Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs


Abstract
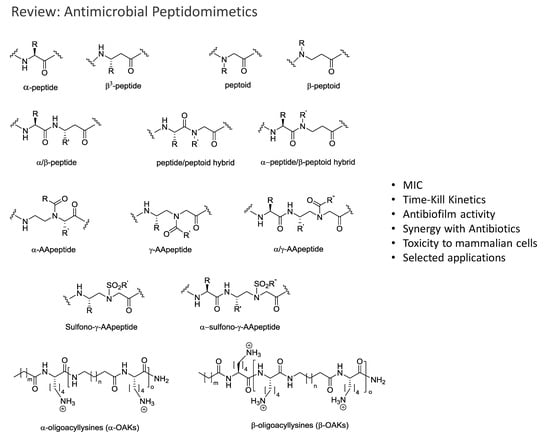
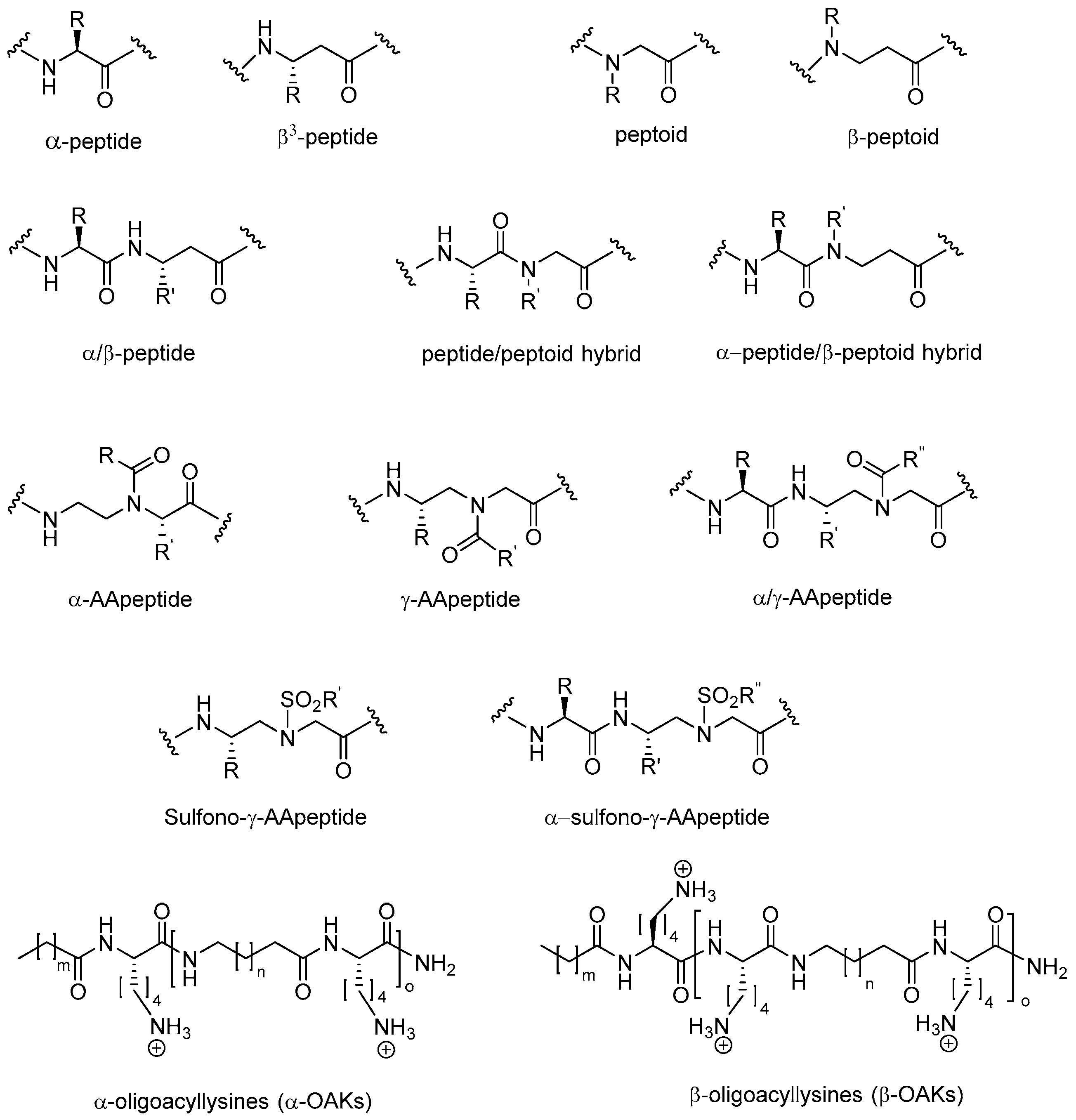
:
1. Introduction
1.1. Antimicrobial Resistance
1.2. Development of Antibiotics in Recent Years
1.3. Antimicrobial Peptides (AMPs)
1.4. AMPs as Therapeutic Agents
Resistance to AMPs and Peptidomimetics
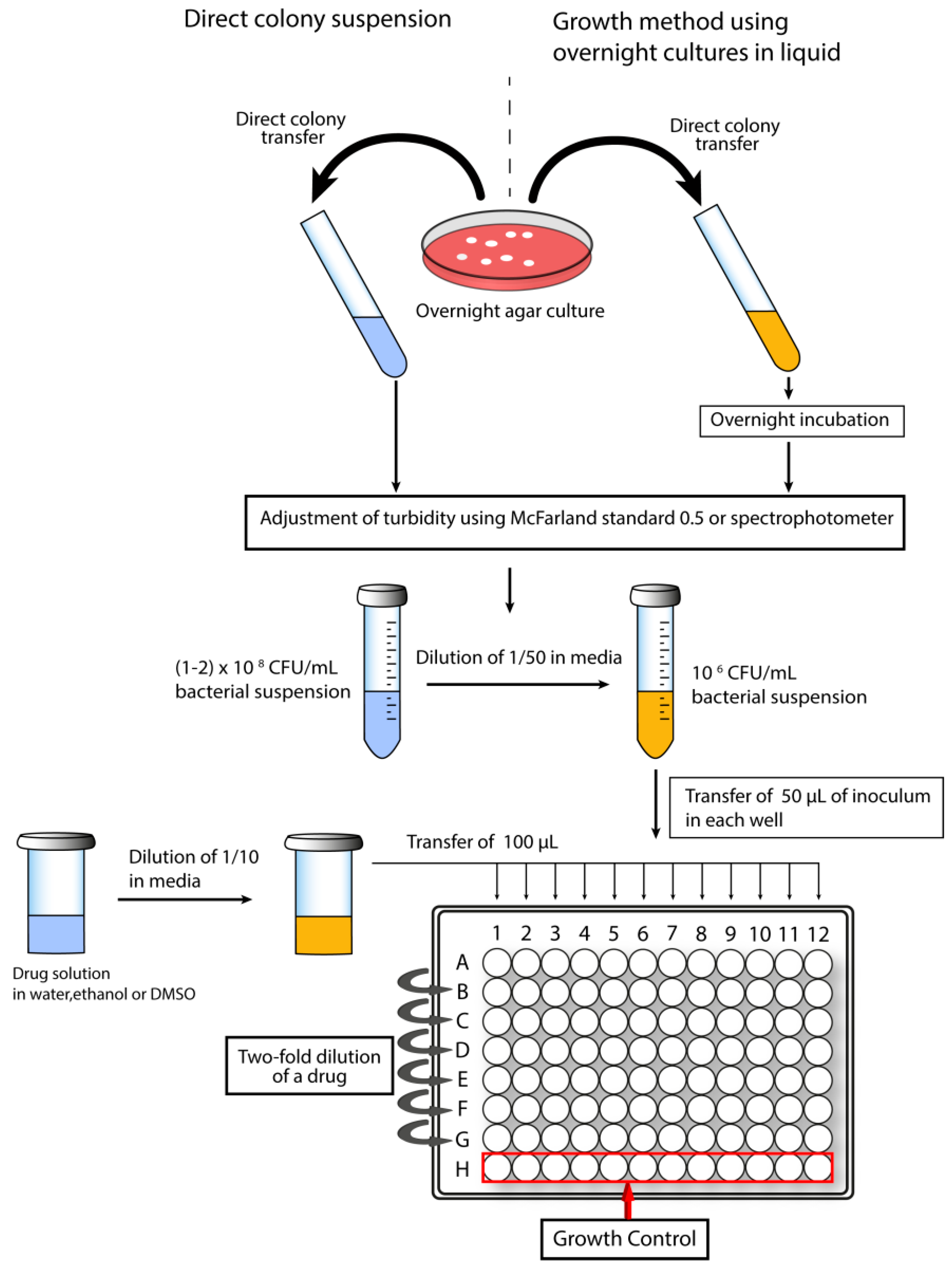
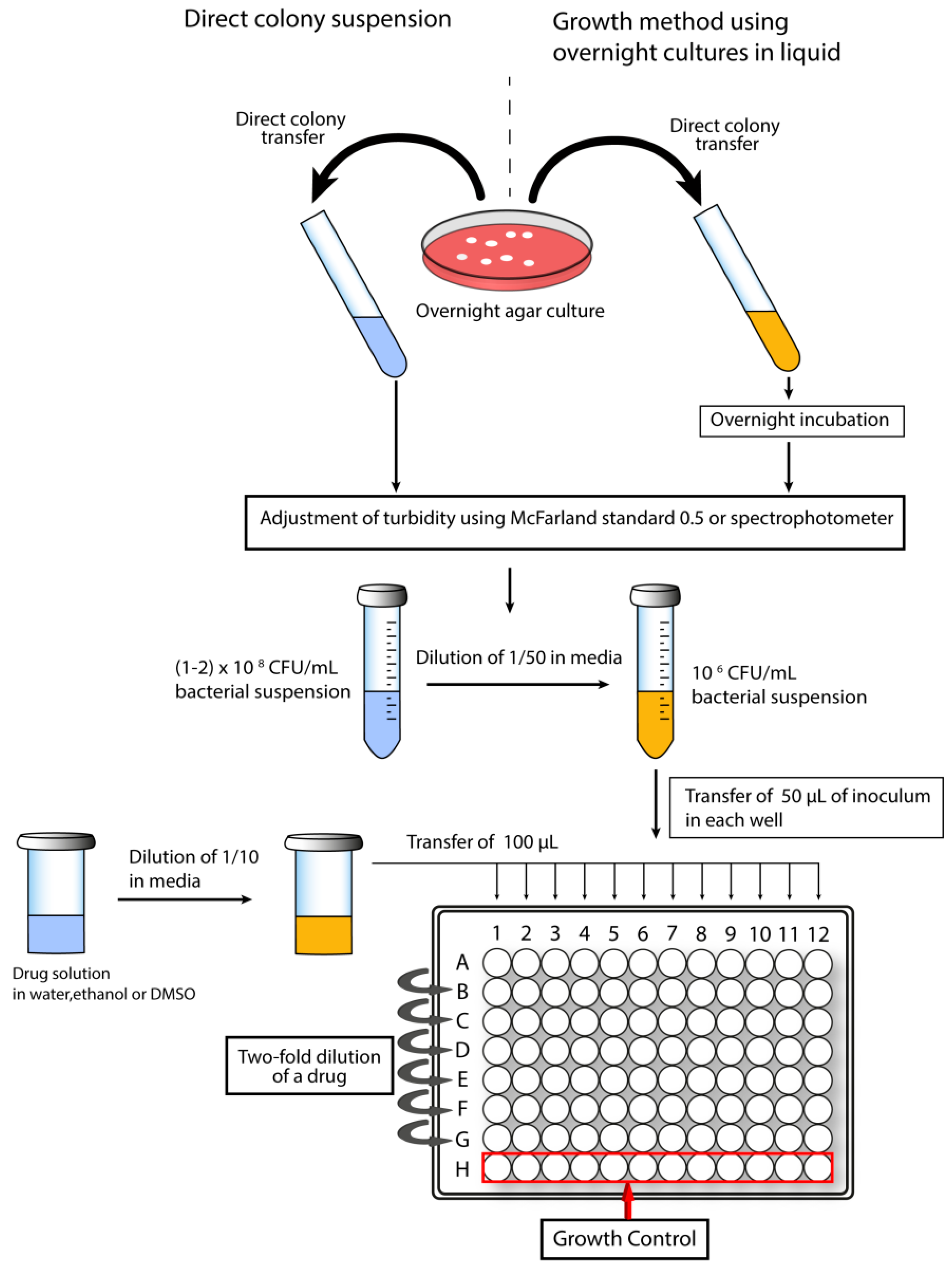
1.5. Screening for Antimicrobial Activity and Cytotoxicity of Peptidomimetics
1.5.1. Determination of Minimal Inhibitory Concentration
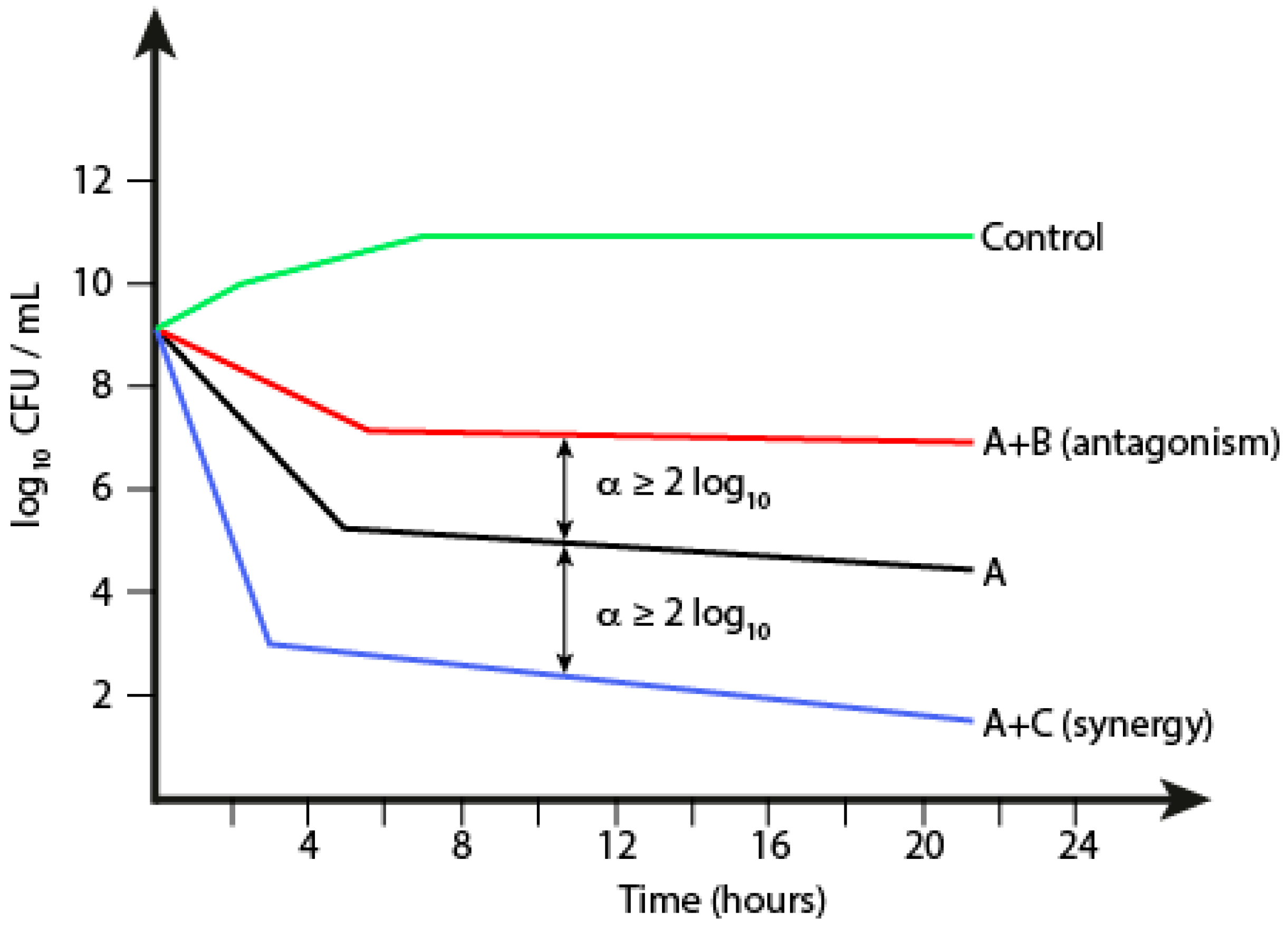
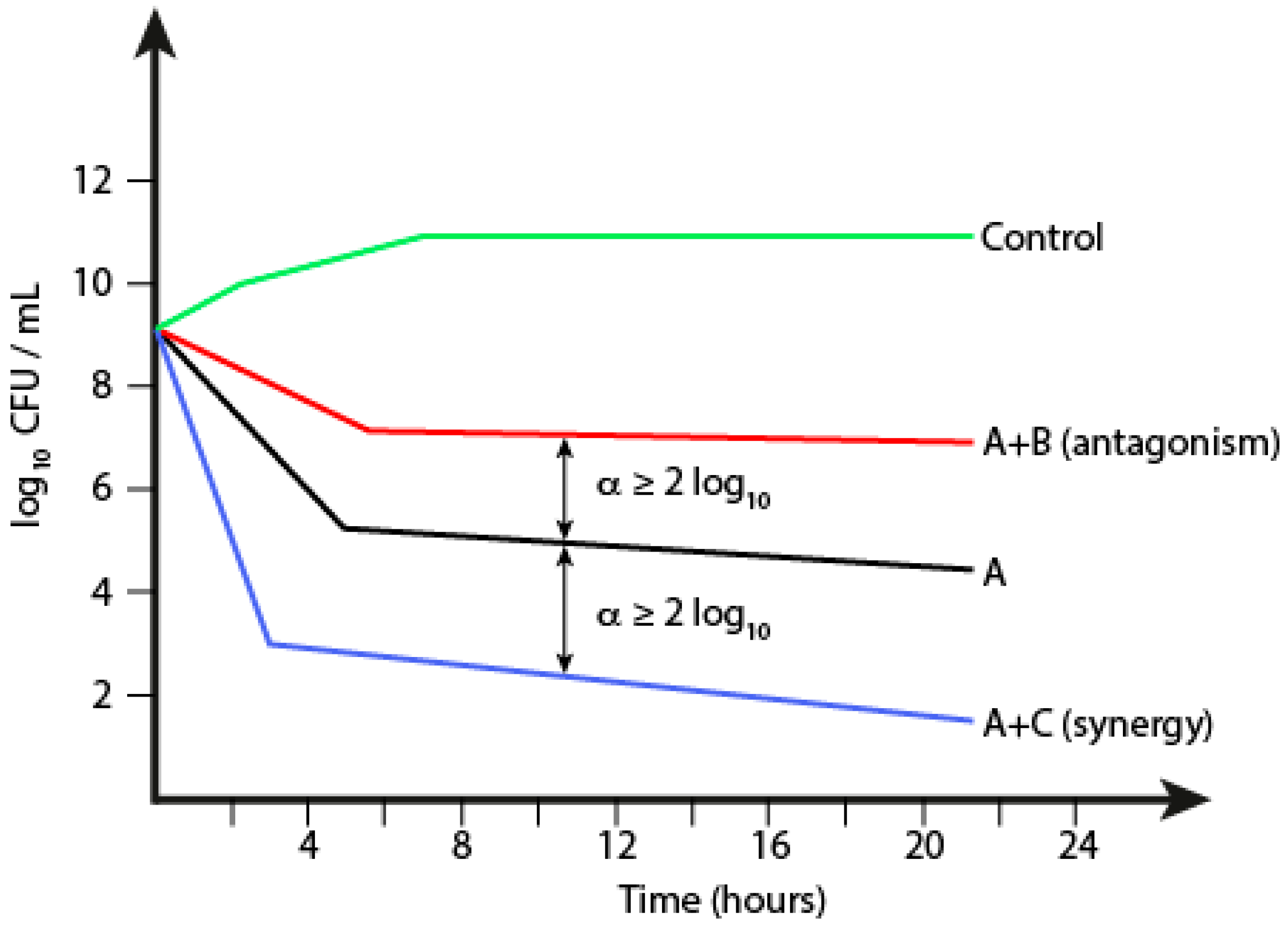
1.5.2. Time-Kill Kinetics
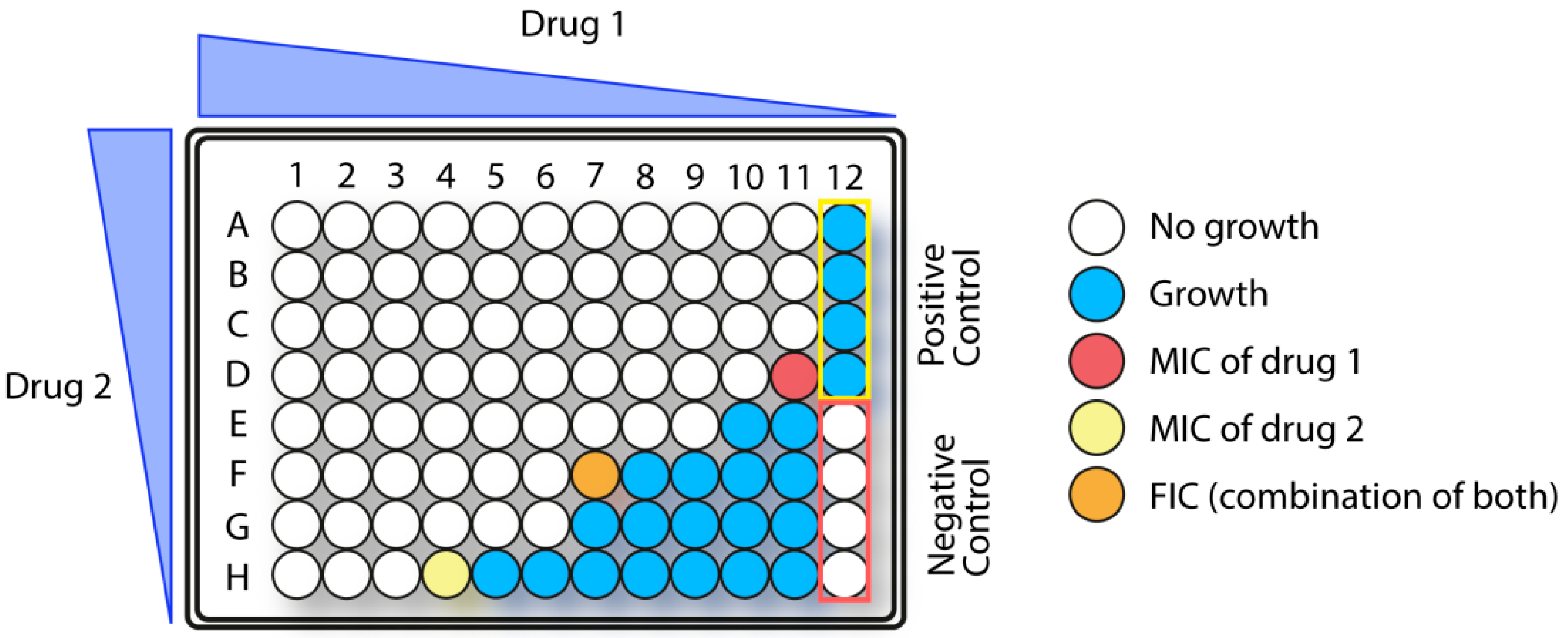
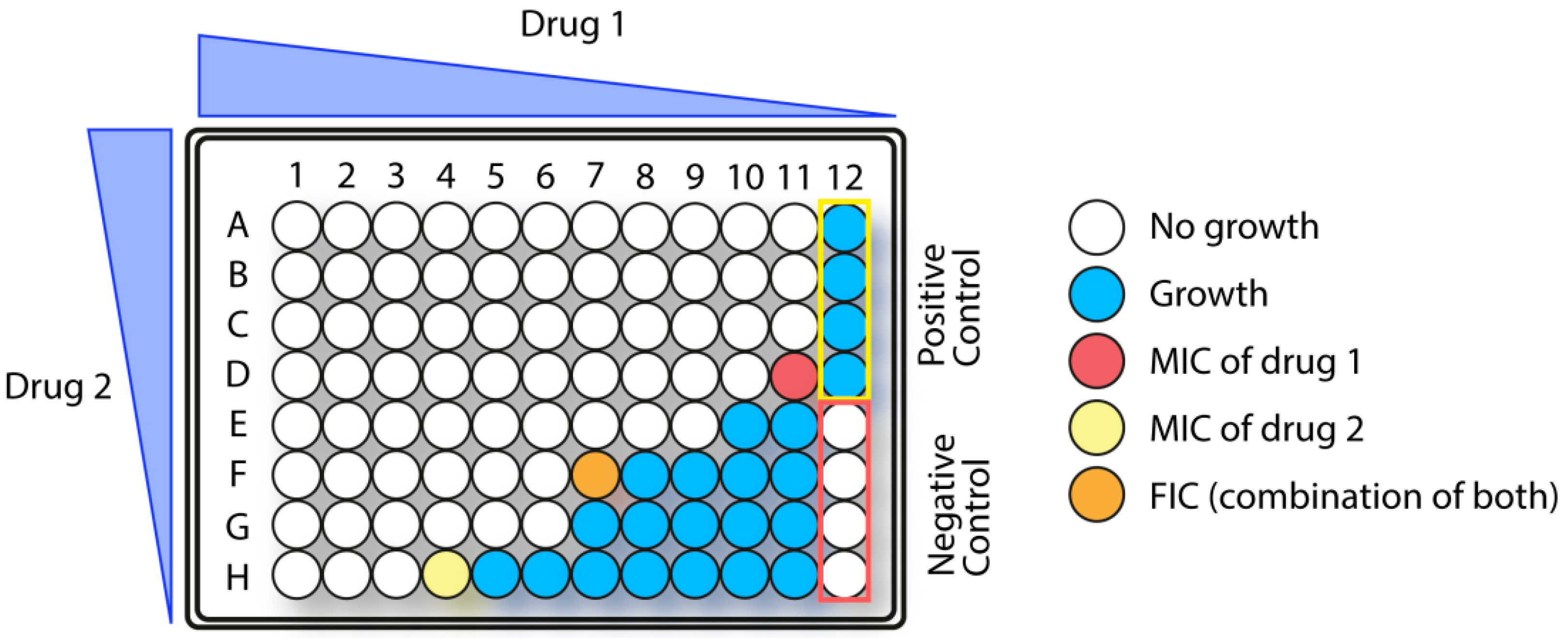
1.5.3. Synergistic Effects in Combination Therapy
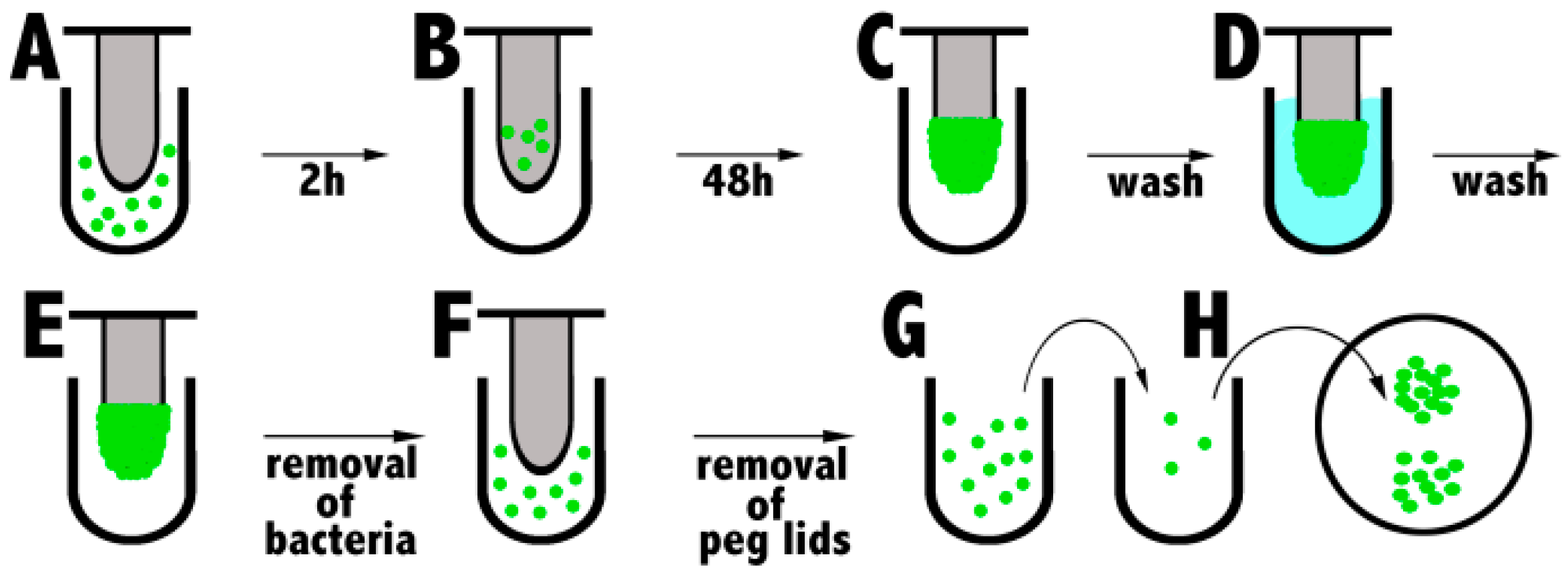
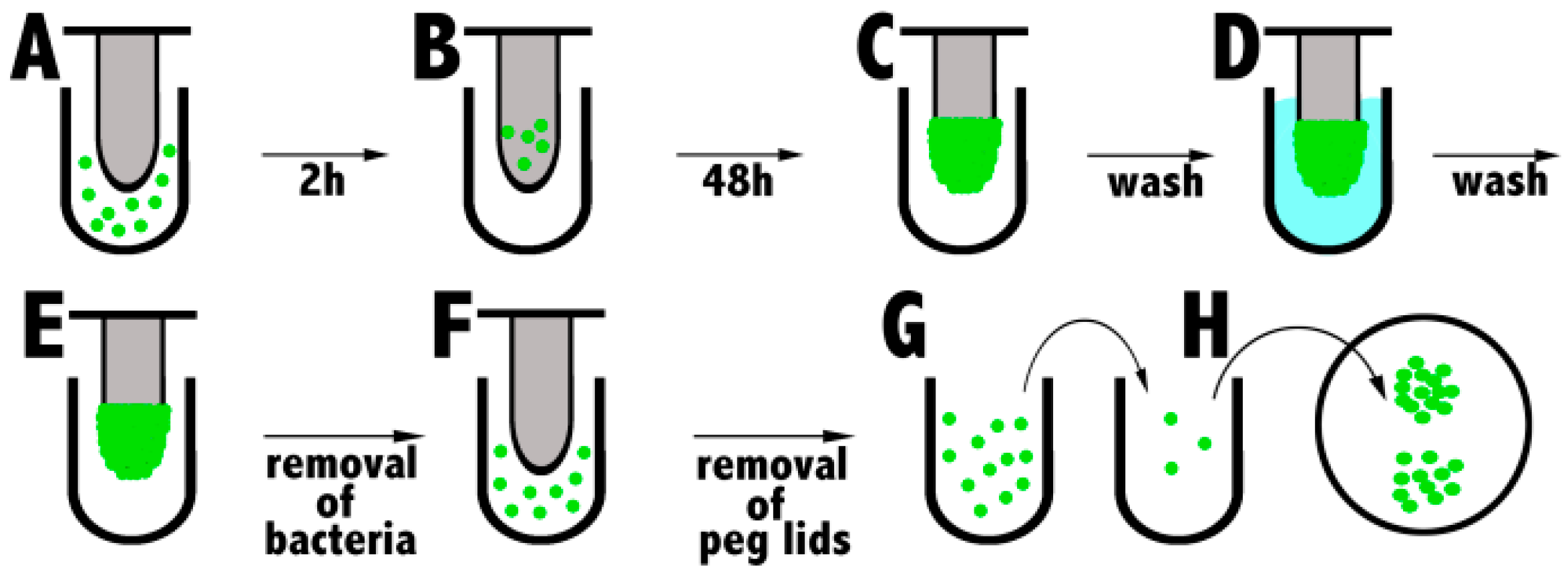
1.5.4. Biofilm Eradication
1.5.5. Cytotoxic Activity
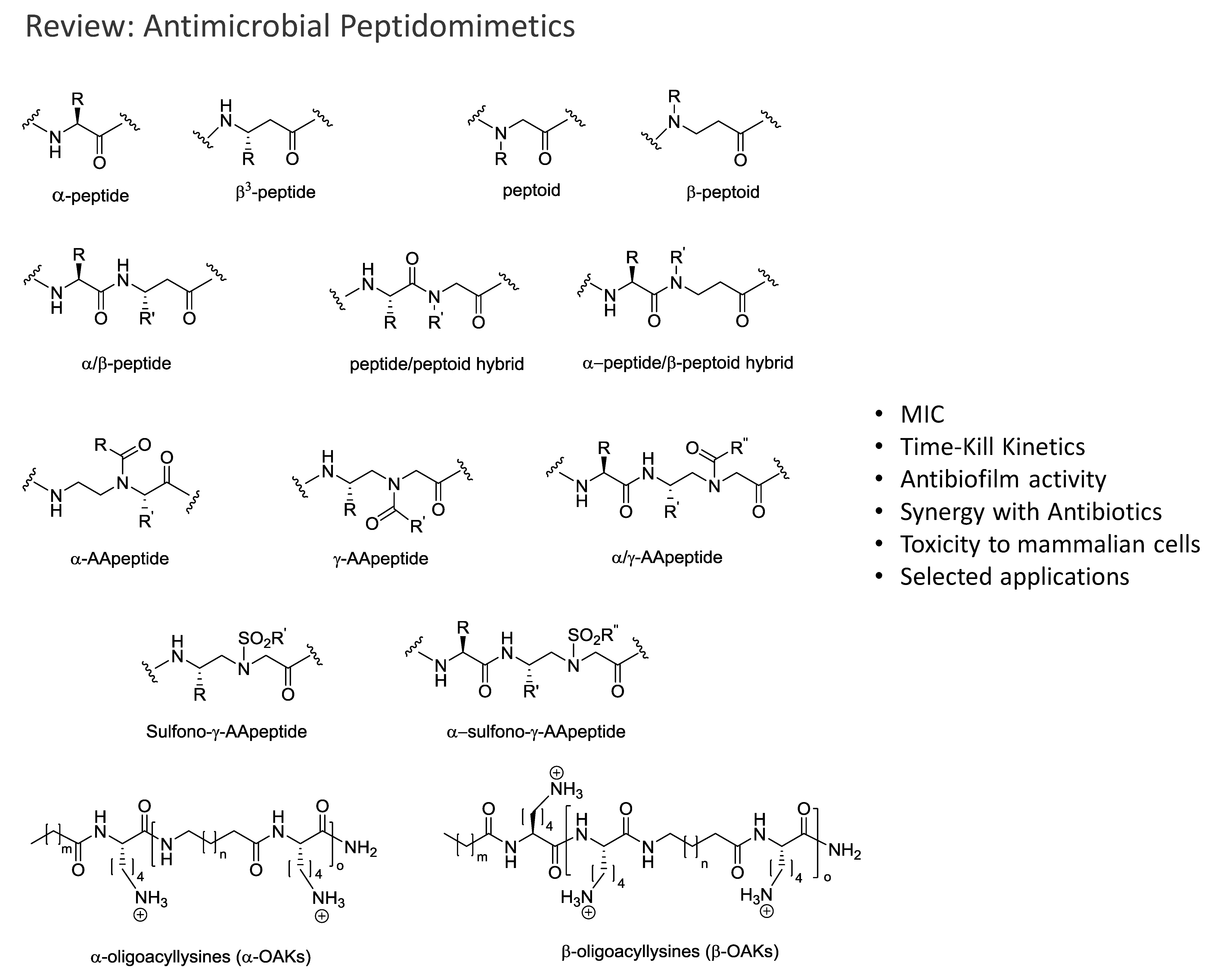
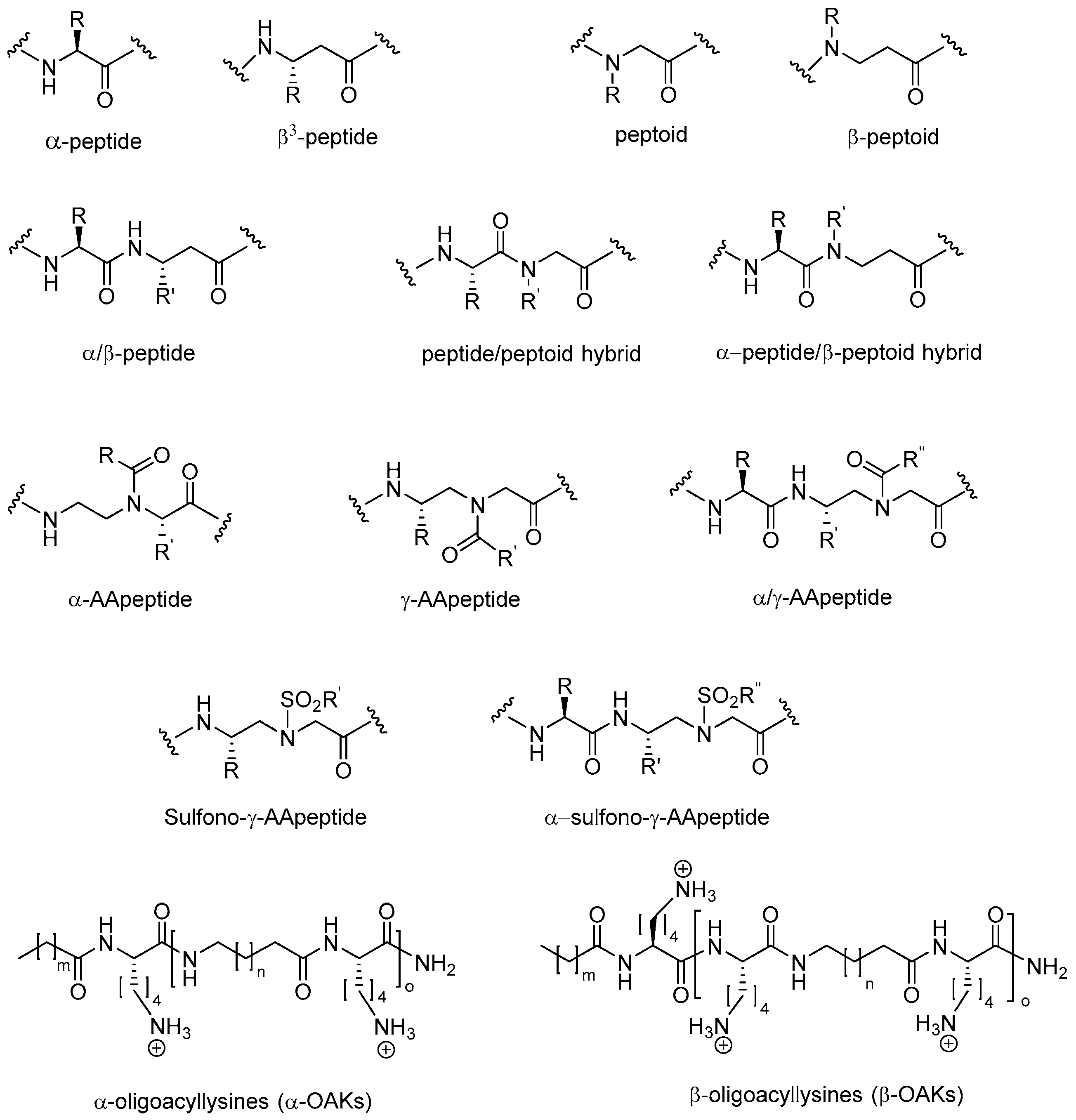
2. Peptidomimetics
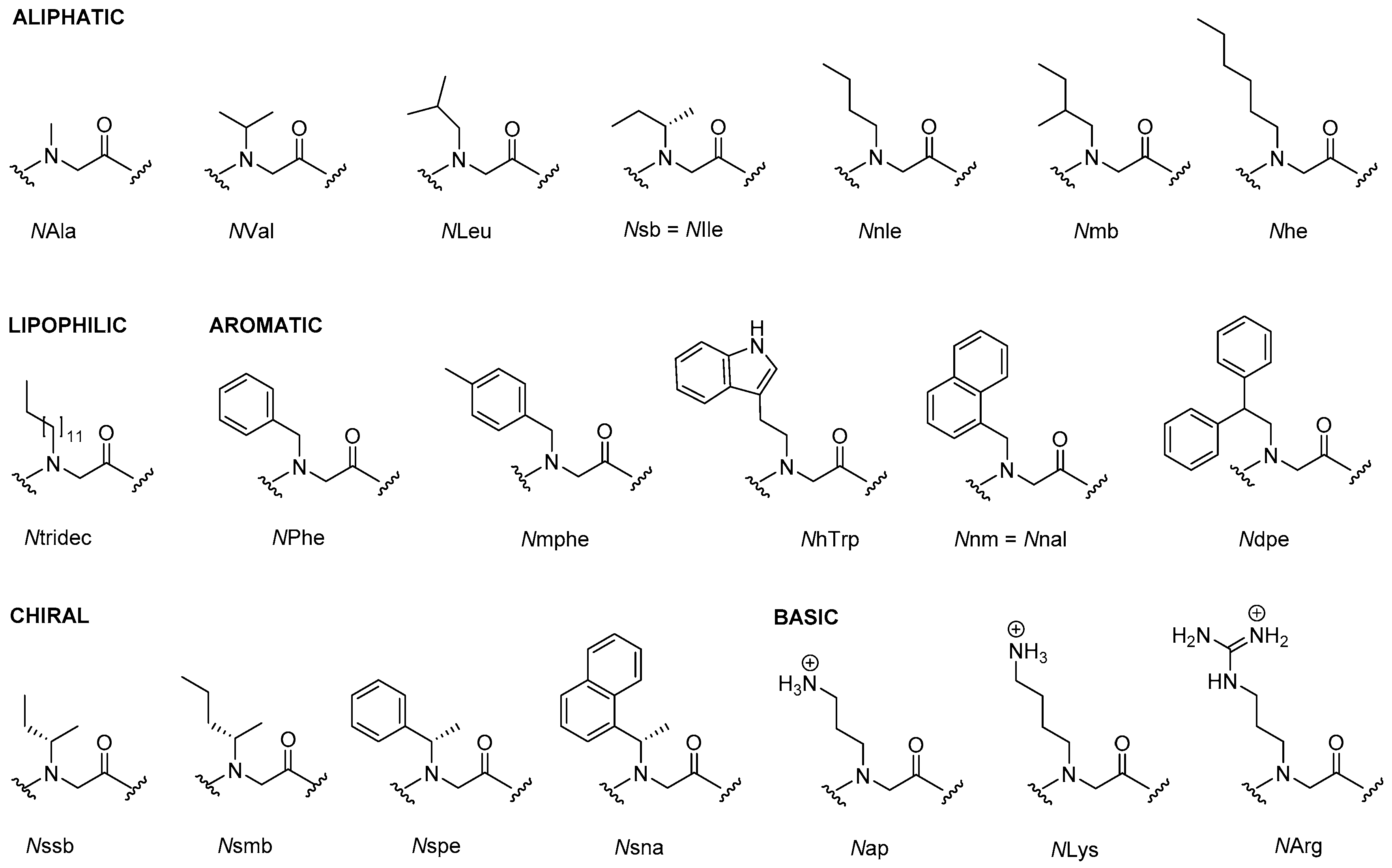
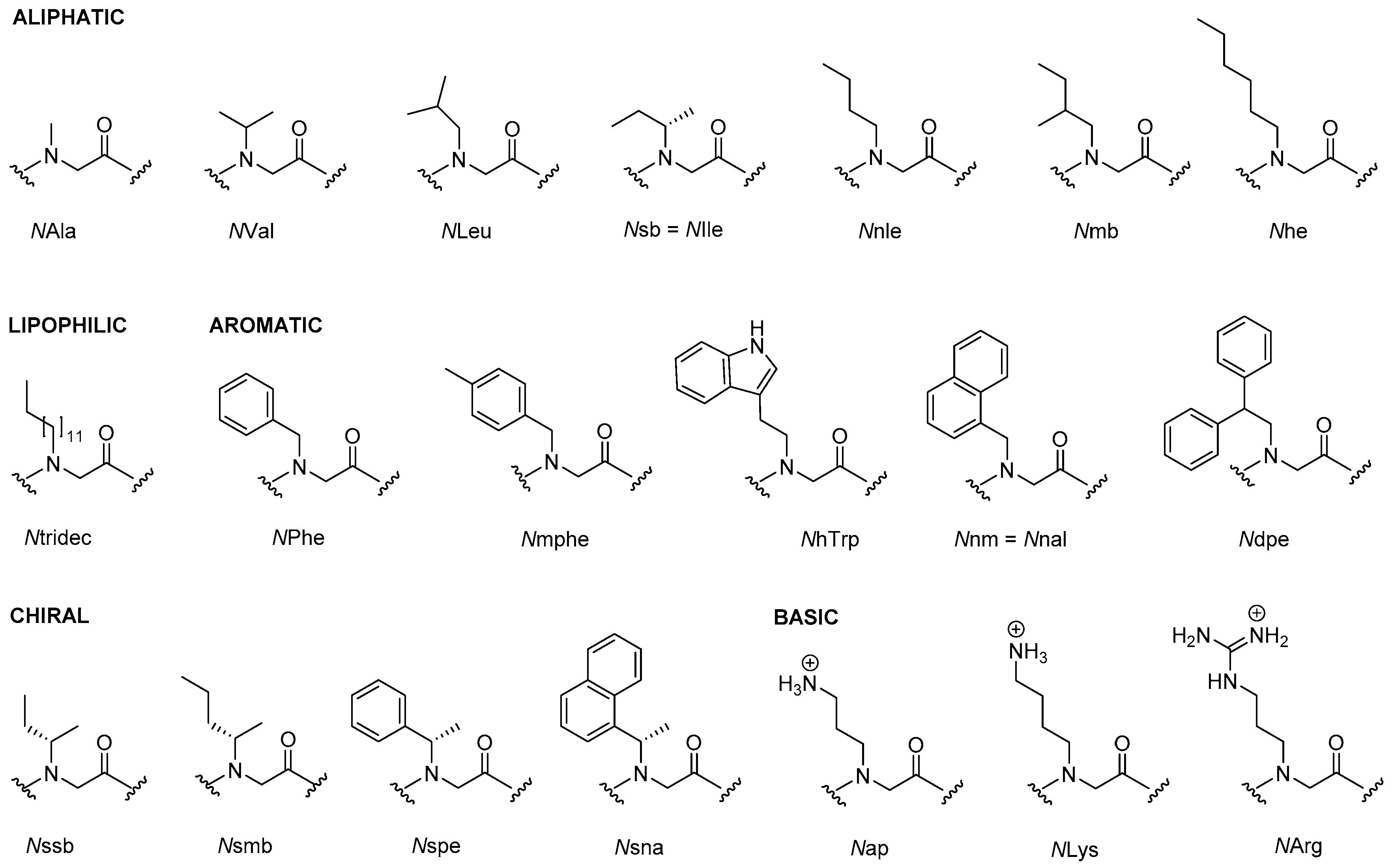
2.1. Peptoids
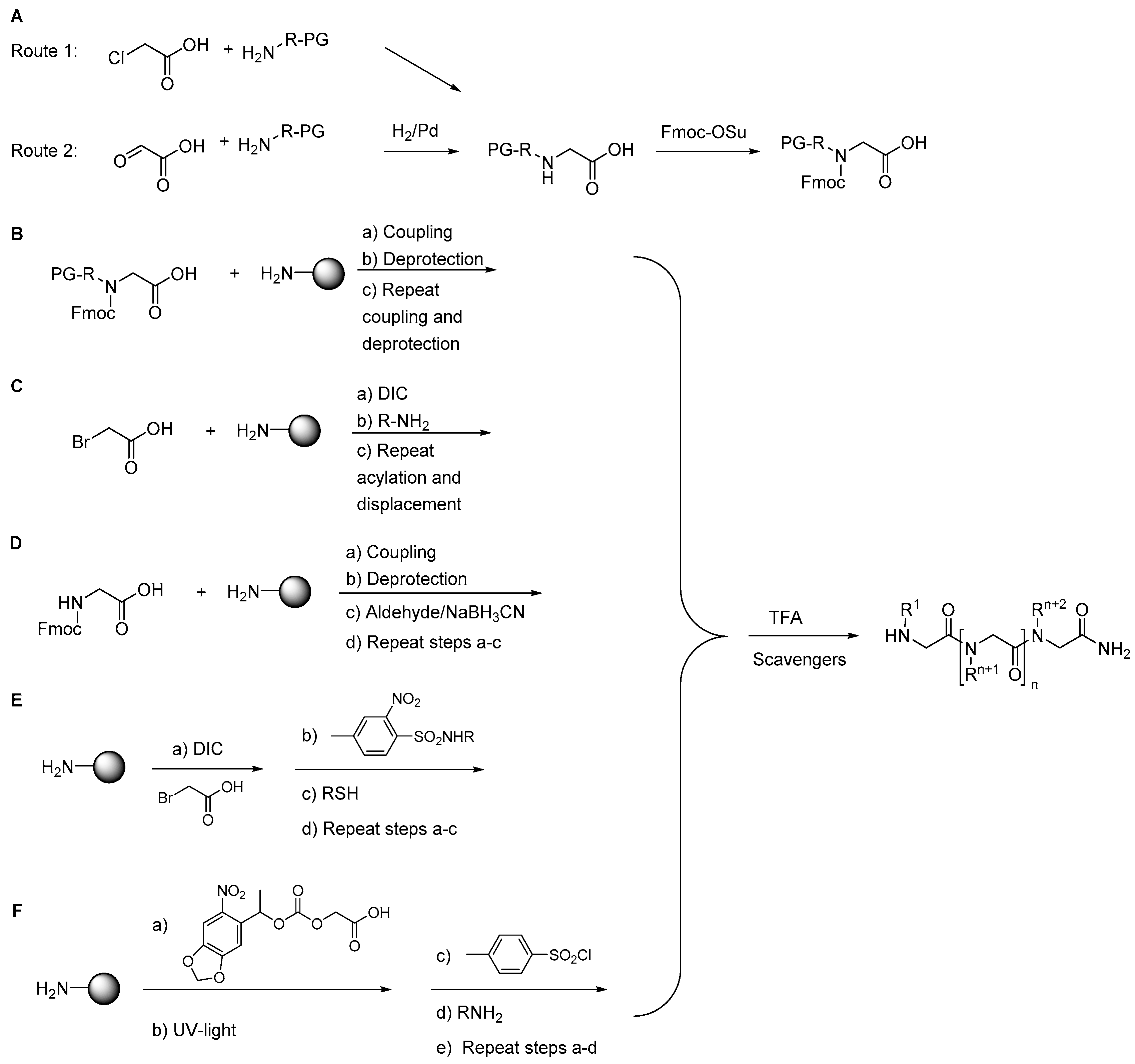
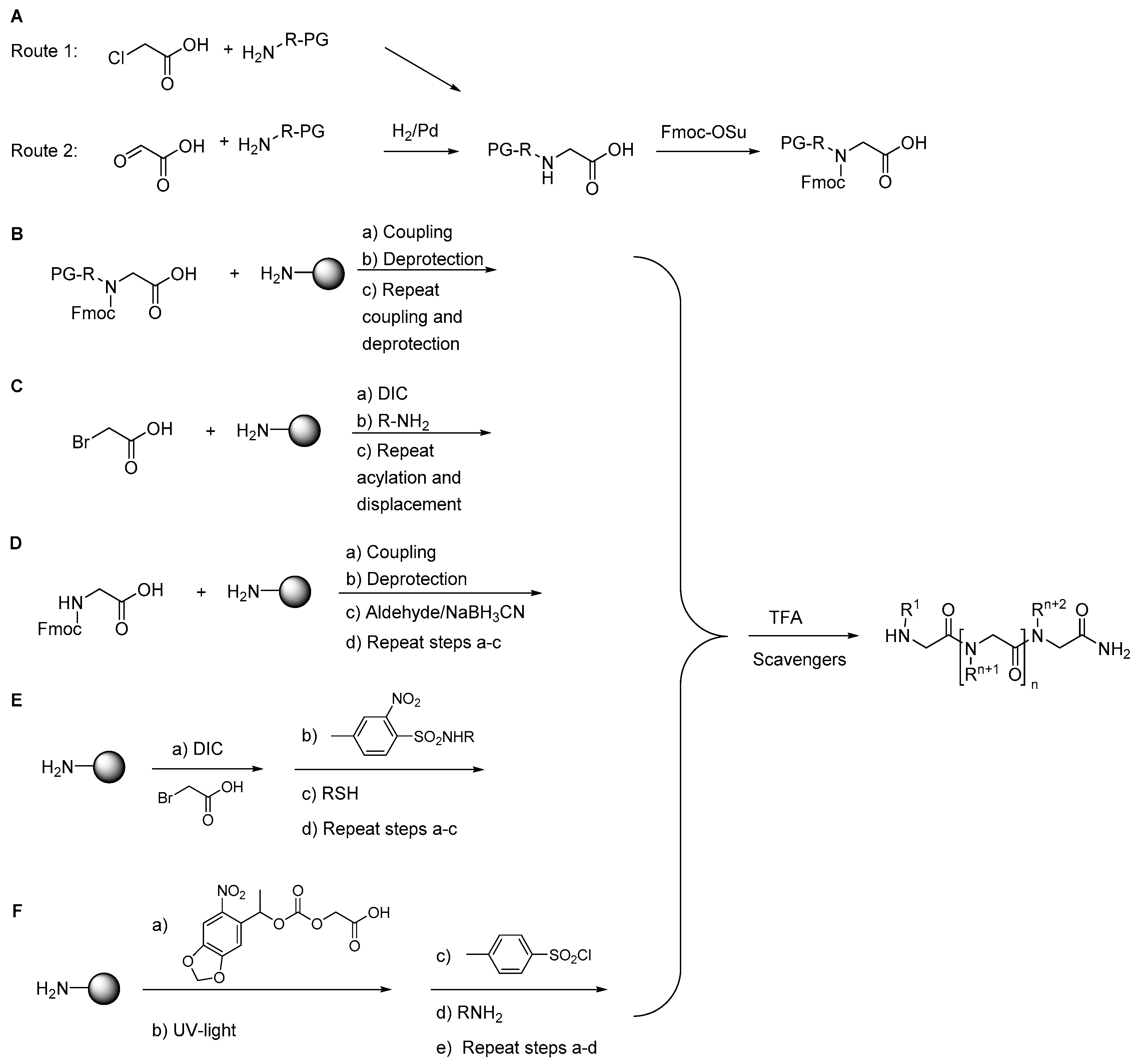
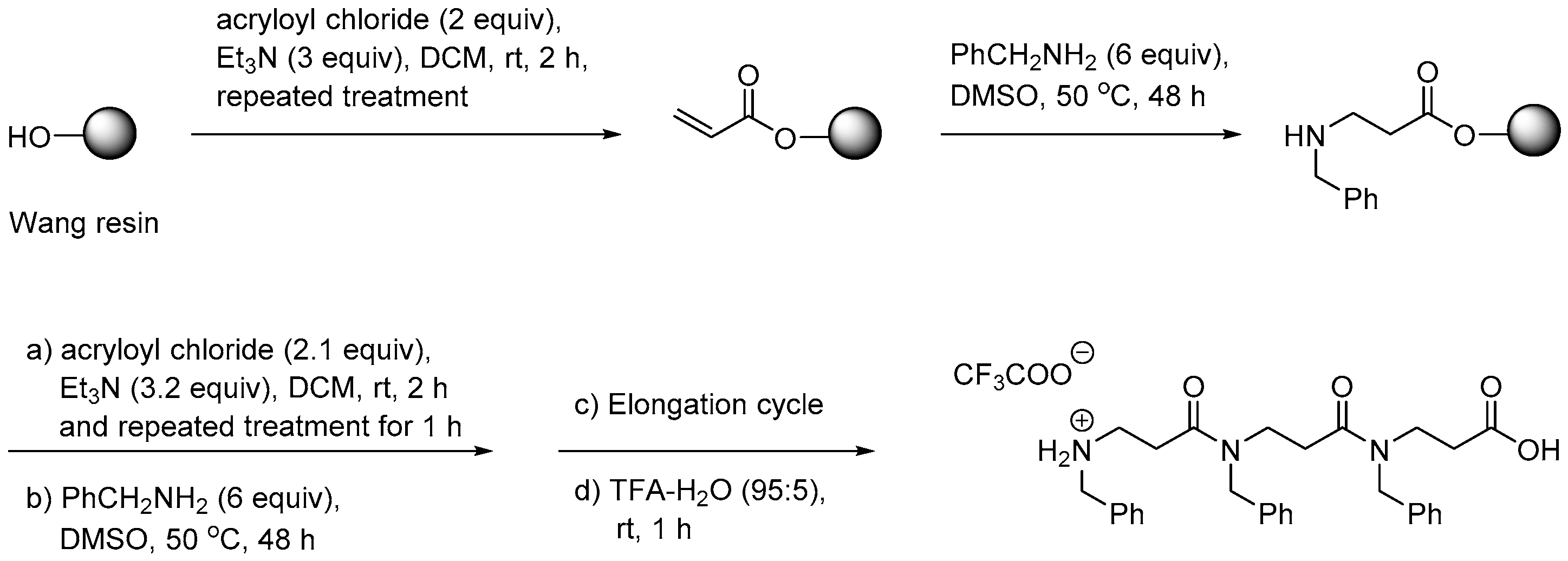
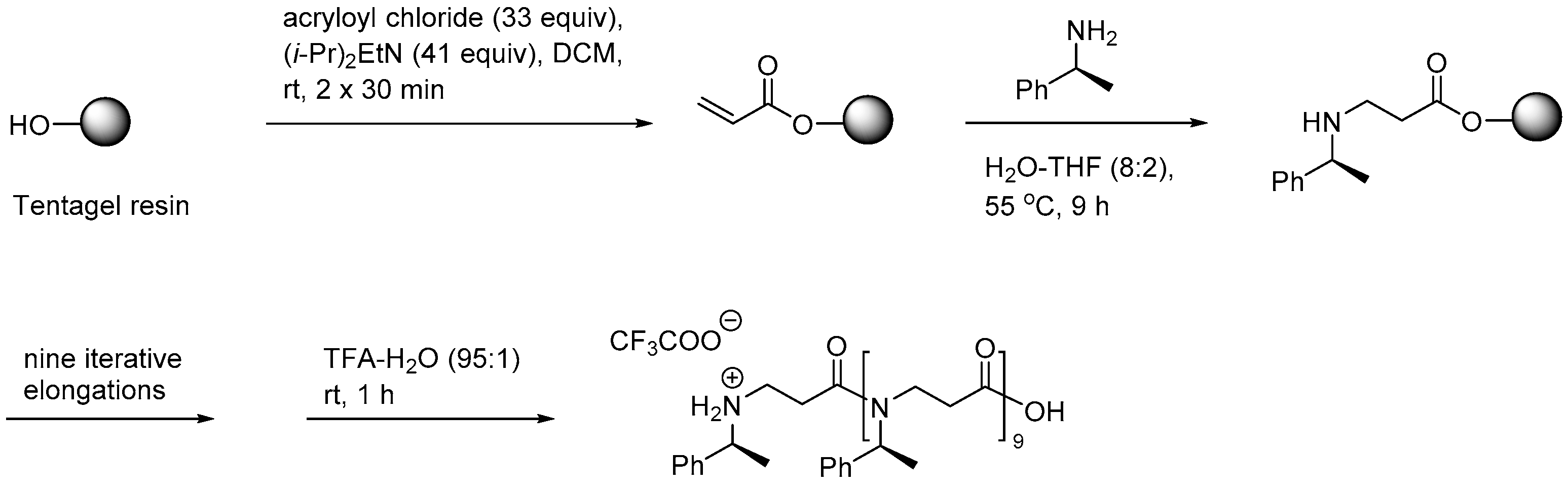
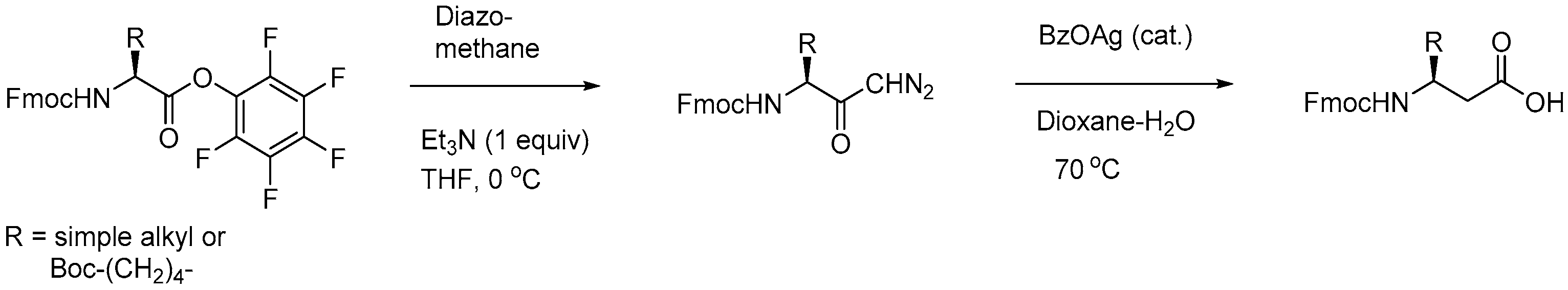
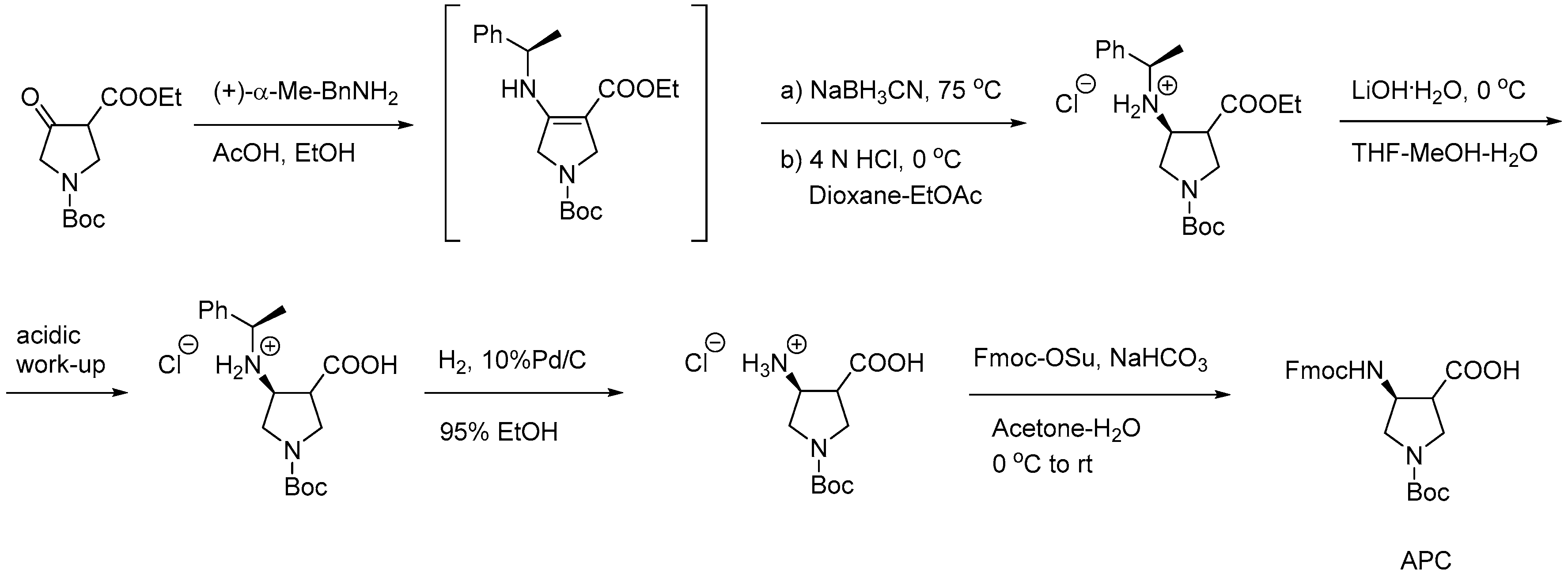
2.1.1. Synthesis
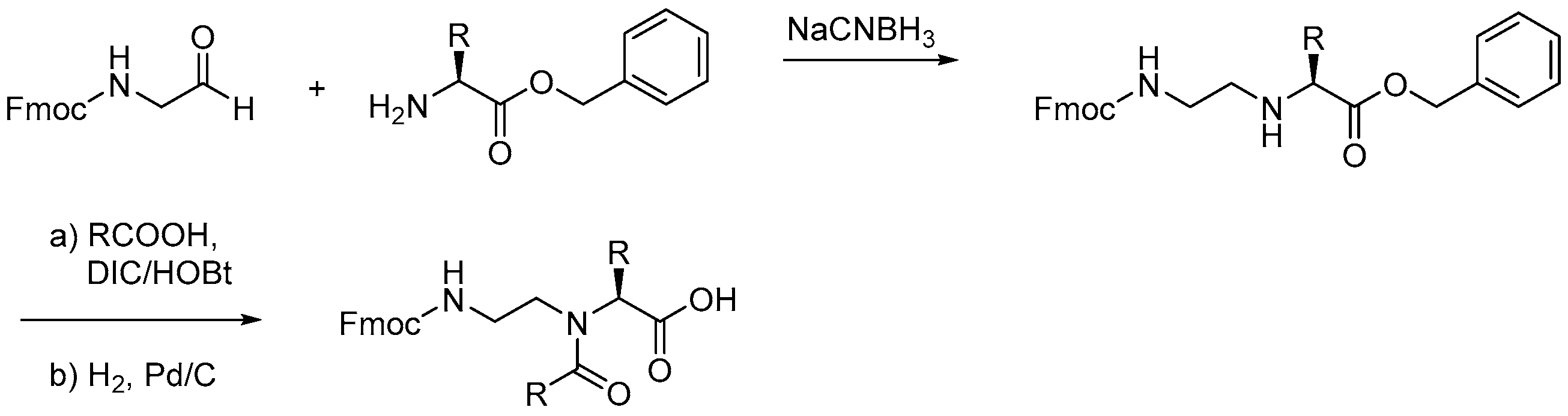
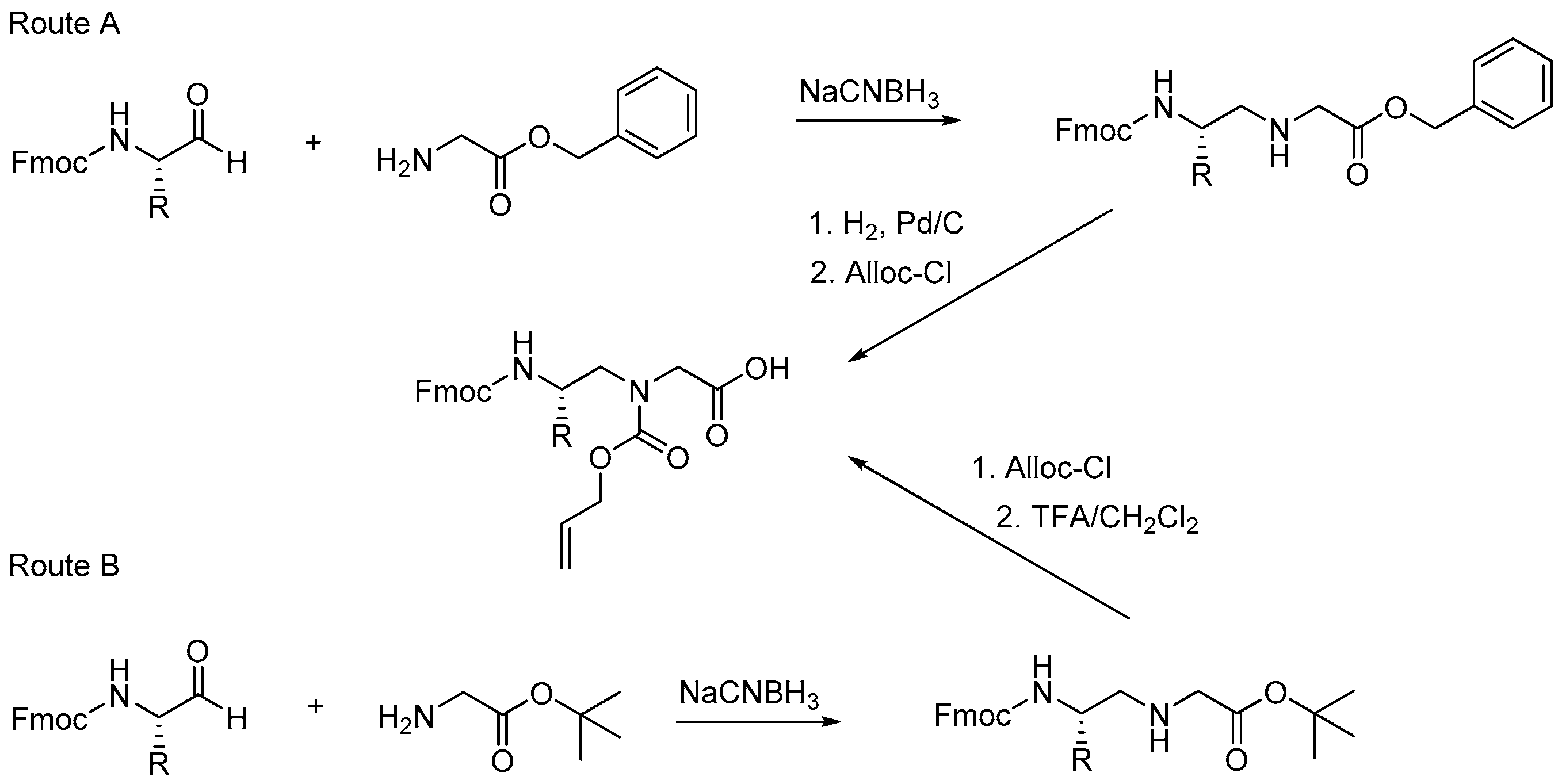
2.1.2. Other Synthetic Developments
2.1.3. Side Reactions
2.1.4. Antimicrobial Activity
2.1.5. Structure-Activity Studies of Peptoids
2.1.6. Libraries
2.1.7. Cyclic Peptoids
2.2. Peptides Displaying Substitution(s) with Peptoid Residues and Peptide-Peptoid Hybrids
2.2.1. AMPs Displaying Substitution(s) with Peptoid Residues
2.2.2. Peptide-Peptoid Hybrids
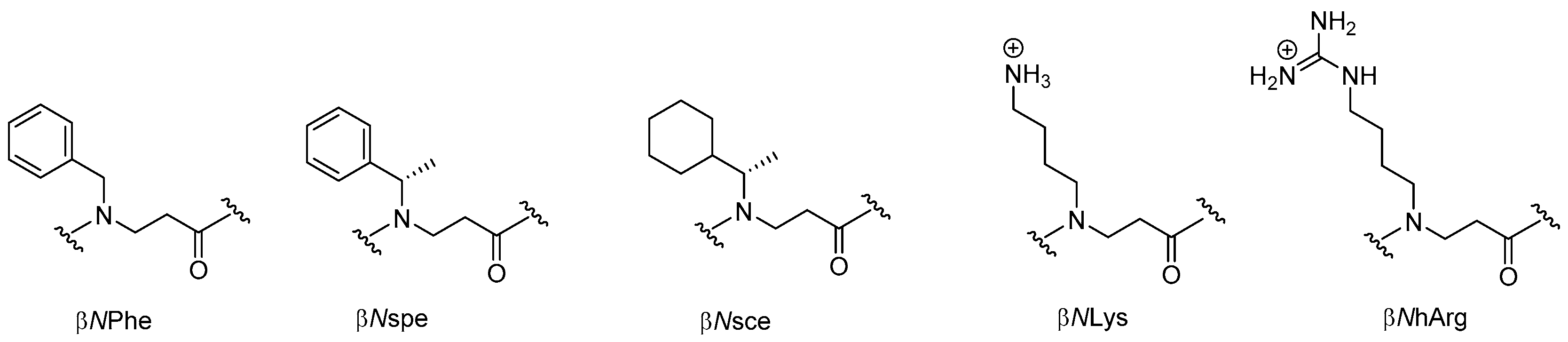
2.3. β-Peptoids
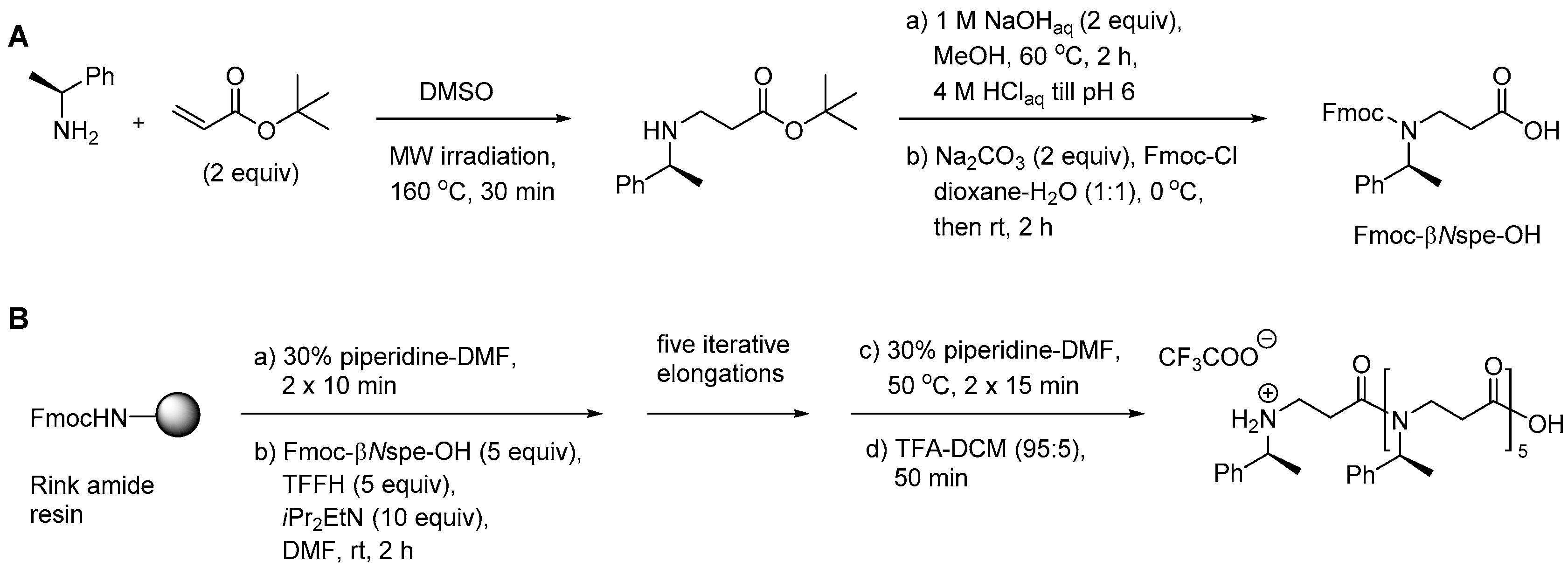
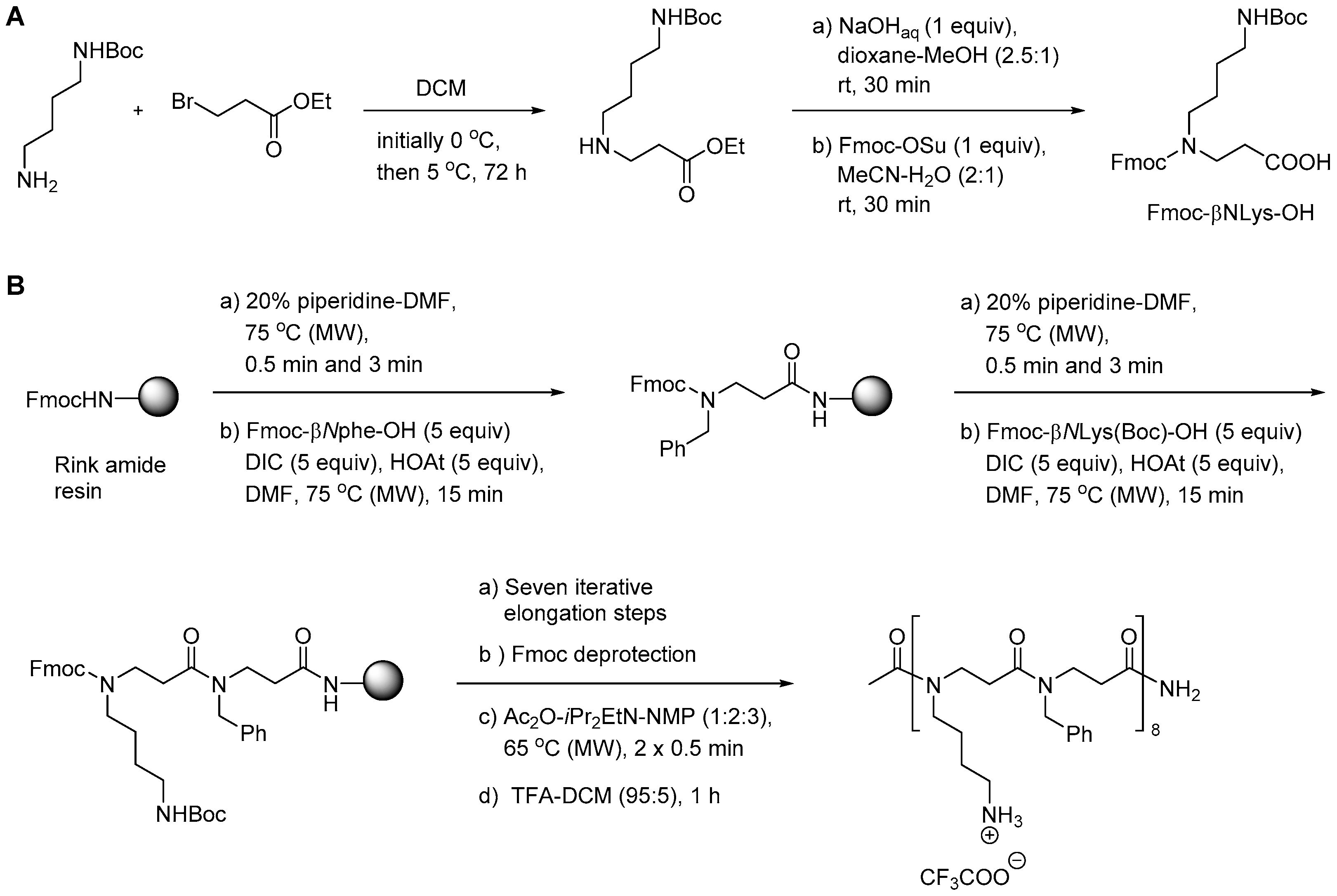
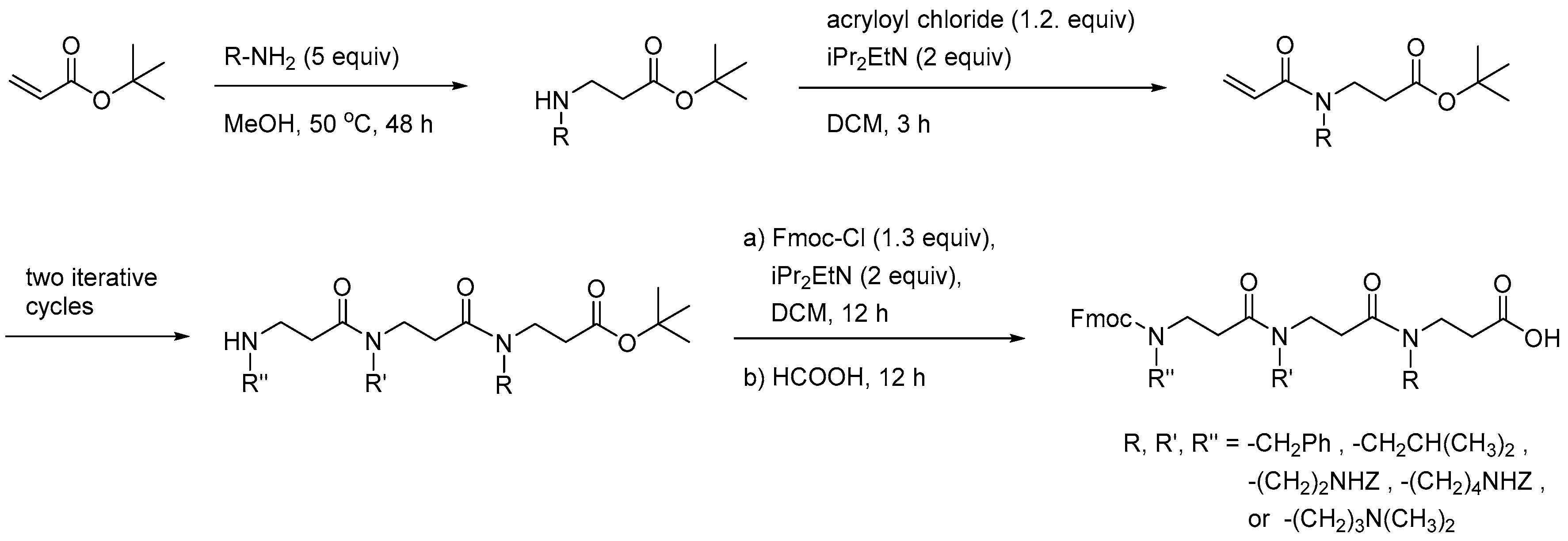
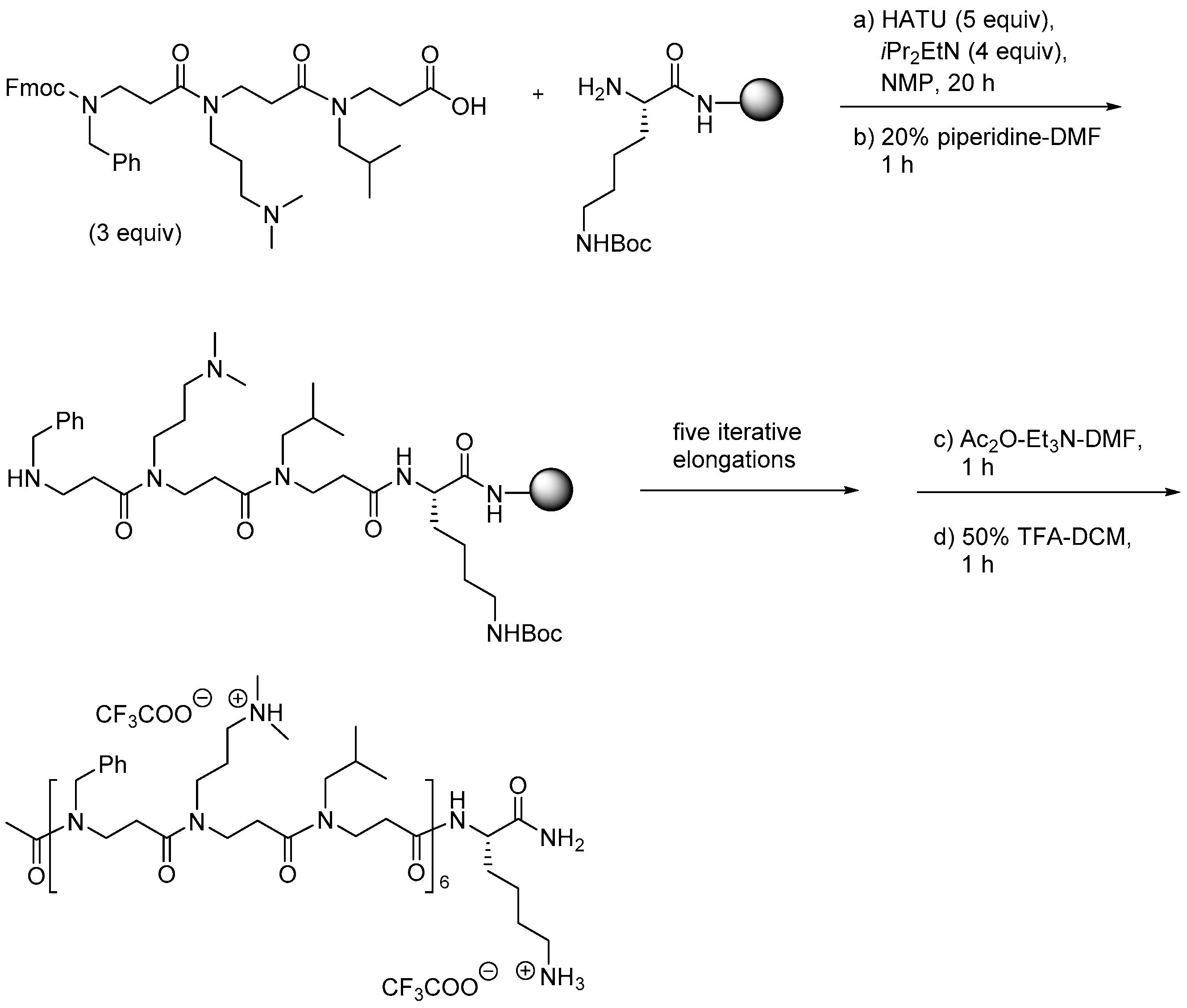
2.3.1. Synthesis of β-Peptoid Oligomers
2.3.2. Antimicrobial Activity
2.4. α-Peptide/β-Peptoid Hybrids
