Anti-HIV Activities and Mechanism of 12-O-Tricosanoylphorbol-20-acetate, a Novel Phorbol Ester from Ostodes katharinae


Abstract
:1. Introduction
2. Results
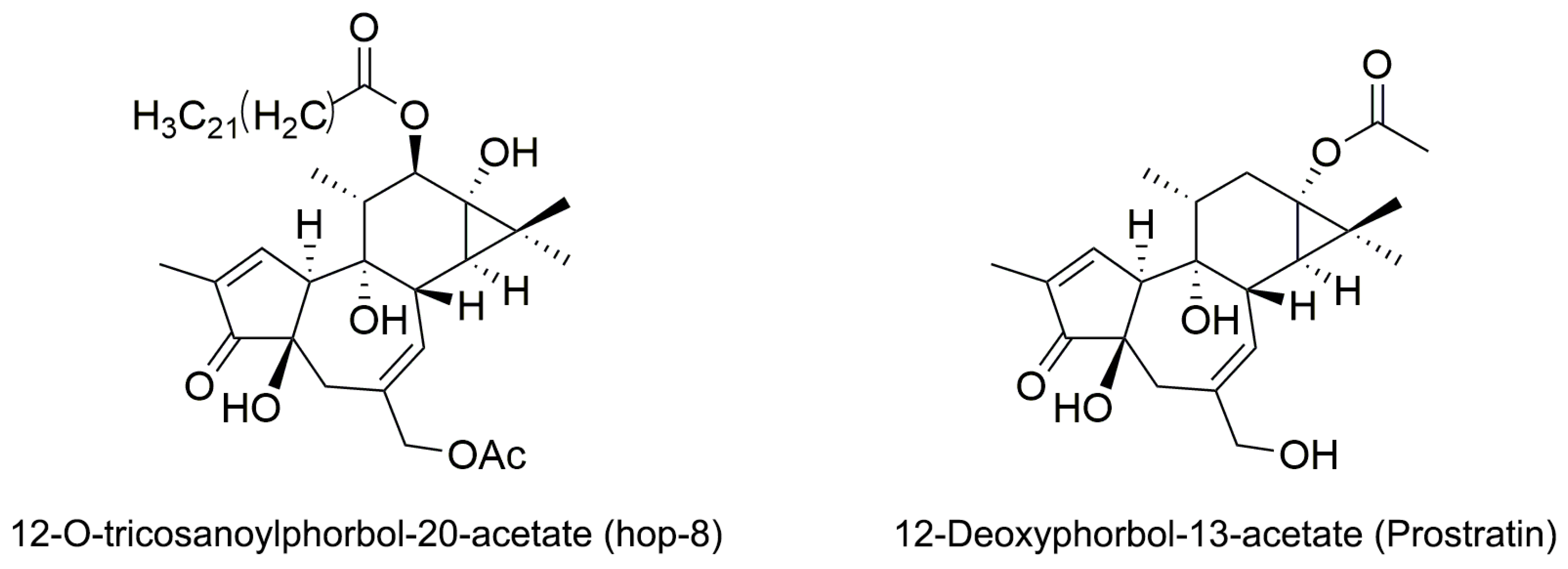
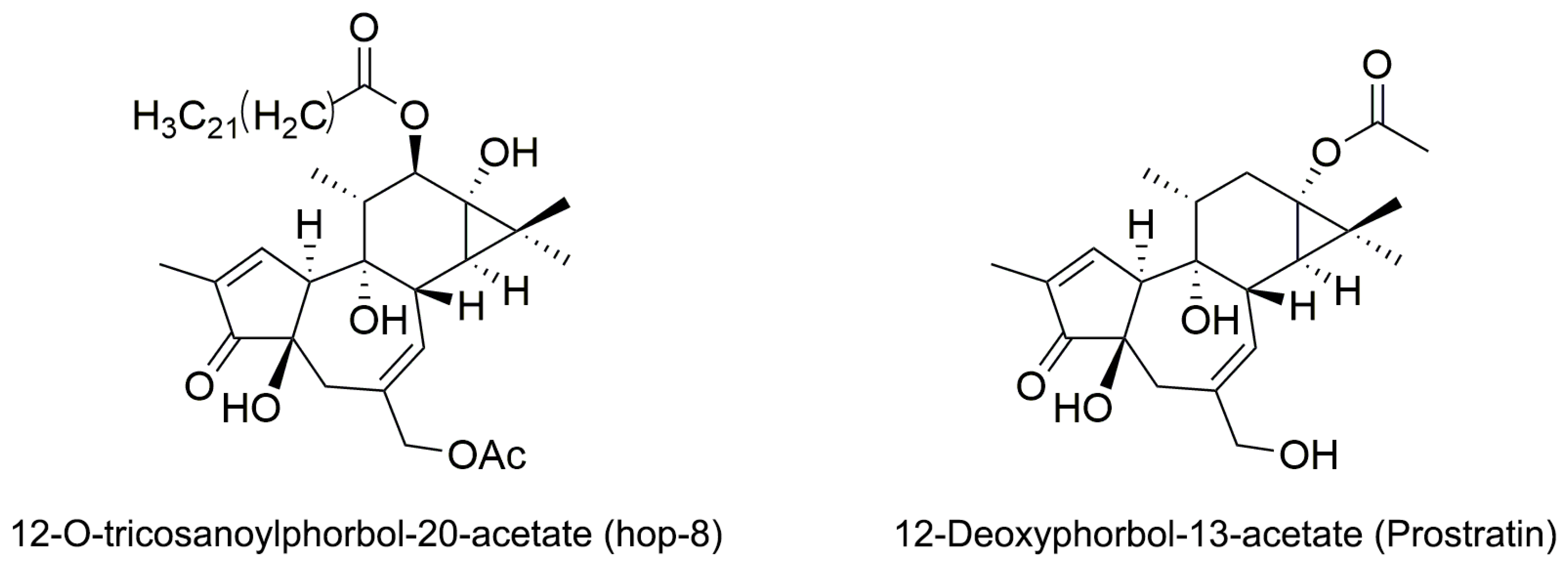
2.1. The Structure Elucidation of Hop-8
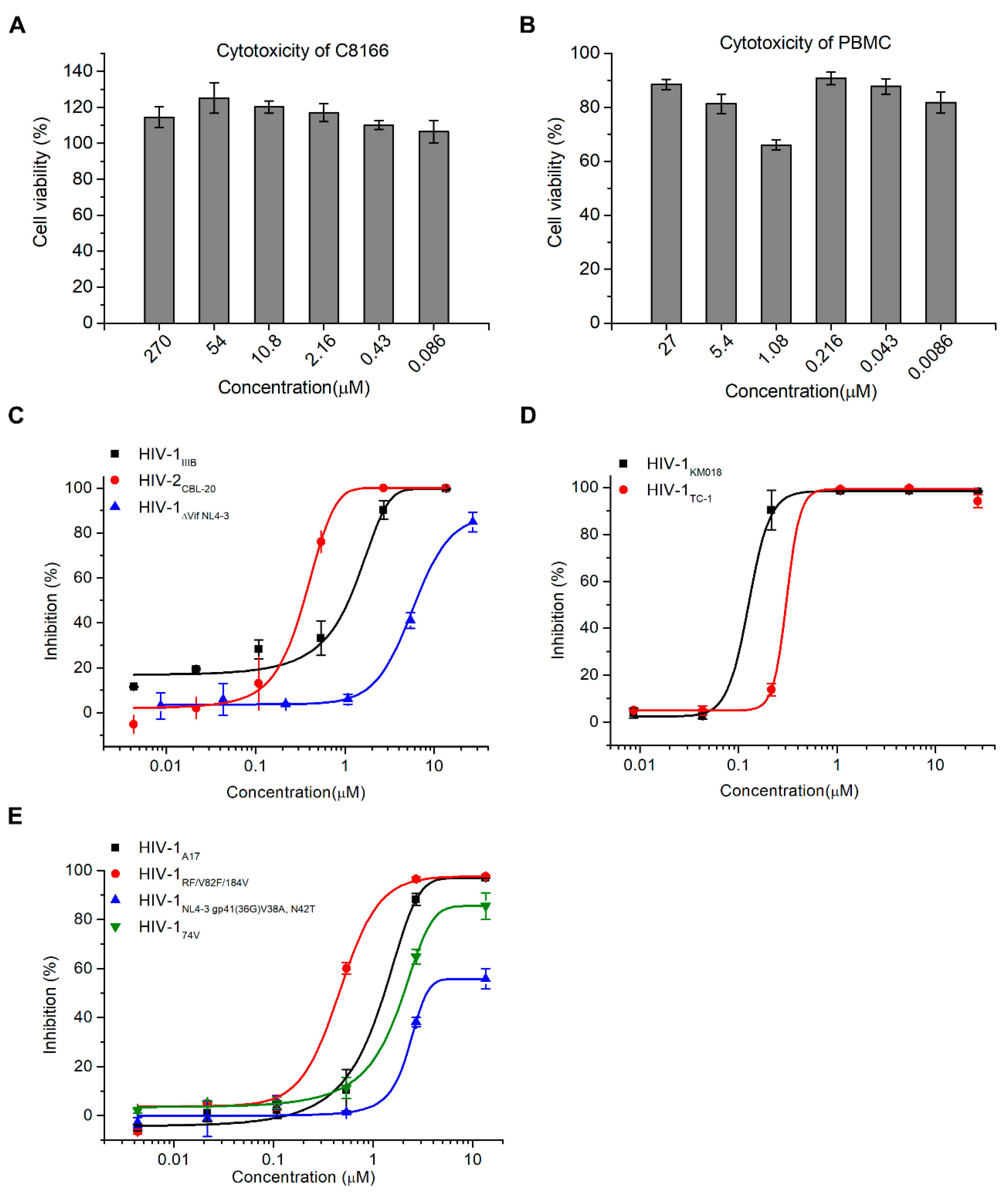
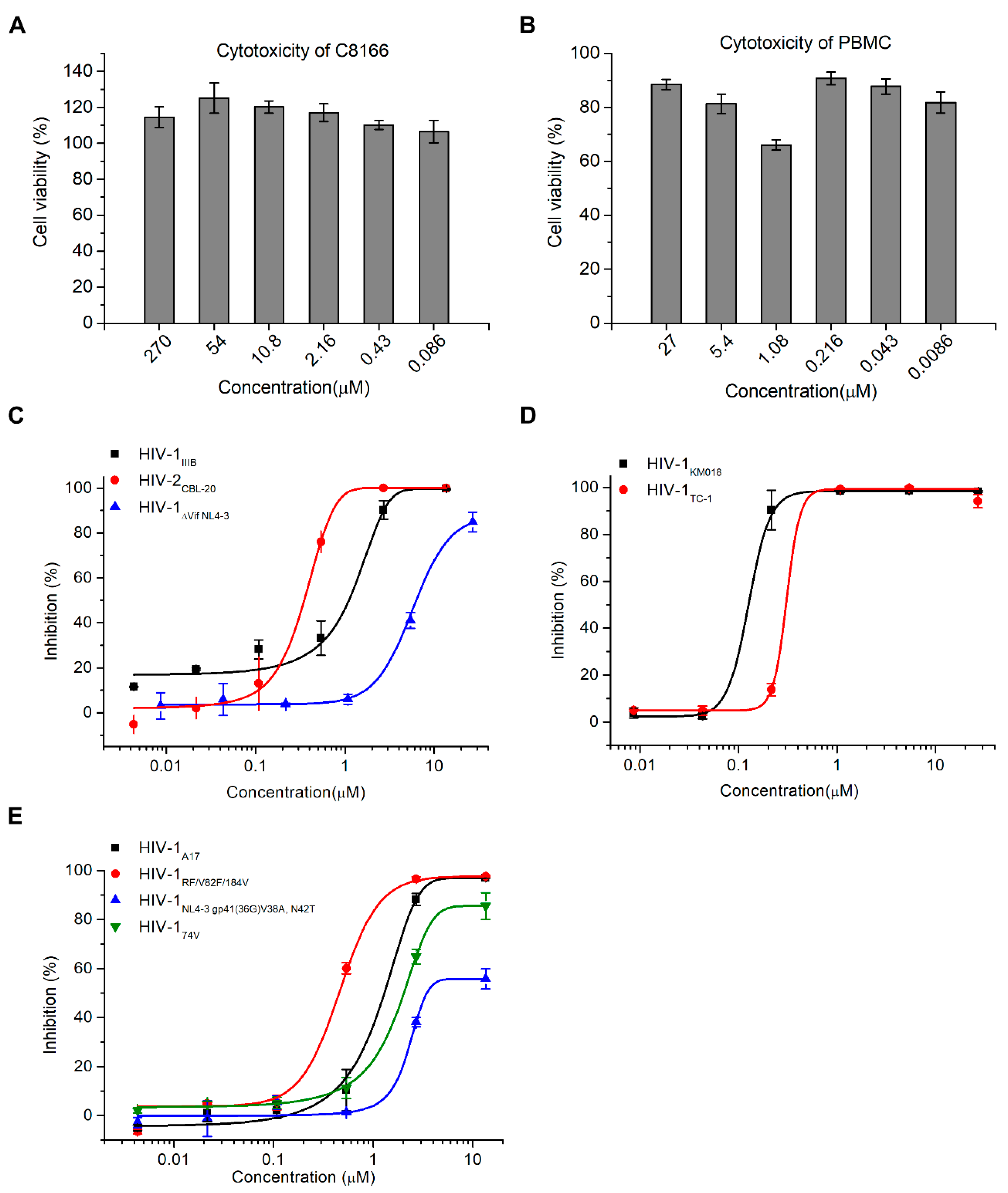
2.2. Hop-8 Significantly Inhibited the Replication of Lab-Adaped HIV-1 and HIV-2, Clinical Isolate Strains, and Drug-Resistant Strains with Low Cytotoxicity
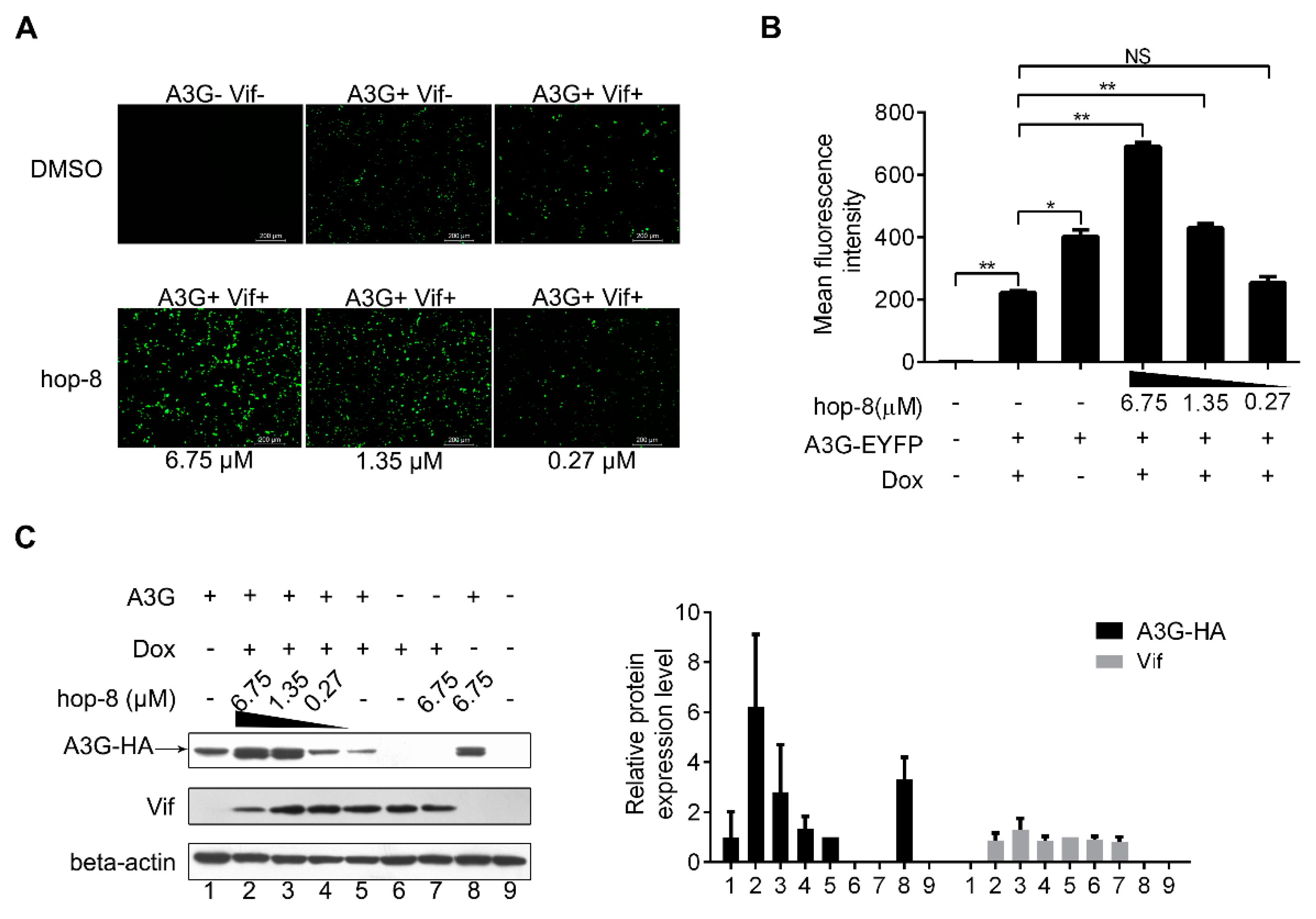
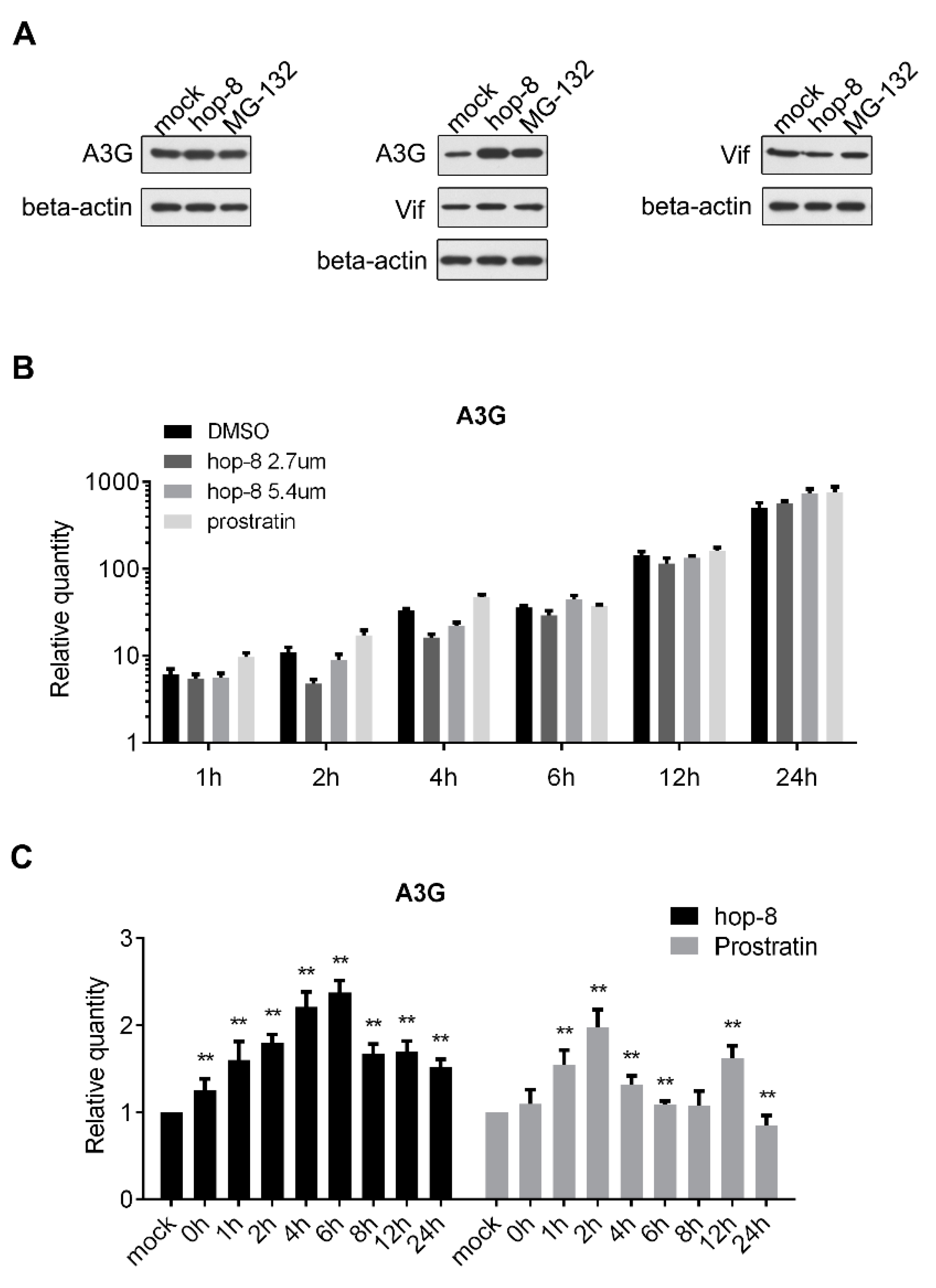
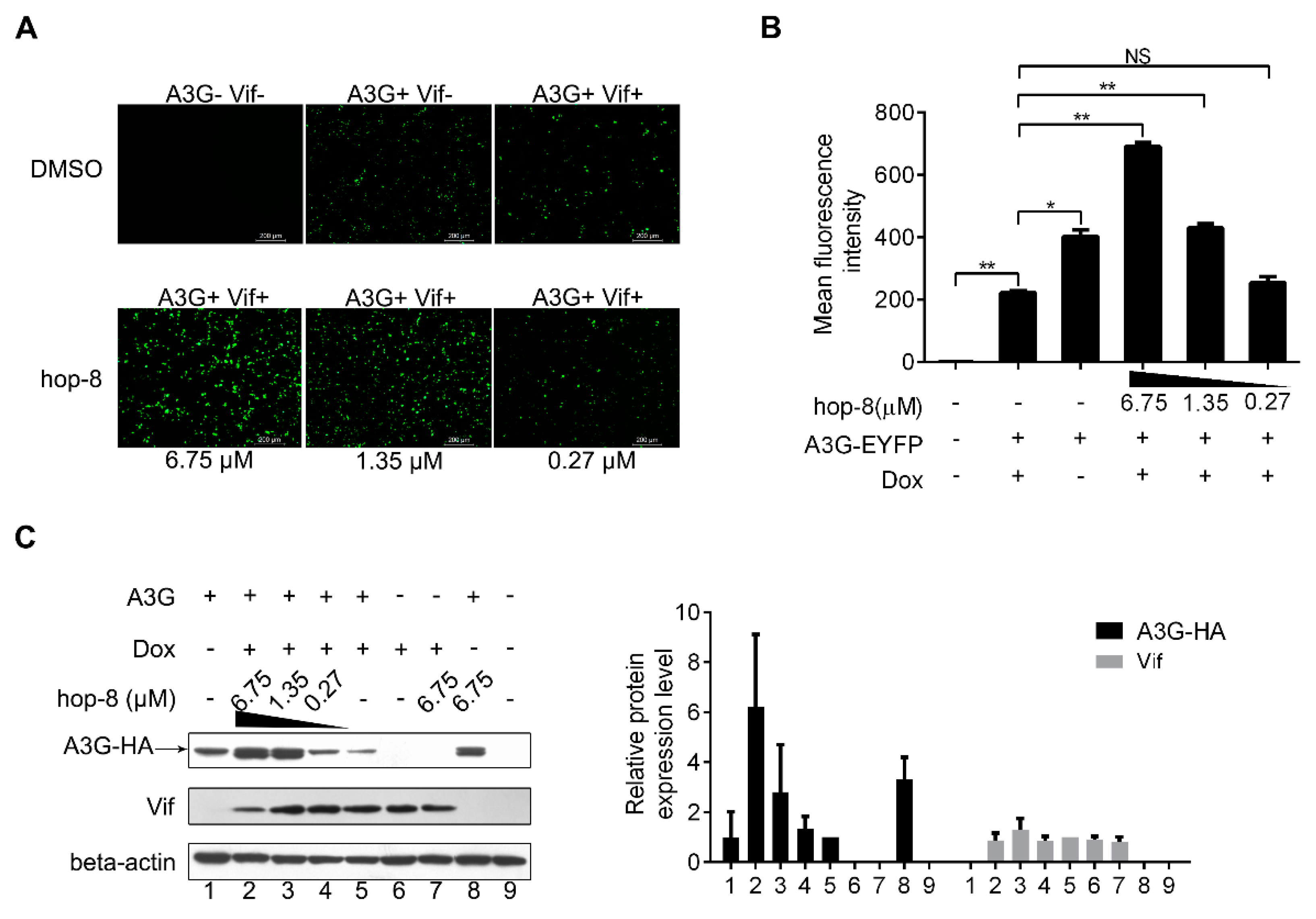
2.3. Hop-8 Restored A3G Levels in Cells Undergoing Vif-Mediated A3G Degradation
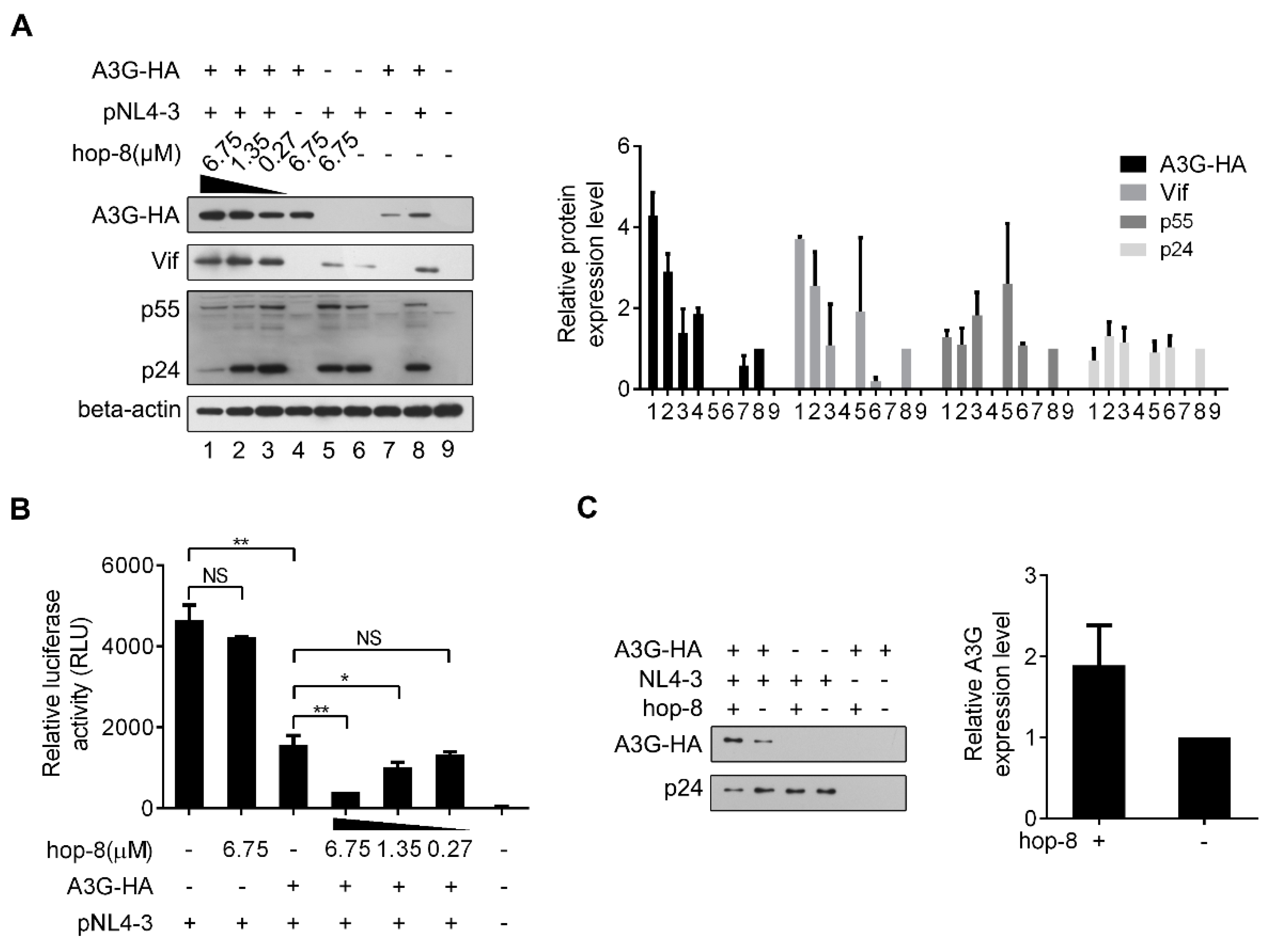
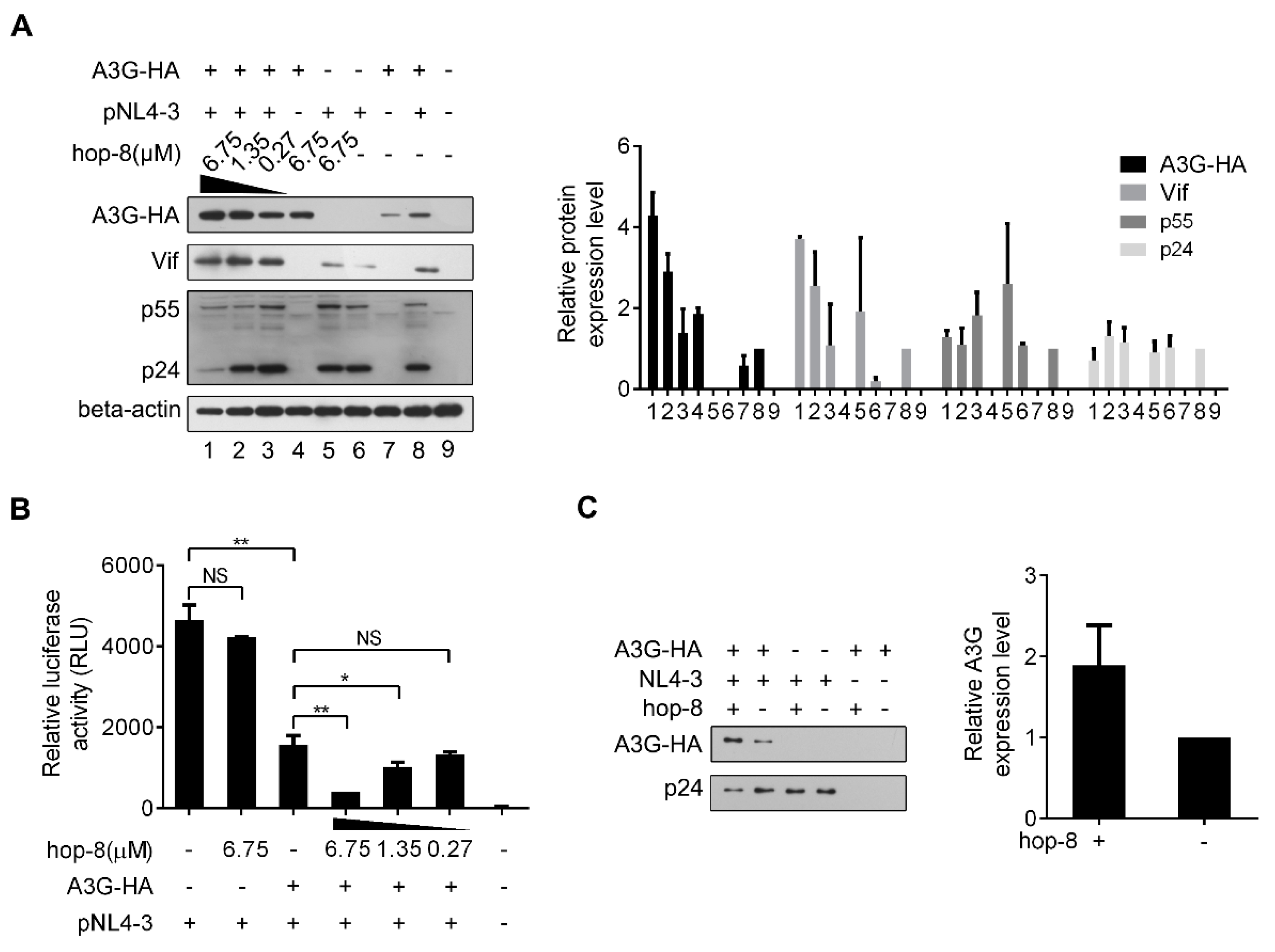
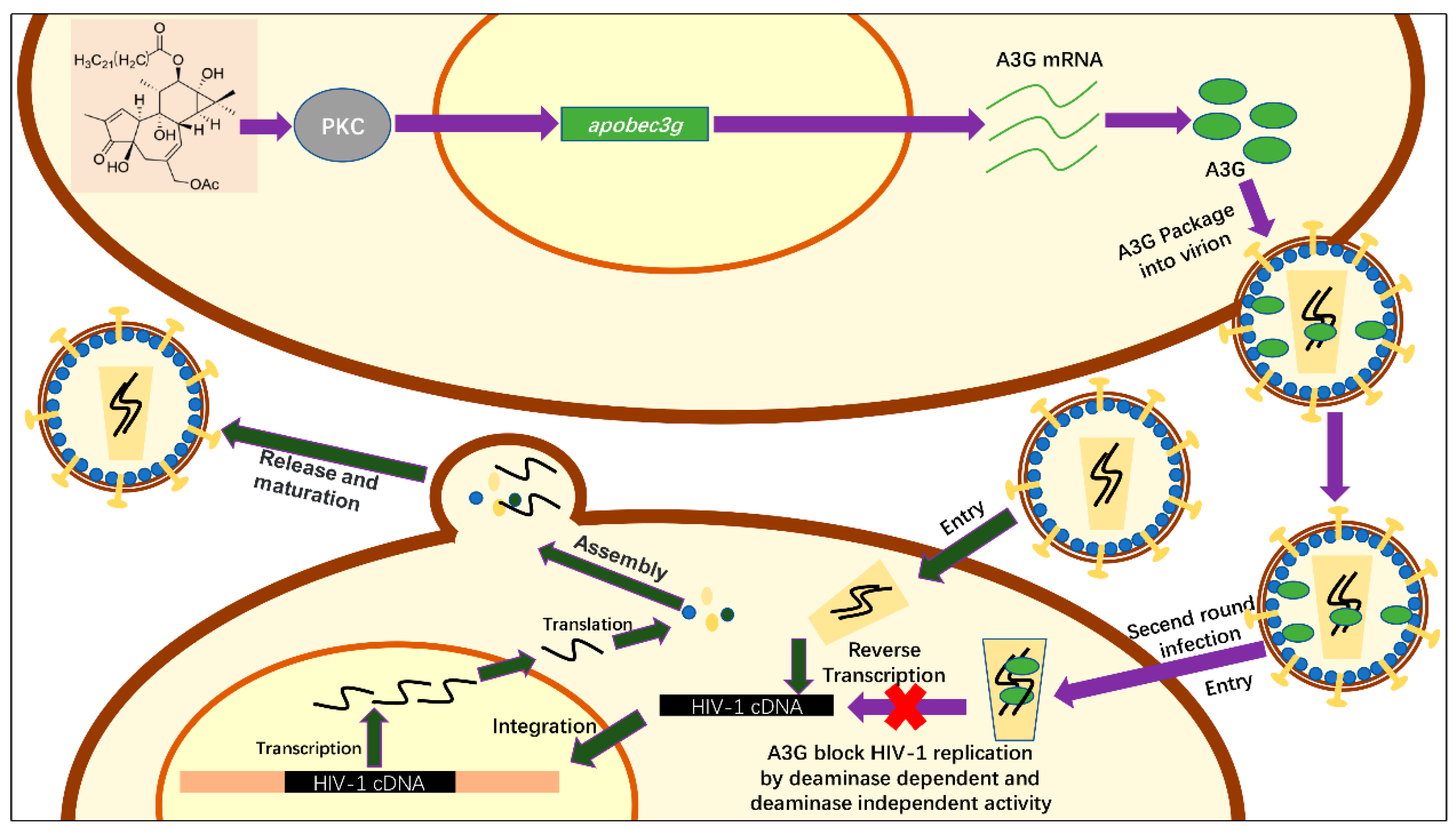
2.4. Hop-8 Upregulated A3G Expression and its Incorporation in the Progeny Virus Reducing its Infectivity
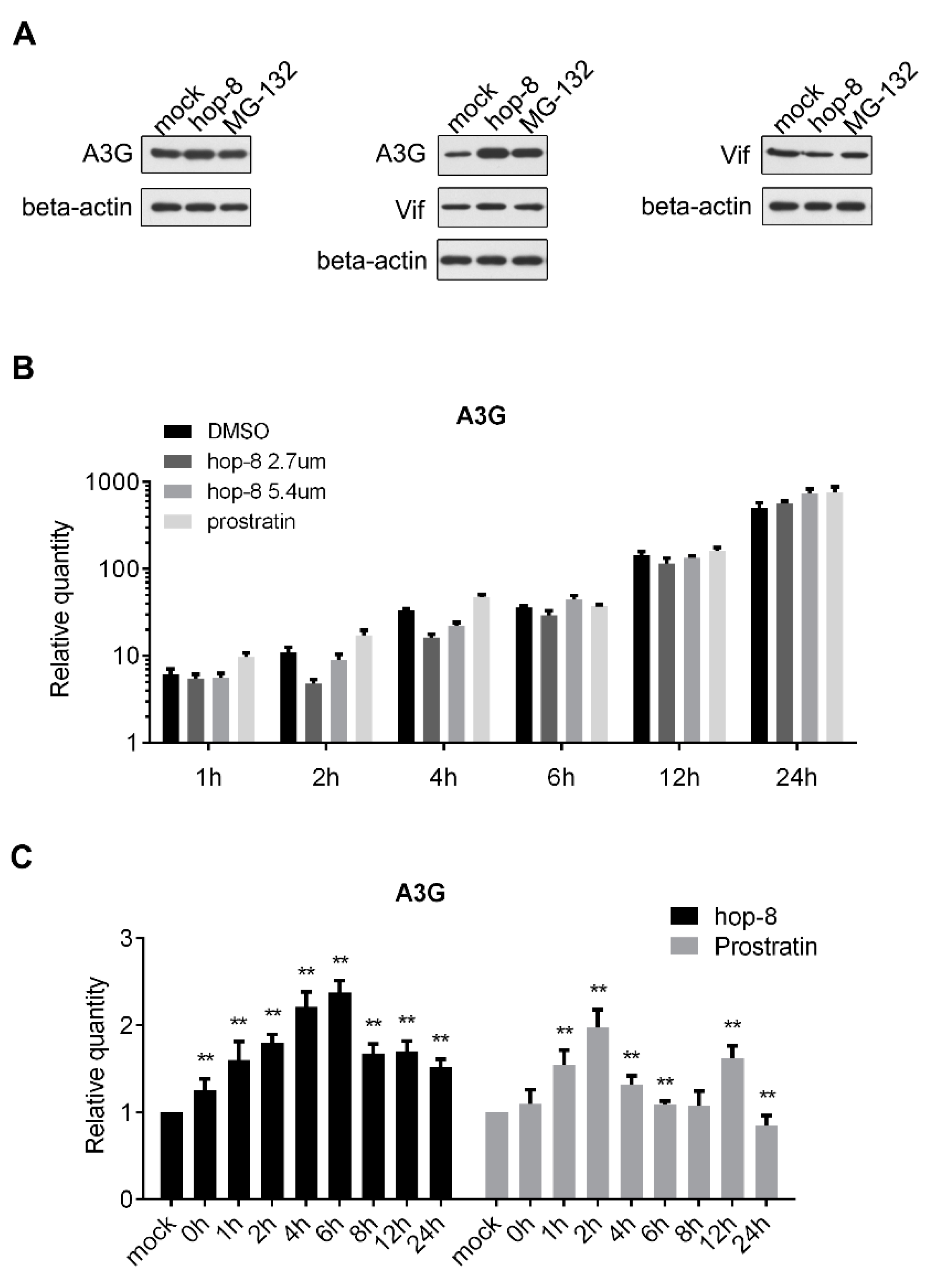
2.5. Hop-8 Upregulated the Expression of A3G at both the Protein and mRNA Levels
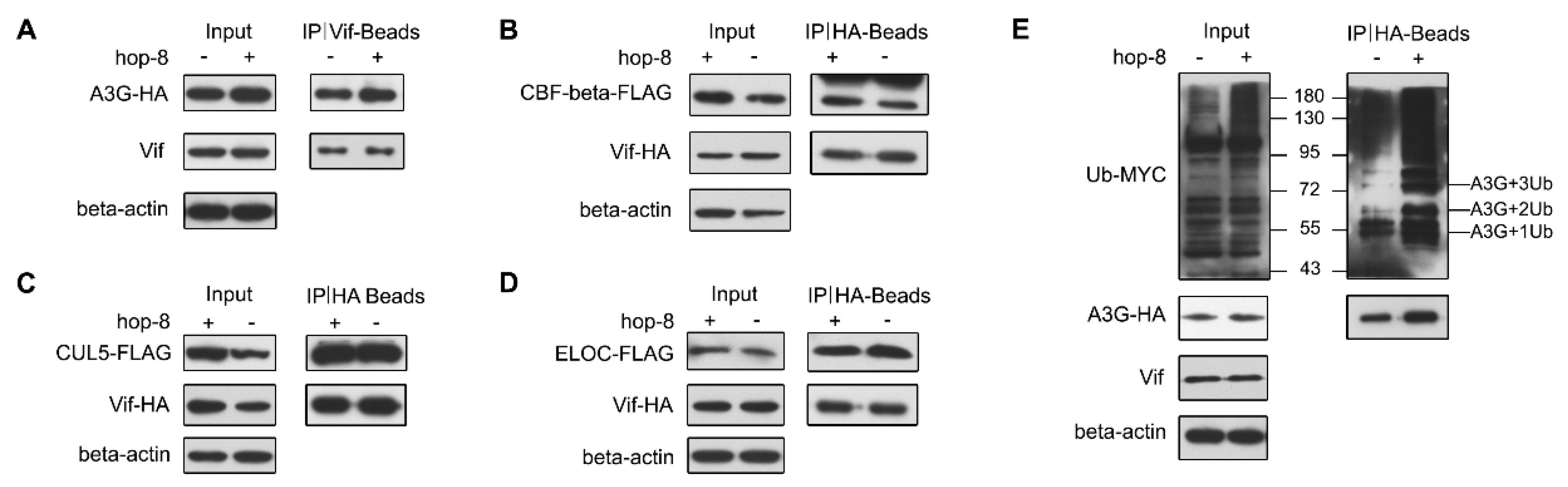
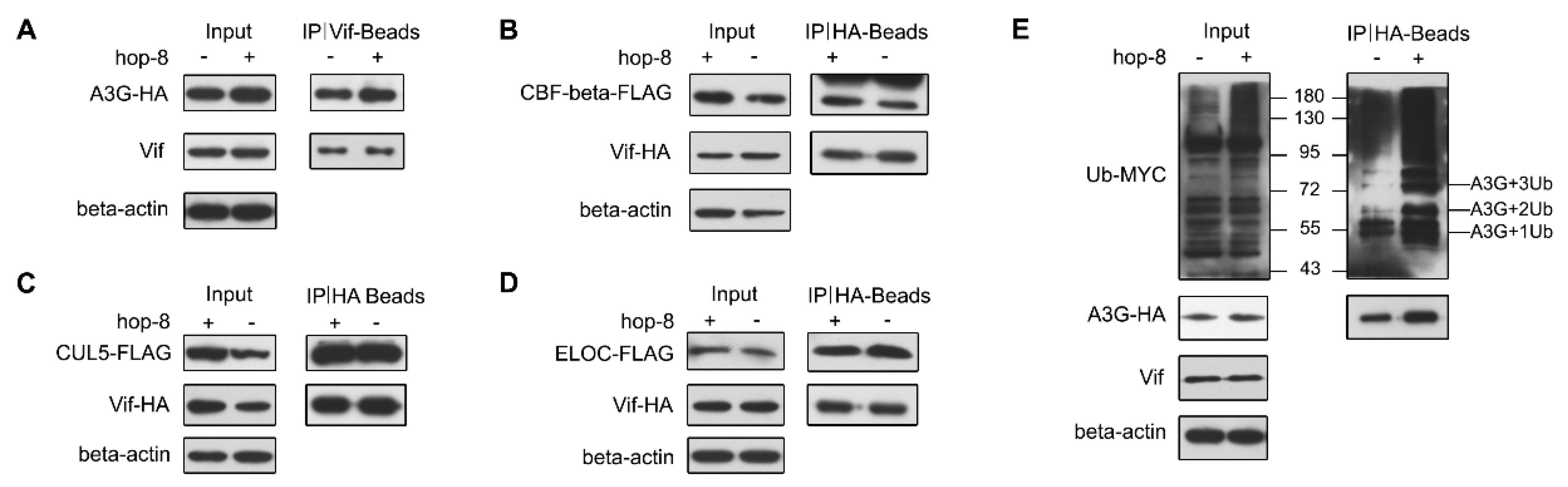
2.6. Hop-8 did not Interfere in Vif Binding with A3G and Recruiting the Cellular ElonginC/B-Cullin 5 E3 Ubiquitin Ligase Complex
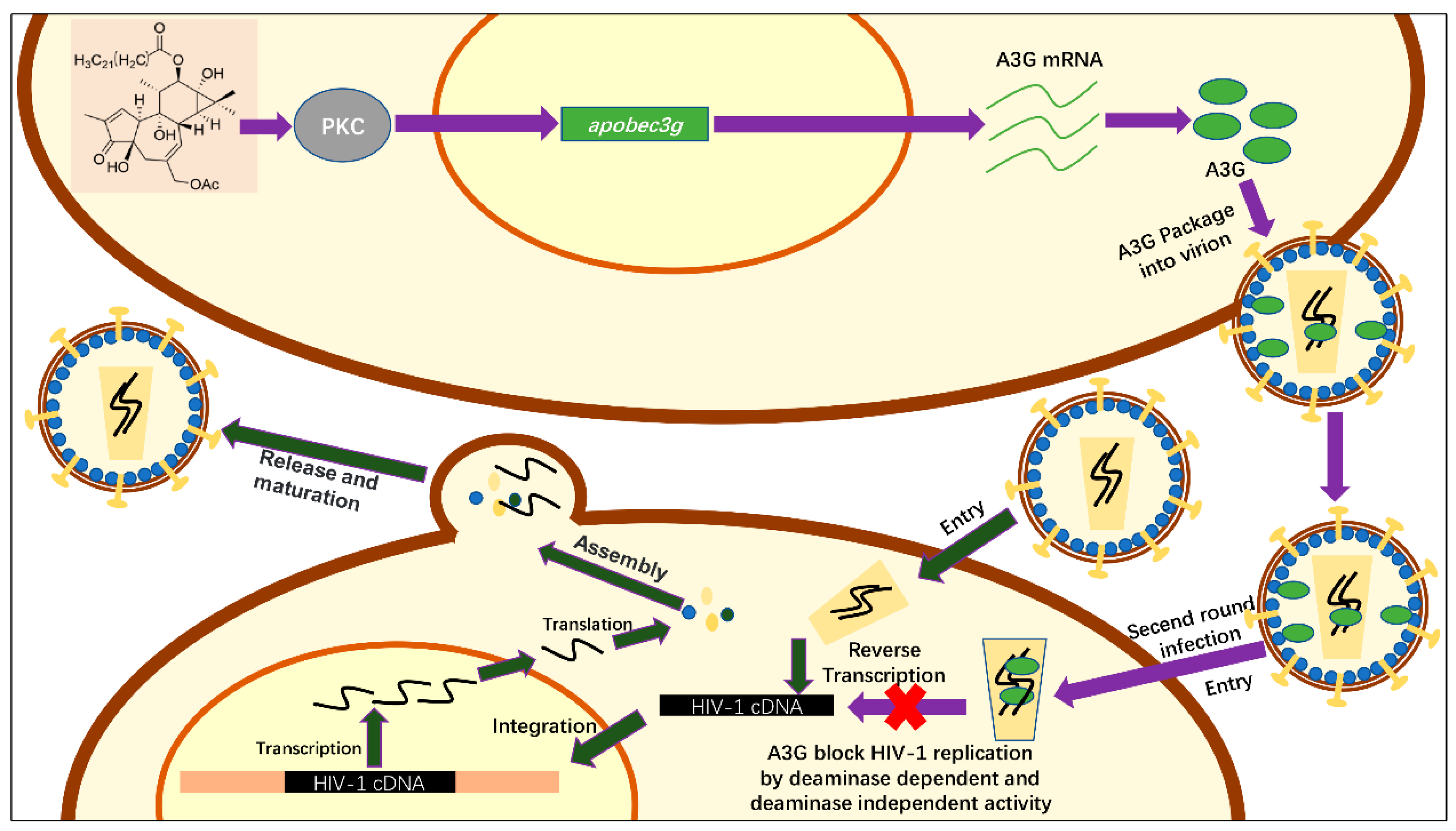
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Compound
4.2.1. Plant Material
4.2.2. Extraction and Isolation
4.2.3. Identification of 12-O-Tricosanoylphorbol-20-acetate (hop-8)
4.3. Cells, Virus, and Plasmids
4.4. Cytotoxicity Assays
4.5. Anti-HIV Activity Assay
4.6. Fluorescence-Based Screening Assay
4.7. HIV-1 Production, Infection, and A3G Incorporation Assay
4.8. Real-Time qPCR
4.9. Western Blot
4.10. Co-Immunoprecipitation (co-IP)
4.11. Information of the Antibodies
4.12. Reverse Transcriptase, Protease, Integrase, and gp41 Inhibition Assays
