A New Erythrinan Alkaloid Glycoside from the Seeds of Erythrina crista-galli


1
Key Laboratory of Bio-Pesticide and Chemistry-Biology, Ministry of Education, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China
2
Key Laboratory of Plant Virology of Fujian Province, Institute of Plant Virology, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China
*
Authors to whom correspondence should be addressed.
Molecules 2017, 22(9), 1558; https://doi.org/10.3390/molecules22091558
Submission received: 3 August 2017
/
Revised: 7 September 2017
/
Accepted: 13 September 2017
/
Published: 16 September 2017
(This article belongs to the Section Natural Products Chemistry)
Abstract
:A new Erythrina alkaloid glycoside, named erythraline-11β-O-glucopyranoside, was isolated from the seeds of Erythrina crista-galli L., together with five known Erythrina alkaloids and an indole alkaloid. The structure of the new alkaloid glycoside was elucidated by spectroscopic methods, and all of the compounds were evaluated for their antiviral activity against tobacco mosaic virus.
1. Introduction
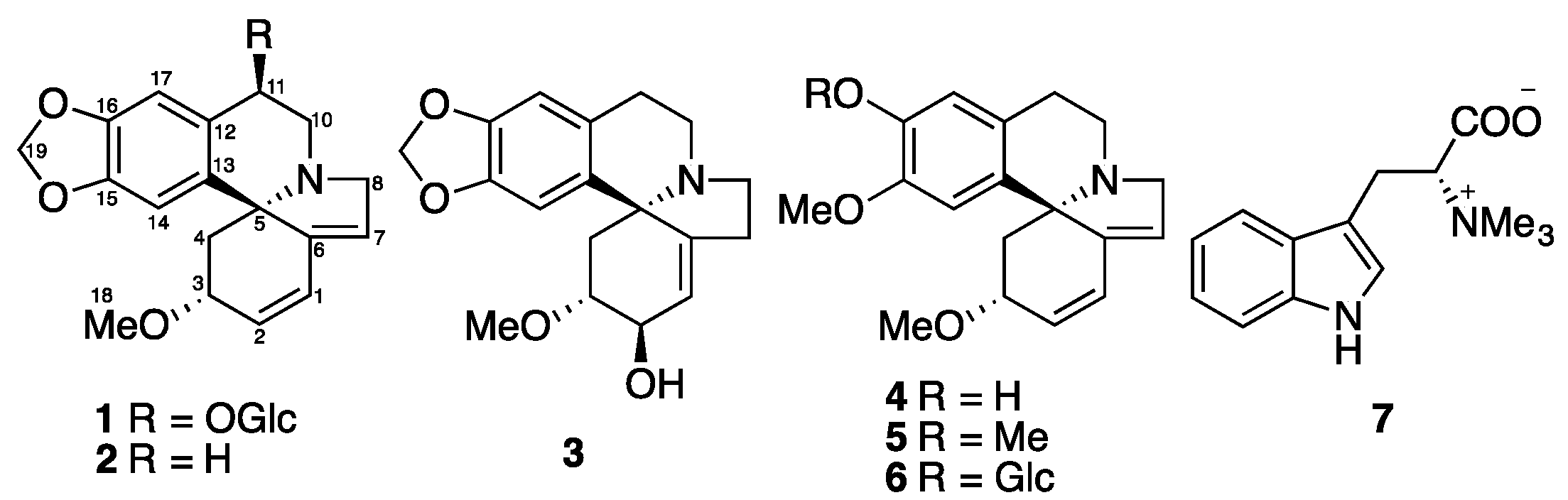
The Erythrina genus, including approximately 200 species, is mainly distributed in tropical and subtropical areas. Four local and six introduced Erythrina species are distributed in China [1]. Erythrina crista-galli L. (Fabaceae), known as cockspur coral tree, is a popular ornamental plant in South America and tropical and subtropical regions of South Asia. Pharmacological investigations have demonstrated that E. crista-galli seed extracts possess sedative, hypertensive, laxative, and diuretic activities [2]. Phytochemical studies on this plant showed the presence of Erythrina [3,4] and benzylisoquinoline alkaloids [5,6]. The alkaloids of the Erythrina type, which possess a unique spirocyclic structure, are responsible for these activities. Great efforts have been made to reveal the biosynthetic pathway and mechanism for the Erythrina alkaloid biosynthesis in the past few decades [2]. Furthermore, there are chemical studies on its non-alkaloidal constituents, in which flavonoids, cinnamylphenols and pterocarpans were reported functioning as phytoalexins and possessing antimicrobial activity [7,8,9,10,11]. In this paper, we describe the isolation and structural characterization of a new Erythrina alkaloids glucoside, erythraline-11β-O-glucopyranoside (1), together with six known alkaloids (2–7) (Figure 1) from the seeds of Erythrina crista-galli, and these isolated compounds were evaluated for their inhibitory activity against tobacco mosaic virus (TMV) using the leaf-disc method.
2. Results and Discussion
Phytochemical study of MeOH extract of the seeds of Erythrina crista-galli led to the isolation of a new Erythrinan alkaloid glucoside, erythraline-11β-O-glucopyranoside (1), together with five known Erythrinan alkaloids, erythraline (2) [12], erythratine (3) [13], erysodine (4) [14], erysotrine (5) [13], (+)-16β-d-glucoerysopine (6) [15], and an indole alkaloid, (−)-hypaphorine (7) [16] (Figure 1). The known structures were identified by comparison of their spectroscopic data with those reported in the literature.
