COX Inhibition Profile and Molecular Docking Studies of Some 2-(Trimethoxyphenyl)-Thiazoles

 , , ,
, , , 
Abstract
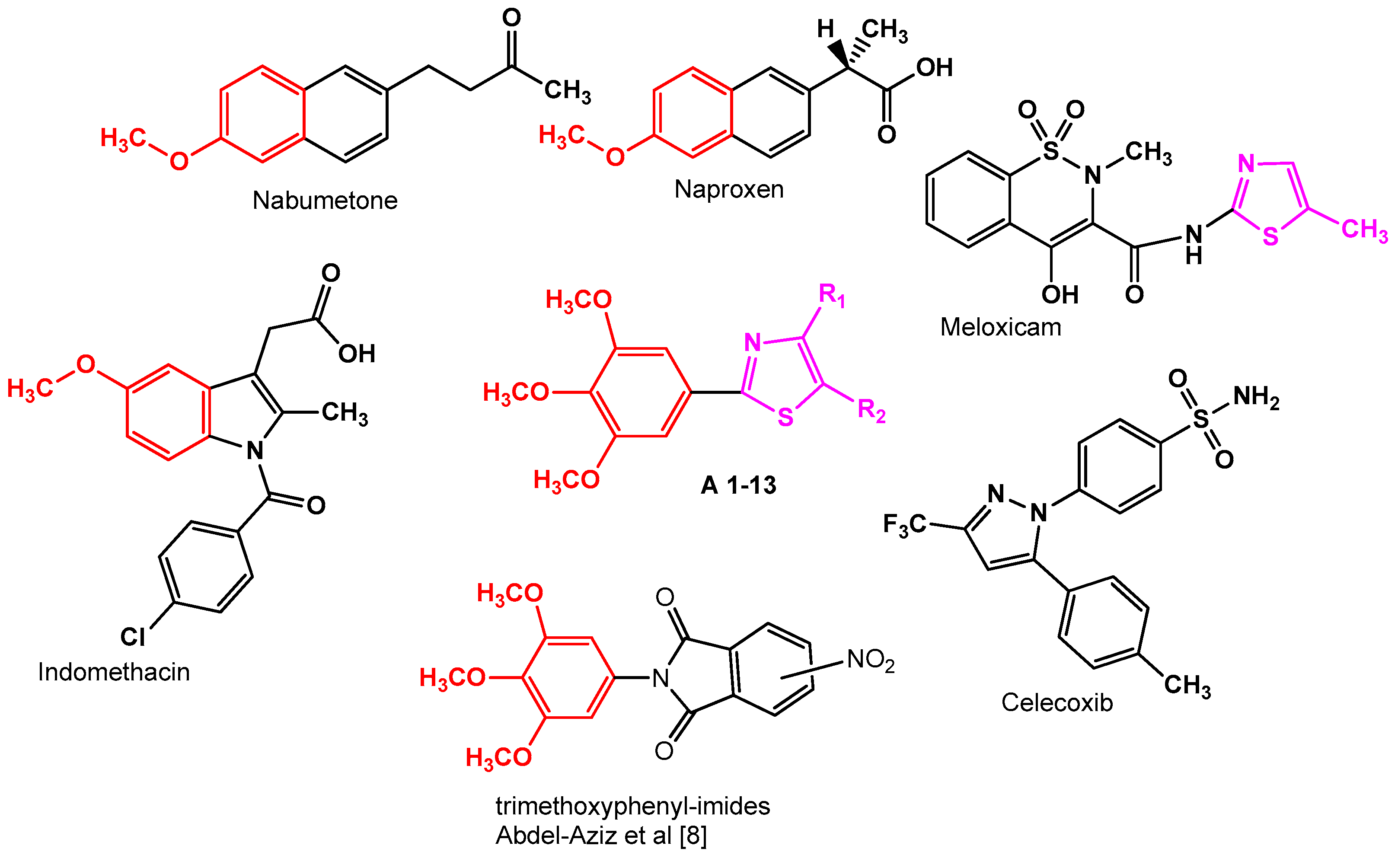
:1. Introduction
2. Results and Discussion
2.1. In Vitro Cyclooxygenase Inhibition Assay
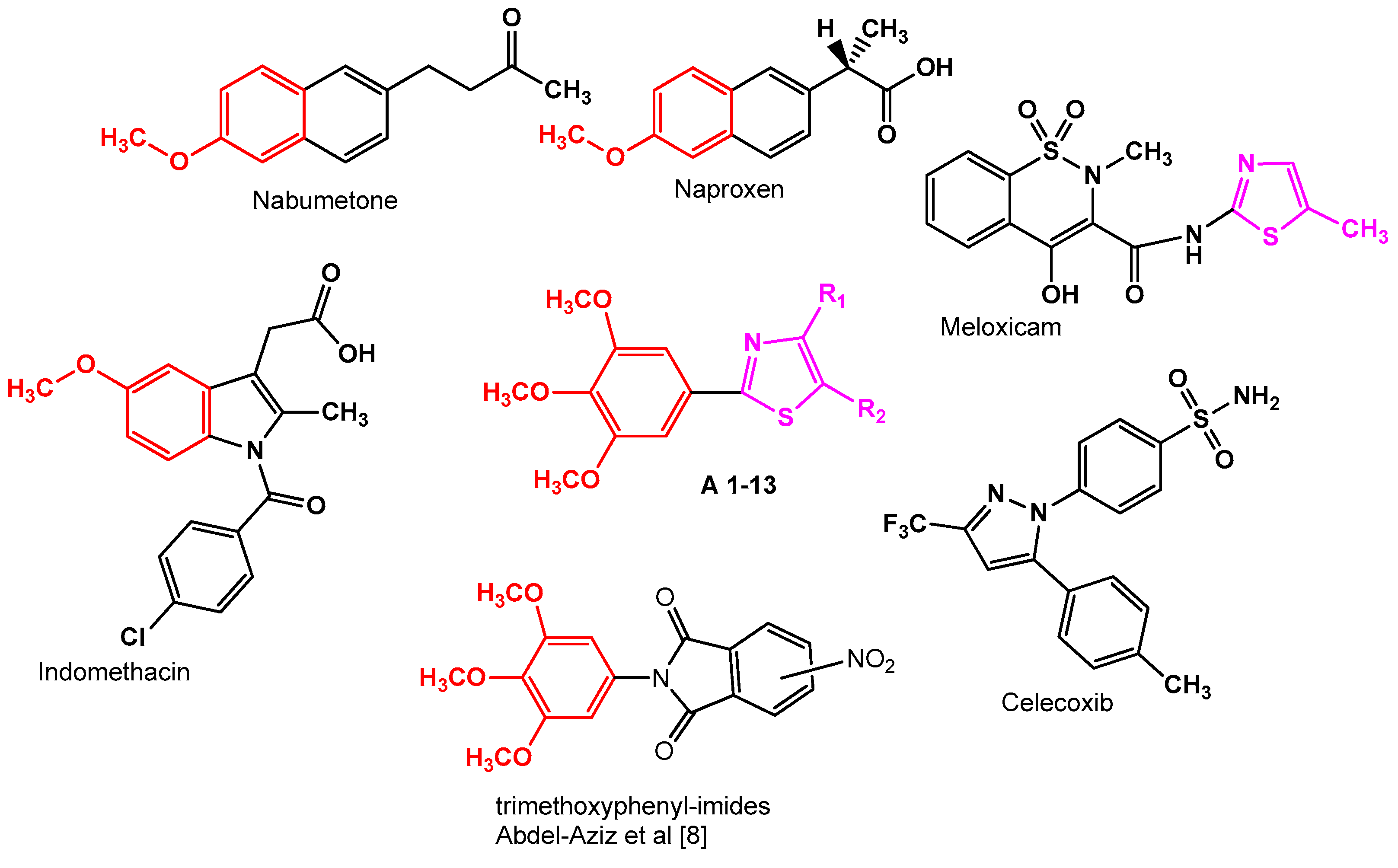
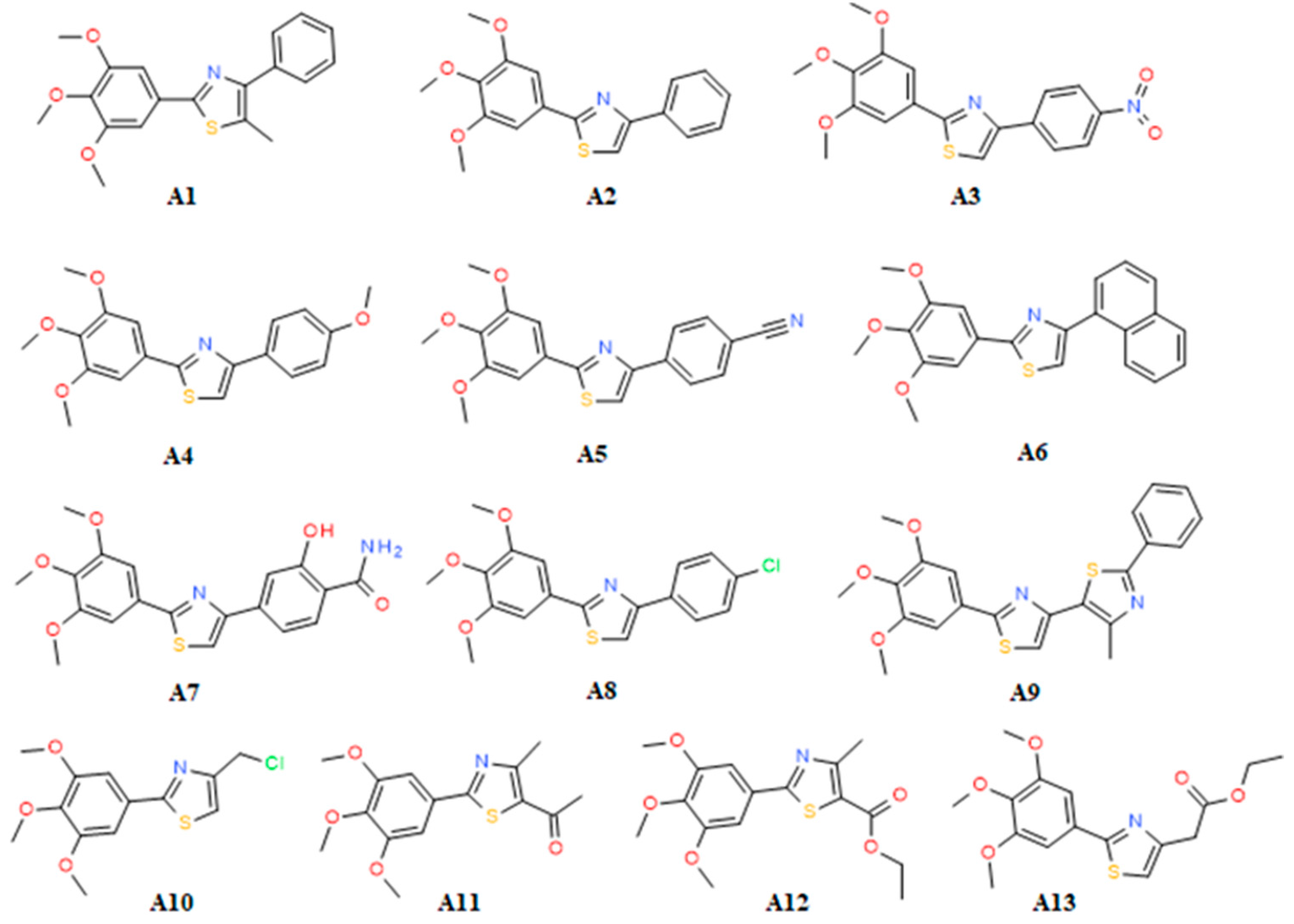
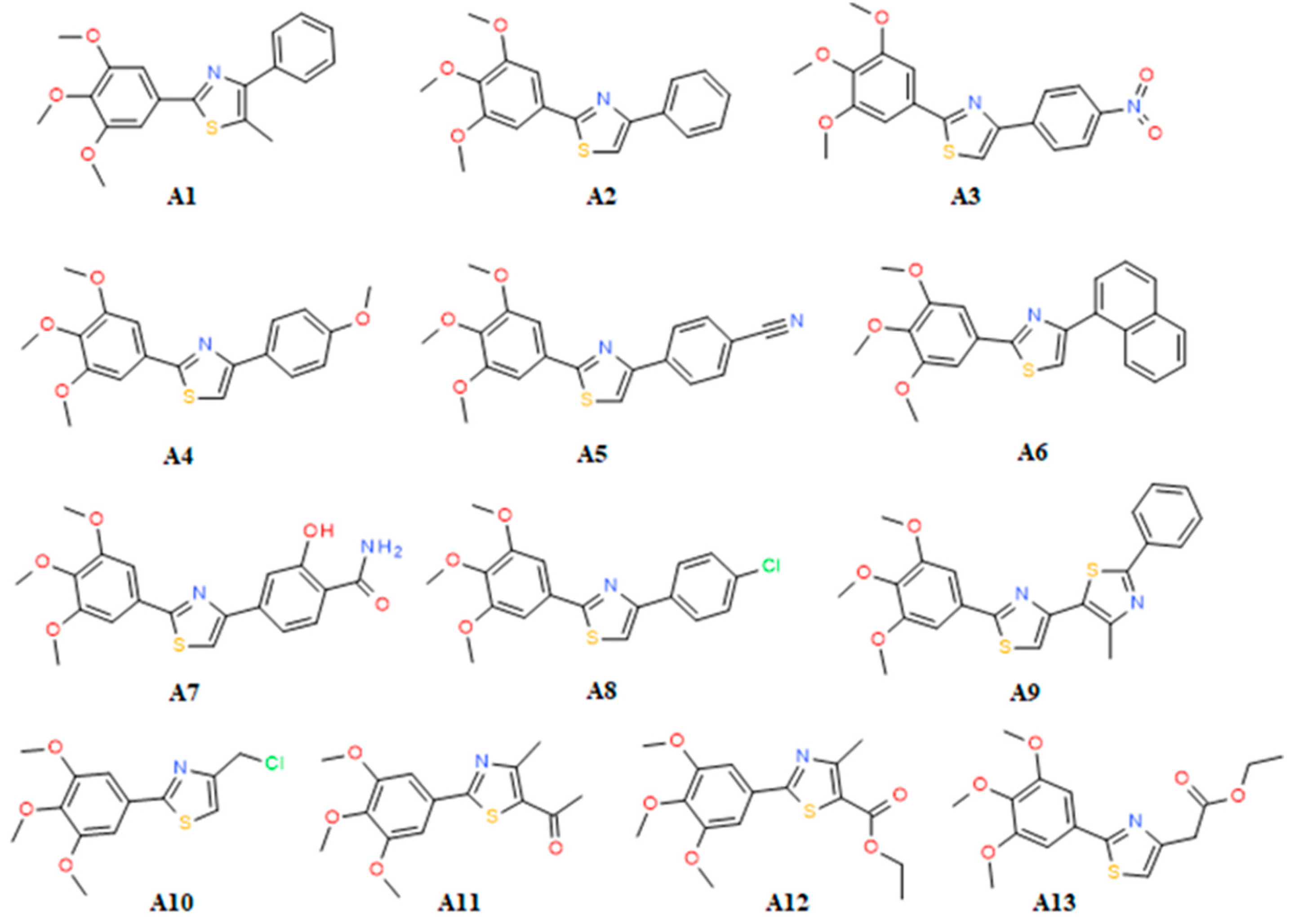
2.2. SAR
2.3. Docking
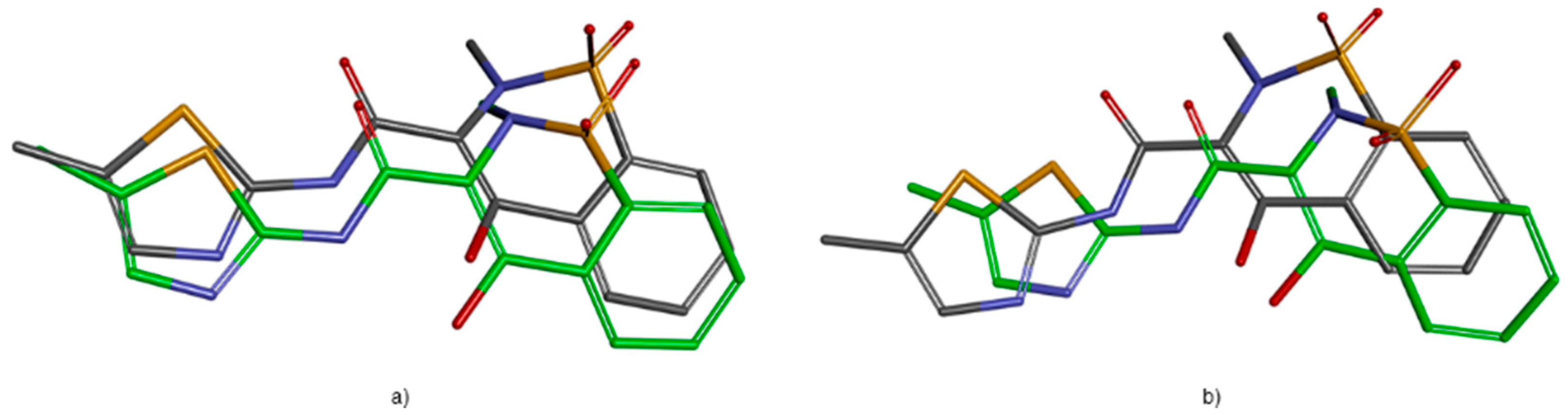
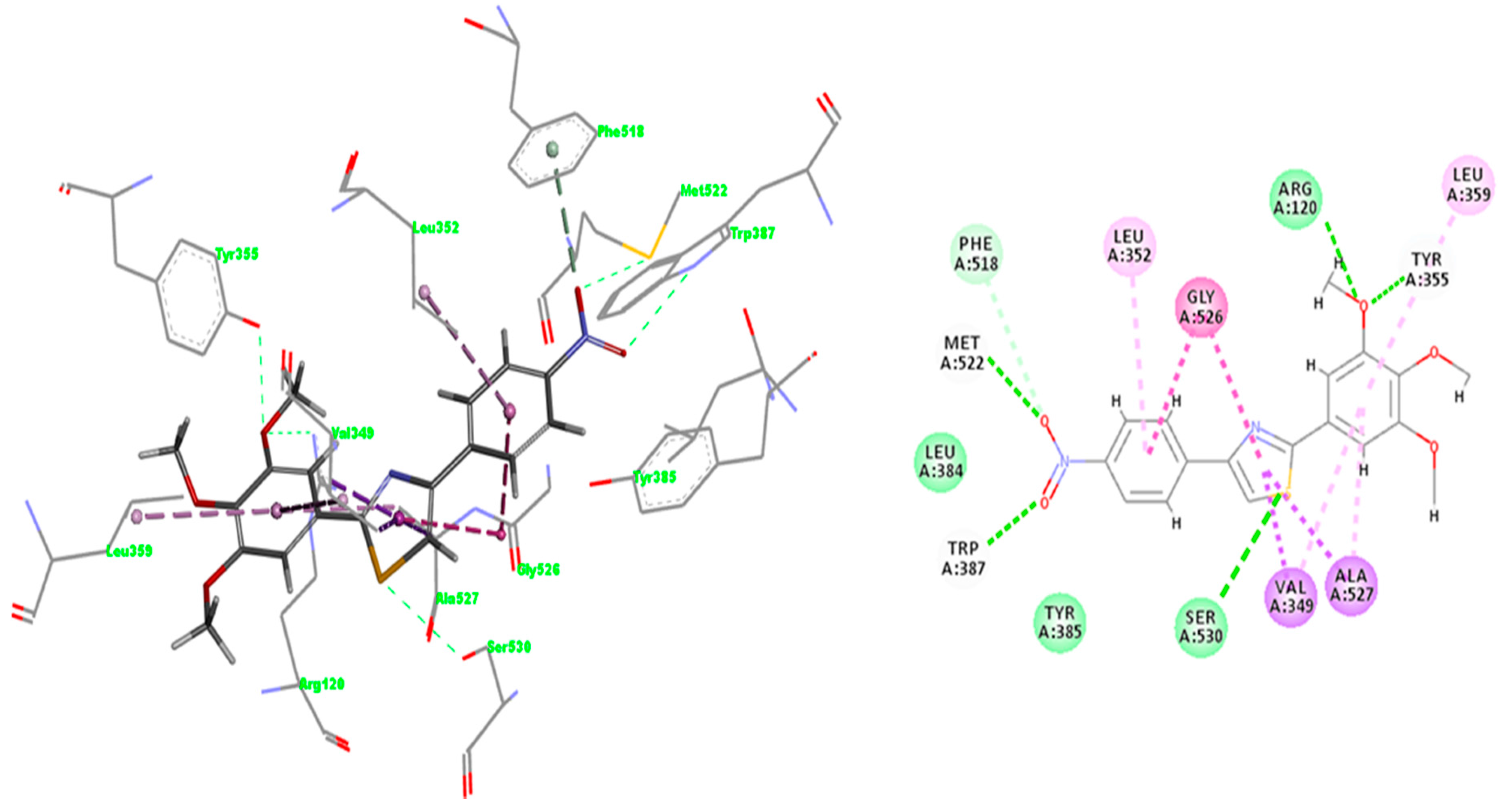
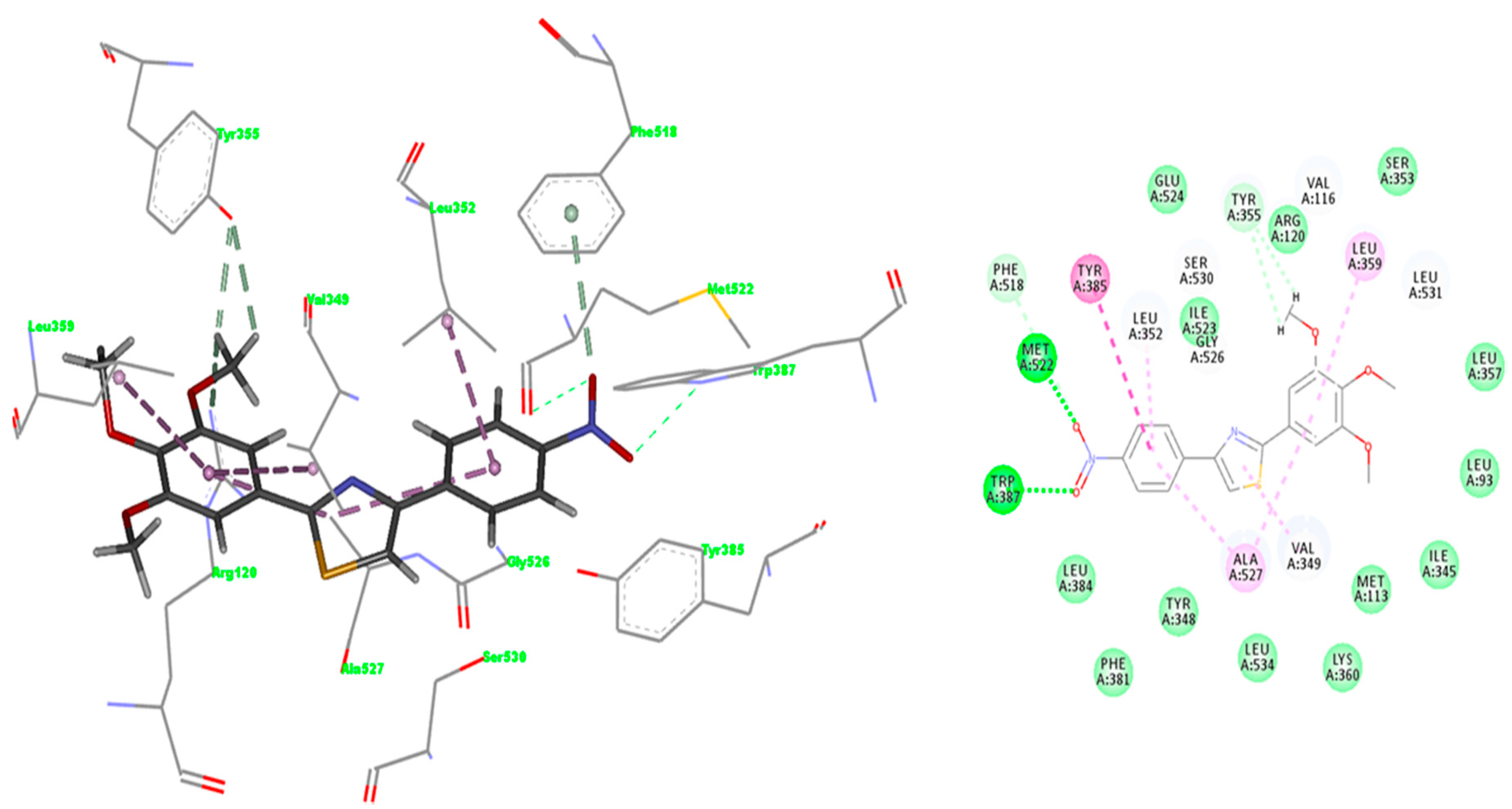
2.3.1. Validation of the Docking Protocol
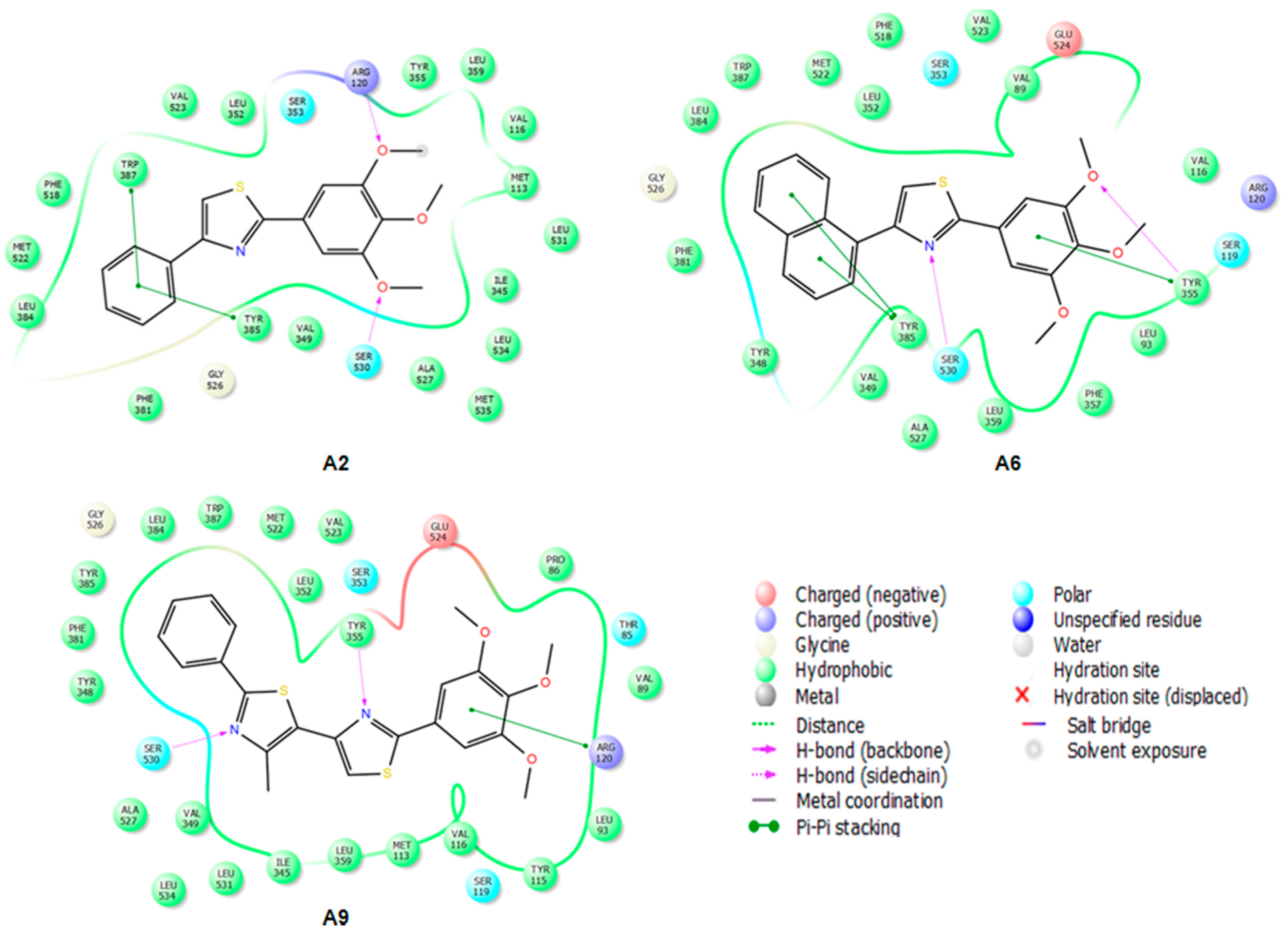
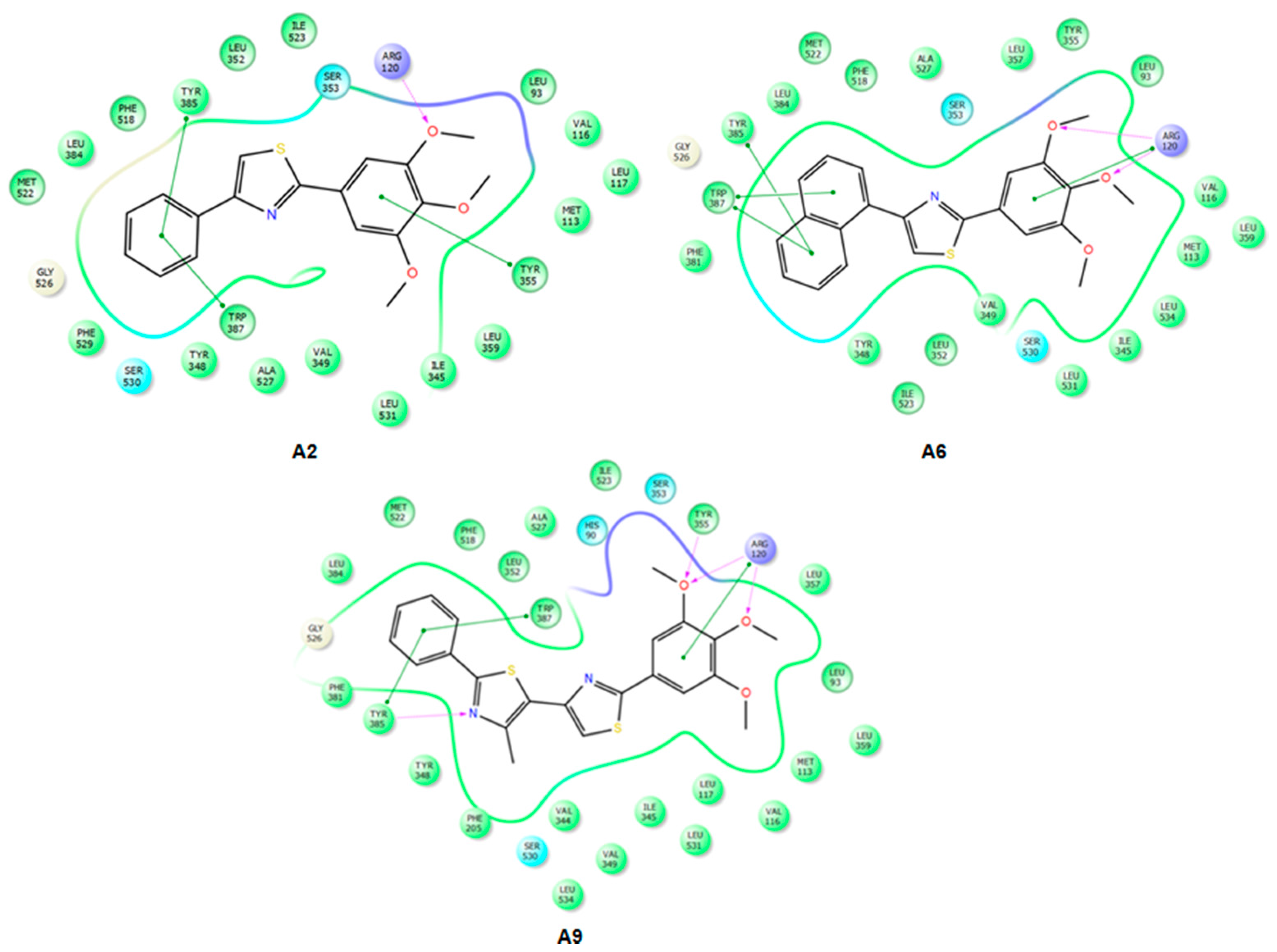
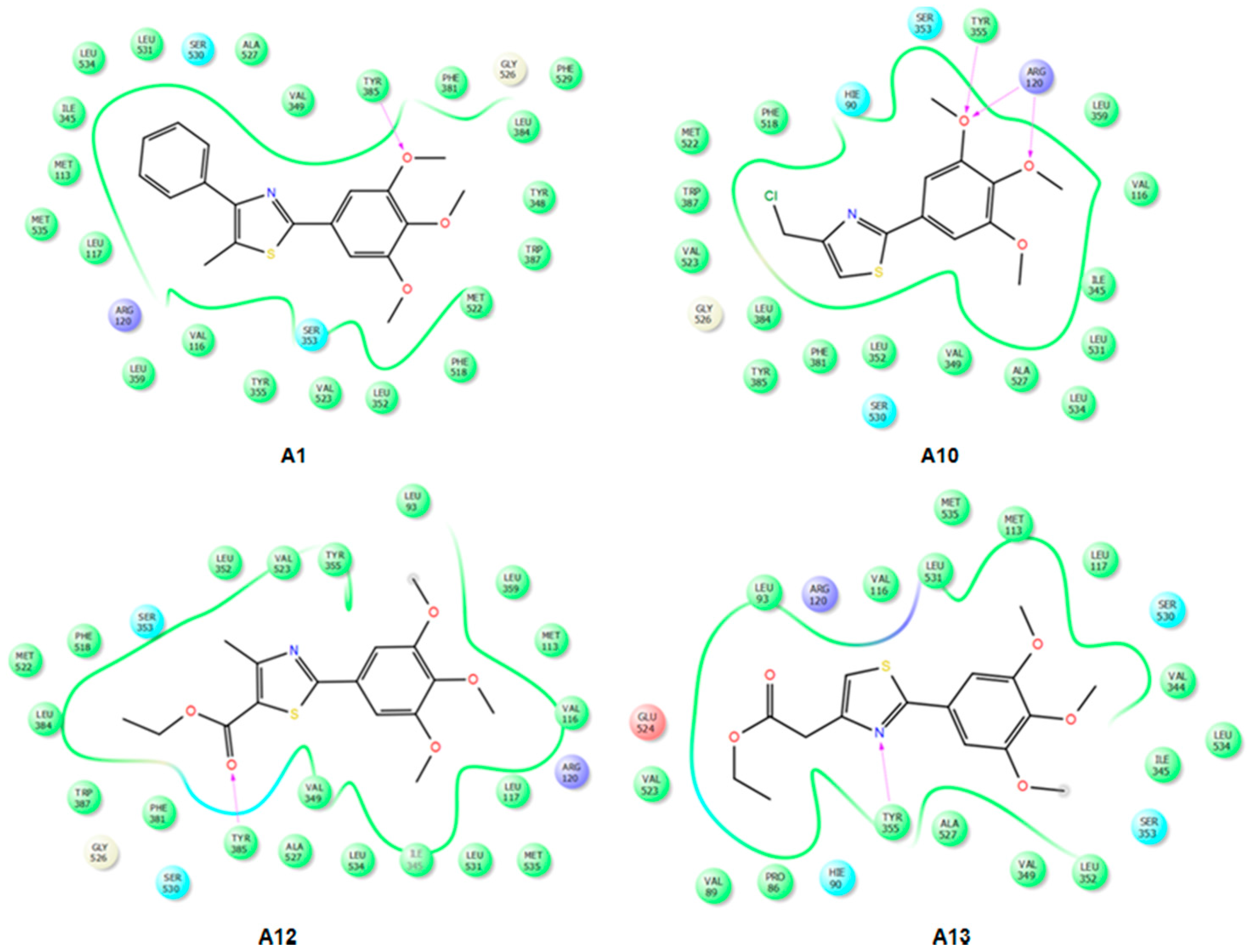
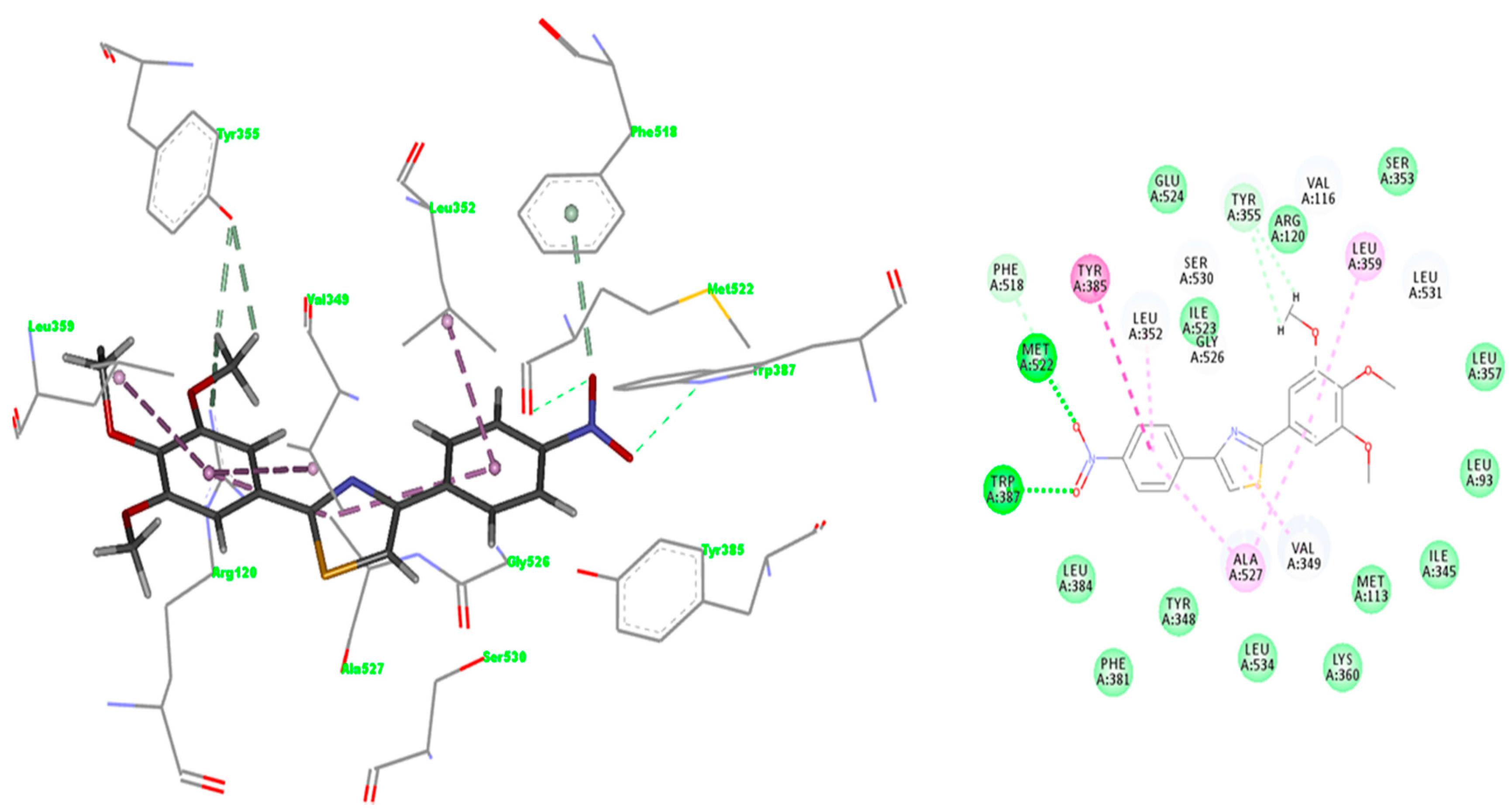
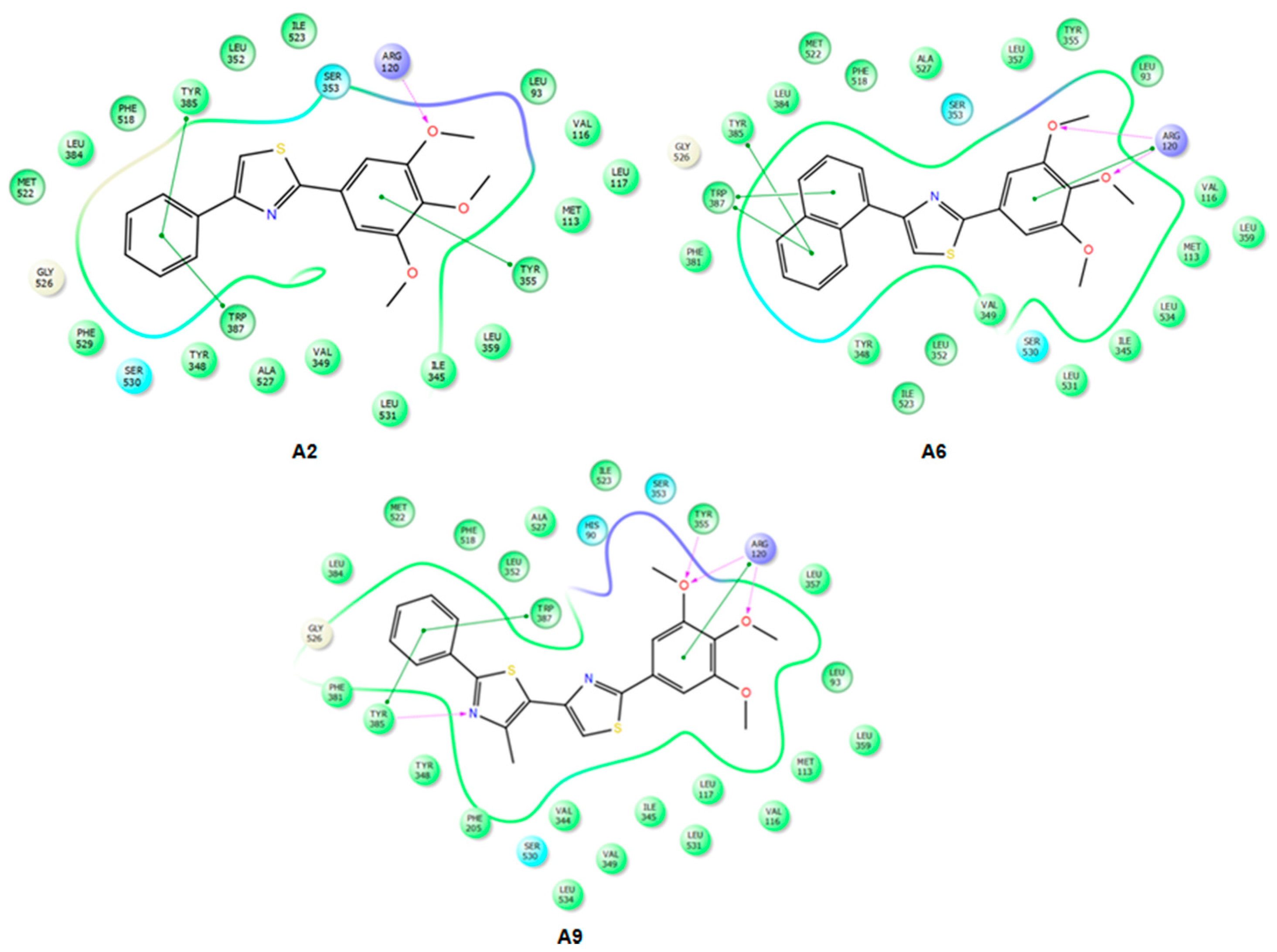
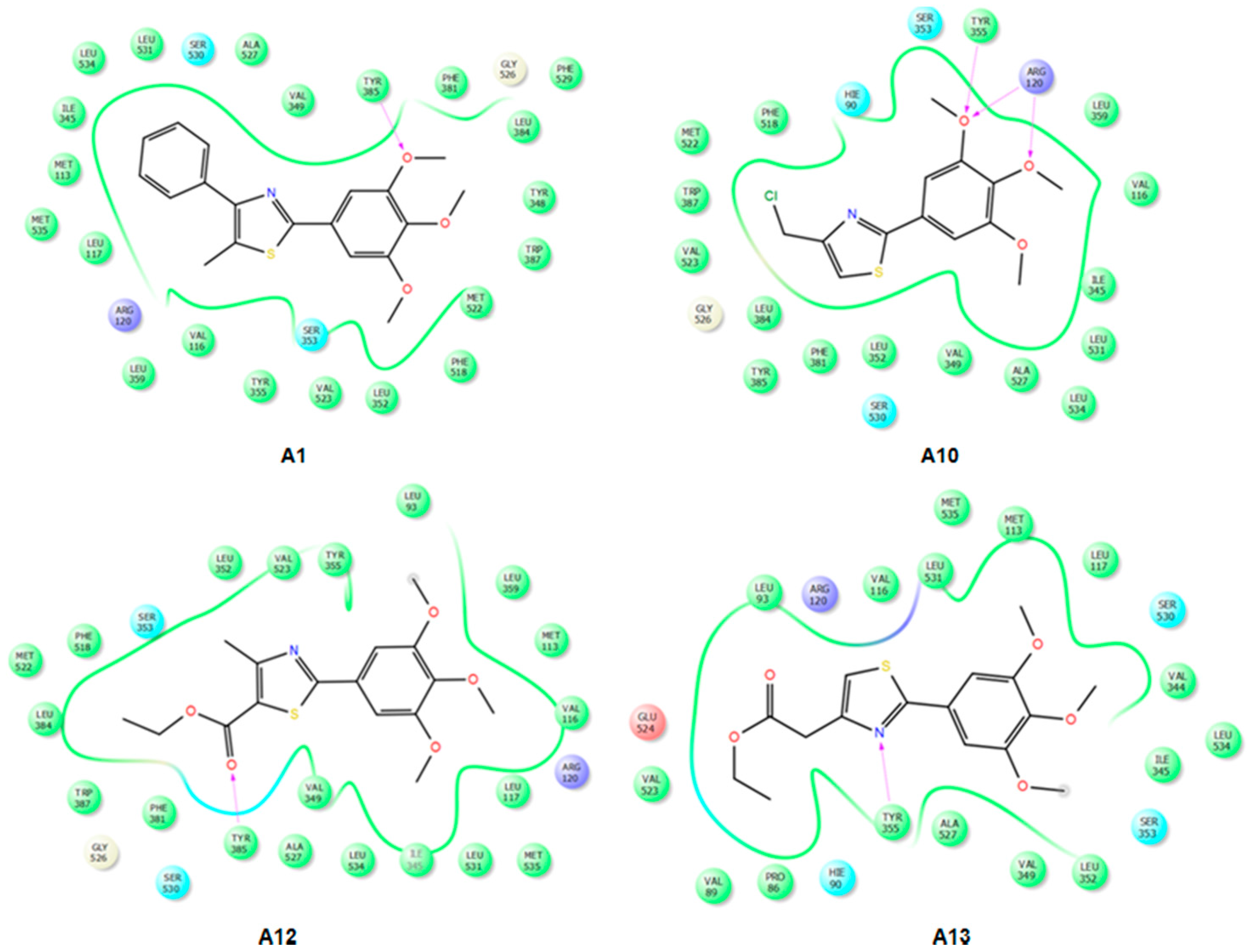
2.3.2. Mode of Binding
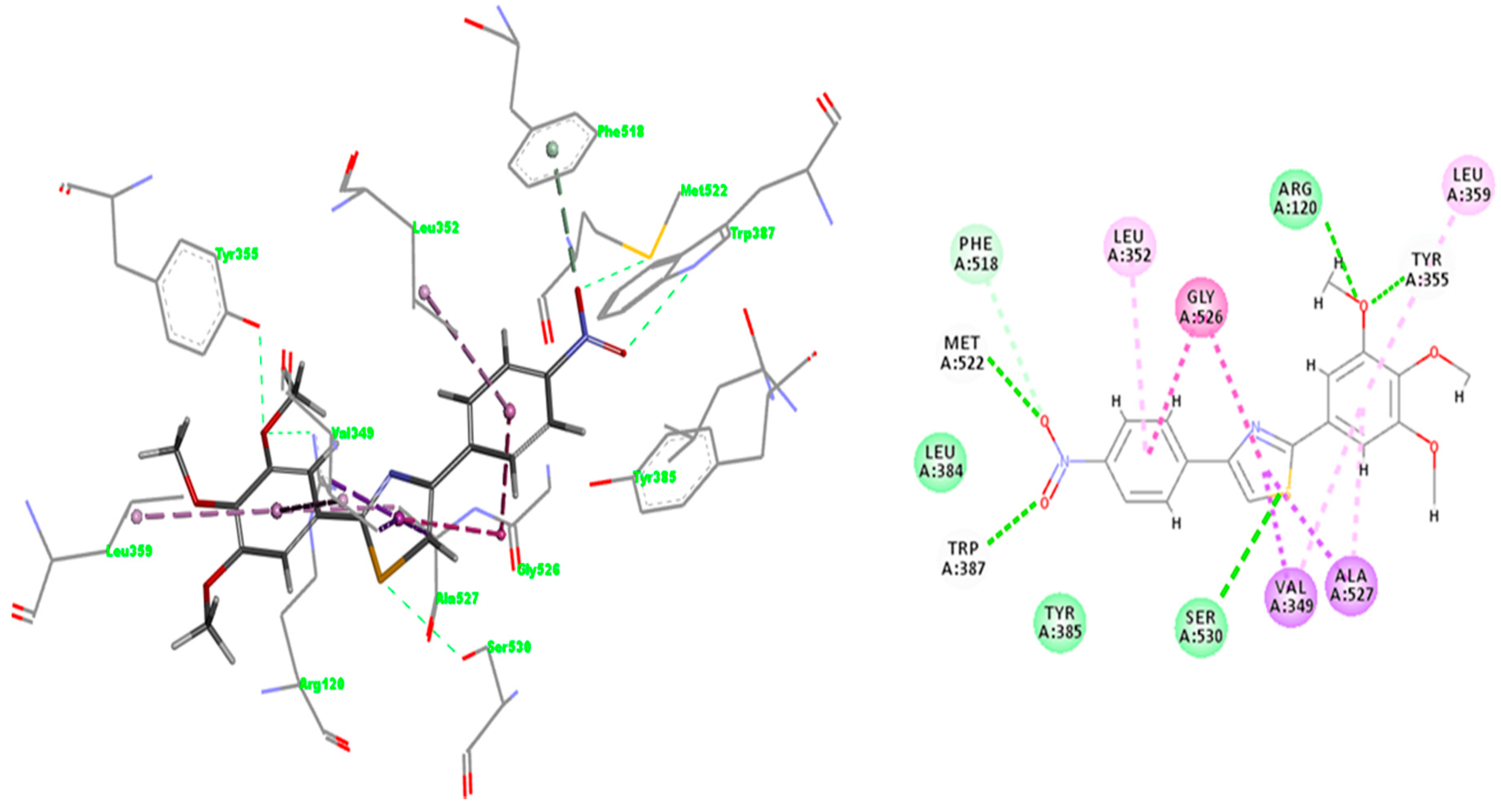
2.3.3. Docking of Compound A3
2.4. Prediction of Pharmacokinetic Properties
