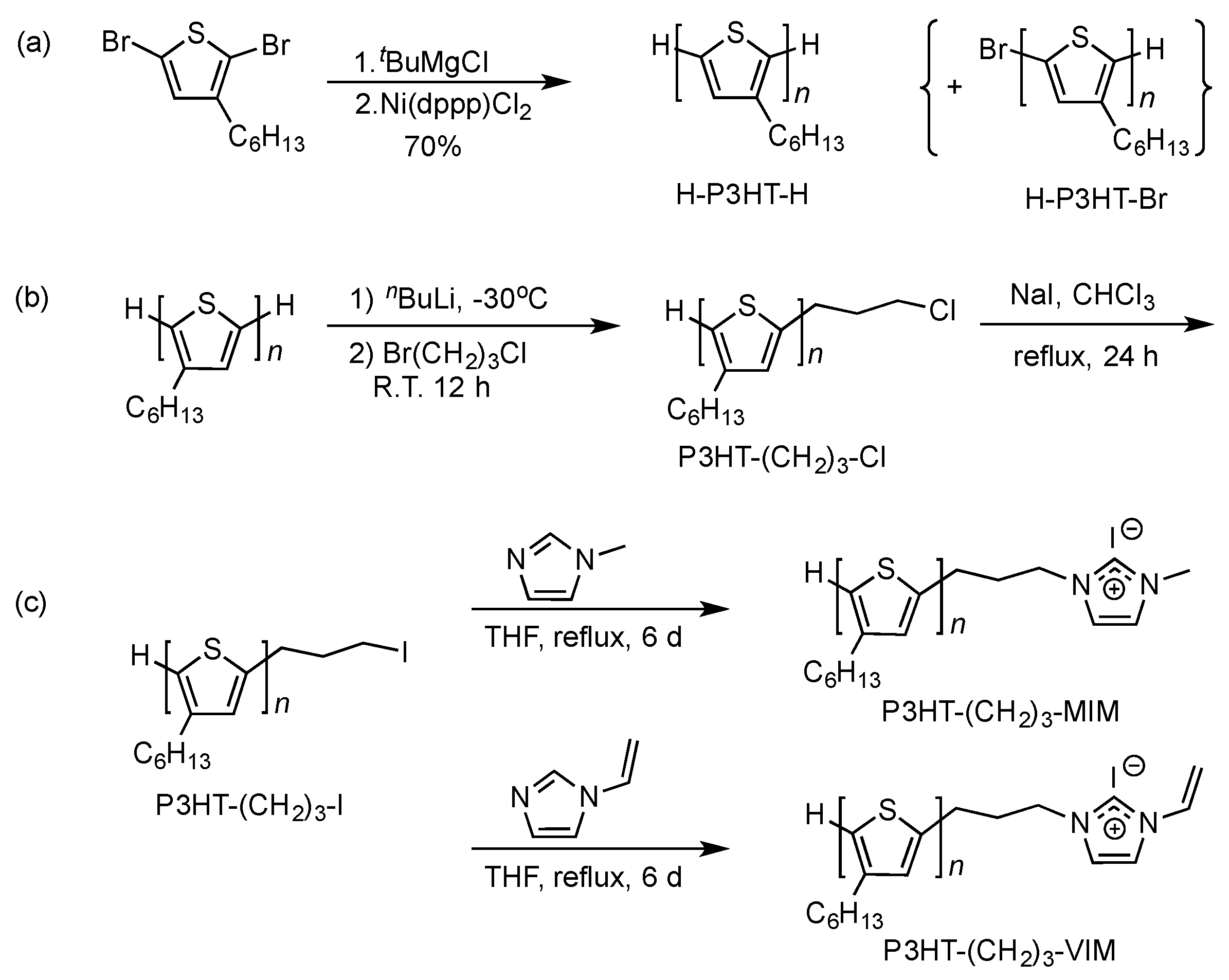
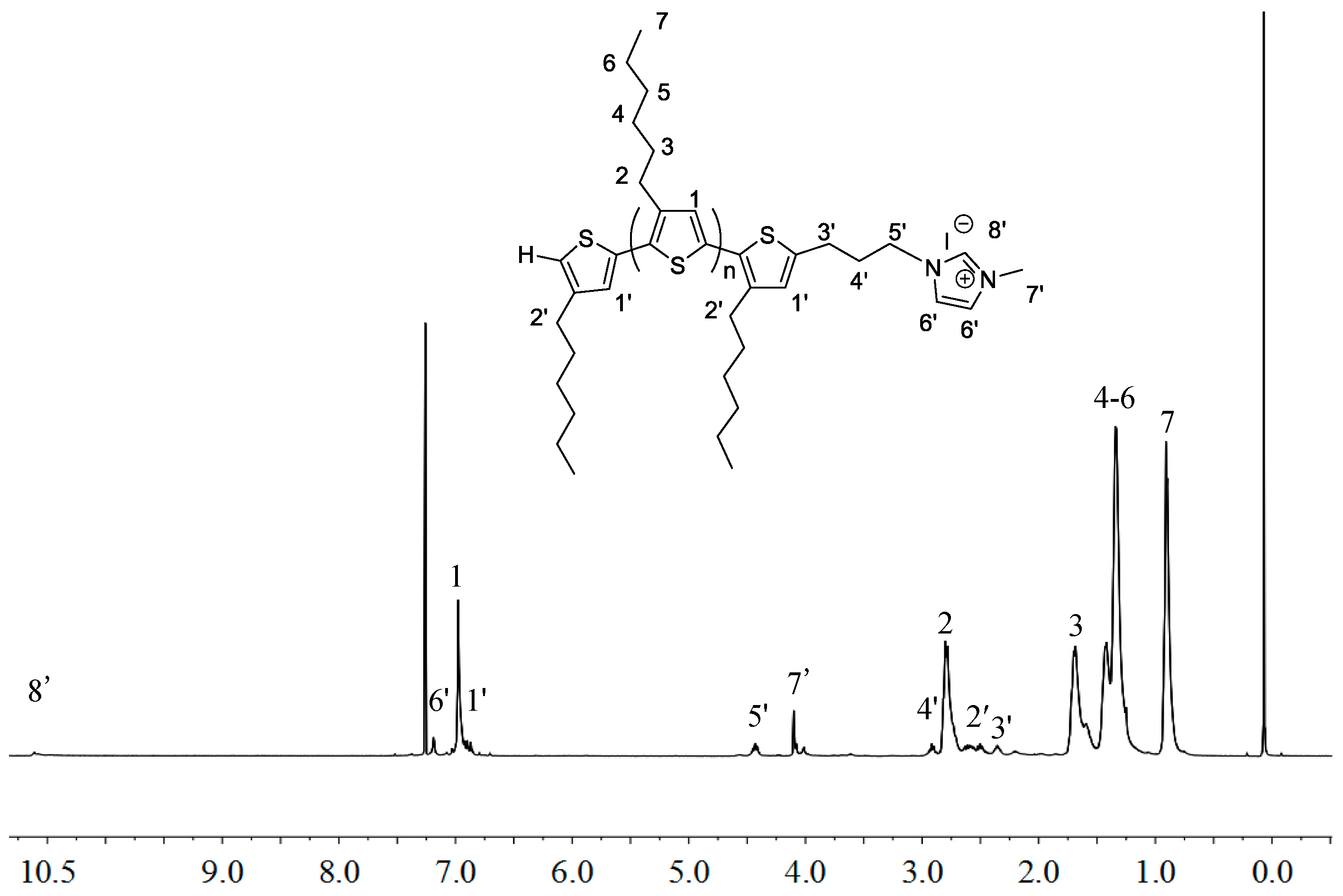
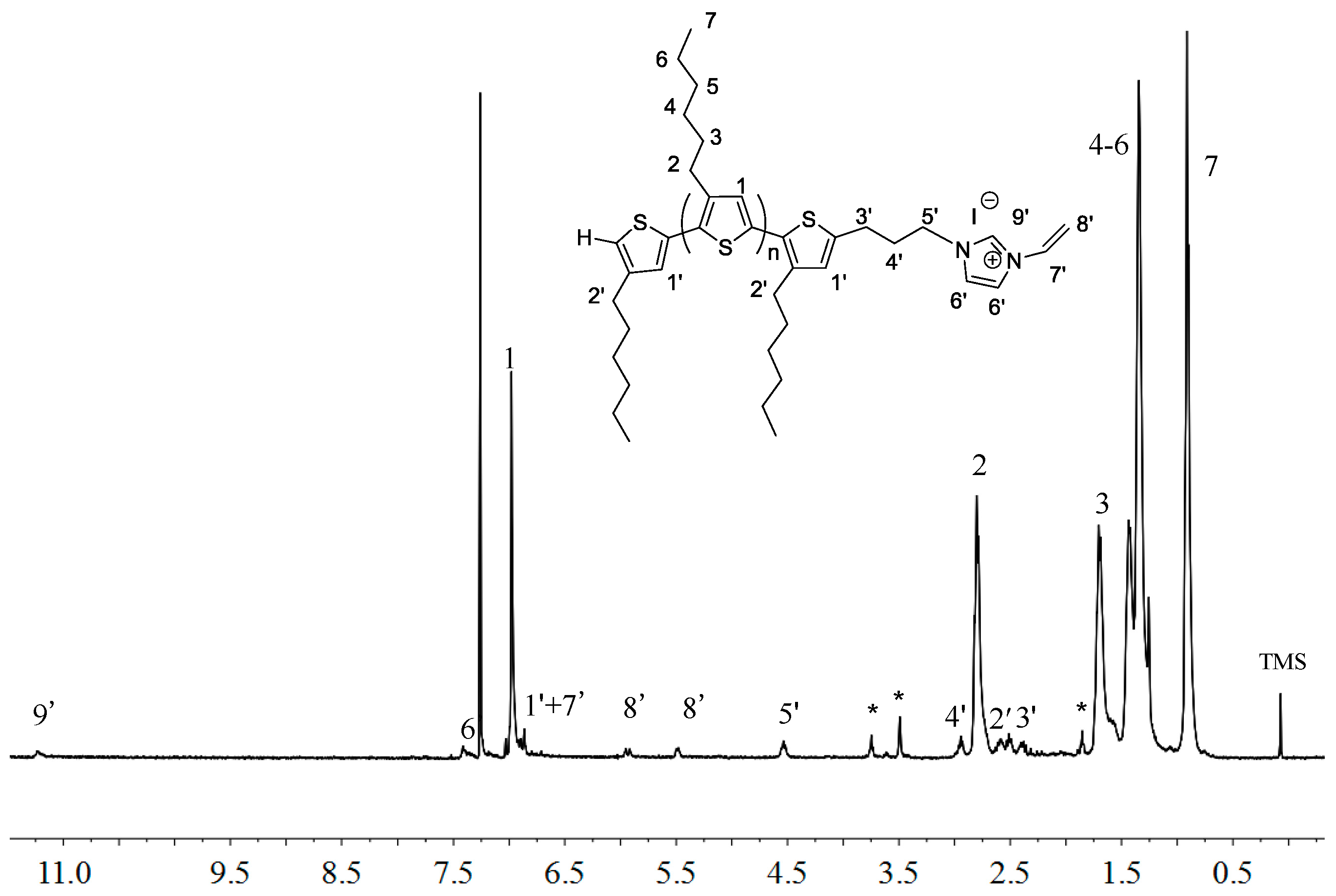
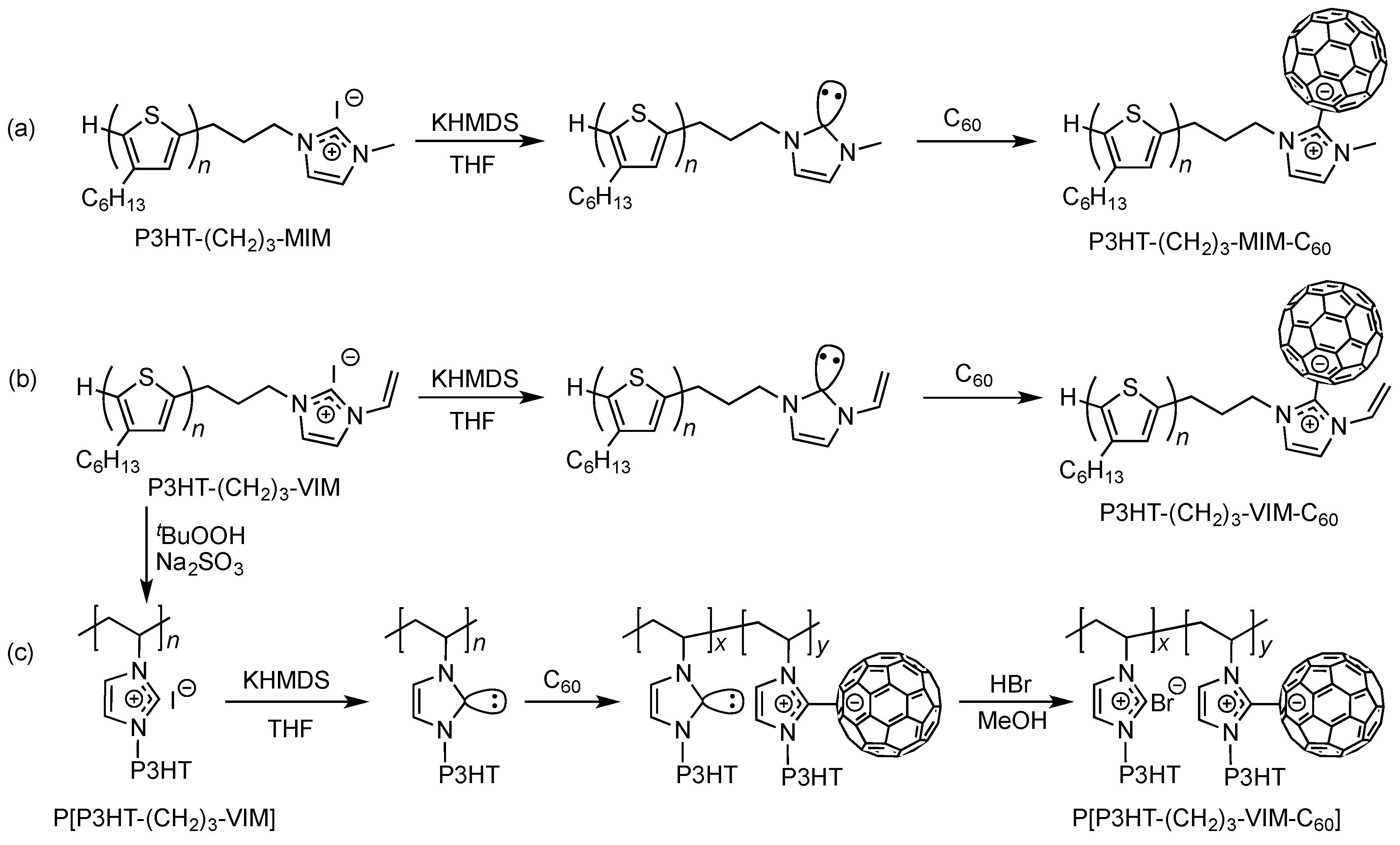
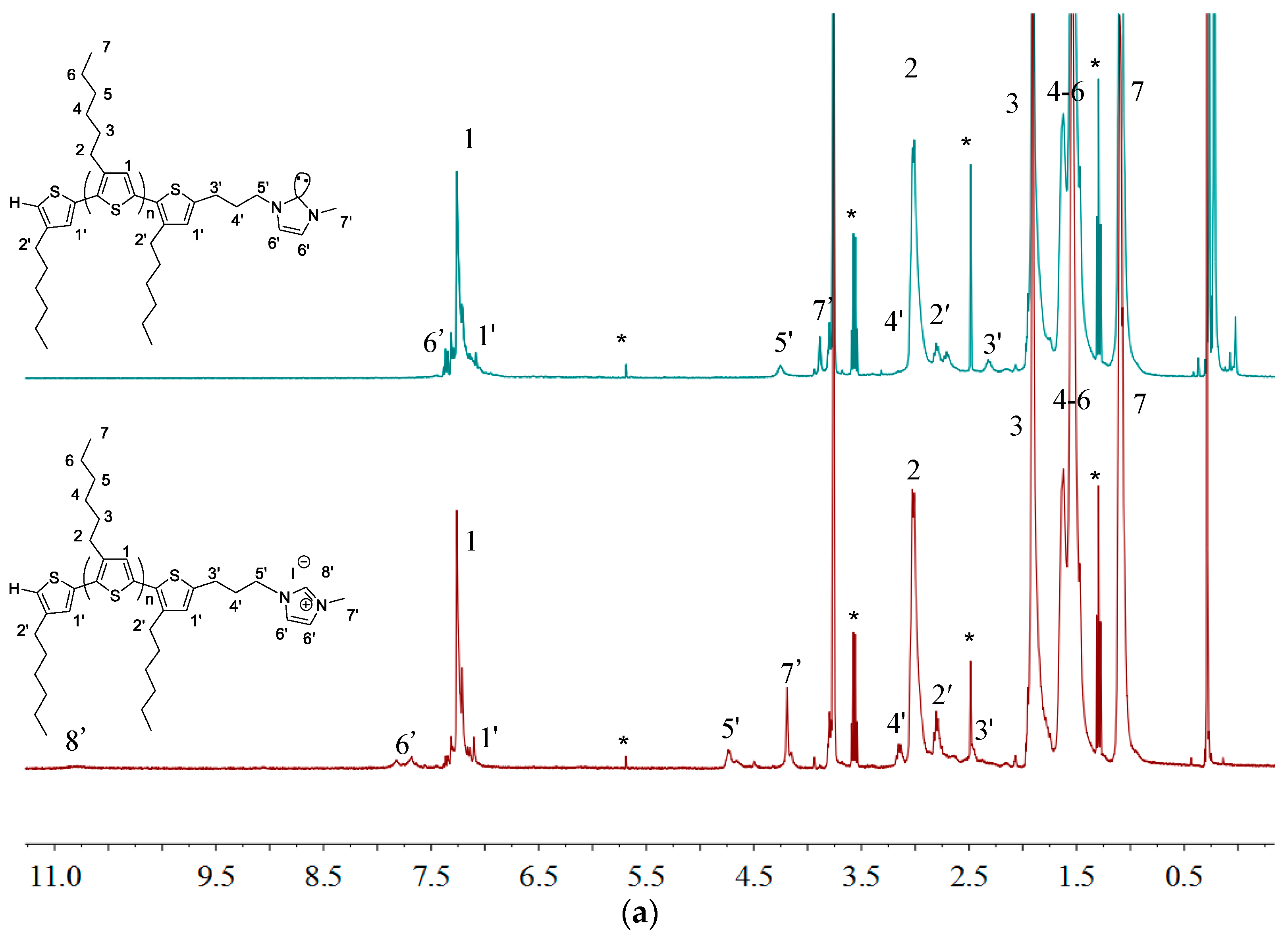
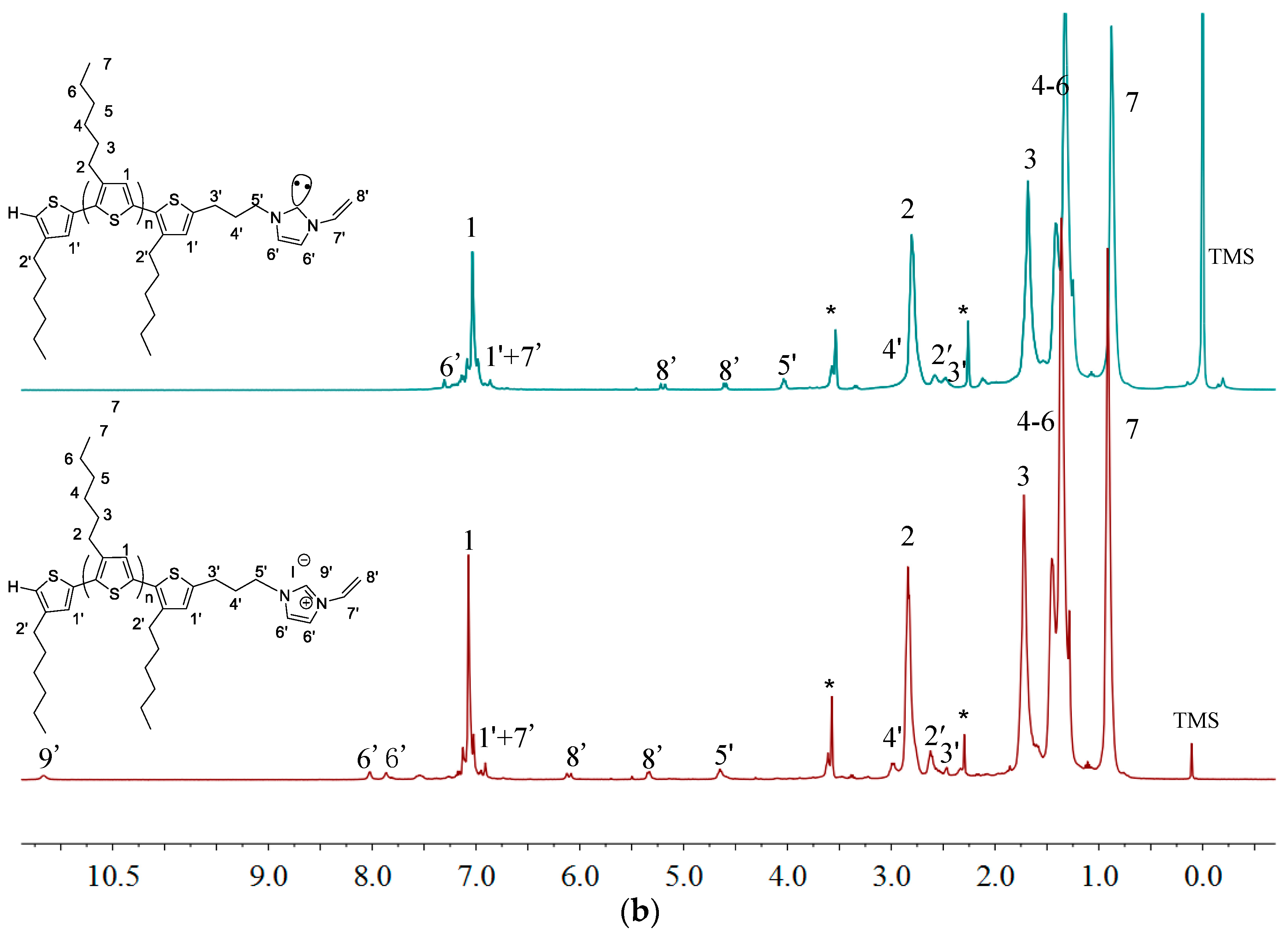
2.1. Synthesis of Macromers P3HT-(CH2)3-MIM and P3HT-(CH2)3-VIM
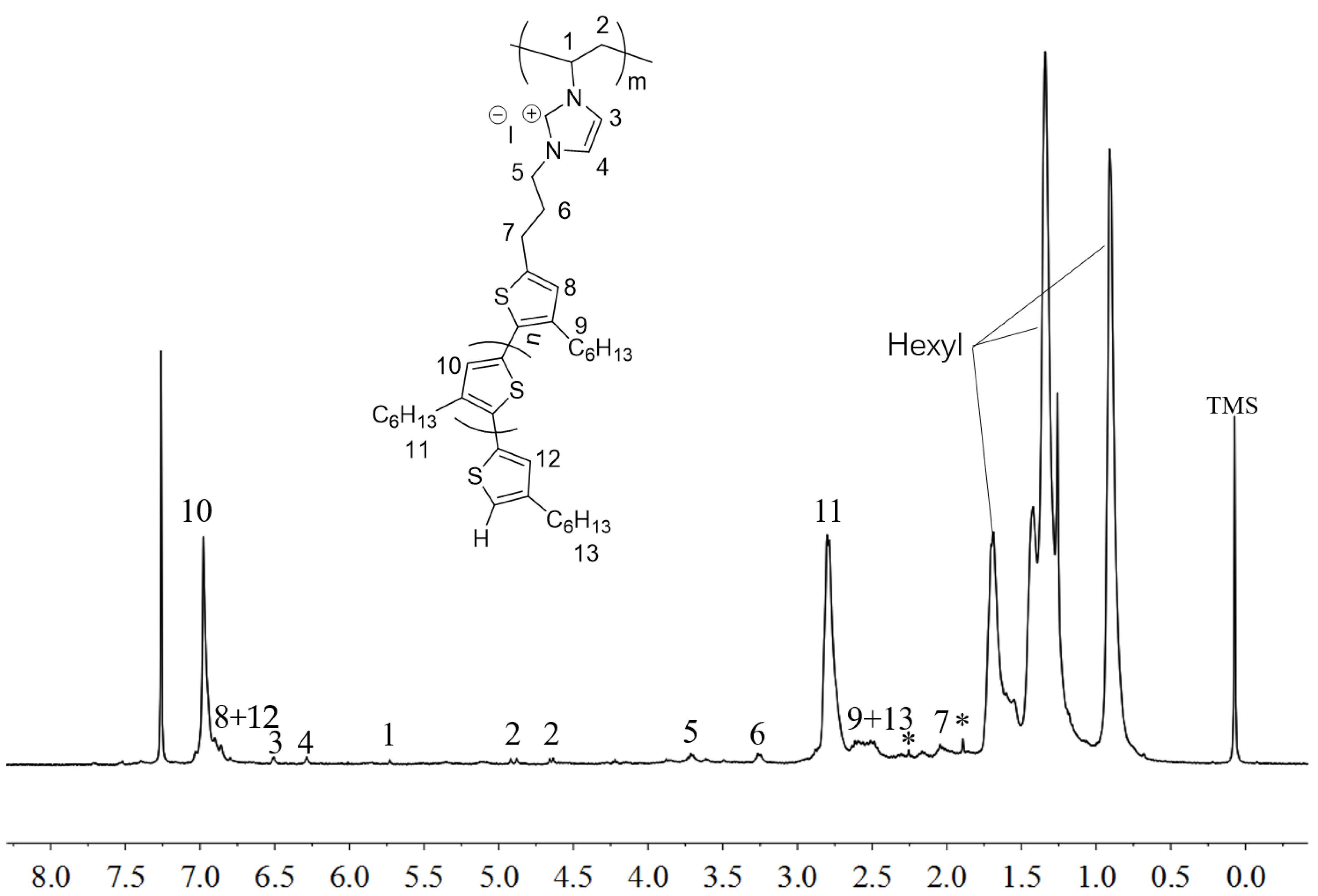
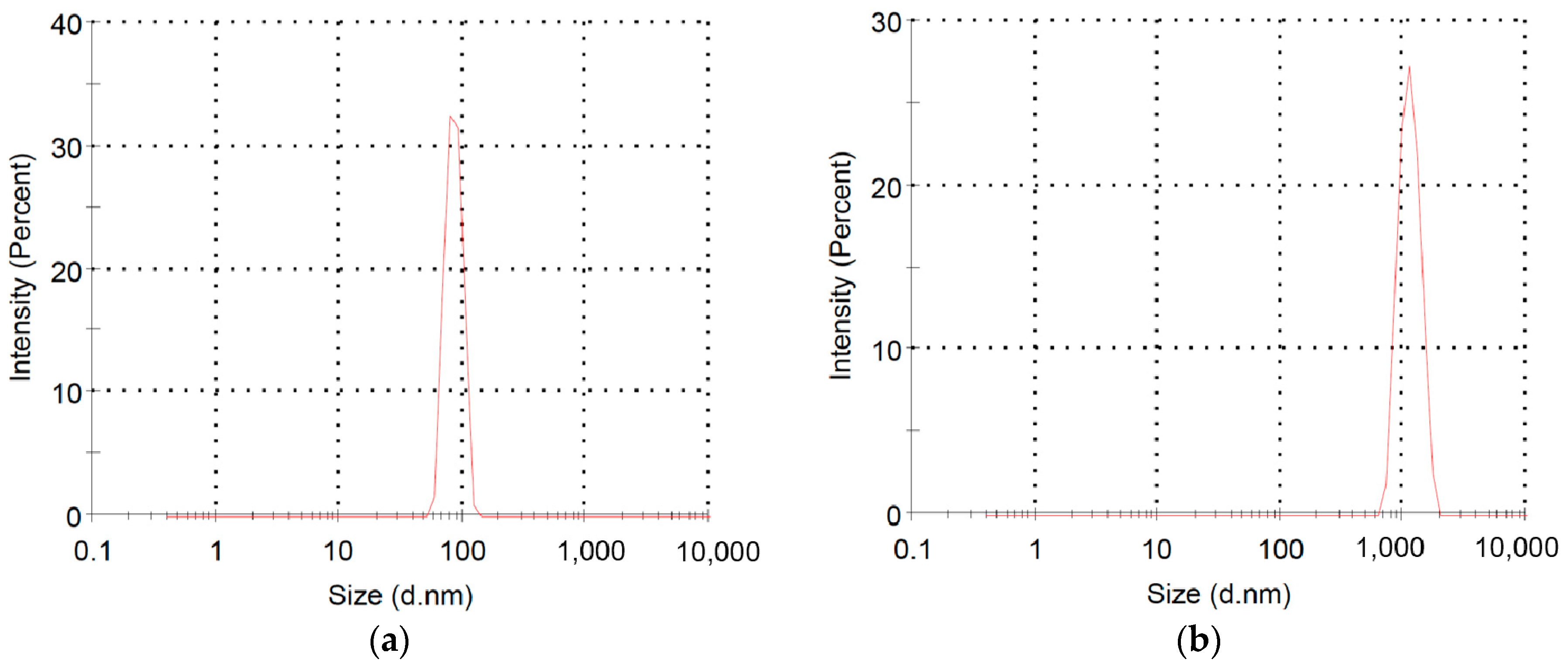
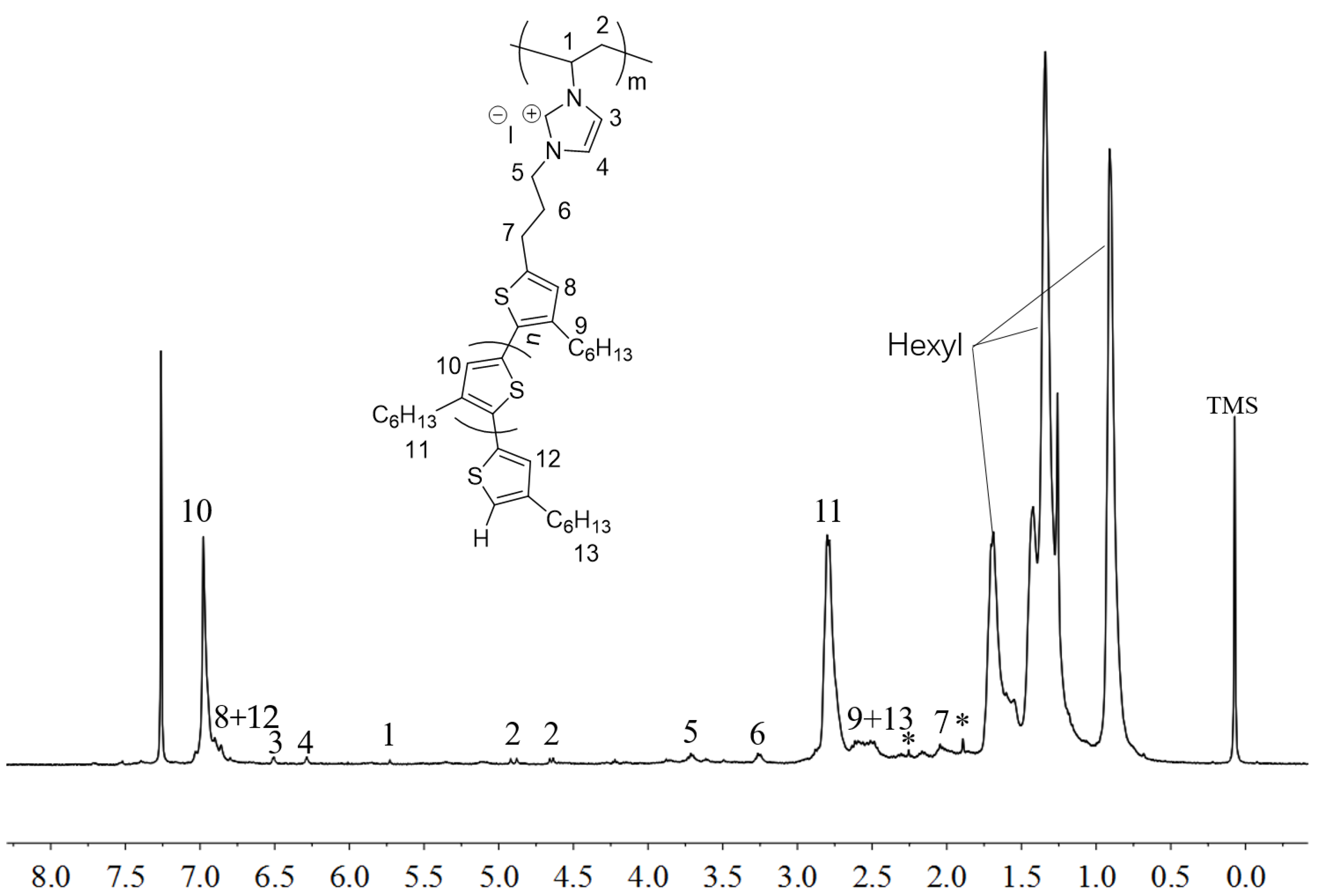
2.2. Polymerization of Macromer P3HT-(CH2)3-VIM
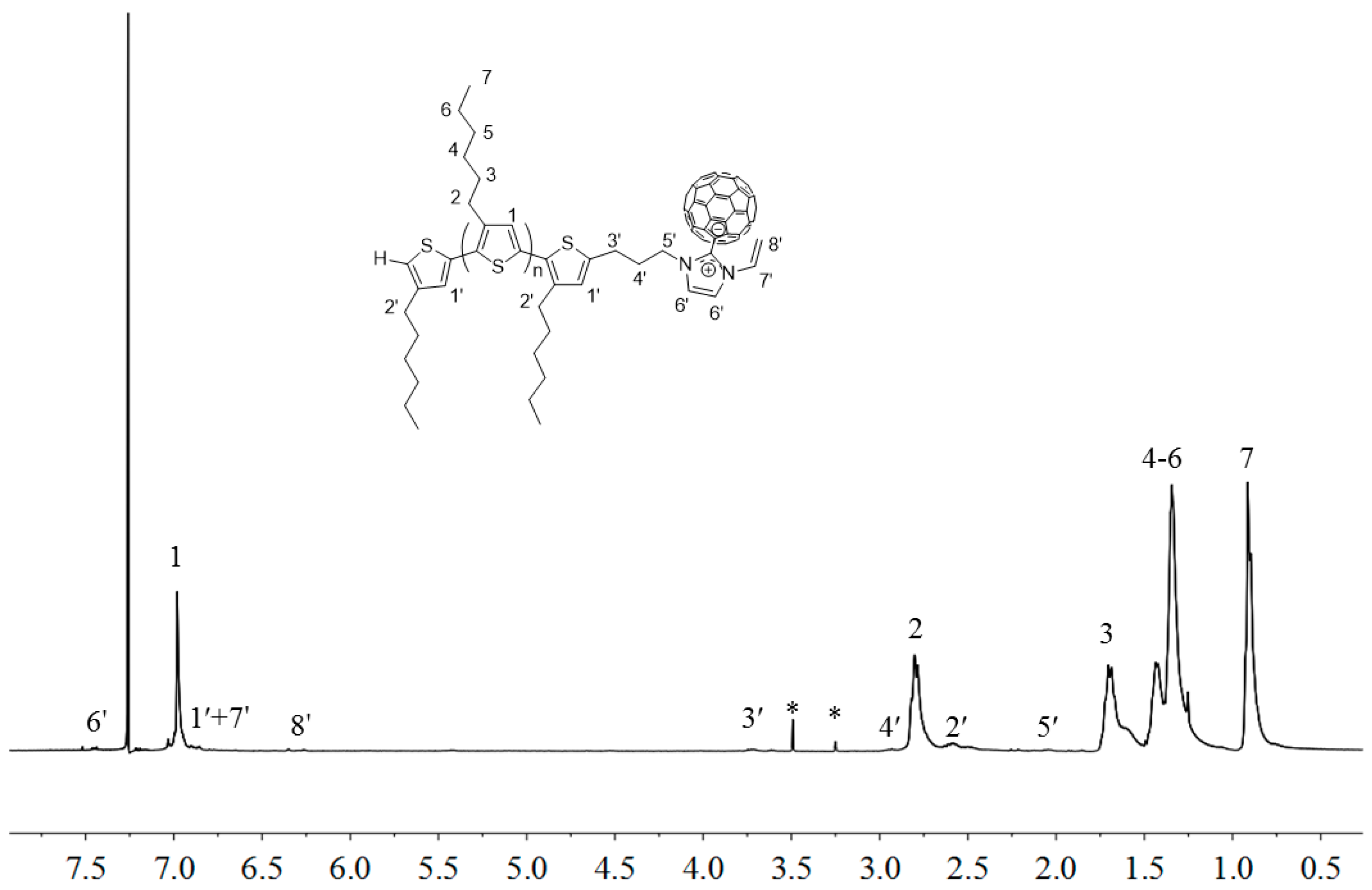
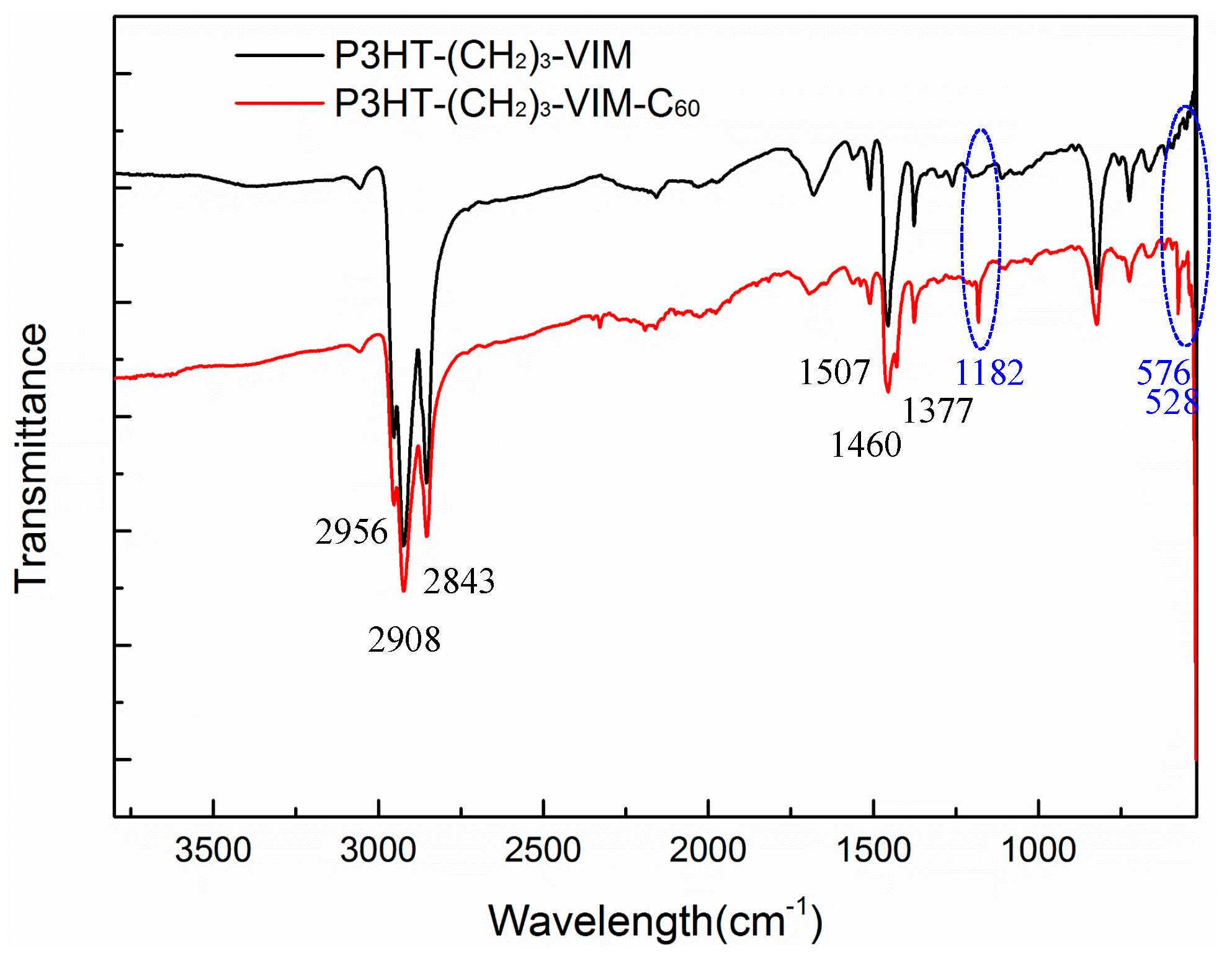
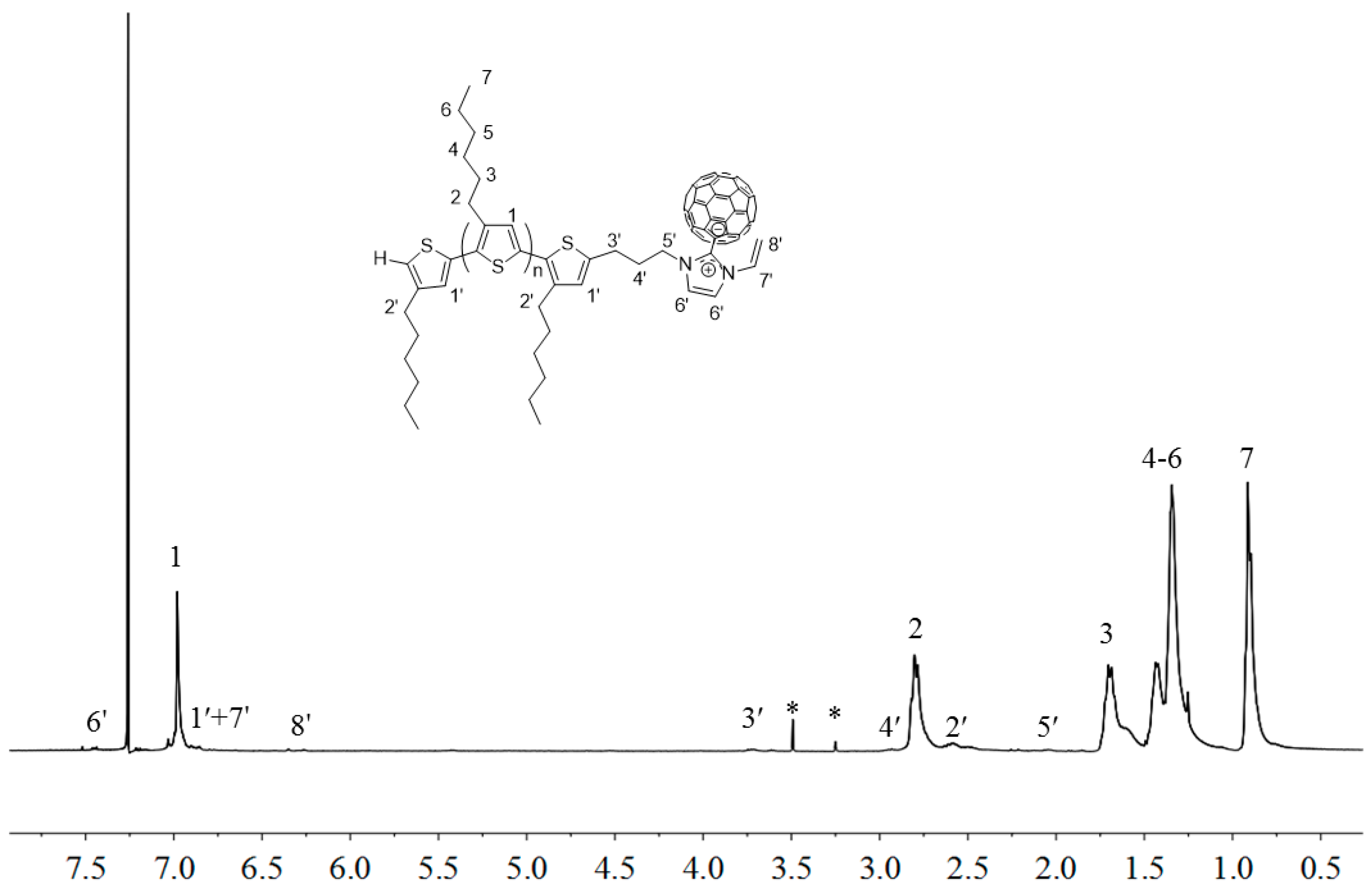
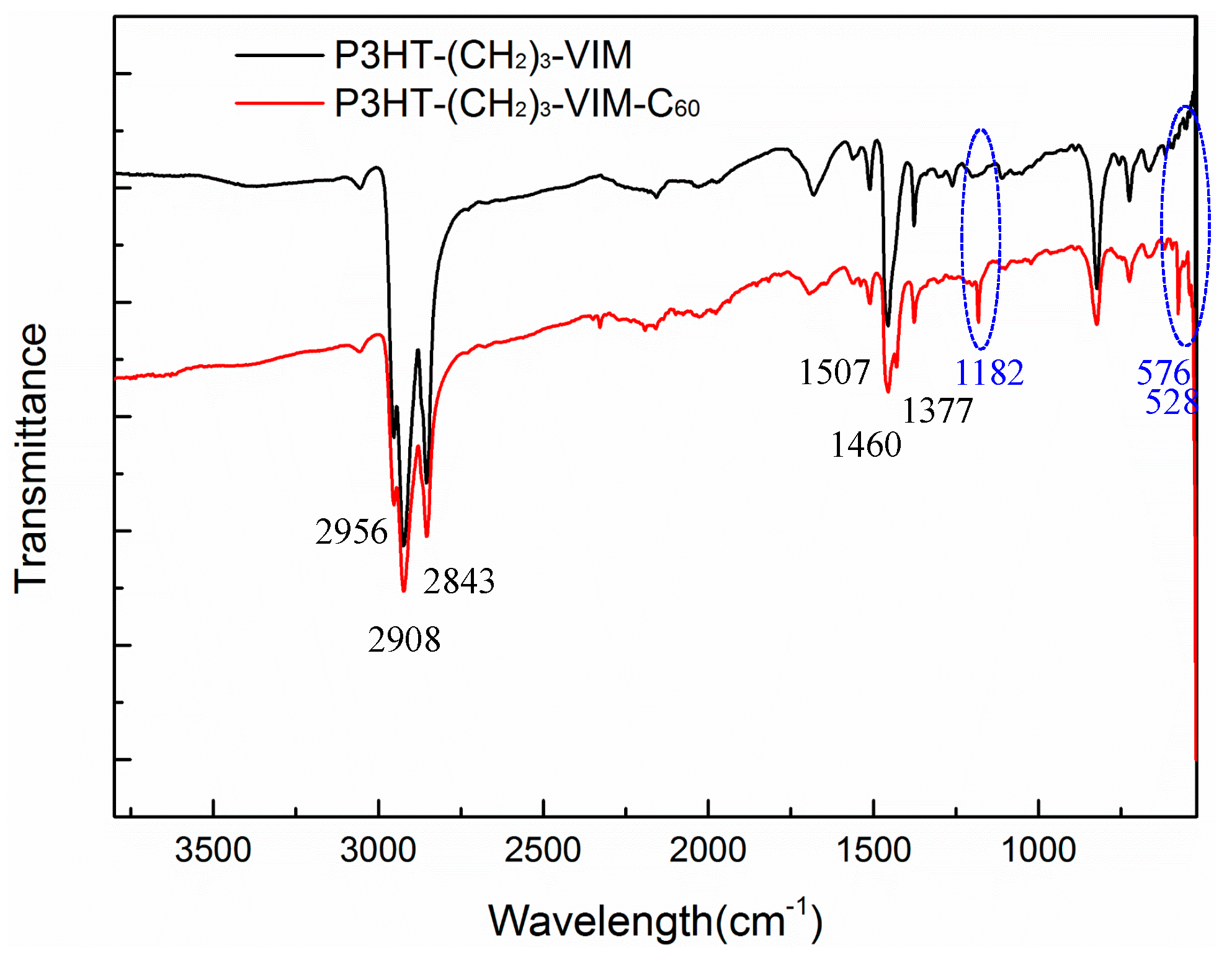
2.3. Synthesis of Linked Donor Polymer-C60 Adduct Dyads
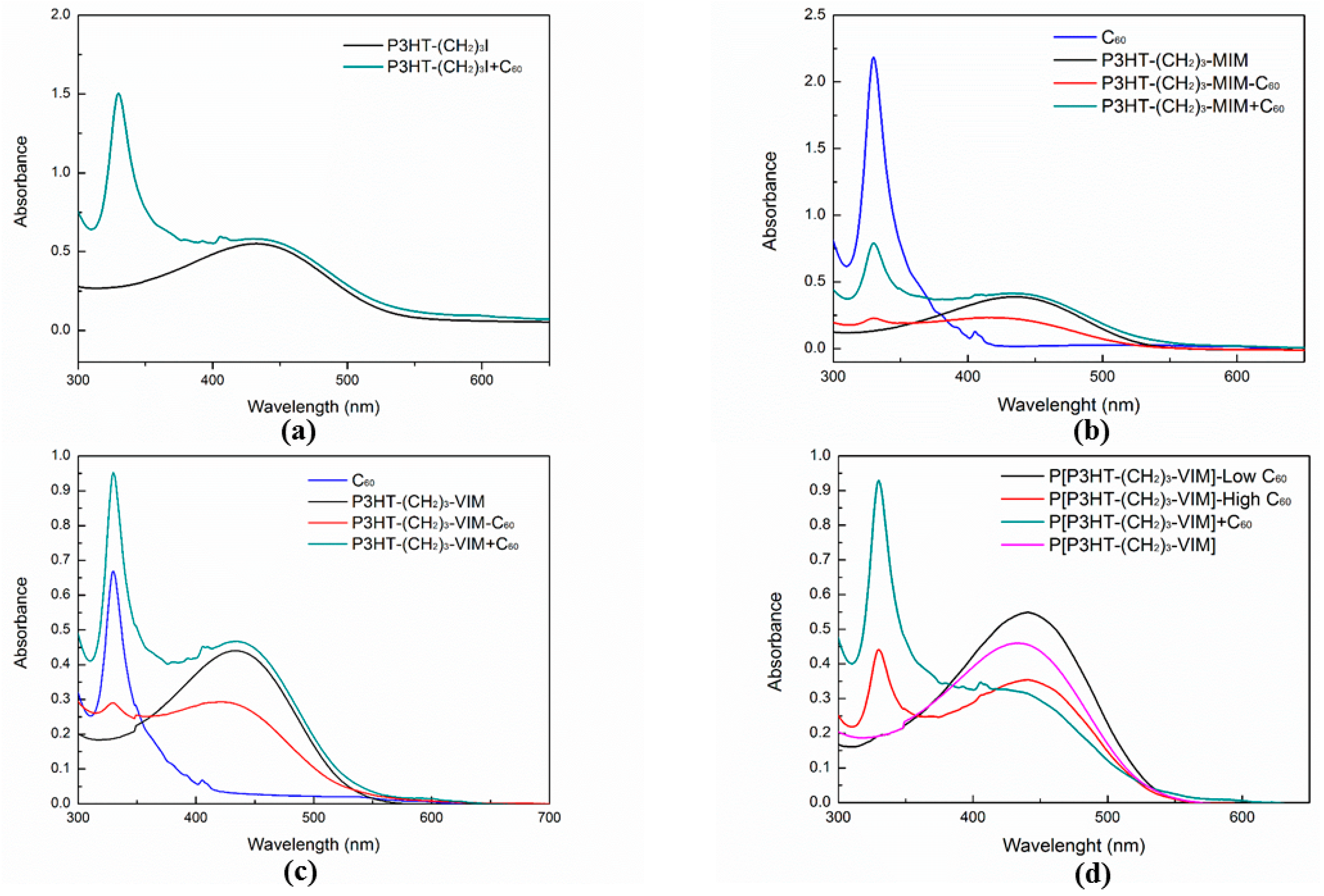
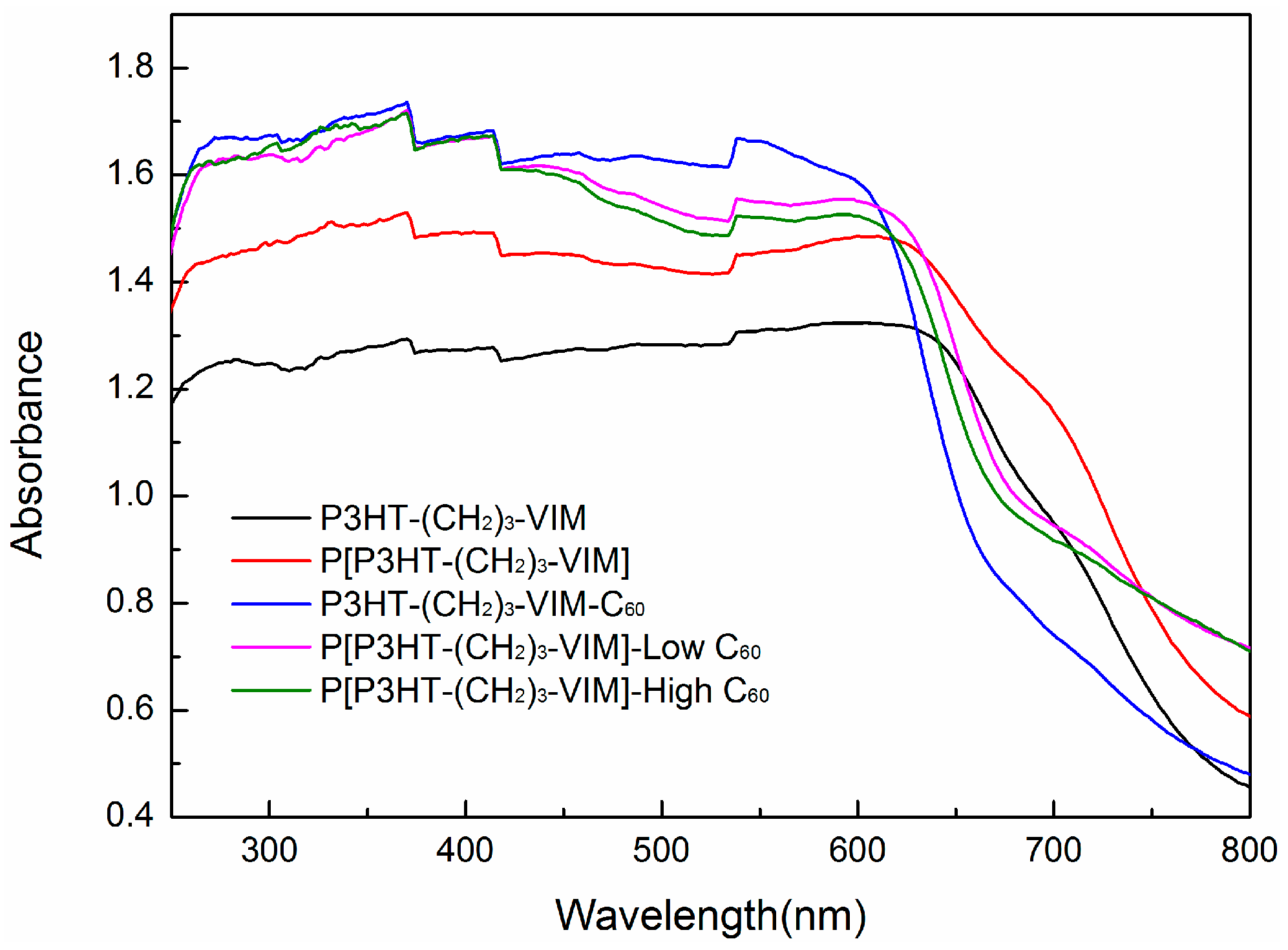
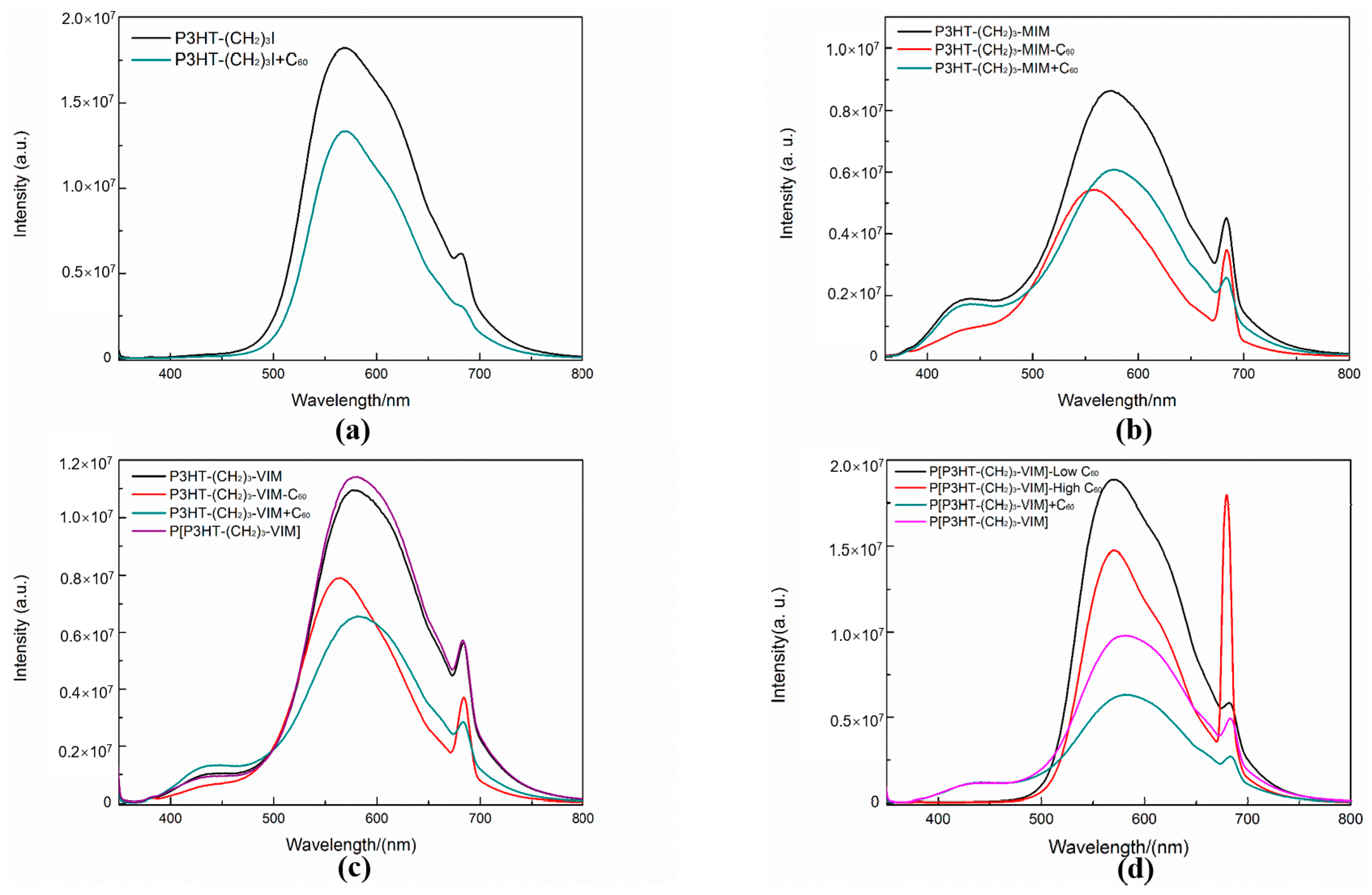
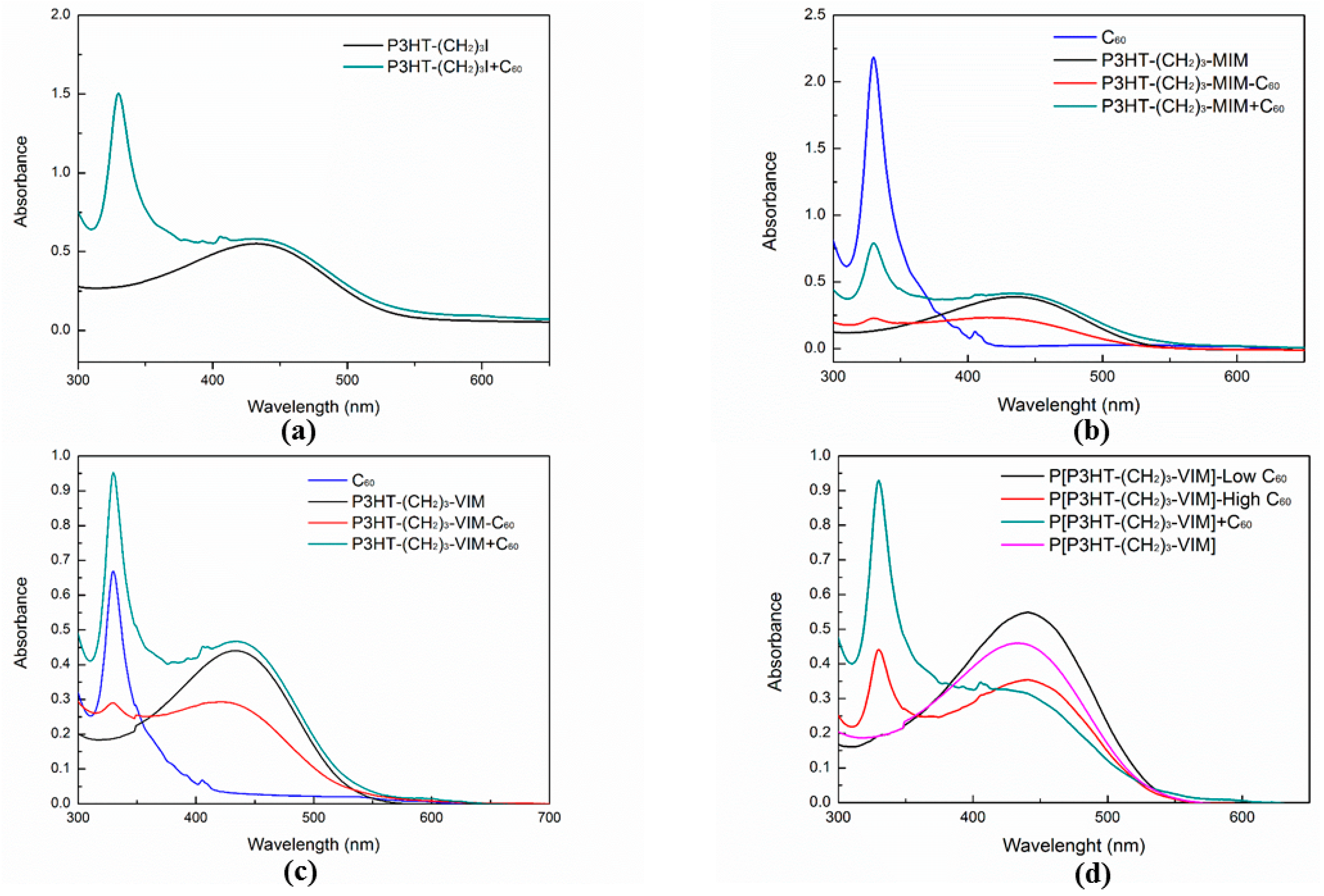
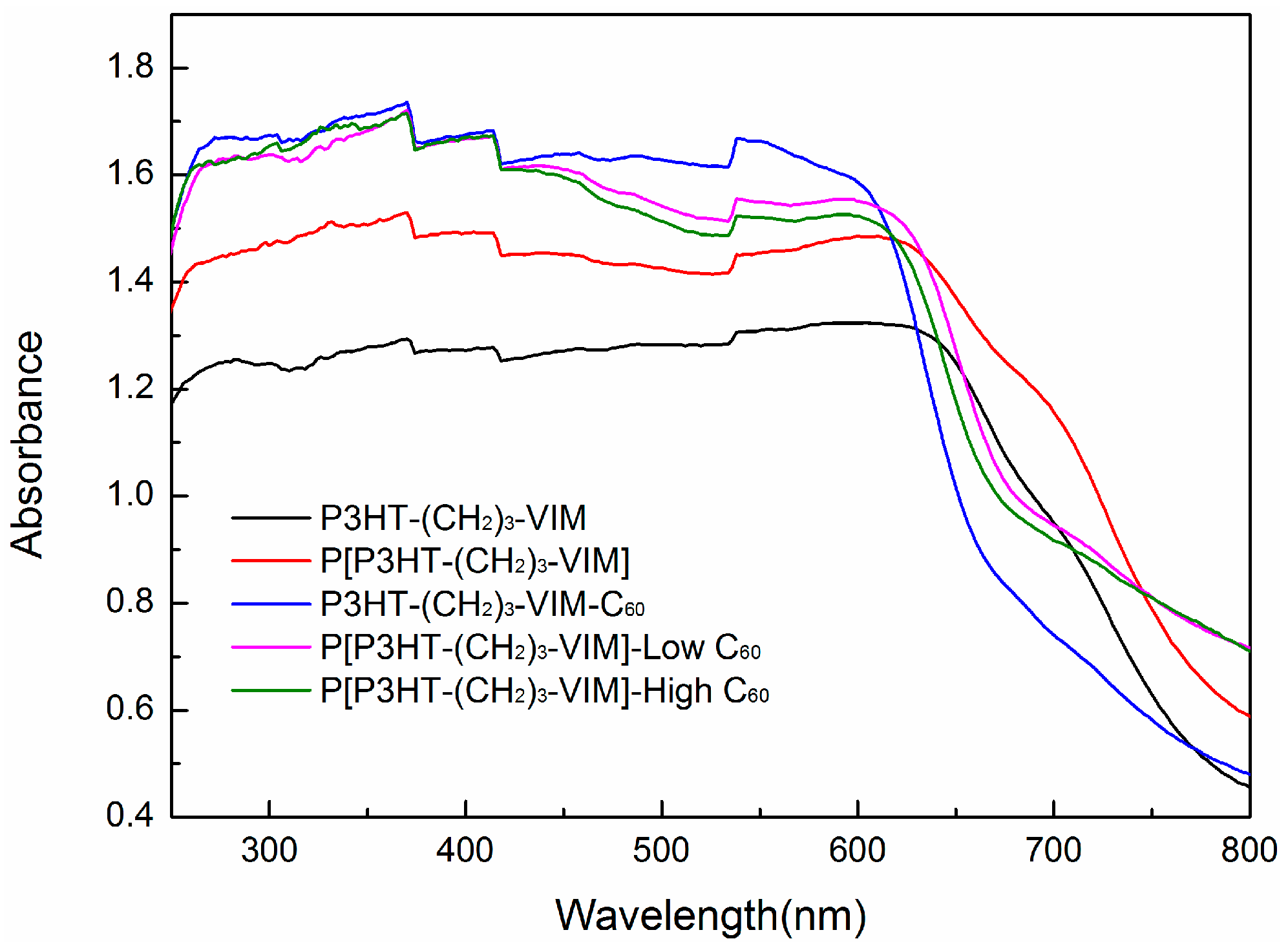
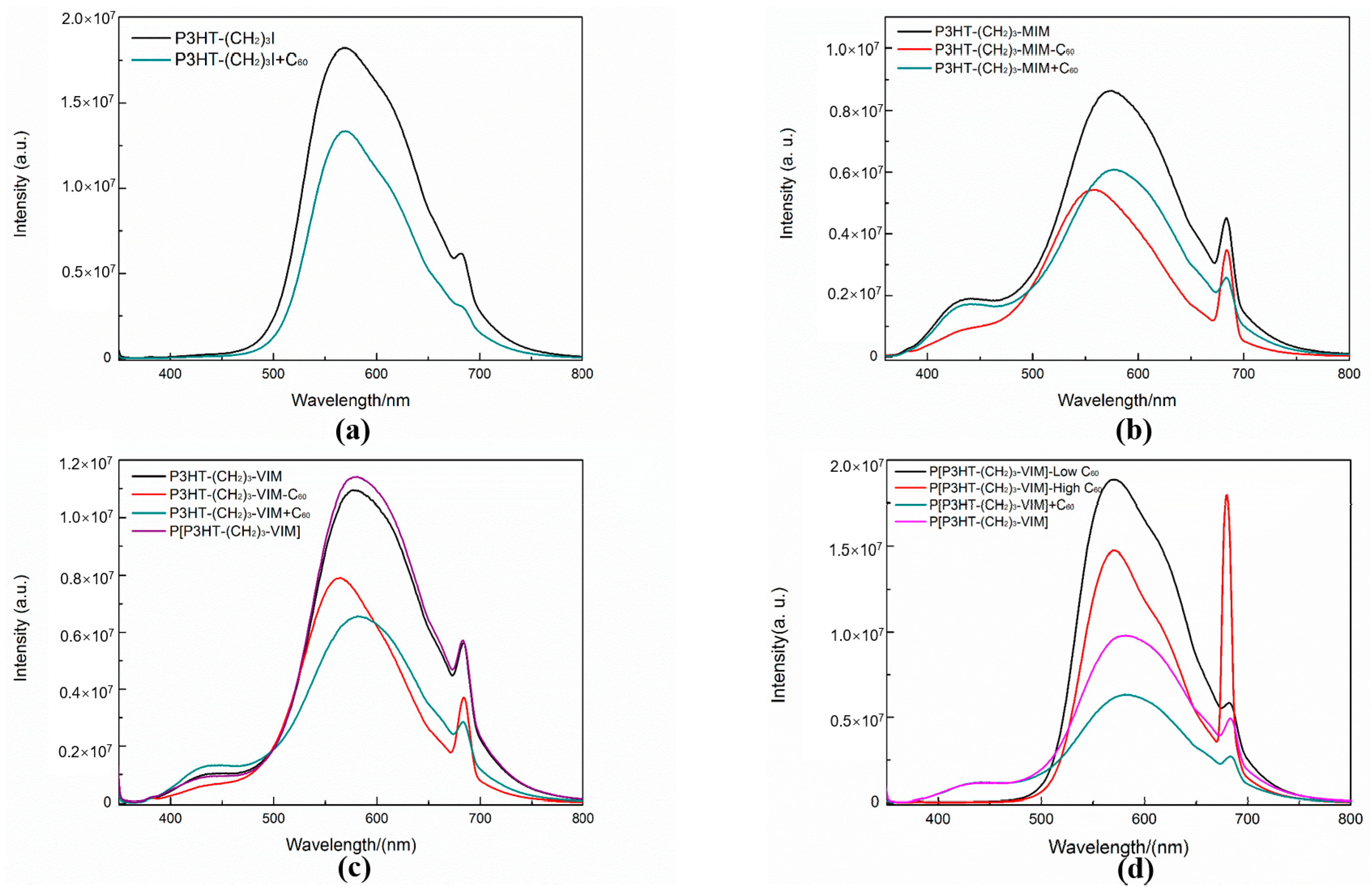
2.4. Electronic Absorption and Emission Properties
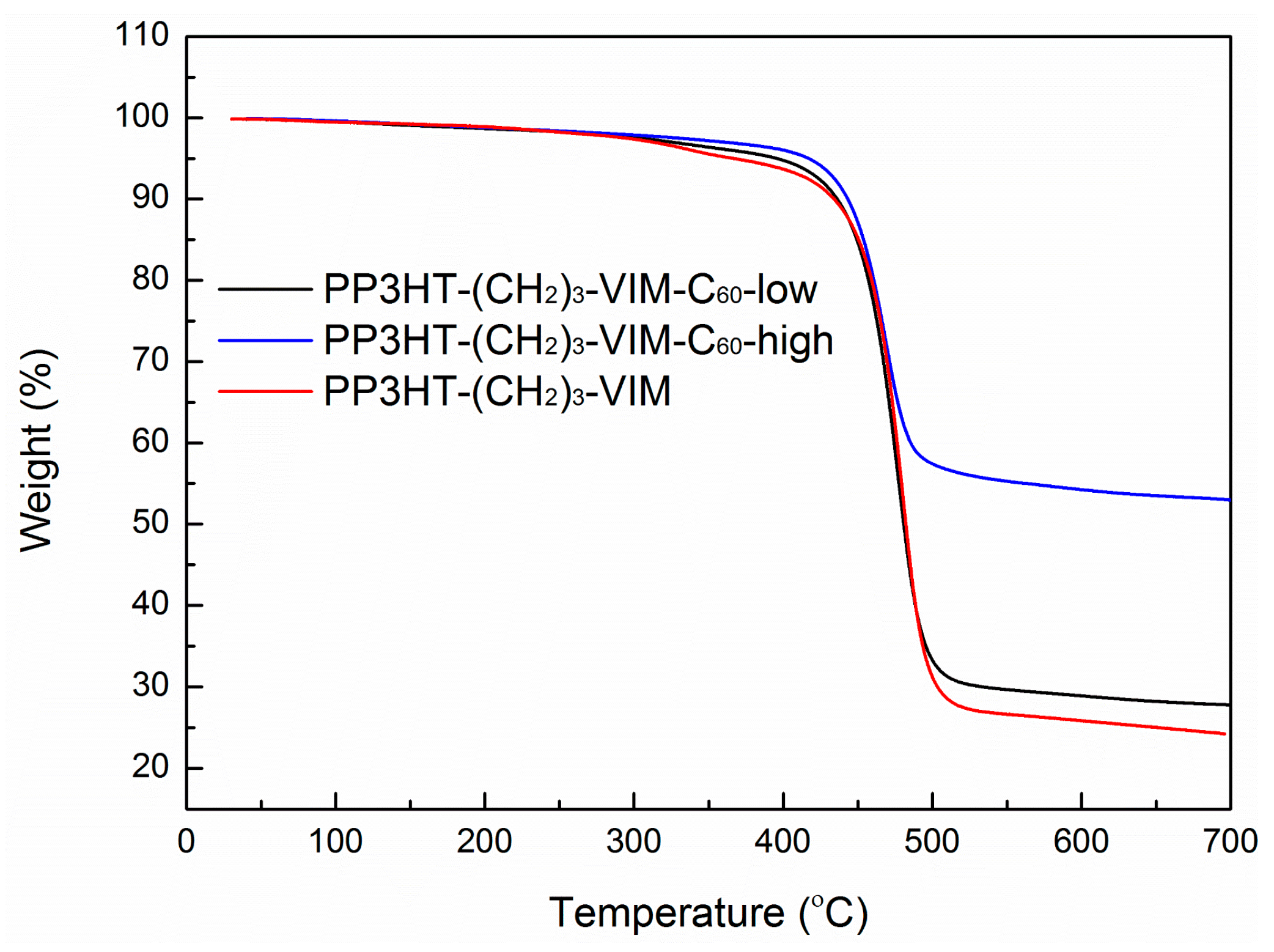
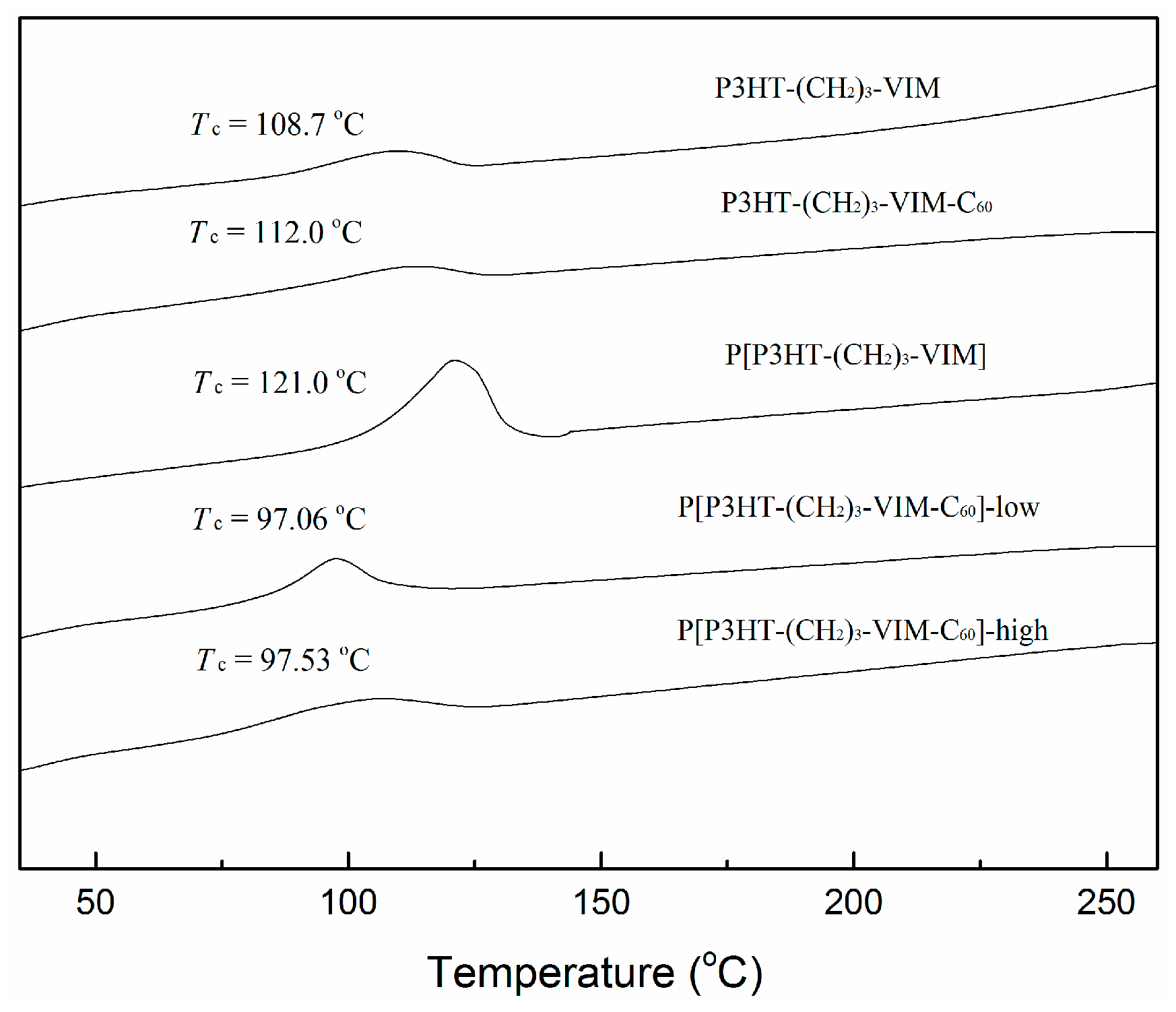
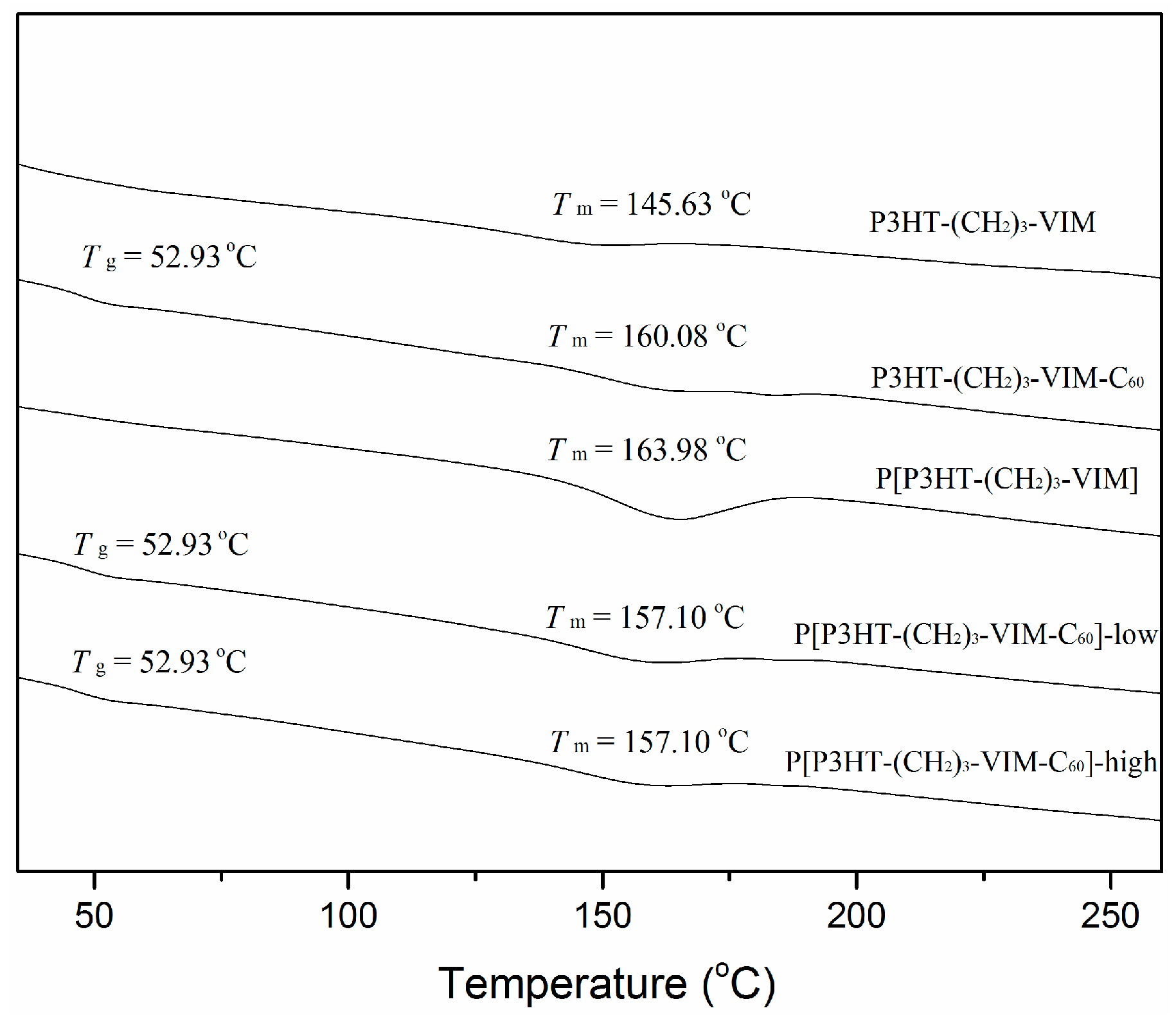
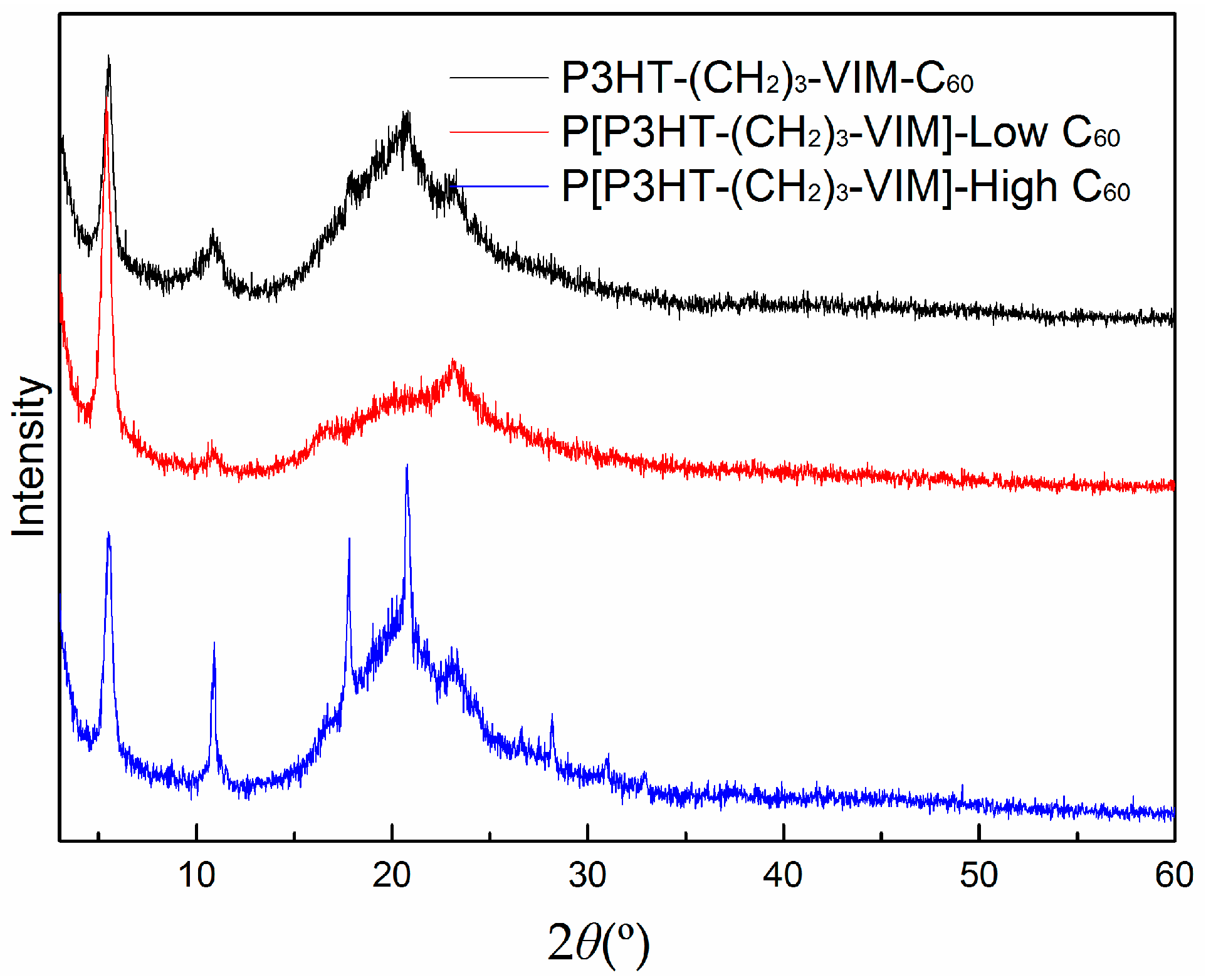
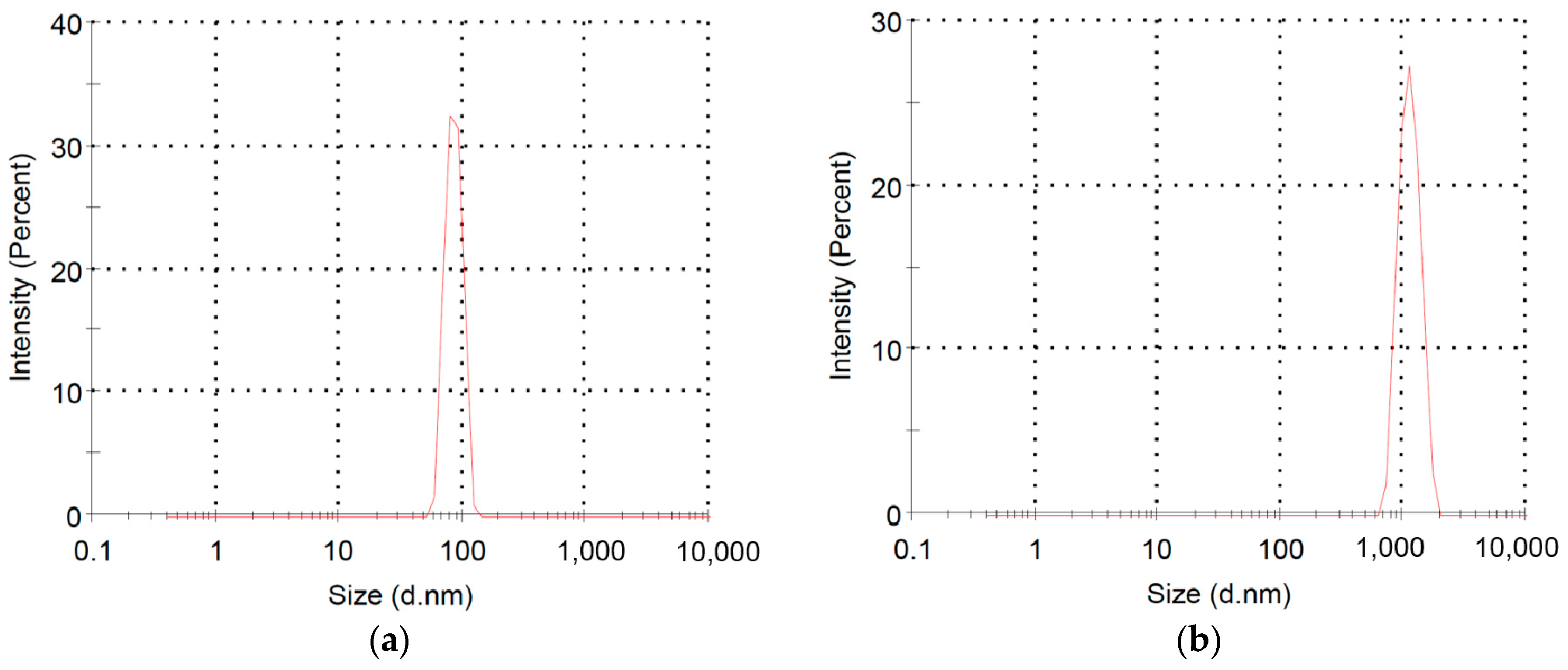
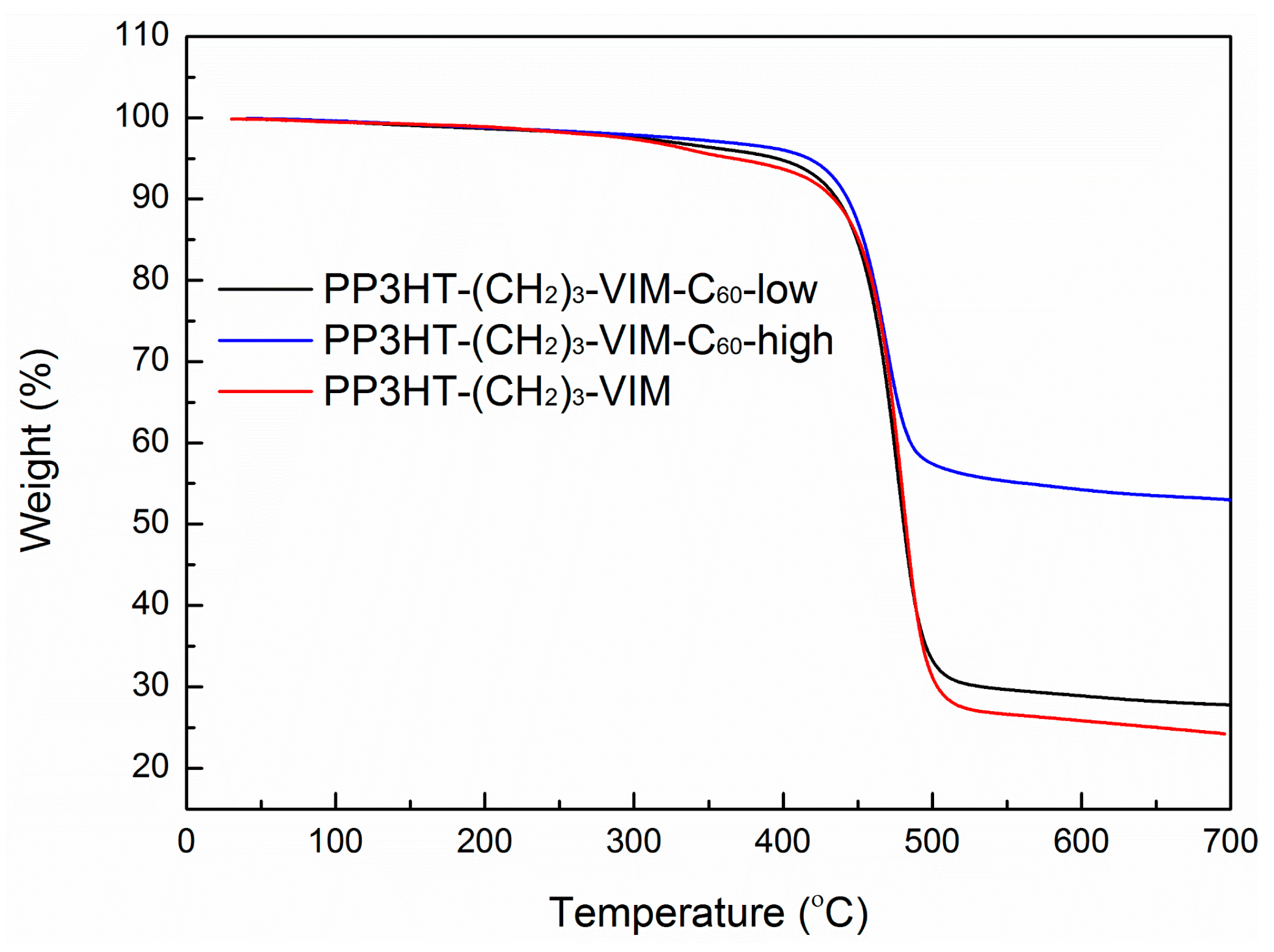
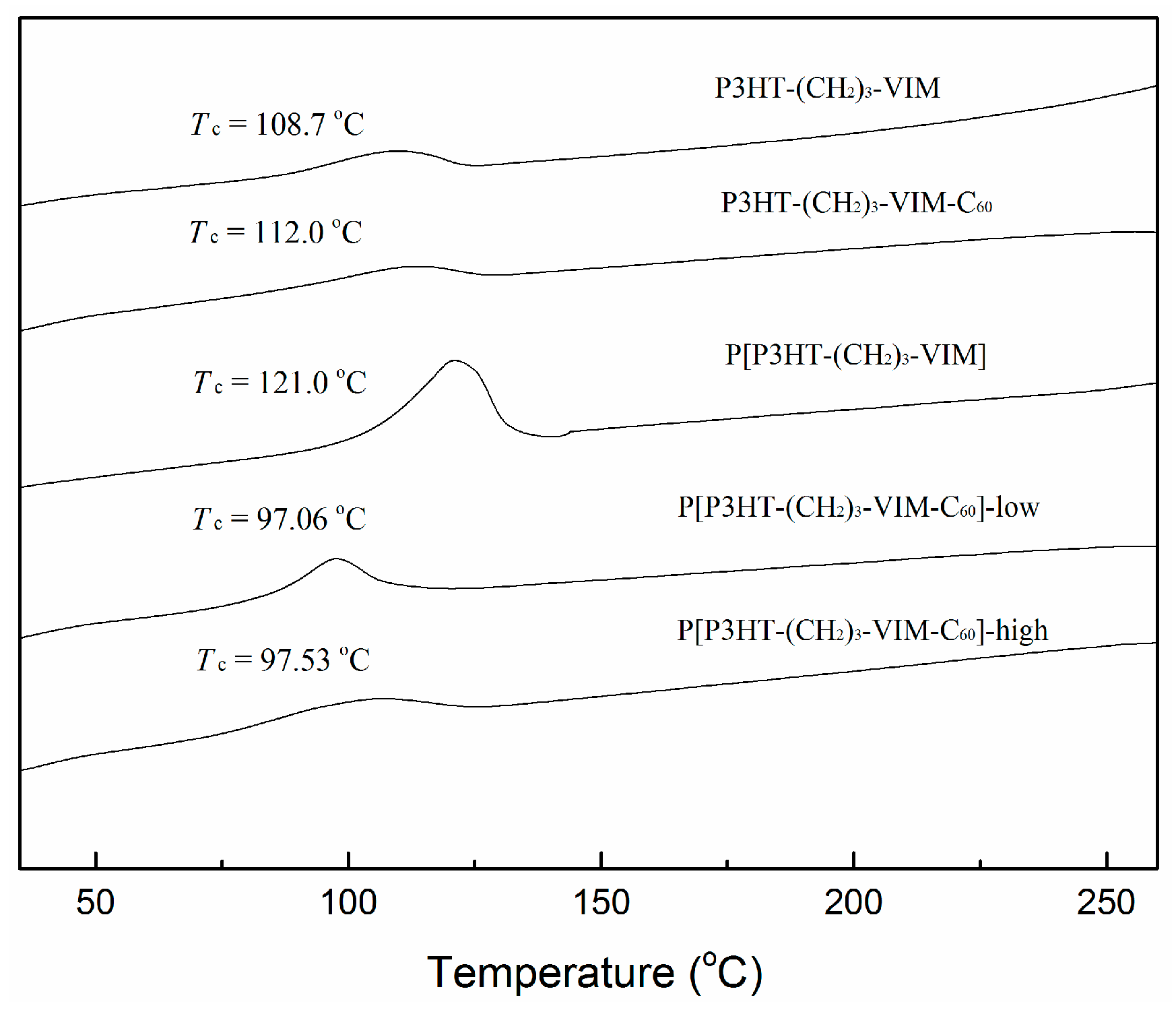
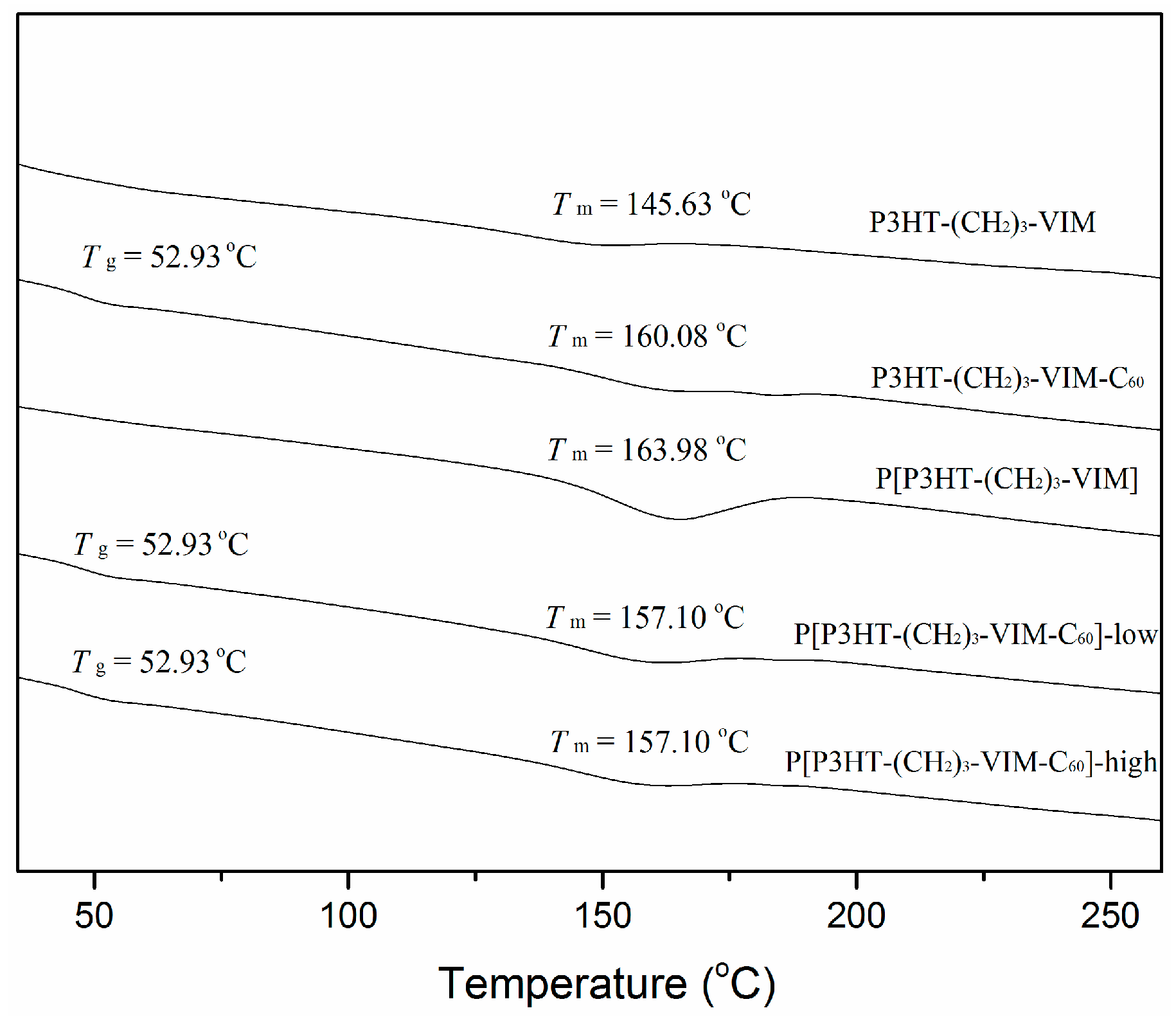
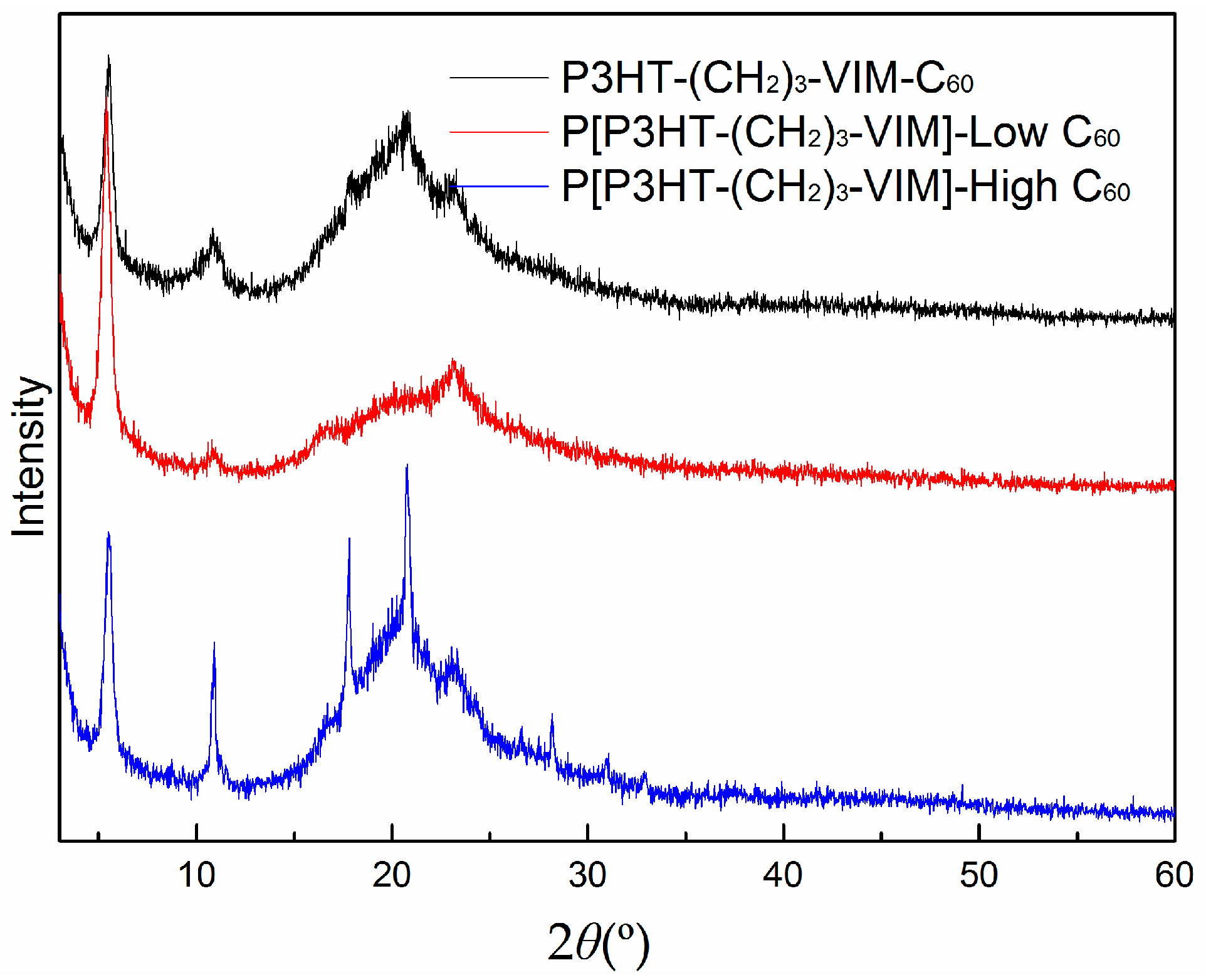
2.5. Thermal Behaviors of Macromer, Brush Polymer, and Their C60 Adducts
3. Experimental Section
3.1. Materials, Reagents, and Methods
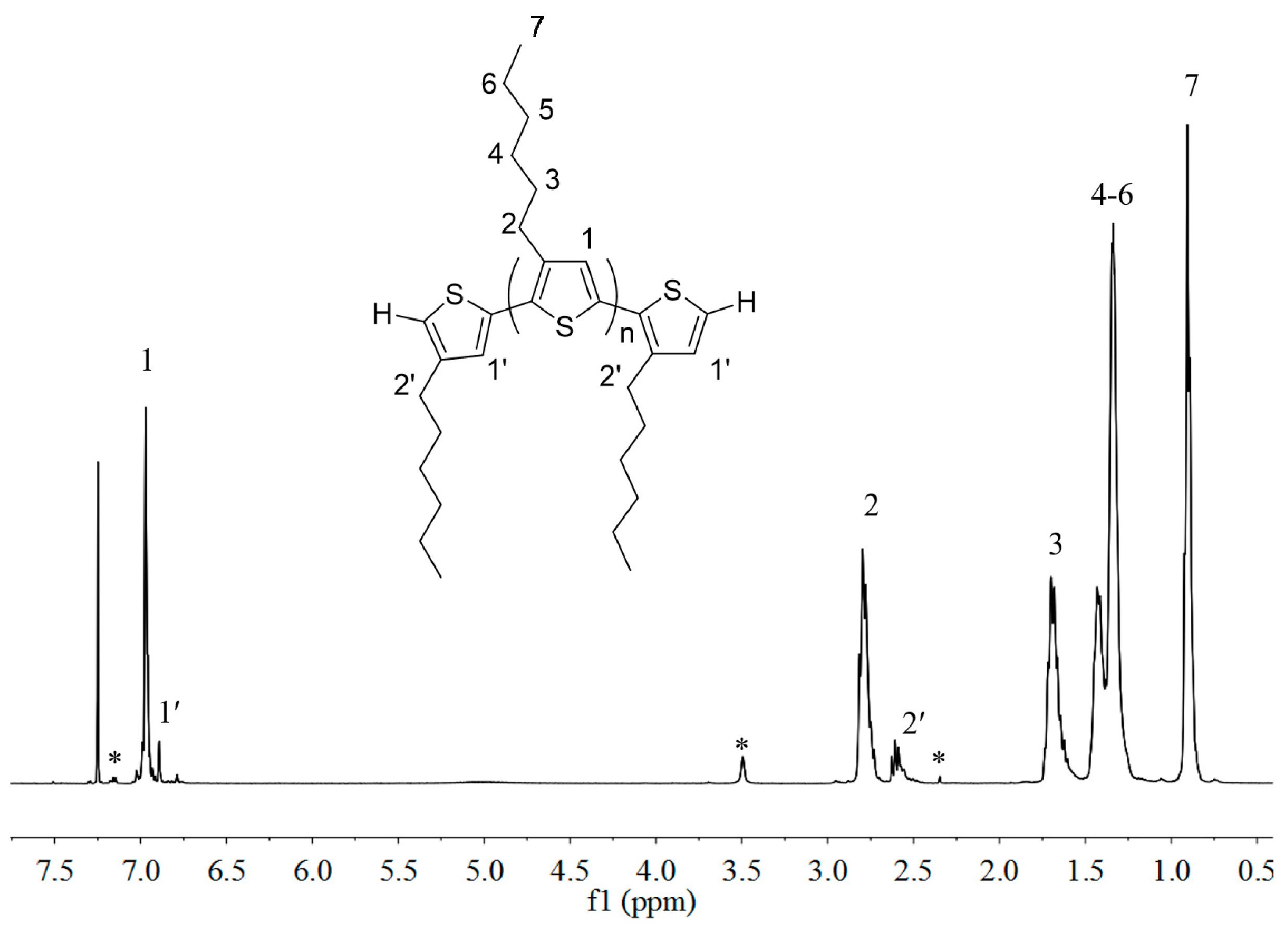
3.2. Preparation of P3HT with H/H Chain-Ends, H-P3HT-H
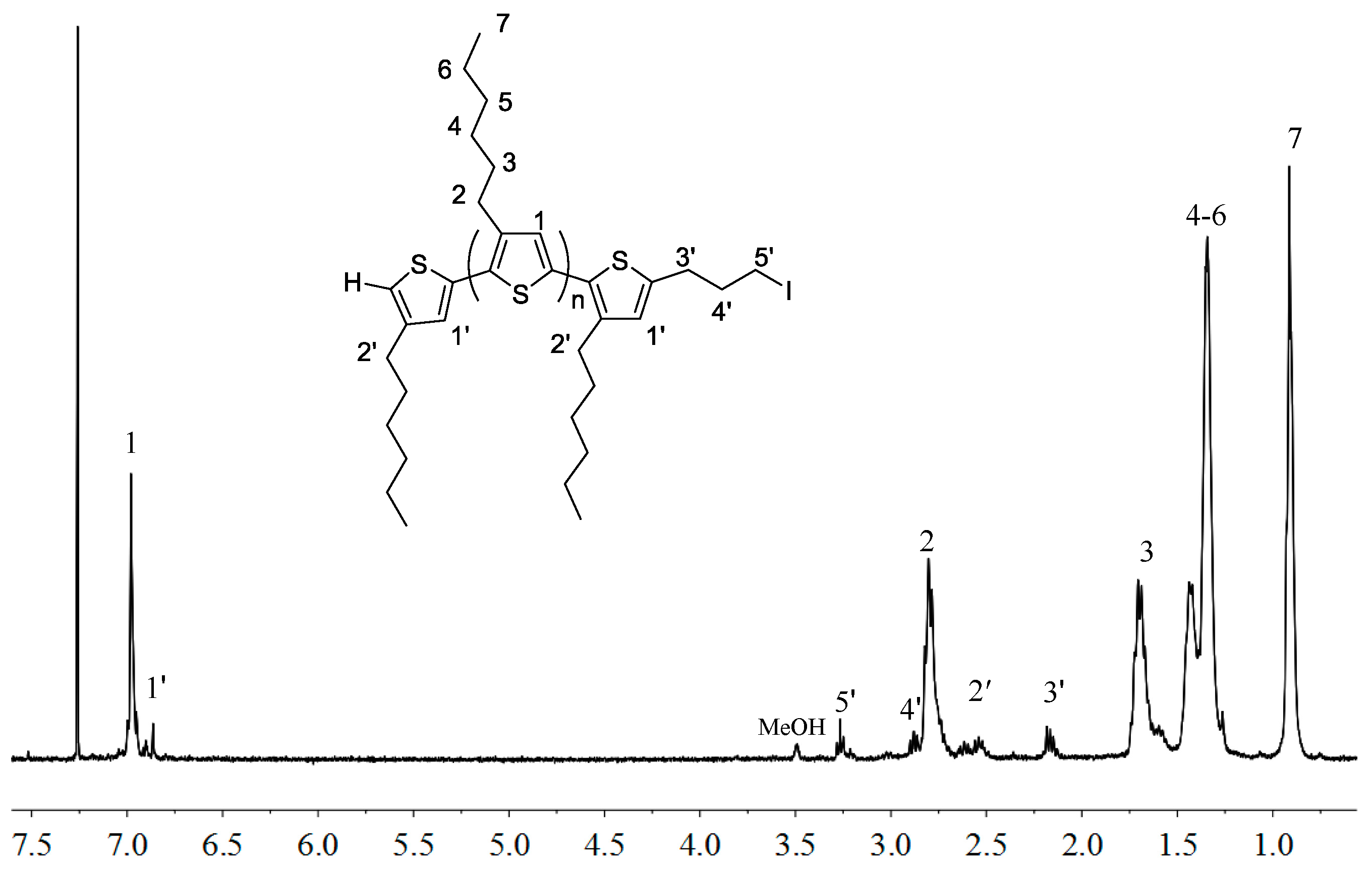
3.3. Synthesis of P3HT-(CH2)3I
3.4. Synthesis of P3HT-(CH2)3-MIM
3.5. Synthesis of P3HT-(CH2)3-VIM
3.6. Polymerization of P3HT-(CH2)3-VIM
3.7. Synthesis of Polymer-C60 Adducts: P3HT-(CH2)3-MIM-C60, P3HT-(CH2)3-VIM-C60 and P[P3HT-(CH2)3-VIM-C60]