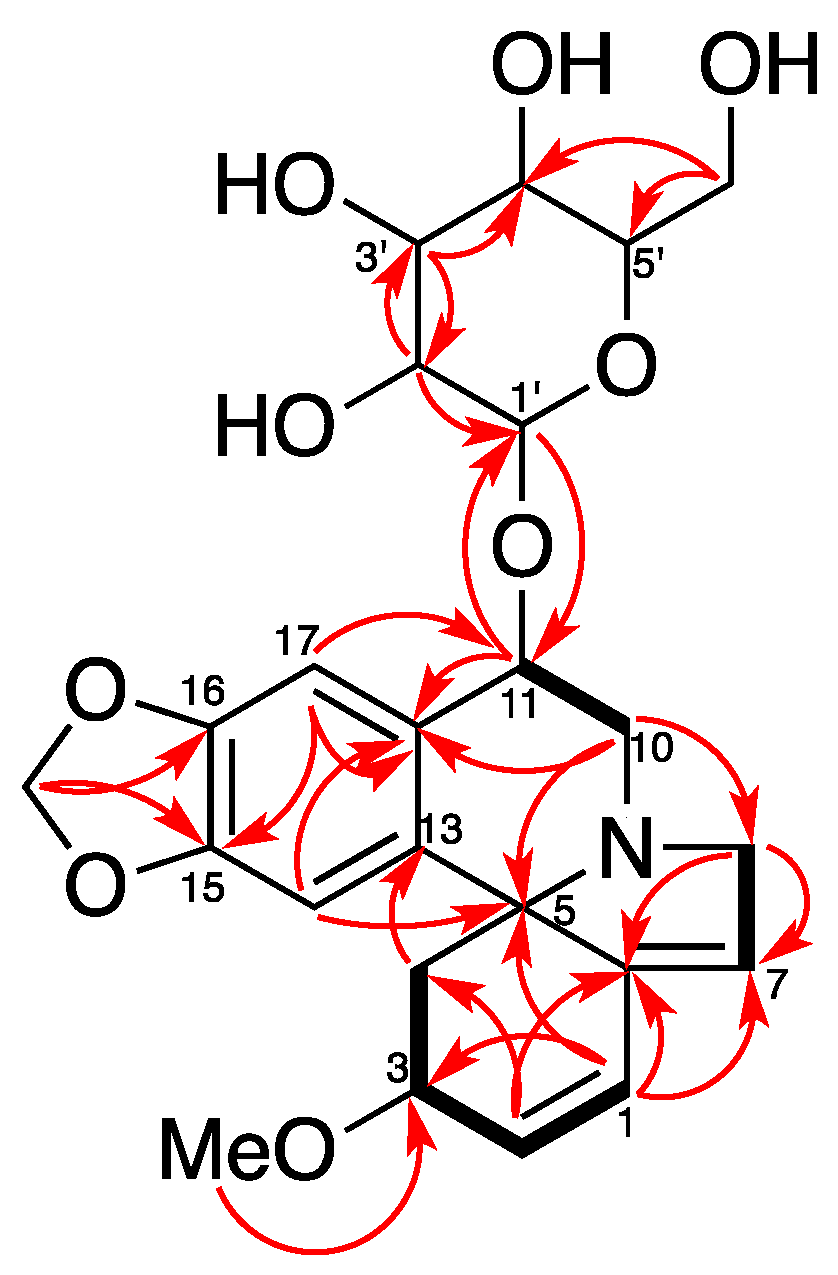
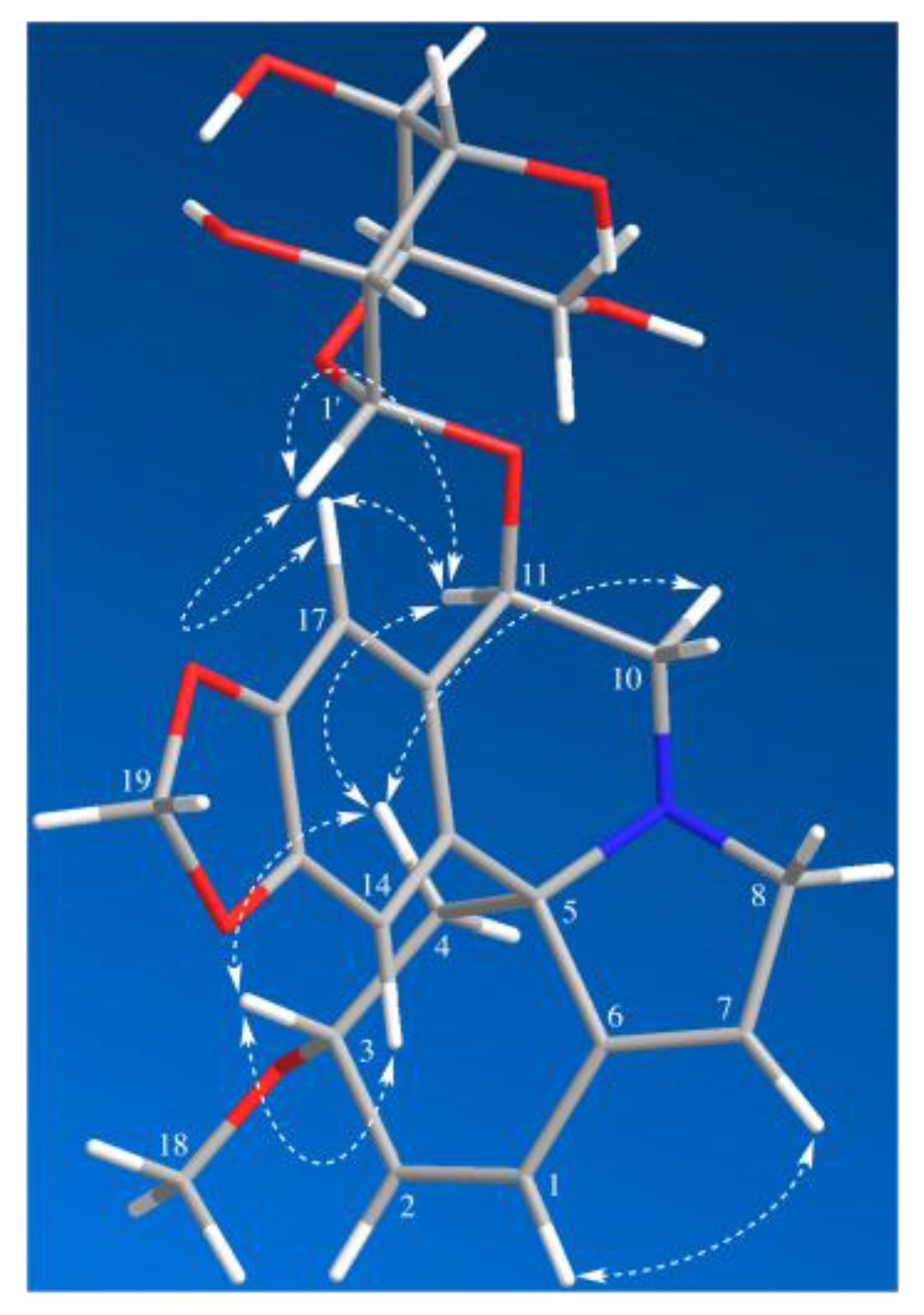
Compound 1 was isolated as a white amorphous powder. It was assigned with a molecular formula of C24H29NO9 by HR-ESI-MS at m/z = 510.1537 [M + Cl]− (Calcd for C24H29ClNO9, 510.1536). The IR spectrum (Supplementary Materials) exhibited absorption bands due to an aromatic moiety and a conjugate olefin (1627, 1503, 1482 cm−1). The 1H-NMR and HSQC spectrum of 1 indicated the presence of a tetrasubstituted aromatic ring [δH 6.73 (1H, s) and 7.15 (1H, s)], a dioxy-methylene [δH 5.91 (1H, d, J = 1.2 Hz), 5.90 (1H, d, J = 1.2 Hz)], two olefins including a trisubstituted one [δH 6.59 (1H, dd, J = 10.2, 2.1 Hz), 6.04 (1H, d, J = 10.2 Hz), 5.78 (1H, br s)], three methylene groups [δH 3.89 (1H, br d, J = 15.5 Hz), 3.70 (1H, dd, J = 15.5, 3.0 Hz), 3.57 (1H, dd, J = 14.1, 4.8 Hz), 3.37–3.30 (1H, overlap), 2.46 (1H, dd, J = 11.5, 5.5 Hz), 1.73 (1H, dd, J = 11.5, 10.4 Hz)], a methoxy group [δH 3.34 (3H, s)], and a glucopyranosyl moiety [δH 4.61 (1H, d, J = 7.8 Hz), 3.92 (1H, dd, J = 11.8, 2.0 Hz), 3.73 (1H, dd, J = 11.8, 5.4 Hz), 3.42 (1H, t, J = 8.7 Hz), 3.39–3.28 (2H, overlap), 3.23 (1H, dd, J = 9.2, 7.8 Hz)]. Its 13C-NMR and DEPT spectra showed 24 carbon resonances, including one methoxy, five methylenes, twelve methines, and six quaternary carbons (including one olefinic). The 13C-NMR data of 1 were similar to those of erythraline (2), except for the additional signals for a glucopyranosyl