2.4.1. Synthesis
2.4.2. Antimicrobial Activity of α-Peptide/Peptoid Hybrids
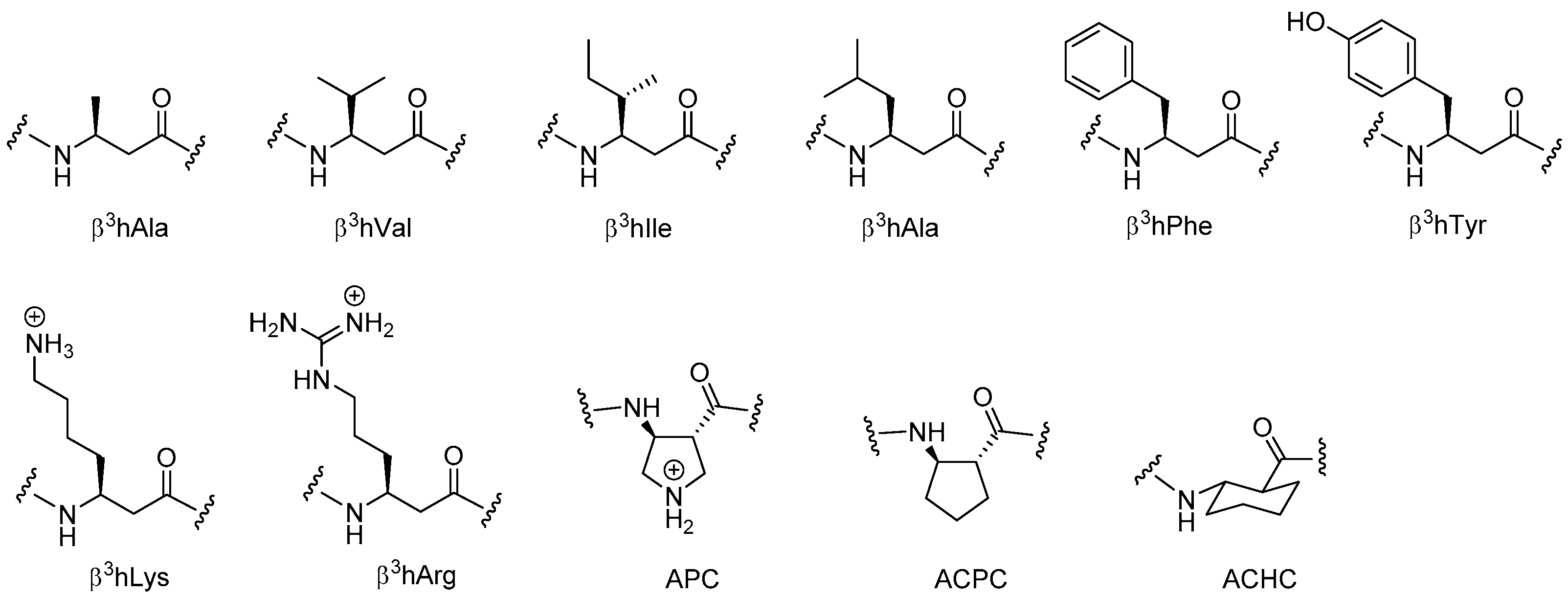
2.5. β3-Peptides
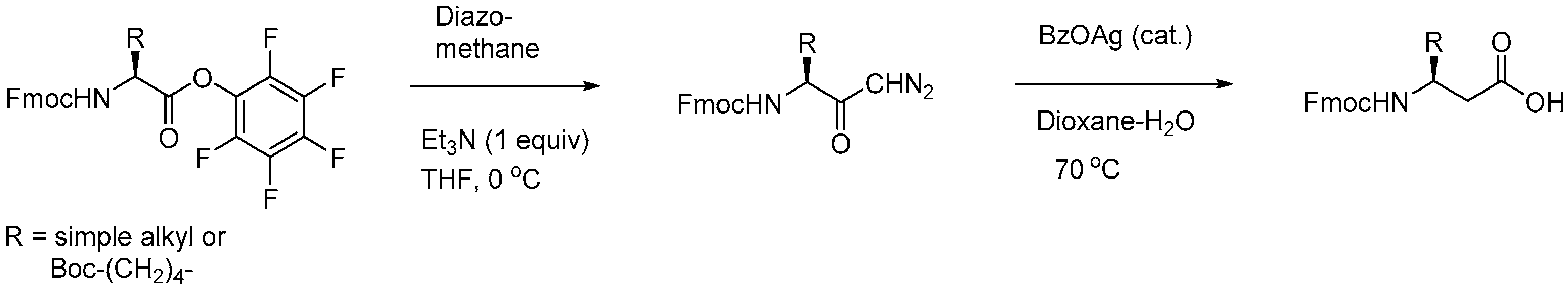
2.5.1. Synthesis of β3-Amino Acid Building Blocks
2.5.2. Antibacterial Activity of β3-Peptides
2.5.3. Antifungal Activity of Helical β3-Peptides
2.6. α-Peptide/β3-Peptide Hybrids
2.7. AApeptides
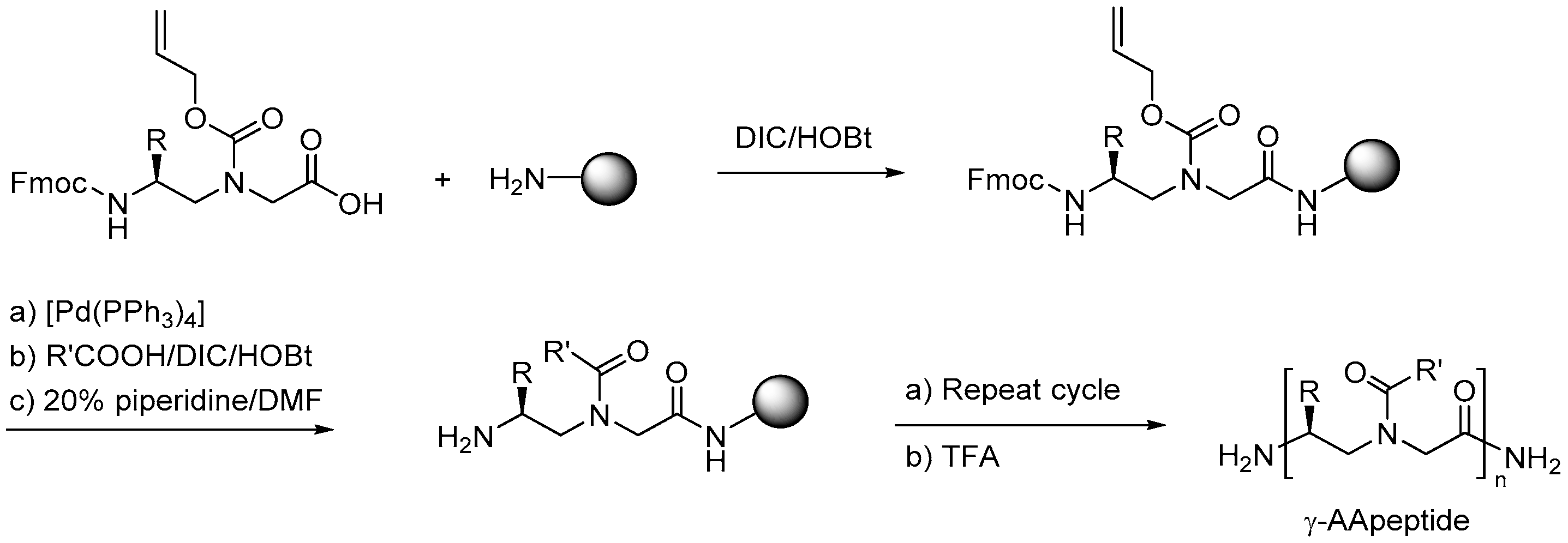
2.7.1. Synthesis
2.7.2. Synthesis of α-AApeptides
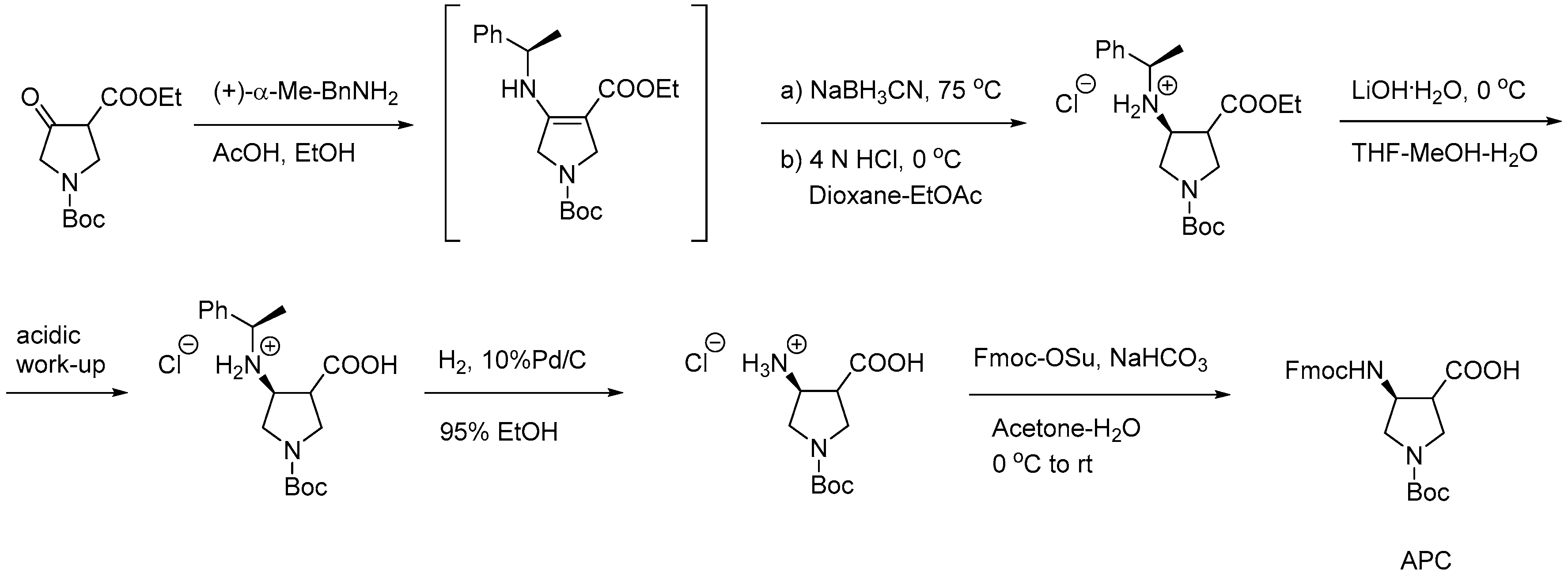
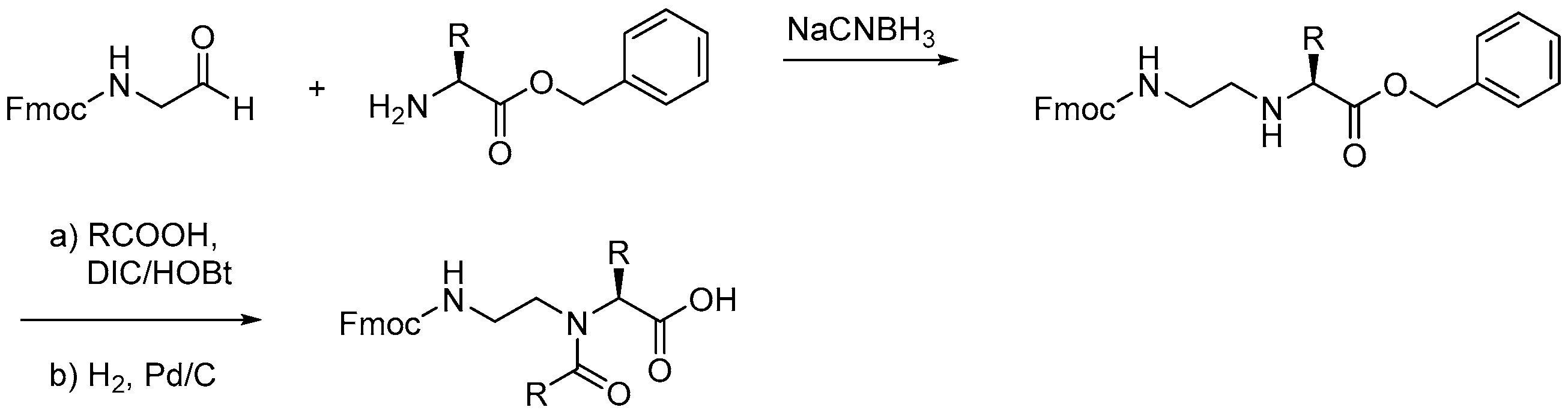
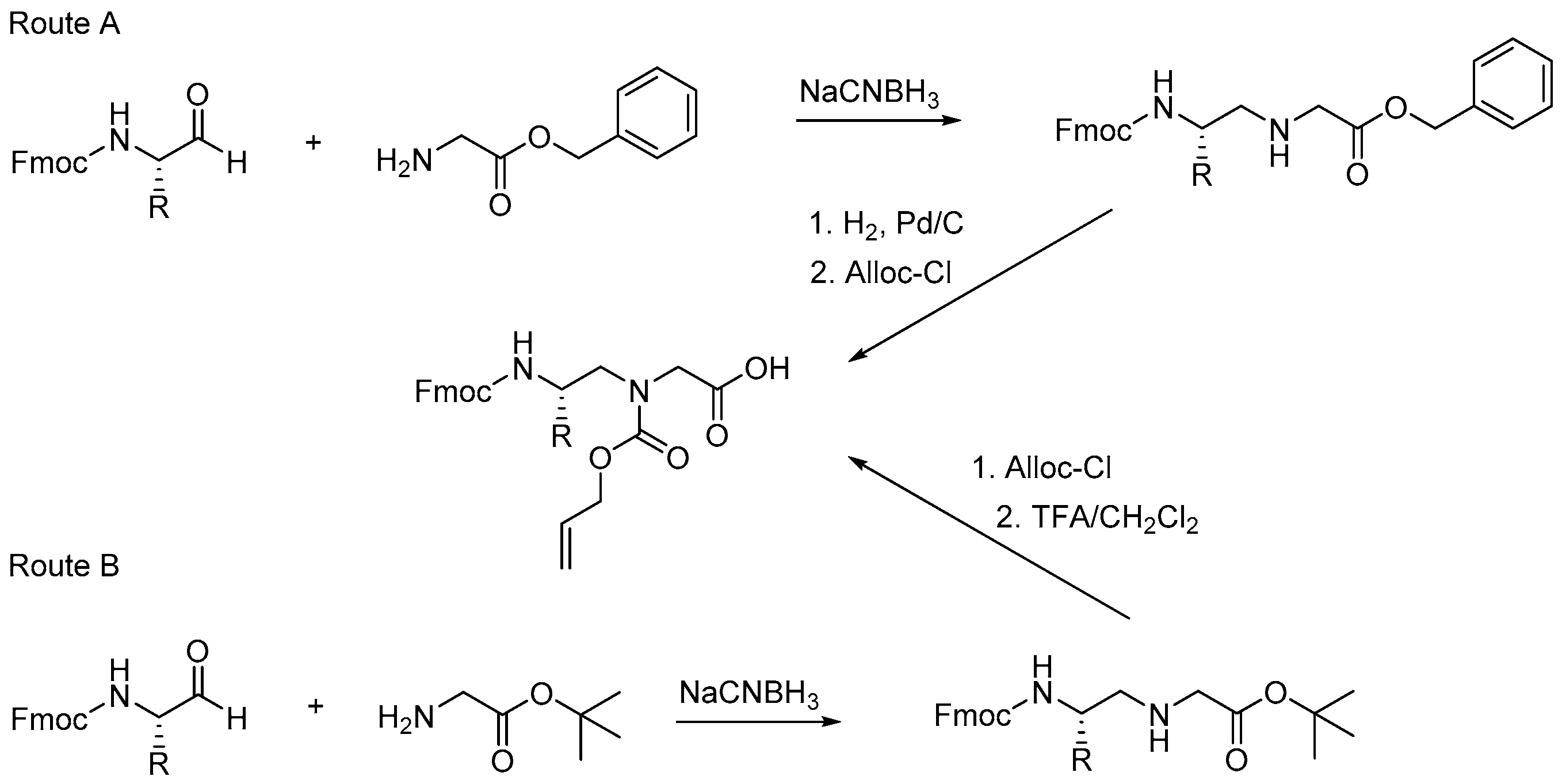
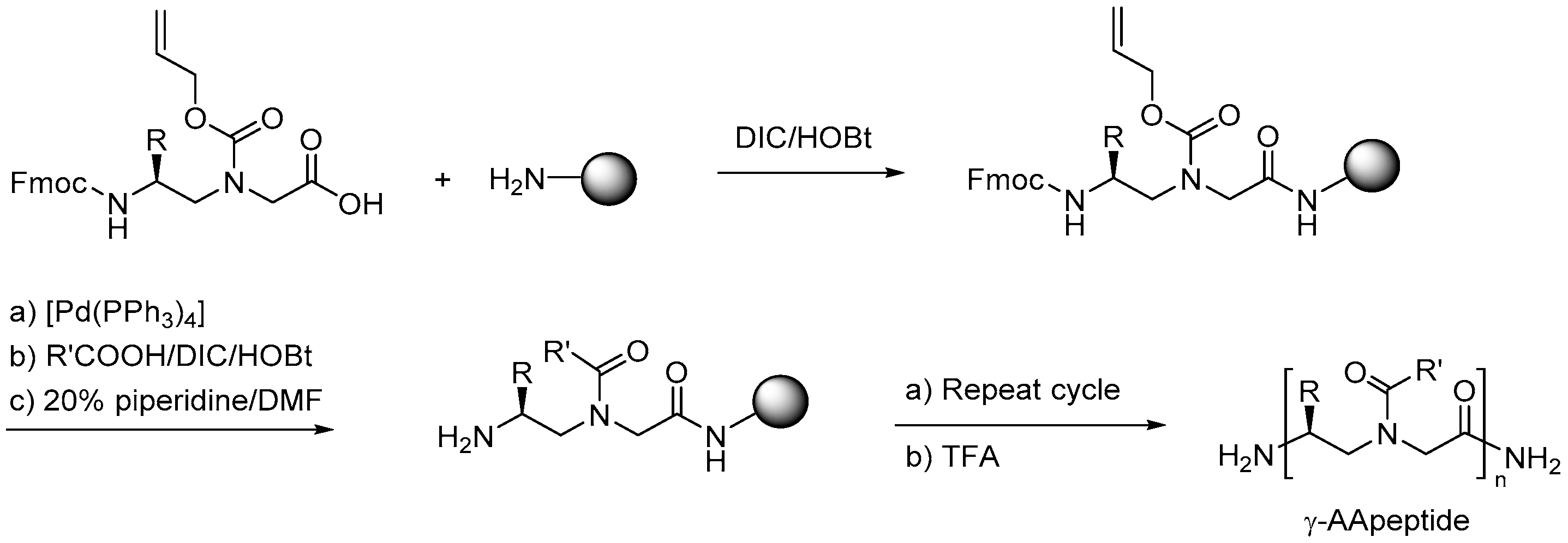
2.7.3. Synthesis of γ-AApeptides
2.7.4. Antimicrobial Activity
α-AApeptides and γ-AApeptides
Lipidated Linear AApeptides and Lipocyclic AApeptides
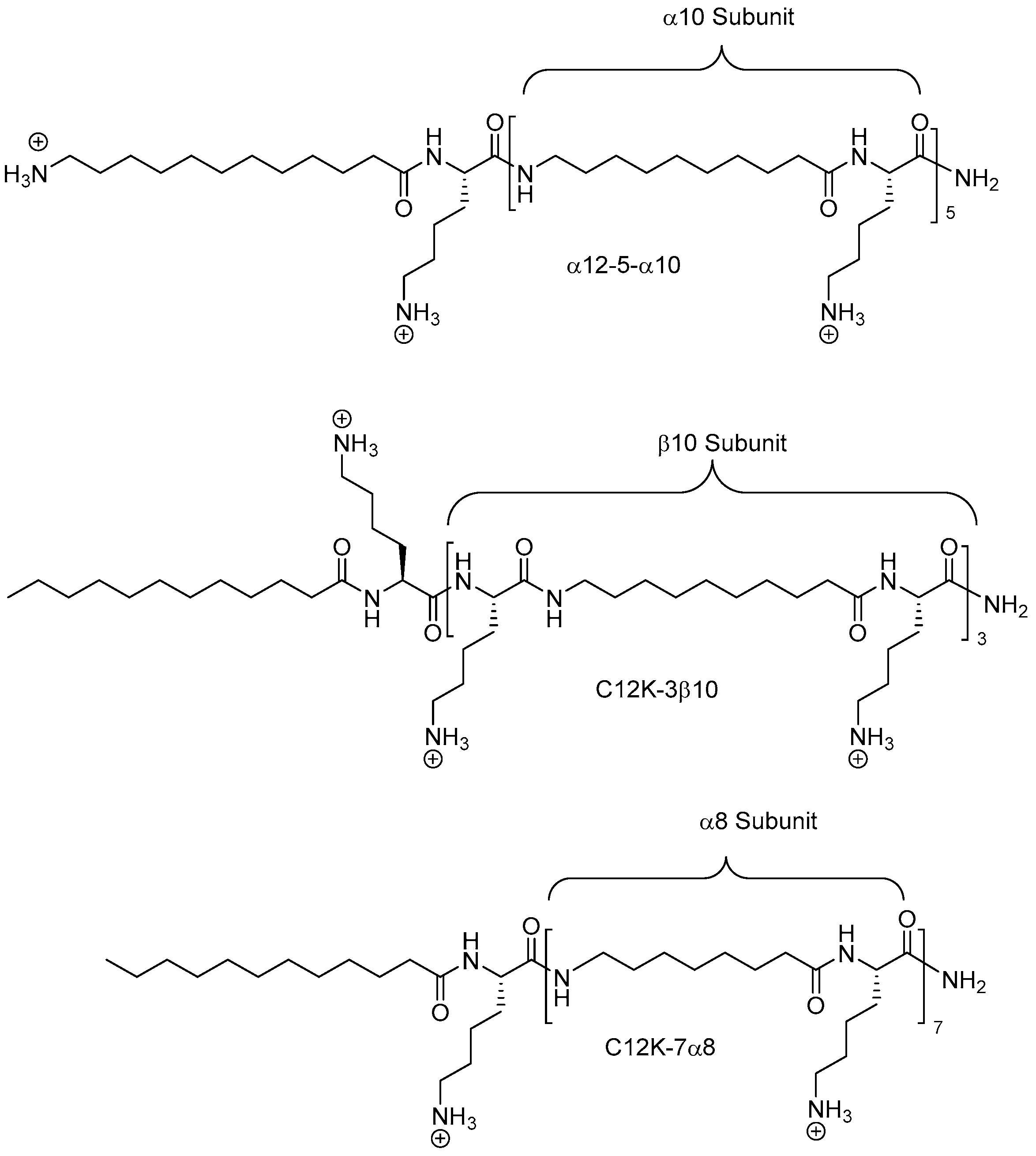
2.8. Oligo-Acyl-Lysyl Peptidomimetics (OAKs)
2.8.1. Synthesis
2.8.2. Antimicrobial Activity
3. Conclusions and Outlook
Author Contributions
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 18 August 2017).
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the eskape pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism mcr-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Xavier, B.B.; Lammens, C.; Ruhal, R.; Kumar-Singh, S.; Butaye, P.; Goossens, H.; Malhotra-Kumar, S. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Eurosurveillance 2016, 21, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Righi, E.; Peri, A.M.; Harris, P.N.A.; Wailan, A.M.; Liborio, M.; Lane, S.W.; Paterson, D.L. Global prevalence of carbapenem resistance in neutropenic patients and association with mortality and carbapenem use: Systematic review and meta-analysis. J. Antimicrob. Chemother. 2017, 72, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Endres, B.T.; Basseres, E.; Alam, M.J.; Garey, K.W. Cadazolid for the treatment of Clostridium difficile. Expert Opin. Investig. Drugs 2017, 26, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Flokas, M.E.; Karageorgos, S.A.; Detsis, M.; Alevizakos, M.; Mylonakis, E. Vancomycin-resistant Enterococci colonisation, risk factors and risk for infection among hospitalised paediatric patients: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2017, 49, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D. Time to deal with antibiotics. Science 2013, 342, 777. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.R.; Smith, T.C.; Bonomo, R.A. On the path to untreatable infections: Colistin use in agriculture and the end of “last resort’ antibiotics. Expert Rev. Anti-Infect. Ther. 2016, 14, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.; Clasen, J.; Hansen, J.E.; Paulander, W.; Petersen, A.; Larsen, A.R.; Frees, D. Copresence of tet(k) and tet(m) in livestock-associated methicillin-resistant Staphylococcus aureus clonal complex 398 is associated with increased fitness during exposure to sublethal concentrations of tetracycline. Antimicrob. Agents Chemother. 2016, 60, 4401–4403. [Google Scholar] [CrossRef] [PubMed]
- Collignon, P. The importance of a one health approach to preventing the development and spread of antibiotic resistance. In One Health: The Human-Animal-Environment Interfaces in Emerging Infectious Diseases; Mackenzie, J.S., Jeggo, M., Daszak, P., Richt, J.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 366, pp. 19–36. [Google Scholar]
- Van Duijkeren, E.; Catry, B.; Greko, C.; Moreno, M.A.; Pomba, M.C.; Pyörälä, S.; Ružauskas, M.; Sanders, P.; Threlfall, E.J.; Torren-Edo, J.; et al. Review on methicillin-resistant Staphylococcus pseudintermedius. J. Antimicrob. Chemother. 2011, 66, 2705–2714. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, R.; Burnens, A.; Maranta, C.A.; Perreten, V. Human infection associated with methicillin-resistant Staphylococcus pseudintermedius st71. J. Antimicrob. Chemother. 2010, 65, 2047–2048. [Google Scholar] [CrossRef] [PubMed]
- Starlander, G.; Borjesson, S.; Gronlund-Andersson, U.; Tellgren-Roth, C.; Melhusa, A. Cluster of infections caused by methicillin-resistant Staphylococcus pseudintermedius in humans in a tertiary hospital. J. Clin. Microbiol. 2014, 52, 3118–3120. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics—A pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Dodds, D.R. Antibiotic resistance: A current epilogue. Biochem. Pharmacol. 2017, 134, 139–146. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Action Plan for Antimicrobial Resistance; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Sinha, M.S.; Kesselheim, A.S. Regulatory incentives for antibiotic drug development: A review of recent proposals. Bioorg. Med. Chem. 2016, 24, 6446–6451. [Google Scholar] [CrossRef] [PubMed]
- Arzanlou, M.; Chai, W.C.; Venter, H. Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 2017, 61, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Palmer, J.D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Tangden, T.; Adler, M.; Cars, O.; Sandegren, L.; Lowdin, E. Frequent emergence of porin-deficient subpopulations with reduced carbapenem susceptibility in ESBL-producing Escherichia coli during exposure to ertapenem in an in vitro pharmacokinetic model. J. Antimicrob. Chemother. 2013, 68, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A new class of genetic element, Staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Yang, S.J.; Chen, L.; Muller, C.; Saleh-Mghir, A.; Kuhn, S.; Peschel, A.; Yeaman, M.R.; Nast, C.C.; Kreiswirth, B.N.; et al. Emergence of daptomycin resistance in daptomycin-naive rabbits with methicillin-resistant Staphylococcus aureus prosthetic joint infection is associated with resistance to host defense cationic peptides and mprf polymorphisms. PLoS ONE 2013, 8, e71151. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.P.; Woodford, N. Global spread of antibiotic resistance: The example of new delhi metallo-beta-lactamase (NDM)-mediated carbapenem resistance. J. Med. Microbiol. 2013, 62, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 2017, 70, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Tsiodras, S.; Gold, H.S.; Sakoulas, G.; Eliopoulos, G.M.; Wennersten, C.; Venkataraman, L.; Moellering, R.C.; Ferraro, M.J. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 2001, 358, 207–208. [Google Scholar] [CrossRef]
- Bush, K. Investigational agents for the treatment of Gram-negative bacterial infections: A reality check. ACS Infect. Dis. 2015, 1, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E.W. Antimicrobial peptides: An introduction. In Antimicrobial Peptides: Methods and Protocols; Hansen, P.R., Ed.; Springer: New York, NY, USA, 2017; pp. 3–22. [Google Scholar]