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCullough, R.D. The chemistry of conducting polythiophenes. Adv. Mater. 1998, 10, 93–116. [Google Scholar] [CrossRef]
- Ludwigs, S. P3HT Revisited—From molecular scale to solar cell devices. In Advances in Polymer Science Series; Springer: Berlin/Heidelberg, Germany, 2014; Volume 265, ISBN 978-3-662-45145-8. [Google Scholar]
- Boudouris, B.W.; Frisbie, C.D.; Hillmyer, M.A. Nanoporous poly(3-alkylthiophene) thin films generated from block copolymer templates. Macromolecules 2008, 41, 67–75. [Google Scholar] [CrossRef]
- Botiz, I.; Darling, S.B. Self-assembly of poly(3-hexylthiophene)-block-polylactide block copolymer and subsequent incorporation of electron acceptor material. Macromolecules 2009, 42, 8211–8217. [Google Scholar] [CrossRef]
- Sivanandan, K.; Chatterjee, T.; Treat, N.; Kramer, E.J.; Hawker, C.J. High surface area poly(3-hexylthiophenes) thin films from cleavable graft copolymers. Macromolecules 2010, 43, 233–241. [Google Scholar] [CrossRef]
- Iovu, M.C.; Craley, C.R.; Jeffries-El, M.; Krankowski, A.B.; Zhang, R.; McCullough, R.D. Conducting regioregular polythiophene block copolymer nanofibrils synthesized by reversible addition fragmentation chain transfer polymerization (RAFT) and nitroxide mediated polymerization (NMP). Macromolecules 2007, 40, 4733–4735. [Google Scholar] [CrossRef]
- Iovu, M.C.; Jeffries-El, M.; Sheina, E.E.; Cooper, J.R.; McCullough, R.D. Regioregular poly(3-alkylthiophene) conducting block copolymers. Polymer 2005, 46, 8582–8586. [Google Scholar] [CrossRef]
- Higashihara, T.; Ohshimizu, K.; Hirao, A.; Ueda, M. Facile synthesis of ABA triblock copolymer containing regioregular poly(3-hexylthiophene) and polystyrene segments via linking reaction of poly(styryl)lithium. Macromolecules 2008, 41, 9505–9507. [Google Scholar] [CrossRef]