group at δC 105.9 (C-1’), 78.2 (C-3’), 78.0 (C-5’), 75.4 (C-2’), 71.6 (C-4′), and 62.8 (C-6’), as well as an oxygenated methine appearing at δC 74.5 instead of a methylene carbon appearing at δC 25.3 (C-11) in that of 2. The data above suggested a glucose moiety attached through an oxygen to C-11. The glucoside must have a β attachment, since the anomeric proton H-1′ (δH 4.61) exhibits a diaxial coupling (J = 7.8 Hz) with H-2’ (δH 3.23). The HMBC and 1H-1H COSY correlations (Figure 2) confirmed that 1 was a typical Erythrinan alkaloid having a characteristic spiro-carbon positioned in the center of rings A, B, and C with two olefins of ∆ 1,2 and ∆ 6,7 and a dioxy-methylene group attached to the C-15 and C-16. The presence of a glucose moiety at C-11 was further confirmed by HMBC correlation observed from the anomeric proton H-1’ to C-11. The HMBC correlation from methoxy protons H-18 (δH 3.32) to C-3 (δC 77.4) indicated that C-3 was substituted with a methoxy group. The relative configuration of 1 was determined by NOESY experiment. The observed cross-peaks (Figure 3) between H-3/H-14, H-4β, and correlations between H-4β/H-11, H-10α implied that the protons at C-3 and C-11 were placed at β- and α-orientation, respectively. Therefore, the structure of 1 was established as erythraline-11β-O-glucopyranoside. Biogenetic considerations on Erythrina alkaloids and the positive optical rotation value suggested that 1 has an S configuration at C-5 [6,14,17].