4.13. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, J.; Maselli, L.M.; Stern, A.C.; Spada, C.; Bydlowski, S.P. Impact of antiretroviral therapy on lipid metabolism of human immunodeficiency virus-infected patients: Old and new drugs. World J. Virol. 2015, 4, 56–77. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.M.; Moecklinghoff, C.; DeMasi, R. When can HIV clinical trials detect treatment effects on drug resistance? Int. J. STD AIDS 2015, 26, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Stolbach, A.; Paziana, K.; Heverling, H.; Pham, P. A Review of the Toxicity of HIV Medications II: Interactions with Drugs and Complementary and Alternative Medicine Products. J. Med. Toxicol. 2015, 11, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host factors mediating HIV-1 replication. Virus Res. 2011, 161, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Giese, S.; Marsh, M. Tetherin Can Restrict Cell-Free and Cell-Cell Transmission of HIV from Primary Macrophages to T Cells. PLoS Pathog. 2014, 10, e1004189. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. CHF Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Yang, H.; Zhu, J.W.; Liu, F.L.; Tian, R.R.; Zheng, H.Y.; Han, J.B.; Shi, P.; Zheng, Y.T. Independent birth of a novel TRIMCyp in Tupaia belangeri with a divergent function from its paralog TRIM5. Mol. Biol. Evol. 2014, 31, 2985–2997. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Liu, F.L.; Mu, D.; Deng, D.Y.; Zheng, Y.T. Increased expression and dysregulated association of restriction factors and type I interferon in HIV, HCV mono- and co-infected patients. J. Med. Virol. 2016, 88, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Rosler, C.; Malim, M.H.; von Weizsacker, F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology 2007, 46, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Wiegand, H.L.; Hulme, A.E.; Garcia-Perez, J.L.; O’Shea, K.S.; Moran, J.V.; Cullen, B.R. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Chaipan, C.; Smith, J.L.; Hu, W.S.; Pathak, V.K. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 2013, 87, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Iwatani, Y. APOBEC3G-Mediated G-to-A Hypermutation of the HIV-1 Genome: The Missing Link in Antiviral Molecular Mechanisms. Front. Microbiol. 2016, 7, 2027. [Google Scholar] [CrossRef] [PubMed]