- Higashihara, T.; Ueda, M. Living anionic polymerization of 4-vinyltriphenylamine for synthesis of novel block copolymers containing low-polydisperse poly(4-vinyltriphenylamine) and regioregular poly(3-hexylthiophene) segments. Macromolecules 2009, 42, 8794–8800. [Google Scholar] [CrossRef]
- Higashihara, T.; Ueda, M. Synthesis and characterization of a novel coil–rod–coil triblock copolymers comprised of regioregular poly(3-hexylthiophene) and poly(methyl methacrylate) segments. React. Funct. Polym. 2009, 69, 457–462. [Google Scholar] [CrossRef]
- Dai, C.A.; Yen, W.C.; Ho, C.C.; Su, W.F. Facile synthesis of well-defined block copolymers containing regioregular poly(3-hexyl thiophene) via anionic macroinitiation method and their self-assembly behavior. J. Am. Chem. Soc. 2007, 129, 11036–11038. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Rho, Y.; Higashihara, T.; Ahn, B.; Ree, M.; Ueda, M. Preparation of nanoporous poly(3-hexylthiophene) films based on a template system of block copolymers via ionic interaction. Macromolecules 2010, 43, 4843–4852. [Google Scholar] [CrossRef]
- Ji, E.; Pellerin, V.; Rubatat, L.; Grelet, E.; Bousquet, A.; Billon, L. Self-assembly of ionizable “clicked” P3HT-b-PMMA copolymers: Ionic bonding group/counterion effects on morphology. Macromolecules 2017, 50, 235–243. [Google Scholar] [CrossRef]
- Wang, Y.; Bailey, T.S.; Hong, M.; Chen, E.Y.-X. Stereoregular brush polymers and graft copolymers by chiral zirconocene-mediated coordination polymerization of P3HT macromers. Polymers 2017, 9, 139. [Google Scholar] [CrossRef]
- Alonzo, J.; Kochemba, M.; Pickel, D.L.; Ramanathan, M.; Sun, Z.; Li, D.; Chen, J.; Sumpter, B.D.; Heller, W.T.; Kilbey, S.M. Assembly and organization of poly(3-hexylthiophene) brushes and their potential use as novel anode buffer layers for organic photovoltaics. Nanoscale 2013, 5, 9357–9364. [Google Scholar] [CrossRef] [PubMed]