- Fan, L.L.; Sun, J.; Zhou, M.F.; Zhou, J.; Lao, X.Z.; Zheng, H.; Xu, H.M. Dramp: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Jenssen, H. Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Torcato, I.M.; Huang, Y.-H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.R.B.; Troeira Henriques, S. Design and characterization of novel antimicrobial peptides, r-bp100 and rw-bp100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta Biomembr. 2013, 1828, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y.; Oren, Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides 2001, 22, 1629–1641. [Google Scholar] [CrossRef]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.; Andersson, M.; Karshikoff, A.; Otting, G. Molecular electroporation: A unifying concept for the description of membrane pore formation by antibacterial peptides, exemplified with nk-lysin. FEBS Lett. 1999, 462, 155–158. [Google Scholar] [CrossRef]
- Van’t Hof, W.; Veerman, E.C.I.; Helmerhorst, E.J.; Amerongen, A.V.N. Antimicrobial peptides: Properties and applicability. Biol. Chem. 2001, 382, 597–619. [Google Scholar] [CrossRef]
- Pokorny, A.; Birkbeck, T.H.; Almeida, P.F.F. Mechanism and kinetics of delta-lysin interaction with phospholipid vesicles. Biochemistry 2002, 41, 11044–11056. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W. Action of antimicrobial peptides: Two-state model. Biochemistry 2000, 39, 8347–8352. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, G.; Gimenez, D.; Esteban-Martin, S.; Sanchez-Munoz, O.L.; Salgado, J. A lipocentric view of peptide-induced pores. Eur. Biophys. J. 2011, 40, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [PubMed]
- Sumi, C.D.; Yang, B.W.; Yeo, I.-C.; Hahm, Y.T. Antimicrobial peptides of the genus Bacillus: A new era for antibiotics. Can. J. Microbiol. 2014, 61, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, C.L. Advances in peptide pharmaceuticals. Curr. Pharm. Biotechnol. 2009, 10, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.A.; Citron, D.M.; Wang, C.C. Development of Tyrocidine A analogues with improved antibacterial activity. Bioorg. Med. Chem. 2007, 15, 6667–6677. [Google Scholar] [CrossRef] [PubMed]