3. Materials and Methods
3.1. In Vitro Cyclooxygenase Inhibitor Assay
3.2. Molecular Modeling
3.2.1. Protein Preparation
3.2.2. Ligand Preparations
3.2.3. Induced Fit Docking
3.3. Prediction of Drug-Likeness and Pharmacokinetic Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Back, M.; Yin, L.; Ingelsson, E. Cyclooxygenase-2 inhibitors and cardiovascular risk in a nation-wide cohort study after the withdrawal of rofecoxib. Eur. Heart J. 2012, 33, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- García Rodríguez, L.A.; González-Pérez, A.; Bueno, H.; Hwa, J. NSAID Use Selectively Increases the Risk of Non-Fatal Myocardial Infarction: A Systematic Review of Randomised Trials and Observational Studies. PLoS ONE 2011, 6, e16780. [Google Scholar] [CrossRef] [PubMed]
- Al-Saeed, A. Gastrointestinal and Cardiovascular Risk of Nonsteroidal Anti-inflammatory Drugs. Oman Med. J. 2011, 26, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Knights, K.M.; Mangoni, A.A.; Miners, J.O. Defining the COX inhibitor selectivity of NSAIDs: Implications for understanding toxicity. Expert Rev. Clin. Pharmacol. 2010, 3, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, M. A systematic review on the role of eicosanoid pathways in rheumatoid arthritis. Adv. Med. Sci. 2018, 63, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.; Bombardier, C.; Cannon, C. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Borne, R.; Mark, L.; Wilson, N. Nonsteroidal Anti-Inflammatory Drugs. In Foye’s Principles of Medicinal Chemistry; Thomas, L.L., David, A.W., Victoria, F.R., Zito, W., Eds.; Wolters Kluwer. Lippincott Williams & Wilkins: Baltimore, MD, USA, 2013; p. 1021. ISBN 9781609133450. [Google Scholar]