- Paoprasert, P.; Spalenka, J.W.; Peterson, D.L.; Ruther, R.E.; Hamers, R.J.; Evansa, P.G.; Gopalan, P. Grafting of poly(3-hexylthiophene) brushes on oxides using click chemistry. J. Mater. Chem. 2010, 20, 2651–2658. [Google Scholar] [CrossRef]
- Meng, D.; Sun, J.; Jiang, S.; Zeng, Y.; Li, Y.; Yan, S.; Geng, J.; Huang, Y. Grafting P3HT brushes on GO sheets: Distinctive properties of the GO/P3HT composites due to different grafting approaches. J. Mater. Chem. 2012, 22, 21583–21591. [Google Scholar] [CrossRef]
- Pang, X.; Zhan, L.; Feng, C.; Wu, R.; Ma, H.; Lin, Z. Functional copolymer brushes composed of a hydrophobic backbone and densely grafted conjugated side chains via a combination of living polymerization with click chemistry. Polym. Chem. 2013, 4, 2025–2032. [Google Scholar] [CrossRef]
- Heinrich, C.D.; Thelakkat, M. Poly-(3-hexylthiophene) bottlebrush copolymers with tailored side-chain lengths and high charge carrier mobilities. J. Mater. Chem. C 2016, 4, 5370–5378. [Google Scholar] [CrossRef]
- Khanduyeva, N.; Senkovskyy, V.; Beryozkina, T.; Bocharova, V.; Simon, F.; Nitschke, M.; Stamm, M.; Grötzschel, R.; Kiriy, A. Grafting of poly(3-hexylthiophene) from poly(4-bromostyrene) films by Kumada catalyst-transfer polycondensation: Revealing of the composite films structure. Macromolecules 2008, 41, 7383–7389. [Google Scholar] [CrossRef]
- Khanduyeva, N.; Senkovskyy, V.; Beryozkina, T.; Horecha, M.; Stamm, M.; Uhrich, C.; Riede, M.; Leo, K.; Kiriy, A. Surface engineering using Kumada catalyst-transfer polycondensation (KCTP): Preparation and structuring of poly(3-hexylthiophene)-based graft copolymer brushes. J. Am. Chem. Soc. 2009, 131, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-K.; Pickel, D.L.; Kochemba, W.M.; Chen, J.; Uhrig, D.; Hinestrosa, J.P.; Carrillo, J.-M.; Shao, M.; Do, C.; Messman, J.M.; et al. Poly(3-hexylthiophene) molecular bottlebrushes via ring-opening metathesis polymerization: Macromolecular architecture enhanced aggregation. ACS Macro Lett. 2013, 2, 761–765. [Google Scholar] [CrossRef]