- Opi, S.; Kao, S.; Goila-Gaur, R.; Khan, M.A.; Miyagi, E.; Takeuchi, H.; Strebel, K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J. Virol. 2007, 81, 8236–8246. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Ode, H.; Iwatani, Y. Structural Features of Antiviral APOBEC3 Proteins are Linked to Their Functional Activities. Front. Microbiol. 2011, 2, 258. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Prabhu, P.; Kenig, E.; Smith, Y.; Britan-Rosich, E.; Kotler, M. APOBEC3G inhibits HIV-1 RNA elongation by inactivating the viral trans-activation response element. J. Mol. Biol. 2014, 426, 2840–2853. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhtenko, L.S.; Dutta, S.; Banga, J.; Li, M.; Harris, R.S.; Lyubchenko, Y.L. APOBEC3G Interacts with ssDNA by Two Modes: AFM Studies. Sci. Rep. 2015, 5, 15648. [Google Scholar] [CrossRef] [PubMed]
- Bouyac, M.; Courcoul, M.; Bertoia, G.; Baudat, Y.; Gabuzda, D.; Blanc, D.; Chazal, N.; Boulanger, P.; Sire, J.; Vigne, R.; et al. Human immunodeficiency virus type 1 Vif protein binds to the Pr55Gag precursor. J. Virol. 1997, 71, 9358–9365. [Google Scholar] [PubMed]
- Goncalves, J.; Shi, B.; Yang, X.; Gabuzda, D. Biological activity of human immunodeficiency virus type 1 Vif requires membrane targeting by C-terminal basic domains. J. Virol. 1995, 69, 7196–7204. [Google Scholar] [PubMed]
- Kao, S.; Goila-Gaur, R.; Miyagi, E.; Khan, M.A.; Opi, S.; Takeuchi, H.; Strebel, K. Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology 2007, 369, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, D.; Bernacchi, S.; Xavier Guerrero, S.; Brachet, F.; Larue, V.; Paillart, J.C.; Tisne, C. Characterization of RNA binding and chaperoning activities of HIV-1 Vif protein. Importance of the C-terminal unstructured tail. RNA Biol. 2014, 11, 906–920. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, K.S.; Leal, E.; dos Santos, A.M.; Lima e Lima, A.H.; Alves, C.N.; Lameira, J. Structural analysis of viral infectivity factor of HIV type 1 and its interaction with A3G, EloC and EloB. PLoS ONE 2014, 9, e89116. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Booiman, T.; Kootstra, N.; Simon, V.; Ooms, M. Identification of the HIV-1 Vif and Human APOBEC3G Protein Interface. Cell Rep. 2015, 13, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, A.M.; Yu, X.F. Host restriction of HIV-1 by APOBEC3 and viral evasion through Vif. Curr. Top. Microbiol. 2009, 339, 1–25. [Google Scholar]
- Bohn, M.F.; Shandilya, S.M.; Albin, J.S.; Kouno, T.; Anderson, B.D.; McDougle, R.M.; Carpenter, M.A.; Rathore, A.; Evans, L.; Davis, A.N.; et al. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding domain. Structure 2013, 21, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Cen, S.; Peng, Z.G.; Li, X.Y.; Li, Z.R.; Ma, J.; Wang, Y.M.; Fan, B.; You, X.F.; Wang, Y.P.; Liu, F.; et al. Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J. Biol. Chem. 2010, 285, 16546–16552. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zuo, T.; Luo, X.; Jin, H.; Liu, Z.; Yang, Z.; Yu, X.; Zhang, L.; Zhang, L. Indolizine derivatives as HIV-1 VIF-ElonginC interaction inhibitors. Chem. Biol. Drug Des. 2013, 81, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Nathans, R.; Cao, H.; Sharova, N.; Ali, A.; Sharkey, M.; Stranska, R.; Stevenson, M.; Rana, T.M. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 2008, 26, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Pery, E.; Sheehy, A.; Nebane, N.M.; Misra, V.; Mankowski, M.K.; Rasmussen, L.; White, E.L.; Ptak, R.G.; Gabuzda, D. Redoxal, an inhibitor of de novo pyrimidine biosynthesis, augments APOBEC3G antiviral activity against