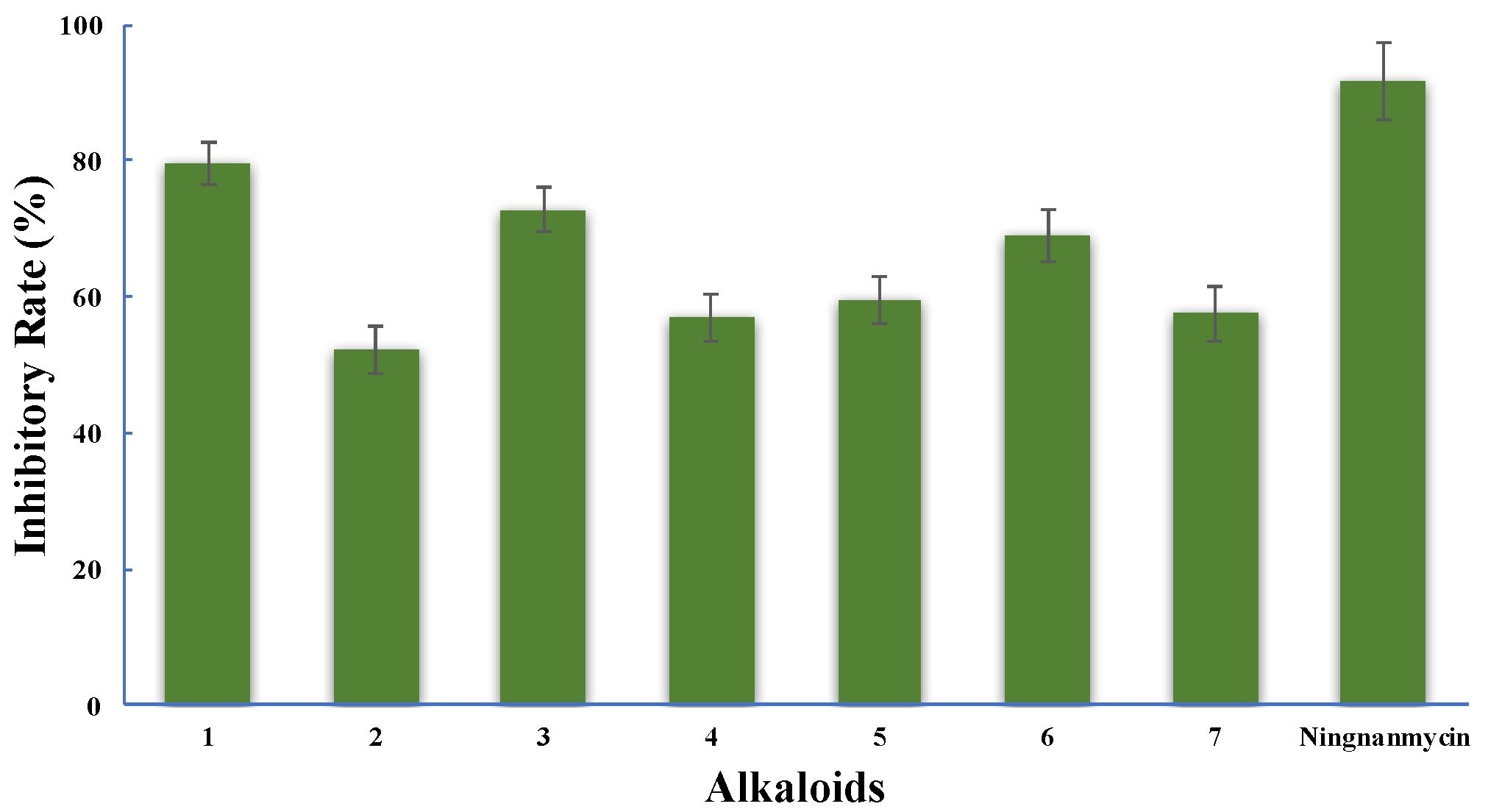
Alkaloids 1–7 showed potent inhibition against the replication of TMV at a concentration of 500 μg/mL, with inhibitory rates varying from 52.1% to 79.7% (Figure 4). A commercial antiviral agent, ningnanmycin, exhibited 91.7% inhibition as tested under the same condition. Furthermore, the IC50 values of alkaloids 1–7 were determined as 0.59, 1.52, 1.04, 1.48, 1.28, 0.74, and 1.69 mM using the leaf-disc method, while the positive control, ningnanmycin, possessed an IC50 of 0.18 mM during a test under the same condition. The new alkaloid glycoside, erythraline-11β-O-glucopyranoside (1), showed a noticeable increase in the inhibition against TMV as compared with its aglycone, erythraline (2).
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a JASCO-1020 polarimeter (JASCO, Tokyo, Japan). UV spectra were measured using a Perkin Elmer Lambda 800 UV/VIS spectrometer (PerkinElmer, Waltham, MA, USA). IR spectra were obtained with a Thermo Nicolet Avatar 360 FT-IR spectrometer (Thermo Nicolet Corporation, Madison, WI, USA). 1H- and 13C-NMR spectra were obtained with a Bruker AVANCE III 500 spectrometer (Bruker BioSpin, Rheinstetten, Germany) using tetramethylsilane as an internal standard. HRESIMS were obtained with a Thermo Scientific™ TSQ Quantum Access MAX triple stage quadrupole mass spectrometer (Thermo Scientific, San Jose, CA, USA). Sephadex LH–20 (25–100 μm, Pharmacia Fine Chemical Co., Ltd., Uppsala, Sweden), Lichroprep RP-18 gel (40–63 μm, Merck, Darmstade, Germany), Silica gel (200–300 mesh) and Silica gel H (Qingdao Oceanic Chemical Co., Qingdao, China) were used for column chromatography. Thin-layer chromatography (TLC) was performed on glass-backed plates coated with 0.25 mm layers of Silica gel H (Qingdao Oceanic Chemical Co., Qingdao, China). Fractions were monitored by TLC and spots were visualized by heating silica gel plates sprayed with 5% H2SO4 in EtOH. All solvents and chemicals used were of analytical reagent grade (Sinopharm Chemical Reagent Co., Ltd., Beijing, China), and water was doubly distilled before use.