- As, D.V.; Subbiah, J.; Jones, D.J.; Wong, W.W.H. Controlled synthesis of well-defined semiconducting brush polymers. Macromol. Chem. Phys. 2016, 217, 403–413. [Google Scholar]
- Wan, M.; Wu, W.; Sang, G.; Zou, Y.; Liu, Y.; Li, Y. Poly(thienylene-vinylene-thienylene) with cyano substituent: Synthesis and application in field-effect transistor and polymer solar cell. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 4028–4036. [Google Scholar] [CrossRef]
- Zou, Y.; Wu, W.; Sang, G.; Yang, Y.; Liu, Y.; Li, Y. Polythiophene derivative with phenothiazine—Vinylene conjugated side chain: Synthesis and its application in field-effect transistors. Macromolecules 2007, 40, 7231–7237. [Google Scholar] [CrossRef]
- Zou, Y.; Sang, G.; Wu, W.; Liu, Y.; Li, Y. A polythiophene derivative with octyloxyl triphenylamine-vinylene conjugated side chain: Synthesis and its applications in field-effect transistor and polymer solar cell. Synth. Met. 2009, 159, 182–187. [Google Scholar] [CrossRef]
- Clarke, T.M.; Ballantyne, A.M.; Nelson, J.; Bradley, D.D.C.; Durrant, J.R. Free energy control of charge photogeneration in polythiophene/fullerene solar cells: The influence of thermal annealing on P3HT/PCBM blends. Adv. Funct. Mater. 2008, 18, 4029–4035. [Google Scholar] [CrossRef]
- Sharma, G.D.; Suresh, P.; Sharma, S.S.; Vijay, Y.K.; Mikroyannidis, J.A. Effect of solvent and subsequent thermal annealing on the performance of phenylenevinylene copolymer: PCBM solar cells. ACS Appl. Mater. Interfaces 2010, 2, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yao, Y.; Yang, H.; Shrotriya, V.; Yang, G.; Yang, Y. “Solvent annealing” effect in polymer solar cells based on poly(3-hexylthiophene) and methanofullerenes. Adv. Funct. Mater. 2007, 17, 1636–1644. [Google Scholar] [CrossRef]