- Mosges, R.; Baues, C.M.; Schroder, T.; Sahin, K. Acute bacterial otitis externa: Efficacy and safety of topical treatment with an antibiotic ear drop formulation in comparison to glycerol treatment. Curr. Med. Res. Opin. 2011, 27, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Awais, M.; Shah, A.A.; Hameed, A.; Hasan, F. Isolation, identification and optimization of Bacitracin produced by Bacillus sp. Pak. J. Bot. 2007, 39, 1303–1312. [Google Scholar]
- Bacitracin. Available online: http://www.drugbank.ca/drugs/DB00626. (accessed on 18 August 2017).
- Deegan, L.H.; Cotter, P.D.; Hill, C.; Ross, P. Bacterlocins: Biological tools for bio-preservation and shelf-life extension. Int. Dairy J. 2006, 16, 1058–1071. [Google Scholar] [CrossRef]
- Humphries, R.M.; Pollett, S.; Sakoulas, G. A current perspective on daptomycin for the clinical microbiologist. Clin. Microbiol. Rev. 2013, 26, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Drug-drug Interaction Study to Investigate Interaction Between Amikacin and POL7080. Available online: https://clinicaltrials.gov/ct2/show/NCT02897869?term=pol7080&rank=4 (accessed on 18 August 2017).
- A First in Human Study of the Safety and Tolerability of Single and Multiple Doses of SPR741 in Healthy Volunteers. Available online: https://clinicaltrials.gov/ct2/show/NCT03022175?term=spr741&rank=1 (accessed on 18 August 2017).
- Spero Therapeutics to Unveil Data on Potentiator Platform at ASM Microbe 2016. Available online: https://sperotherapeutics.com/news/press-releases/spero-therapeutics-unveil-data-potentiator-platform-asm-microbe-2016/ (accessed on 18 August 2017).
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.M.; Fusté, E.; Rabanal, F.; Vinuesa, T.; Viñas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.S.; Baines, S.D.; Todhunter, S.L.; Freeman, J.; Chilton, C.H.; Wilcox, M.H. Evaluation of NVB302 versus vancomycin activity in an in vitro human gut model of Clostridium difficile infection. J. Antimicrob. Chemother. 