3.2. Plant Materials
The seeds of Erythrina crista-galli L. were collected in July 2016 at the Fujian Agriculture and Forestry University (Fuzhou, China), and identified by Professor Chun-Mei Huang. Herbarium specimens were deposited at the Key Laboratory of Bio-Pesticide and Chemistry-Biology, Ministry of Education, Fujian Agriculture and Forestry University, Fuzhou, China (Specimen number: EC16701S).
3.3. Extraction and Isolation
The seeds of E. crista-galli L. (dried and powdered, 950 g) was extracted with MeOH (2 L, 3 d) at room temperature three times. The MeOH extracts (150 g) were dissolved in H2O and successively partitioned between petroleum ether, EtOAc, and n-BuOH. The EtOAc-soluble materials (7.8 g) were subjected to silica gel (200–300 mesh) column chromatography and eluted with acetone in petroleum ether (from 100:0 to 0:100) to obtained 13 fractions (Fractions 1–13). Fraction 6 (145 mg) and Fraction 7 (2.5 g) were purified with RP–18 gel column chromatography, eluting with MeOH in H2O (80%), and then subjected to silica gel (H, eluted with CHCl3/MeOH, 99:1) column chromatography, respectively, to yield Compound 2 (11.5 mg) and Compound 5 (93.8 mg). Fraction 9 (205.0 mg) was first separated with an ODS column chromatography, eluting with MeOH in H2O (70%), and then subjected to silica gel (H, eluted with CHCl3/MeOH, 98:2) column chromatography, to yield Compound 4 (35.8 mg). Fraction 11 (250 mg) was purified with RP–18 gel column chromatography, eluting with MeOH in H2O (60%), and then subjected to silica gel (H, eluted with CHCl3/MeOH, 95:5) column chromatography, to yield Compound 3 (28.1 mg). Fraction 12 (1.05 g) was purified with ODS column chromatography, eluting with MeOH in H2O (60%), and then subjected to silica gel (H, eluted with CHCl3/MeOH, 90:10) column chromatography, to yield Compound 1 (17.1 mg) and Compound 6 (23.1 mg). Fraction 13 (424 mg) was purified with ODS column chromatography, eluting with MeOH in H2O (40%) to yield Compound 7 (39.2 mg).
Erythraline-11β-O-glucopyranoside (1). White amorphous powder. + 130.6 (c 0.1 MeOH); UV (MeOH) λmax (log ε): 205 (1.97), 226 (0.89), 287 (0.22); IR (KBr) υmax: 1627, 1503, 1482 cm−1; HR-ESI-MS m/z: 510.1537 (Calcd for C24H29ClNO9, 510.1536); 1H-NMR (500 MHz, CD3OD) and 13C-NMR (125 MHz, CD3OD) data (see Table 1).
3.4. Anti-TMV Assay
3.4.1. Virus and Host Plant
Purified TMV (strain U1) was obtained from Institute of Plant Virology, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, whose concentration was determined as 15 mg/mL using an ultraviolet spectrophotometer method (). The purified virus was kept at −20 °C and was diluted to 30 μg/mL with 0.01 M PBS before use. Nicotiana tabacum cv. K326, which were cultivated and grown to a 5–6-leaf stage in an insect-free greenhouse, were used as an anti-TMV assay, as a systemic TMV infection host. Purified compounds were dissolved in DMSO and diluted with 0.01 M PBS to a certain concentration for test. The final concentration of DMSO in the test solution (≤2%) showed no adverse effect on the plants.
3.4.2. Leaf-Disc Method
Growing leaves of N. tabacum cv. K326 were mechanically inoculated with TMV (30 μg/mL in 0.01 M PBS). After 6 h, leaf discs (1 cm diameter) were punched and floated on solutions for test. Discs of healthy and inoculated leaves floated on a solution of 0.01 M PBS with 2% DMSO were used as a mock and negative control, respectively. Three replicates were carried out for each sample. After incubating for 48 h at 25 °C in a culture chamber, the leaf discs were grounded in 0.01 M carbonate coating buffer (pH 9.6), and OD405 values were measured using TAS-ELISA method. TAS-ELISA was performed as described in the literature [18,19]. Virus concentration was calculated from a standard curve constructed using OD405 values of purified TMV at concentrations of 1.0, 0.5, 0.25, 0.125, and 0.0625 μg/mL. The inhibition of test solutions on TMV was calculated as follows: inhibition rate = [1 − (virus concentration of treatment) / (virus concentration of negative control)] × 100%.
4. Conclusions
A new Erythrina alkaloid glycoside together with five known Erythrina alkaloids and an indole alkaloid have been isolated from the MeOH extract of the seeds of Erythrina Crista-galli L. All the isolated alkaloids showed noticeable inhibition against the replication of TMV but in a relatively higher concentration as compared with that of the positive control agent. Further investigations are needed to reveal the biological action mechanism of Erythrina alkaloids.