- Peet, J.; Kim, J.Y.; Coates, N.E.; Ma, W.L.; Moses, D.; Heeger, A.J.; Bazan, G.C. Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat. Mater. 2007, 6, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Frisbie, C.D. Correlation of phase behavior and charge transport in conjugated polymer/fullerene blends. J. Phys. Chem. C 2008, 112, 17726–17736. [Google Scholar] [CrossRef]
- Yamashiro, T.; Aso, Y.; Otsubo, T.; Tang, H.; Harima, Y.; Yamashita, K. Intramolecular energy transfer of [60]fullerene-linked oligothiophenes. Chem. Lett. 1999, 443–444. [Google Scholar] [CrossRef]
- Saravanan, C.; Liu, C.-L.; Chang, Y.-M.; Lu, J.-D.; Hsieh, Y.-J.; Rwei, S.-P.; Wang, L. [60]Fulleropyrrolidines bearing π-conjugated moiety for polymer solar cells: Contribution of the chromophoric substituent on C60 to the photocurrent. Appl. Mater. Interfaces 2012, 4, 6133–6141. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, M.; Salatelli, E.; Benelli, T.; Caretti, D.; Giorgini, L.; Di-Nicola, F.P. A regioregular polythiophene–fullerene for polymeric solar cells. J. Appl. Polym. Sci. 2015, 132, 42121/1–42121/10. [Google Scholar] [CrossRef]
- Nisic, F.; Colombo, A.; Dragonetti, C.; Cominetti, A.; Pellegrino, A.; Perin, N.; Po, R.; Tacca, A. Novel terthiophene-substituted fullerene derivatives as easily accessible acceptor molecules for bulk-heterojunction polymer solar cells. Int. J. Photoenergy 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Murata, Y.; Suzuki, M.; Komatsu, K. Synthesis and electropolymerization of fullerene-terthiophene dyads. Org. Biomol. Chem. 2003, 1, 2624–2625. [Google Scholar] [CrossRef] [PubMed]
- Sivula, K.; Ball, Z.T.; Watanabe, N.; Fréchet, J.M.J. Amphiphilic diblock copolymer compatibilizers and their effect on the morphology and performance of polythiophene: Fullerene solar cells. Adv. Mater. 2006, 18, 206–210. [Google Scholar] [CrossRef]