2013, 68, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Velden, W.J.; van Iersel, T.M.; Blijlevens, N.M.; Donnelly, J.P. Safety and tolerability of the antimicrobial peptide human lactoferrin 1-11 (hLF1-11). BMC Med. 2009, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, Y.; Qian, G.; Wang, Y.; Chen, H.; Li, Y.Z.; Liu, F.; Shen, Y.; Du, L. Identification and characterization of the anti-methicillin-resistant Staphylococcus aureus WAP-8294a2 biosynthetic gene cluster from Lysobacter enzymogenes OH11. Antimicrob. Agents Chemother. 2011, 55, 5581–5589. [Google Scholar] [CrossRef] [PubMed]
- C16G2 STAMP Program. Available online: http://www.c3jtherapeutics.com/technologies/c16g2/ (accessed on 18 August 2017).
- Kaplan, C.W.; Sim, J.H.; Shah, K.R.; Kolesnikova-Kaplan, A.; Shi, W.Y.; Eckert, R. Selective membrane disruption: Mode of action of C16G2, a specifically targeted antimicrobial peptide. Antimicrob. Agents Chemother. 2011, 55, 3446–3452. [Google Scholar] [CrossRef] [PubMed]
- Soligenix Inc. SGX9 executive summary. Available online: http://www.soligenix.com/wp-content/uploads/sgx94_executive_summary_032216.pdf (accessed on 18 August 2017).
- A Study of DPK-060 to Investigate Clinical Safety and Efficacy in Patients With Acute External Otitis. Available online: https://clinicaltrials.gov/ct2/show/NCT01447017?term=DPK-060&rank=1 (accessed on 18 August 2017).
- Study of PXL01 Versus Placebo to Inhibit Adhesion Formation After Flexor Tendon Surgery (PHSU02). Available online: https://clinicaltrials.gov/ct2/show/NCT01022242?term=PXL01&rank=1 (accessed on 18 August 2017).
- Study of the Effects of Brilacidin Oral Rinse on Radiation-induced Oral Mucositis in Patients with Head and Neck Cancer (Brilacidin). Available online: https://clinicaltrials.gov/ct2/show/NCT02324335?term=Brilacidin&rank=1 (accessed on 18 August 2017).
- Phase 2B Dose-Ranging Study of PAC113 Mouthrinse in HIV Seropositive Individuals With Oral Candidiasis. Available online: https://clinicaltrials.gov/ct2/show/NCT00659971?term=PAC113&rank=1 (accessed on 18 August 2017).
- Murepavavadin (POL7980). Available online: http://www.polyphor.com/products/pol7080 (accessed on 18 August 2017).