human immunodeficiency virus type 1. Virology 2015, 484, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Zhong, L.M.; Chen, B.; Pan, T.; Zhang, X.; Liang, L.T.; Li, Q.W.; Zhang, Z.Y.; Chen, H.; Zhou, J.; et al. Identification of an HIV-1 replication inhibitor which rescues host restriction factor APOBEC3G in Vif-APOBEC3G complex. Antivir. Res. 2015, 122, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Sarkis, P.T.N.; Ying, S.; Xu, R.; Yu, X.F. STAT1-Independent Cell Type-Specific Regulation of Antiviral APOBEC3G by IFN-alpha. J. Immunol. 2006, 177, 4530–4540. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.M.; Marin, M.; Kozak, S.L.; Kabat, D. Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J. Biol. Chem. 2004, 279, 41744–41749. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, H.; Zhang, X.; Shang, J.; Kang, Y. Basal transcription of APOBEC3G is regulated by USF1 gene in hepatocyte. Biochem. Biophys. Res. Commun. 2016, 470, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Bogi, K.; Lorenzo, P.S.; Szallasi, Z.; Acs, P.; Wagner, G.S.; Blumberg, P.M. Differential selectivity of ligands for the C1a and C1b phorbol ester binding domains of protein kinase Cdelta: Possible correlation with tumor-promoting activity. Cancer Res. 1998, 58, 1423–1428. [Google Scholar] [PubMed]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [PubMed]
- El-Mekkawy, S.; Meselhy, M.R.; Abdel-Hafez, A.A.; Nakamura, N.; Hattori, M.; Kawahata, T.; Otake, T. Inhibition of cytopathic effect of human immunodeficiency virus type-1 by various phorbol derivatives. Chem. Pharm. Bull. (Tokyo) 2002, 50, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Kee, J.M.; Warrington, J.M. Practical synthesis of prostratin, DPP, and their analogs, adjuvant leads against latent HIV. Science 2008, 320, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Blumberg, P.; Hamer, D. Activation of latent HIV-1 expression by the potent anti-tumor promoter 12-deoxyphorbol 13-phenylacetate. Antivir. Res. 2003, 59, 89–98. [Google Scholar] [CrossRef]