- Abdel-Aziz, A.A.-M.; ElTahir, K.E.H.; Asiri, Y.A. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. Eur. J. Med. Chem. 2011, 46, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Araniciu, C.; Pârvu, A.E.; Tiperciuc, B.; Palage, M.; Oniga, S.; Verité, P.; Oniga, O. Synthesis and evaluation of the anti-inflammatory activity of some 2-(Trimethoxyphenyl)-4-R1-5-R2-Thiazoles. Dig. J. Nanomater. Biostructures 2013, 8, 699–709. [Google Scholar]
- Pola, S. Significance of Thiazole-based Heterocycles for Bioactive Systems. In Scope of Selective Heterocycles from Organic and Pharmaceutical Perspective; InTech: Rijeka, Crotia, 2016; ISBN 978-953-51-2503-7. [Google Scholar]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, C.M.; Oniga, O.; Pârvu, A.; Tiperciuc, B.; Verite, P.; Pîrnău, A.; Crişan, O.; Bojită, M.; Pop, R. Synthesis and anti-inflammatory evaluation of some new acyl-hydrazones bearing 2-aryl-thiazole. Eur. J. Med. Chem. 2011, 46, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Helal, M.H.M.; Salem, M.A.; El-Gaby, M.S.A.; Aljahdali, M. Synthesis