Supplementary Materials
The following are available online. The 1H- and 13C-NMR of all the isolated compounds, and the IR, UV/Vis, HRESIMS, DEPT, HSQC, HMBC, and NOESY of Compound 1.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 31371987 and 31501687).
Author Contributions
Qing-Wei Tan and Qi-Jian Chen conceived and designed the experiments. Jian-Cheng Ni and Pei-Hua Fang performed the experiments and test. Qing-Wei Tan analyzed the data and composed the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, B.J.; Wu, B.; Bao, M.F.; Ni, L.; Cai, X.H. New dimeric and trimeric Erythrina alkaloids from Erythrina variegata. RSC Adv. 2016, 6, 87863–87868. [Google Scholar] [CrossRef]
- Maier, U.H.; Rödl, W.; Deus-Neumann, B.; Zenk, M.H. Biosynthesis of Erythrina alkaloids in Erythrina crista-galli. Phytochemistry 1999, 52, 372–382. [Google Scholar] [CrossRef]
- Ito, K.; Haruna, M.; Jinno, Y.; Furukawa, H. Studies on the Erythrina alkaloids. XI. Alkaloids of Erythrina crysta-galli. Linn. Structure of new alkaloids, Crystamidine. Chem. Pharm. Bull. 2008, 24, 52–55. [Google Scholar] [CrossRef]
- Chawla, A.S.; Gupta, M.P.; Jackson, A.H. Alkaloidal constituents of Erythrina crista-galli flowers. J. Nat. Prod. 1987, 50, 1146–1148. [Google Scholar] [CrossRef]
- Ju-Ichi, M.; Fujitani, Y.; Furukawa, H. Structure of cristadine—A new benzylisoqunoline alkaloid. Heterocycles 1982, 19, 849–850. [Google Scholar] [CrossRef]
- Ozawa, M.; Kawamata, S.; Etoh, T.; Hayashi, M.; Komiyama, K.; Kishida, A.; Kuroda, C.; Ohsaki, A. Structures of new Erythrinan alkaloids and nitric oxide production inhibitors from Erythrina crista-galli. Chem. Pharm. Bull. 2010, 58, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Tanaka, T.; Etoh, H. Three pterocarpans from Erythrina crista-galli. Phytochemistry 1997, 45, 835–838. [Google Scholar] [CrossRef]
- Mitscher, L.A.; Ward, J.A.; Drake, S.; Rao, G.S. Antimicrobial agents from higher plants. Erycristagallin, a new pterocarpene from the roots of the bolivian coral tree, Erythrina crista-galli. Heterocycles 1984, 22, 1673–1675. [Google Scholar] [CrossRef]
- Mitscher, L.A.; Gollapudi, S.R.; Gerlach, D.C.; Drake, S.D.; Véliz, E.A.; Ward, J.A. Erycristin, a new antimicrobial petrocarpan from Erythrina crista-galli. Phytochemistry 1988, 27, 381–385. [Google Scholar] [CrossRef]
- Iinuma, M.; Okawa, Y.; Tanaka, T. Three new cinnamylphenols in heartwood of Erythrina crista-galli. Phytochemistry 1994, 37, 1153–1155. [Google Scholar] [CrossRef]
- Ingham, J.L.; Markham, K.R. Identification of the Erythrina phytoalexin cristacarpin and a note on the chirality of other 6α-hydroxypterocarpans. Phytochemistry 1980, 19, 1203–1207. [Google Scholar] [CrossRef]
- Chawla, A.S.; Chunchatprasert, S.; Jackson, A.H. Studies of Erythrina alkaloids: VII–13C-NMR spectral studies of some Erythina alkaloids. Org. Magn. Reson. 1983, 21, 39–41. [Google Scholar] [CrossRef]
- Barton, D.H.; James, R.; Kirby, G.W.; Turner, D.W.; Widdowson, D.A. Phenol oxidation and biosynthesis. Part XVIII. The structure and biosynthesis of Erythrina alkaloids. J. Chem. Soc. C Org. 1968, 12, 1529–1537. [Google Scholar] [CrossRef]
- Amer, M.E.; El-Masry, S. Three novel glycodienoid alkaloids from Erythrina lysistemon. J. Nat. Prod. 1991, 54, 161–166. [Google Scholar] [CrossRef]
- Wanjala, C.C.W.; Majinda, R.T. Two novel glucodienoid alkaloids from Erythrina latissima seeds. J. Nat. Prod. 2000, 63, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Honda, K.; Nakai, I.; Kishida, A.; Ohsaki, A. Hypaphorine, an indole alkaloid from Erythrina velutina, induced sleep on normal mice. Bioorg. Med. Chem. Lett. 2008, 18, 3992–3994. [Google Scholar] [CrossRef] [PubMed]
- Rukachaisirikul, T.; Innok, P.; Suksamrarn, A. Erythrina alkaloids and a pterocarpan from the bark of Erythrina subumbrans. J. Nat. Prod. 2008, 71, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yan, X.H.; Dong, J.H.; Sang, P.; Fang, X.; Di, Y.T.; Zhang, Z.K.; Hao, X.J. Tobacco mosaic virus (TMV) inhibitors from Picrasma quassioides Benn. J. Agric. Food Chem. 2009, 57, 6590–6595. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Fan, H.J.; Li, Y.; Shi, Z.L.; Pan, Y.; Lu, C.P. Development of a multi-mimotope peptide as a vaccine immunogen for infectious bursal disease virus. Vaccine 2007, 25, 4447–4455. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Structure of Compounds 1–7.