- Witvrouw, M.; Pannecouque, C.; Fikkert, V.; Hantson, A.; Van Remoortel, B.; Hezareh, M.; De Clercq, E.; Brown, S.J. Potent and selective inhibition of HIV and SIV by prostratin interacting with viral entry. Antivir. Chem. Chemother. 2003, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Miana, G.A.; Riaz, M.; Shahzad-ul-Hussan, S.; Paracha, R.Z.; Paracha, U.Z. Prostratin: An Overview. Mini Rev. Med. Chem. 2015, 15, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Chu, H.; Felding, J.; Baran, P.S. Nineteen-step total synthesis of (+)-phorbol. Nature 2016, 532, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Parai, M.K.; Jiang, X.; Sharova, N.; Singh, G.; Stevenson, M.; Rana, T.M. SAR and Lead Optimization of an HIV-1 Vif-APOBEC3G Axis Inhibitor. ACS Med. Chem. Lett. 2012, 3, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.P.; Stewart, R.A.; Hogan, P.A.; Ptak, R.G.; Mankowski, M.K.; Hartman, T.L.; Buckheit, R.W., Jr.; Snyder, B.A.; Salter, J.D.; Morales, G.A.; et al. An analog of camptothecin inactive against Topoisomerase I is broadly neutralizing of HIV-1 through inhibition of Vif-dependent APOBEC3G degradation. Antivir. Res. 2016, 136, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Shindo, K.; Izumi, T.; Io, K.; Shinohara, M.; Komano, J.; Kobayashi, M.; Kadowaki, N.; Harris, R.S.; Takaori-Kondo, A. Small molecules that inhibit Vif-induced degradation of APOBEC3G. Virol. J. 2014, 11, 122. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Liang, W.; Hua, D.; Zhou, X.; Du, J.; Evans, S.L.; Gao, Q.; Wang, H.; Viqueira, R.; Wei, W.; et al. Evolutionarily conserved requirement for core binding factor beta in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-cullin 5-RING E3 ubiquitin ligase. J. Virol. 2014, 88, 3320–3328. [Google Scholar] [CrossRef] [PubMed]