and biological evaluation of some novel thiazole compounds as potential anti-inflammatory agents. Eur. J. Med. Chem. 2013, 65, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Kumar, S.; Kaushik, P.; Kaushik, D.; Gupta, G.K. Synthesis and pharmacological evaluation of some novel 2-(5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazol-1-yl)-4-(coumarin-3-yl)thiazoles. Eur. J. Med. Chem. 2013, 62, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Abdelazeem, A.H.; El-Saadi, M.T.; Safi El-Din, A.G.; Omar, H.A.; El-Moghazy, S.M. Design, synthesis and analgesic/anti-inflammatory evaluation of novel diarylthiazole and diarylimidazole derivatives towards selective COX-1 inhibitors with better gastric profile. Bioorg. Med. Chem. 2017, 25, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Araniciu, C.; Palage, M.; Oniga, S.; Pirnau, A.; Verité, P.; Oniga, O. Synthesis and characterization of some novel 5,2 -and 4,2-bisthiazoles derivatives. Rev. Chim. 2013, 64, 1067–1071. [Google Scholar]
- Araniciu, C.; Pârvu, A.E.; Palage, M.D.; Oniga, S.D.; Benedec, D.; Oniga, I.; Oniga, O. The effect of some 4,2 and 5,2 bisthiazole derivatives on nitro-oxidative stress and phagocytosis in acute experimental inflammation. Molecules 2014, 19, 9240–9256. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.I.; Crisan, L.; Bora, A.; Pacureanu, L.M.; Avram, S.; Kurunczi, L. Retrospective group fusion similarity search based on eROCE evaluation metric. Bioorg. Med. Chem. 2013, 21, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Pacureanu, L.; Crisan, L.; Bora, A.; Avram, S.; Kurunczi, L. In silico classification and virtual screening of maleimide derivatives using projection to latent structures discriminant analysis (PLS-DA) and hybrid docking. Monatshefte für Chemie-Chem. Mon. 2012, 143, 1559–1573. [Google Scholar] [CrossRef]
- Tudor, I.O. Chemoinformatics in Drug Discovery; Mannhold, R., Kubinyi, H., Folkers, G., Eds.; Wiley VCH: Weinheim, Germany, 2005; ISBN 9783527307531. [Google Scholar]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams Bind in a Novel Mode to the Cyclooxygenase Active Site via a Two-water-mediated H-bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Iyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, A.J.; Malkowski, M.G. The structure of NS-398 bound to cyclooxygenase-2. J. Struct. Biol. 2011, 176, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Bonomi, M.; Marinelli, L.; Gervasio, F.L.; Cavalli, A.; Novellino, E.; Parrinello, M. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 5411–5416. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio Modeling Environment, Version 4.5, Release 2017, Dassault Systèmes BIOVIA: San Diego, CA, USA, 2016.