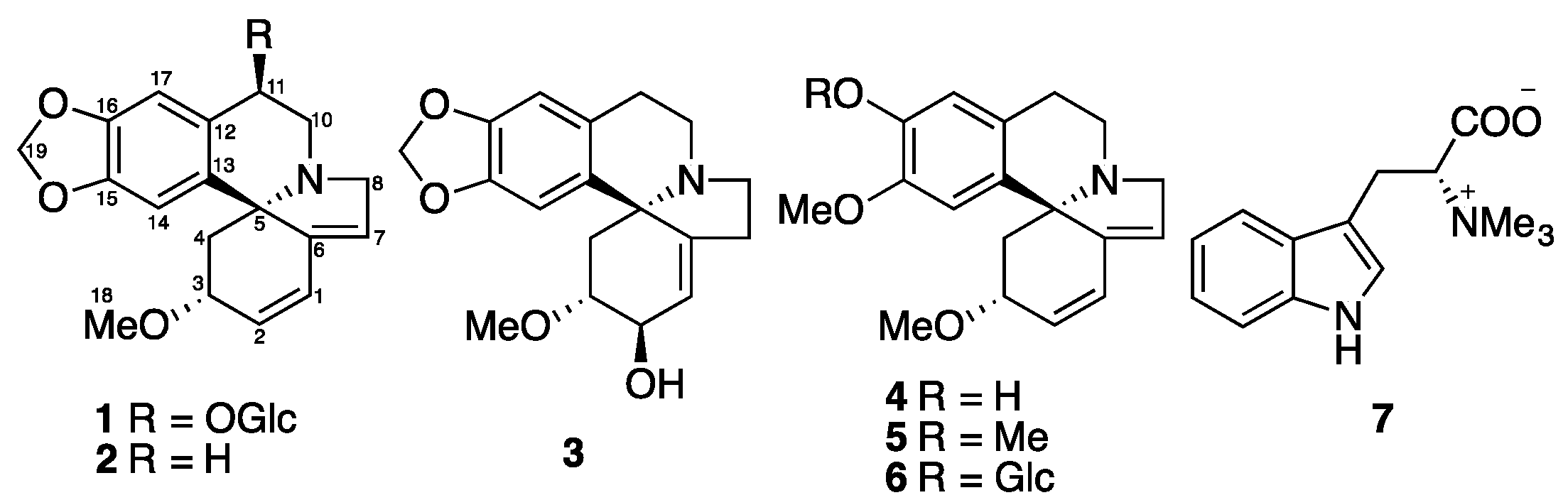
Figure 2.
1H-1H COSY (−) and key HMBC (→) correlations of Compound 1.

Figure 3.
Key NOESY correlations of Compound 1.
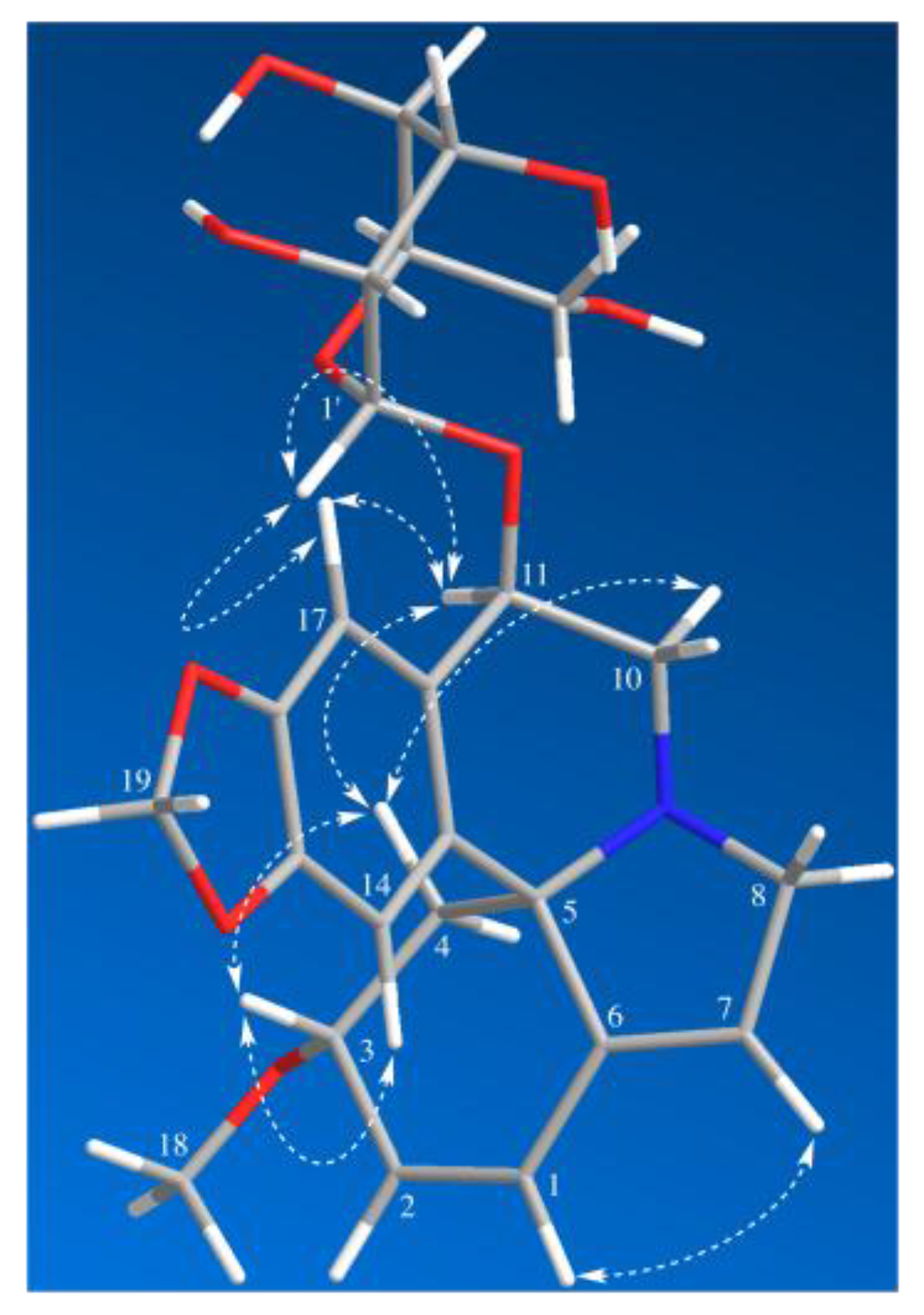
Figure 4.