- Ruybal, P.; Gravisaco, M.J.; Barcala, V.; Escalada, A.; Cremaschi, G.; Taboga, O.; Waldner, C.; Mongini, C. Transgene expression enhancement in T-lymphoma cell lines. Int. Immunopharmacol. 2005, 5, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Neudorf, S.; Jones, M.; Parker, S.; Papes, R.; Lattier, D. Phorbol esters down-regulate transcription and translation of the CD4 gene. J. Immunol. 1991, 146, 2836–2840. [Google Scholar] [PubMed]
- Nakamura, K.; Sasada, T.; Sono, H.; Yodoi, J. Inhibition of protein kinase C-mediated CD4 down-regulation by oxidative stress in T lymphocytes. J. Immunol. 1996, 157, 5339–5349. [Google Scholar] [PubMed]
- Bai, R.; Zhang, X.J.; Li, Y.L.; Liu, J.P.; Zhang, H.B.; Xiao, W.L.; Pu, J.X.; Sun, H.D.; Zheng, Y.T.; Liu, L.X. SJP-L-5, a novel small-molecule compound, inhibits HIV-1 infection by blocking viral DNA nuclear entry. BMC Microbiol. 2015, 15, 274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Wang, Q.; Chen, J.J.; Zhang, X.M.; Tam, S.C.; Zheng, Y.T. The anti-HIV-1 effect of scutellarin. Biochem. Biophys. Res. Commun. 2005, 334, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, L.M.; Liu, G.M.; Liu, Y.J.; Zheng, C.B.; Lv, Y.J.; Li, H.Z.; Zheng, Y.T. Potent anti-HIV activities and mechanisms of action of a pine cone extract from Pinus yunnanensis. Molecules 2012, 17, 6916–6929. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Wang, J.P.; Xiao, J.C.; Zhao, Z.W.; Zheng, Y.T. Preparation and characterization of three monoclonal antibodies against HIV-1 p24 capsid protein. Cell. Mol. Immunol. 2007, 4, 203–208. [Google Scholar] [PubMed]
- Wang, R.R.; Yang, Q.H.; Luo, R.H.; Peng, Y.M.; Dai, S.X.; Zhang, X.J.; Chen, H.; Cui, X.Q.; Liu, Y.J.; Huang, J.F.; et al. Azvudine, A Novel Nucleoside Reverse Transcriptase Inhibitor Showed Good Drug Combination Features and Better Inhibition on Drug-Resistant Strains than Lamivudine In Vitro. PLoS ONE 2014, 9, e105617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Luo, R.H.; Hou, X.Y.; Wang, R.R.; Yan, G.Y.; Chen, H.; Zhang, R.H.; Shi, J.Y.; Zheng, Y.T.; Li, R.; et