- Sommer, M.; Huettner, S.; Thelakkat, M. Donor-acceptor block copolymers for photovoltaic applications. J. Mater. Chem. 2010, 20, 10788–10797. [Google Scholar] [CrossRef]
- Li, H.; Risko, C.; Seo, J.H.; Campbell, C.; Wu, G.; Brédas, J.-L.; Bazan, G.C. Fullerene-carbene lewis acid-base adducts. J. Am. Chem. Soc. 2011, 133, 12410–12413. [Google Scholar] [CrossRef] [PubMed]
- Lorbach, A.; Maverick, E.; Carreras, A.; Alemany, P.; Wu, G.; Garcia-Garibay, M.A.; Bazan, G.C. A fullerene-carbene adduct as a crystalline molecular rotor: Remarkable behavior of a spherically-shaped rotator. Phys. Chem. Chem. Phys. 2014, 16, 12980–12986. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Chen, E.Y.-X. Polymeric carbon Lewis base–acid adducts: Poly(NHC-C60). Polym. Chem. 2015, 6, 1741–1750. [Google Scholar] [CrossRef]
- Iovu, M.C.; Sheina, E.E.; McCullough, R.D. Experimental evidence for the quasi-“living” nature of the Grignard metathesis method for the synthesis of regioregular poly(3-alkylthiophenes). Macromolecules 2005, 38, 8649–8656. [Google Scholar] [CrossRef]
- Blicke, F.F.; Burckhalter, J.H. α-Thienylaminoalkanes. J. Am. Chem. Soc. 1942, 64, 477–480. [Google Scholar] [CrossRef]
- Varma, R.S.; Namboodiri, V.V. An expeditious solvent-free route to ionic liquids using microwaves. Chem. Commun. 2001, 643–644. [Google Scholar] [CrossRef]
- Araki, Y.; Luo, H.; Nakamura, T.; Fujitsuka, M.; Ito, O.; Kanato, H.; Aso, Y.; Otsubo, T. Photoinduced charge separation and charge recombination of oligothiophene-viologen dyads in polar solvent. J. Phys. Chem. A 2004, 108, 10649–10655. [Google Scholar] [CrossRef]
- Chen, S.; Wang, T.; Chang, P.; Yang, C.; Lee, Y. Poly(ionic liquid) prepared by photopolymerization of ionic liquid monomers as quasi-solid-state electrolytes for dye-sensitized solar cells. React. Funct. Polym. 2016, 108, 103–112. [Google Scholar] [CrossRef]