Inhibitory activity of the isolated alkaloids 1–7 against the replication of TMV.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
1H- and 13C-NMR data of Compounds 1 and 2 (δ in ppm, CD3OD)
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| C-1 | 126.2 | 6.59 (1H, dd, J = 10.2, 2.1 Hz) | 125.4 | 6.53 (1H, dd, J = 10.1, 2.3 Hz) |
| C-2 | 132.7 | 6.04 (1H, d, J = 10.2 Hz) | 131.6 | 5.97 (1H, d, J = 10.1 Hz) |
| C-3 | 77.4 | 4.01 (1H, m) | 76.2 | 3.96 (1H, m) |
| C-4 | 41.2 | 2.46 (1H, dd, J = 11.5, 5.5 Hz), 1.73 (1H, dd, J = 11.5, 10.4 Hz) | 42.0 | 2.49 (1H, dd, J = 11.5, 5.5 Hz) 1.83 (1H, dd, J = 11.5, 10.4 Hz) |
| C-5 | 67.8 | □ | 67.5 | □ |
| C-6 | 143.7 | □ | 142.4 | □ |
| C-7 | 124.5 | 5.78 (1H, br s) | 123.1 | 5.72 (1H, br s, 1H) |
| C-8 | 59.3 | 3.89 (1H, br d, J = 15.5 Hz) 3.70 (1H, dd, J = 15.5, 3.0 Hz) | 57.7 | 3.72 (1H, dd, J = 14.5, 3.0 Hz) 3.53–3.45 (1H, overlap) |
| C-10 | 50.4 | 3.57 (1H, dd, J = 14.1, 4.8 Hz) 3.37–3.30 (1H, overlap) | 44.6 | 3.53–3.45 (1H, overlap) 2.92–2.83 (1H, overlap) |
| C-11 | 74.5 | 4.70 (1H, t, J = 4.3 Hz) | 25.3 | 2.92–2.83 (1H, overlap) 2.71–2.64 (1H, m) |
| C-12 | 129.1 | □ | 128.1 | □ |
| C-13 | 132.0 | □ | 132.7 | □ |
| C-14 | 106.3 | 6.73 (1H, s) | 106.3 | 6.62 (1H, s) |
| C-15 | 148.9 | □ | 146.2 | □ |
| C-16 | 148.1 | □ | 145.9 | □ |
| C-17 | 109.7 | 7.15 (1H, s) | 108.8 | 6.76 (1H, s) |
| C-18 | 56.6 | 3.34 (3H, s) | 56.1 | 3.32 (3H, s) |
| C-19 | 102.3 | 5.91 (1H, d, J = 1.2 Hz) 5.90 (1H, d, J = 1.2 Hz) | 100.8 | 5.90 (1H, d, J = 1.5 Hz) 5.87 (1H, d, J = 1.5 Hz) |
| C-1′ | 105.9 | 4.61 (1H, d, J = 7.8 Hz) | □ | □ |
| C-2′ | 75.4 | 3.23 (1H, dd, J = 9.2, 7.8 Hz) | □ | □ |
| C-3′ | 78.2 | 3.42 (1H, t, J = 8.8 Hz) | □ | □ |
| C-4′ | 71.6 | 3.39–3.28 (1H, overlap) | □ | □ |
| C-5′ | 78.0 | 3.39–3.28 (1H, overlap) | □ | □ |
| C-6′ | 62.8 | 3.92 (1H, dd, J = 11.8, 2.0 Hz) 3.73 (1H, dd, J = 11.8, 5.4 Hz) | □ | □ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tan, Q.-W.; Ni, J.-C.; Fang, P.-H.; Chen, Q.-J. A New Erythrinan Alkaloid Glycoside from the Seeds of Erythrina crista-galli. Molecules 2017, 22, 1558. https://doi.org/10.3390/molecules22091558
AMA Style
Tan Q-W, Ni J-C, Fang P-H, Chen Q-J. A New Erythrinan Alkaloid Glycoside from the Seeds of Erythrina crista-galli. Molecules. 2017; 22(9):1558. https://doi.org/10.3390/molecules22091558
Chicago/Turabian StyleTan, Qing-Wei, Jian-Cheng Ni, Pei-Hua Fang, and Qi-Jian Chen. 2017. "A New Erythrinan Alkaloid Glycoside from the Seeds of Erythrina crista-galli" Molecules 22, no. 9: 1558. https://doi.org/10.3390/molecules22091558