al. Synthesis, biological evaluation and molecular docking study of N-(2-methoxyphenyl)-6-((4-nitrophenyl)sulfonyl)benzamide derivatives as potent HIV-1 Vif antagonists. Eur. J. Med. Chem. 2017, 129, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Wang, R.R.; Gao, Y.D.; Yang, L.M.; Sun, Y.; Huang, J.F.; Tien, P.; Zheng, Y.T. A novel enzyme-linked immunosorbent assay for screening HIV-1 fusion inhibitors targeting HIV-1 Gp41 core structure. J. Biomol. Screen. 2011, 16, 221–229. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Virus | EC50 (μM) | |
|---|---|---|---|
| Hop-8 | Prostratin | ||
| PBMC | HIV-1KM018 | 0.106 ± 0.003 | 0.948 ± 0.146 |
| HIV-1TC-1 | 0.390 ± 0.038 | ND 2 | |
| C8166 | HIV-1IIIB | 0.873 ± 0.005 | 3.701 ± 0.803 |
| HIV-1ΔVif NL4-3 | 7.987 ± 0.481 | > 10 | |
| HIV-1NL4-3 gp41 (36G) V38A, N42T | 6.915 ± 1.053 | > 10 | |
| HIV-1A17 | 1.303 ± 0.078 | 3.340 ± 2.075 | |
| HIV-1RF/V82F/184V | 0.396 ± 0.005 | 4.069 ± 2.531 | |
| HIV-174V | 1.828 ± 0.104 | 2.457 ± 0.483 | |
| HIV-2CBL-20 1 | 0.255 ± 0.023 | 0.568 ± 0.154 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Zhang, R.; Luo, R.-H.; Yang, L.-M.; Wang, R.-R.; Hao, X.-J.; Zheng, Y.-T. Anti-HIV Activities and Mechanism of 12-O-Tricosanoylphorbol-20-acetate, a Novel Phorbol Ester from Ostodes katharinae. Molecules 2017, 22, 1498. https://doi.org/10.3390/molecules22091498
Chen H, Zhang R, Luo R-H, Yang L-M, Wang R-R, Hao X-J, Zheng Y-T. Anti-HIV Activities and Mechanism of 12-O-Tricosanoylphorbol-20-acetate, a Novel Phorbol Ester from Ostodes katharinae. Molecules. 2017; 22(9):1498. https://doi.org/10.3390/molecules22091498
Chicago/Turabian StyleChen, Huan, Rong Zhang, Rong-Hua Luo, Liu-Meng Yang, Rui-Rui Wang, Xiao-Jiang Hao, and Yong-Tang Zheng. 2017. "Anti-HIV Activities and Mechanism of 12-O-Tricosanoylphorbol-20-acetate, a Novel Phorbol Ester from Ostodes katharinae" Molecules 22, no. 9: 1498. https://doi.org/10.3390/molecules22091498