- Czichy, M.; Wagner, P.; Łapkowski, M.; Officer, D.L. Effect of π-conjugation on electrochemical properties of poly(terthiophene)s 3′-substituted with fullerene C60. J. Electroanal. Chem. 2016, 772, 103–109. [Google Scholar] [CrossRef]
- Khodakarimi, S.; Hekmatshoar, M.H.; Abbasi, F. X-ray reflectivity and topography of the solvent-treated P3HT: PCBM thin films. J. Mater. Sci. Mater. Electron 2016, 27, 182–190. [Google Scholar] [CrossRef]
- Lee, J.U.; Jung, J.W.; Emrick, T.; Russell, T.P.; Jo, W.H. Morphology control of a polythiophene-fullerene bulk heterojunction for enhancement of the high-temperature stability of solar cell performance by a new donor-acceptor diblock copolymer. Nanotechnology 2010, 21, 105201. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Shin, M.; Kim, H.; Kim, Y. Temperature/time-dependent crystallization of polythiophene: Fullerene bulk heterojunction films for polymer solar cells. Nanoscale 2010, 2, 2384–2389. [Google Scholar] [CrossRef] [PubMed]
- Karagiannidis, P.G.; Kassavetis, S.; Pitsalidis, C.; Logothetidis, S. Thermal annealing effect on the nanomechanical properties and structure of P3HT: PCBM thin films. Thin Solid Films 2011, 519, 4105–4109. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Hong, M.; Bailey, T.S.; Chen, E.Y.-X. Brush Polymer of Donor-Accepter Dyads via Adduct Formation between Lewis Base Polymer Donor and All Carbon Lewis Acid Acceptor. Molecules 2017, 22, 1564. https://doi.org/10.3390/molecules22091564
Wang Y, Hong M, Bailey TS, Chen EY-X. Brush Polymer of Donor-Accepter Dyads via Adduct Formation between Lewis Base Polymer Donor and All Carbon Lewis Acid Acceptor. Molecules. 2017; 22(9):1564. https://doi.org/10.3390/molecules22091564
Chicago/Turabian StyleWang, Yang, Miao Hong, Travis S. Bailey, and Eugene Y.-X. Chen. 2017. "Brush Polymer of Donor-Accepter Dyads via Adduct Formation between Lewis Base Polymer Donor and All Carbon Lewis Acid Acceptor" Molecules 22, no. 9: 1564. https://doi.org/10.3390/molecules22091564