- Maestro, Version 10.2, Schrodinger Software: New York, NY, USA, 2015.
- Yan, L.; Pan, M.; Fu, M.; Wang, J.; Huang, W.; Qian, H. Design, synthesis and biological evaluation of novel analgesic agents targeting both cyclooxygenase and TRPV1. Bioorg. Med. Chem. 2016, 24, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Tan, O.; Ozadali, K.; Piskin, K.; Balkan, A. Molecular modeling, synthesis and screening of some new 4-thiazolidinone derivatives with promising selective COX-2 inhibitory activity. Eur. J. Med. Chem. 2012, 57, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Aldawsari, F.S.; Aguiar, R.P.; Wiirzler, L.A.M.; Aguayo-Ortiz, R.; Aljuhani, N.; Cuman, R.K.N.; Medina-Franco, J.L.; Siraki, A.G.; Velázquez-Martínez, C.A. Anti-inflammatory and antioxidant properties of a novel resveratrol–salicylate hybrid analog. Bioorg. Med. Chem. Lett. 2016, 26, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Undare, S.S.; Valekar, N.J.; Patravale, A.A.; Jamale, D.K.; Vibhute, S.S.; Walekar, L.S.; Kolekar, G.B.; Deshmukh, M.B.; Anbhule, P.V. Synthesis, anti-inflammatory, ulcerogenic and cyclooxygenase activities of indenopyrimidine derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Jorgensen, W.; Duffy, E. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds A1–A13 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 | COX-2 | COX-1 | SI | IC50 | COX-2 | COX-1 | SI |
|---|---|---|---|---|---|---|---|
| C* | 0.06 | 30.68 | 511.3 | A6 | 28.87 | 26.88 | 0.93 |
| M* | 12.50 | 137.83 | 11.03 | A7 | 105.67 | 54.72 | 0.51 |
| I* | 25.65 | 1.80 | 0.07 | A8 | 25.64 | 31.46 | 1.22 |
| A1 | >300.00 | 140.10 | 0.007 | A9 | 70.86 | 83.16 | 1.17 |
| A2 | 23.26 | 34.53 | 1.48 | A10 | 107.00 | 609.64 | 5.69 |
| A3 | 25.50 | 235.67 | 9.24 | A11 | 215.99 | 76.03 | 0.35 |
| A4 | 75.00 | 50.37 | 0.67 | A12 | 272.54 | 409.13 | 1.5 |
| A5 | 148.40 | 73.26 | 0.49 | A13 | 229.48 | 108.77 | 0.47 |
| H Bond | 4M11 | 4O1Z |
|---|---|---|
| N-Thiazole .....HOH25/HOH117 | 2.853 | 3.044 |
| HOH25/HOH117.....Tyr385 | 2.740 | 2.182 |
| HOH25/HOH117.....Ser530 | 3.194 | 3.450 |
| 4-OH Benzothiazine.....Ser530 | 3.047 | 2.966 |
| C=O Carboxamide.....HOH84/HOH161 | 3.184 | 3.638 |
| HOH84/HOH161.....Tyr355 | 2.972 | 3.410 |
| HOH84/HOH161.....Arg120 | 2.680 | 2.373 |
| -NH Carboxamide..... 4-OH Benzothiazine | 2.554 | 2.210 |
| -C=O Carboxamide.....N Benzothiazine | 2.613 | 2.711 |
| Molecule | QPlog HERG | QPP Caco | QPlogBB | QPP MDCK | QPlogKp | ROF | ROT | HOA |
|---|---|---|---|---|---|---|---|---|
| A1 | −5.582 | 8697.865 | 0.408 | 7894.031 | −0.473 | 0 | 0 | 3 |
| A2 | −5.698 | 8587.415 | 0.421 | 8547.499 | −0.34 | 0 | 0 | 3 |
| A3 | −5.606 | 949.754 | −0.653 | 790.214 | −2.314 | 0 | 0 | 3 |
| A4 | −5.638 | 8587.208 | 0.359 | 8550.394 | −0.435 | 0 | 0 | 3 |
| A5 | −5.741 | 1727.341 | −0.381 | 1508.647 | −1.726 | 0 | 1 | 3 |
| A6 | −6.158 | 8494.294 | 0.415 | 8471.98 | −0.141 | 1 | 1 | 1 |
| A7 | −5.38 | 354.207 | −1.157 | 272.109 | −3.189 | 0 | 0 | 3 |
| A8 | −5.625 | 8586.23 | 0.595 | 10,000 | −0.508 | 1 | 1 | 3 |
| A9 | −6.239 | 7022.555 | 0.382 | 9485.234 | −0.47 | 1 | 1 | 1 |
| A10 | −4.516 | 8641.559 | 0.604 | 10,000 | −0.992 | 0 | 0 | 3 |
| A11 | −4.406 | 2652.557 | −0.163 | 2164.163 | −2.012 | 0 | 0 | 3 |
| A12 | −4.706 | 1862.779 | −0.445 | 1235.594 | −2.23 | 0 | 0 | 3 |
| A13 | −5.088 | 1860.846 | −0.478 | 1633.249 | −2.022 | 0 | 0 | 3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oniga, S.D.; Pacureanu, L.; Stoica, C.I.; Palage, M.D.; Crăciun, A.; Rusu, L.R.; Crisan, E.-L.; Araniciu, C. COX Inhibition Profile and Molecular Docking Studies of Some 2-(Trimethoxyphenyl)-Thiazoles. Molecules 2017, 22, 1507. https://doi.org/10.3390/molecules22091507
Oniga SD, Pacureanu L, Stoica CI, Palage MD, Crăciun A, Rusu LR, Crisan E-L, Araniciu C. COX Inhibition Profile and Molecular Docking Studies of Some 2-(Trimethoxyphenyl)-Thiazoles. Molecules. 2017; 22(9):1507. https://doi.org/10.3390/molecules22091507
Chicago/Turabian StyleOniga, Smaranda Dafina, Liliana Pacureanu, Cristina Ioana Stoica, Mariana Doina Palage, Alexandra Crăciun, Laurentiu Răzvan Rusu, Elena-Luminita Crisan, and Cătălin Araniciu. 2017. "COX Inhibition Profile and Molecular Docking Studies of Some 2-(Trimethoxyphenyl)-Thiazoles" Molecules 22, no. 9: 1507. https://doi.org/10.3390/molecules22091507