- Pharmacokinetics, Safety and Efficacy of POL7080 in Patients With Ventilator Associated Pseudomonas Aeruginosa Pneumonia. Available online: https://clinicaltrials.gov/ct2/show/NCT02096328?term=pol7080&rank=1 (accessed on 18 August 2017).
- A Phase II Study to Evaluate the Efficacy and Safety of Two Doses of LTX-109 in Impetigo. Available online: https://clinicaltrials.gov/ct2/show/NCT01803035 (accessed on 18 August 2017).
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.; Bonilla, H.; Saravolatz, L.D.; Fakih, M.G.; Fugelli, A.; Olsen, W.M. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 4478–4482. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.C.; Janson, H.; Wold, H.; Fugelli, A.; Andersson, K.; Hakangard, C.; Olsson, P.; Olsen, W.M. LTX-109 is a novel agent for nasal decolonization of methicillin-resistant and -sensitive Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Dr. Reddy’s Laboratories Ltd. Available online: http://www.drreddys.com/ (accessed on 18 August 2017).
- Malanovic, N.; Leber, R.; Schmuck, M.; Kriechbaum, M.; Cordfunke, R.A.; Drijfhout, J.W.; de Breij, A.; Nibbering, P.H.; Kolb, D.; Lohner, K. Phospholipid-driven differences determine the action of the synthetic antimicrobial peptide OP-145 on Gram-positive bacterial and mammalian membrane model systems. Biochem. Biophys. Acta Biomembr. 2015, 1848, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- PXL01—Prevention of Post-Surgical Adhesions. Available online: http://www.promorepharma.com/en/pxl01-prevention-of-post-surgical-adhesions/ (accessed on 18 August 2017).
- Gronberg, A.; Mahlapuu, M.; Stahle, M.; Whately-Smith, C.; Rollman, O. Treatment with LL-37 is safe and effective in enhancing healing of hard-to-heal venous leg ulcers: A randomized, placebo-controlled clinical trial. Wound Repair Regen. 2014, 22, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Novexatin® (NP213) is a Novel Cationic Antifungal Peptide that Has been Formulated as a Brush-on-Treatment for Onychomycosis (Fungal nail Infection). Available online: https://www.novabiotics.co.uk/pipeline/novexatin-np213 (accessed on 18 August 2017).
- Phase III Efficacy and Safety Study of AB103 in the Treatment of Patients With Necrotizing Soft Tissue Infections (ACCUTE). Available online: https://clinicaltrials.gov/ct2/show/NCT02469857?term=p2TA&rank=1 (accessed on 18 August 2017).
- Kollef, M.; Pittet, D.; Sanchez Garcia, M.; Chastre, J.; Fagon, J.Y.; Bonten, M.; Hyzy, R.; Fleming, T.R.; Fuchs, H.; Bellm, L.; et al. A randomized double-blind trial of iseganan in prevention of ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 2006, 173, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Trial of Iseganan in Prevention of Ventilator-Associated Pneumonia. Available online: https://clinicaltrials.gov/ct2/show/NCT00118781?term=Iseganan&rank=1 (accessed on 18 August 2017).
- Lipsky, B.A.; Holroyd, K.J.; Zasloff, M. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: A randomized, controlled, double-blinded, multicenter trial of pexiganan cream. Clin. Infect. Dis. 2008, 47, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.N.; Dugourd, D.; Castanho, M.A. Omiganan pentahydrochloride in the front line of clinical applications of antimicrobial peptides. Recent Pat. Anti-Infect. Drug Discov. 2006, 1, 201–207. [Google Scholar] [CrossRef]
- Knight-Connoni, V.; Mascio, C.; Chesnel, L.; Silverman, J. Discovery and development of surotomycin for the treatment of Clostridium difficile. J. Ind. Microbiol. Biotechnol. 2016, 43, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Nti-851 (RamoplaninTM). Available online: http://www.nanotherapeutics.com/ramoplanin/ (accessed on 18 August 2017).
- Stiefel, U.; Pultz, N.J.; Helfand, M.S.; Donskey, C.J. Efficacy of oral ramoplanin for inhibition of intestinal colonization by vancomycin-resistant Enterococci in mice. Antimicrob. Agents Chemother. 2004, 48, 2144–2148. [Google Scholar] [CrossRef] [PubMed]
- Oragenics-Intrexon Collaboration Announces Significant Progress towards Commercial Production of Lead Lantibiotic MU1140. Available online: http://www.oragenics.com/news-media/press-releases/detail/13/oragenics-intrexon-collaboration-announces-significant (accessed on 18 August 2017).
- Ghobrial, O.G.; Derendorf, H.; Hillman, J.D. Pharmacodynamic activity of the lantibiotic MU1140. Int. J. Antimicrob. Agents 2009, 33, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Anti-Infective. Available online: http://helixbiomedix.com/antiinfective.html (accessed on 18 August 2017).
- Novarifyn® (NP432) is antibacterial peptide with a number of key benefits over conventional antibiotic therapies and the clear potential to succeed where existing treatments for a number of bacterial infections, including those caused by MRSA, P. aeruginosa, and C. difficile, are failing. Available online: http://www.novabiotics.co.uk/pipeline/novarifyn-np432 (accessed on 18 August 2017).
- Arenecin. Available online: https://adeniumbiotech.com/arencin/ (accessed on 18 August 2017).
- Andra, J.; Jakovkin, I.; Grotzinger, J.; Hecht, O.; Krasnosdembskaya, A.D.; Goldmann, T.; Gutsmann, T.; Leippe, M. Structure and mode of action of the antimicrobial peptide arenicin. Biochem. J. 2008, 410, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Adenium Biotech Pipeline. Available online: https://adeniumbiotech.com/pipeline/ (accessed on 18 August 2017).
- AvidocinTM & PurocinTM proteins. Available online: https://avidbiotics.com/technology/avidocin-proteins/ (accessed on 18 August 2017).
- Gebhart, D.; Lok, S.; Clare, S.; Tomas, M.; Stares, M.; Scholl, D.; Donskey, C.J.; Lawley, T.D.; Govoni, G.R. A modified r-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. mBio 2015, 6, e02368-14. [Google Scholar] [CrossRef] [PubMed]
- Avan, I.; Hall, C.D.; Katritzky, A.R. Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev. 2014, 43, 3575–3594. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug. Resist. Updates 2016, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 2006, 8, 11–26. [Google Scholar] [PubMed]
- Maria-Neto, S.; de Almeida, K.C.; Macedo, M.L.R.; Franco, O.L. Understanding bacterial resistance to antimicrobial peptides: From the surface to deep inside. Biochim. Biophys. Acta 2015, 1848, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Habets, M.G.; Brockhurst, M.A. Therapeutic antimicrobial peptides may compromise natural immunity. Biol. Lett. 2012, 8, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial peptides: mechanisms of action and resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, K.L.; Crispell, E.K.; McBride, S.M. Antimicrobial peptide resistance mechanisms of Gram-positive bacteria. Antibiotics 2014, 3, 461–492. [Google Scholar] [CrossRef] [PubMed]
- Gruenheid, S.; Le Moual, H. Resistance to antimicrobial peptides in Gram-negative bacteria. FEMS Microbiol. Lett. 2012, 330, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Kubicek-Sutherland, J.Z.; Lofton, H.; Vestergaard, M.; Hjort, K.; Ingmer, H.; Andersson, D.I. Antimicrobial peptide exposure selects for Staphylococcus aureus resistance to human defence peptides. J. Antimicrob. Chemother. 2017, 72, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Nuri, R.; Shprung, T.; Shai, Y. Defensive remodeling: How bacterial surface properties and biofilm formation promote resistance to antimicrobial peptides. Biochim. Biophys. Acta 2015, 1848, 3089–3100. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, R.D.; Haney, E.F.; Franzyk, H.; Hancock, R.E. Characterization of a proteolytically stable multifunctional host defense peptidomimetic. Chem. Biol. 2013, 20, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Hein-Kristensen, L.; Franzyk, H.; Holch, A.; Gram, L. Adaptive Evolution of Escherichia coli to an alpha-peptide/beta-peptoid peptidomimetic induces stable resistance. PLoS ONE 2013, 8, e73620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard, 9th ed.; CLSI: Wayne, PA, USA, 2012.
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Citterio, L.; Franzyk, H.; Palarasah, Y.; Andersen, T.E.; Mateiu, R.V.; Gram, L. Improved in vitro evaluation of novel antimicrobials: Potential synergy between human plasma and antibacterial peptidomimetics, amps and antibiotics against human pathogenic bacteria. Res. Microbiol. 2016, 167, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 29707. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, C.R.; Bosso, J.A.; Friedrich, L.V.; White, R.L. Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis. 2002, 44, 363–366. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Yu, C.M.; Yu, V.L.; Chow, J.W. Synergy assessed by checkerboard—A critical analysis. Diagn. Microbiol. Infect. Dis. 1993, 16, 343–349. [Google Scholar] [CrossRef]
- Ceri, H.; Olson, M.E.; Stremick, C.; Read, R.R.; Morck, D.; Buret, A. The calgary biofilm device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 1999, 37, 1771–1776. [Google Scholar] [PubMed]
- Harrison, J.J.; Stremick, C.A.; Turner, R.J.; Allan, N.D.; Olson, M.E.; Ceri, H. Microtiter susceptibility testing of microbes growing on peg lids: A miniaturized biofilm model for high-throughput screening. Nat. Protoc. 2010, 5, 1236–1254. [Google Scholar] [CrossRef] [PubMed]
- Oddo, A.; Hansen, P.R. Hemolytic activity of antimicrobial peptides. Methods Mol. Biol. 2017, 1548, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, R.D.; Sandberg-Schaal, A.; Vissing, K.J.; Nielsen, H.M.; Frimodt-Møller, N.; Franzyk, H. Tailoring cytotoxicity of antimicrobial peptidomimetics with high activity against multidrug-resistant Escherichia coli. J. Med. Chem. 2014, 57, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell viability assays. In Assay Guidance Manual; [Internet: Last updated, March 31, 2017]; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium formazan assay for cell-growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [PubMed]
- Pohjala, L.; Tammela, P.; Samanta, S.K.; Yli-Kauhaluoma, J.; Vuorela, P. Assessing the data quality in predictive toxicology using a panel of cell lines and cytotoxicity assays. Anal. Biochem. 2007, 362, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Godoy, A.; Muldoon, C.; Becker, B.; Elliott, A.G.; Lash, L.H.; Huang, J.X.; Butler, M.S.; Pelingon, R.; Kavanagh, A.M.; Ramu, S.; et al. Activity and Predicted Nephrotoxicity of Synthetic Antibiotics Based on Polymyxin B. J. Med. Chem. 2016, 59, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Tew, G.N.; Scott, R.W.; Klein, M.L.; Degrado, W.F. De novo design of antimicrobial polymers, foldamers, and small molecules: From discovery to practical applications. Acc. Chem. Res. 2010, 43, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Thaker, H.D.; Cankaya, A.; Scott, R.W.; Tew, G.N. Role of amphiphilicity in the design of synthetic mimics of antimicrobial peptides with Gram-negative activity. ACS Med. Chem. Lett. 2013, 4, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Thaker, H.D.; Sgolastra, F.; Clements, D.; Scott, R.W.; Tew, G.N. Synthetic mimics of antimicrobial peptides from triaryl scaffolds. J. Med. Chem. 2011, 54, 2241–2254. [Google Scholar] [CrossRef] [PubMed]
- Flemming, K.; Klingenberg, C.; Cavanagh, J.P.; Sletteng, M.; Stensen, W.; Svendsen, J.S.; Flaegstad, T. High in vitro antimicrobial activity of synthetic antimicrobial peptidomimetics against staphylococcal biofilms. J. Antimicrob. Chemother. 2009, 63, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.E.; Stensen, W.; Kalaaji, M.; Rekdal, Ø.; Svendsen, J.S. Synthetic antimicrobial peptidomimetics with therapeutic potential. J. Med. Chem. 2008, 51, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Alst, T.; Havelkova, M.; Strøm, M.B. Antimicrobial activity of small β-peptidomimetics based on the pharmacophore model of short cationic antimicrobial peptides. J. Med. Chem. 2009, 53, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Gunasekaran, P.; Rajasekaran, G.; Kim, E.Y.; Lee, S.J.; Bang, G.; Cho, K.; Hyun, J.K.; Lee, H.J.; Jeon, Y.H.; et al. Pyrazole derived ultra-short antimicrobial peptidomimetics with potent anti-biofilm activity. Eur. J. Med. Chem. 2017, 125, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Jacob, B.; Gunasekaran, P.; Murugan, R.N.; Ryu, E.K.; Lee, G.H.; Hyun, J.K.; Cheong, C.; Kim, N.H.; Shin, S.Y.; et al. Poly-lysine peptidomimetics having potent antimicrobial activity without hemolytic activity. Amino Acids 2014, 46, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Murugan, R.N.; Jacob, B.; Hyun, J.K.; Cheong, C.; Hwang, E.; Park, H.N.; Seo, J.H.; Srinivasrao, G.; Lee, K.S.; et al. Discovery of novel histidine-derived lipo-amino acids: Applied in the synthesis of ultra-short antimicrobial peptidomimetics having potent antimicrobial activity, salt resistance and protease stability. Eur. J. Med. Chem. 2013, 68, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Panda, S.S.; Oliferenko, A.A.; Oliferenko, P.V.; Girgis, A.S.; Elagawany, M.; Kucukbay, F.Z.; Panda, C.S.; Pillai, G.G.; Samir, A.; et al. Macrocyclic peptidomimetics with antimicrobial activity: Synthesis, bioassay, and molecular modeling studies. Org. Biomol. Chem. 2015, 13, 9492–9503. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Moehle, K.; Abou-Hadeed, K.; Obrecht, D.; Robinson, J.A. Biaryl amino acid templates in place of d-pro-l-pro in cyclic β-hairpin cationic antimicrobial peptidomimetics. Org. Biomol. Chem. 2007, 5, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Wales, S.M.; Hammer, K.A.; Somphol, K.; Kemker, I.; Schroder, D.C.; Tague, A.J.; Brkic, Z.; King, A.M.; Lyras, D.; Riley, T.V.; et al. Synthesis and antimicrobial activity of binaphthyl-based, functionalized oxazole and thiazole peptidomimetics. Org. Biomol. Chem. 2015, 13, 10813–10824. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, I.; Albeck, A.; Gellerman, G.; Shatzmiller, S.; Grynszpan, F. 1,4-dihydropyridine cationic peptidomimetics with antibacterial activity. Int. J. Pept. Res. Ther. 2015, 21, 243–247. [Google Scholar] [CrossRef]
- Lohan, S.; Cameotra, S.S.; Bisht, G.S. Antibacterial evaluation of structurally amphipathic, membrane active small cationic peptidomimetics: Synthesized by incorporating 3-amino benzoic acid as peptidomimetic element. Eur. J. Med. Chem. 2014, 83, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Haldar, J. Membrane-active small molecules: Designs inspired by antimicrobial peptides. ChemMedChem 2015, 10, 1606–1624. [Google Scholar] [CrossRef] [PubMed]
- Liskamp, R.M.J.; Rijkers, D.T.S.; Kruijtzer, J.A.W.; Kemmink, J. Peptides and proteins as a continuing exciting source of inspiration for peptidomimetics. ChemBioChem 2011, 12, 1626–1653. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kodadek, T. Peptoids as potential therapeutics. Curr. Opin. Mol. Ther. 2009, 11, 299–307. [Google Scholar] [PubMed]
- Culf, A.S.; Ouellette, R.J. Solid-phase synthesis of N-substituted glycine oligomers (α-peptoids) and derivatives. Molecules 2010, 15, 5282–5335. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.A.; Blackwell, H.E. Structure–function relationships in peptoids: Recent advances toward deciphering the structural requirements for biological function. Org. Biomol. Chem. 2009, 7, 1508–1524. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.R.; Munk, J.K. Synthesis of antimicrobial peptoids. Methods Mol. Biol. 2013, 1047, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zuckermann, R.N. Peptoid polymers: A highly designable bioinspired material. ACS Nano 2013, 7, 4715–4732. [Google Scholar] [CrossRef] [PubMed]
- Trabocchi, A.; Guarna, A. Peptoids. In Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs; John Wiley & Sons, Ltd.: Chichester, UK, 2014. [Google Scholar] [CrossRef]
- Zuckermann, R.N. Peptoid origins. Pept. Sci. 2011, 96, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Corson, A.E.; Armstrong, S.A.; Wright, M.E.; McClelland, E.E.; Bicker, K.L. Discovery and characterization of a peptoid with antifungal activity against Cryptococcus neoformans. ACS Med. Chem. Lett. 2016, 7, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bolt, H.L.; Eggimann, G.A.; McAuley, D.F.; McMullan, R.; Curran, T.; Zhou, M.; Jahoda, C.A.B.; Cobb, S.L.; Lundy, F.T. Peptoid efficacy against polymicrobial biofilms determined by using propidium monoazide-modified quantitative PCR. ChemBioChem 2017, 18, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Eimerman, P.R.; Hardy, J.W.; Cirillo, J.D.; Contag, C.H.; Barron, A.E. Efficacy of antimicrobial peptoids against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Eggimann, G.A.; Bolt, H.L.; Denny, P.W.; Cobb, S.L. Investigating the anti-leishmanial effects of linear peptoids. ChemMedChem 2015, 10, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Seo, J.; Willingham, S.B.; Czyzewski, A.M.; Gonzalgo, M.L.; Weissman, I.L.; Barron, A.E. Learning from host-defense peptides: Cationic, amphipathic peptoids with potent anticancer activity. PLoS ONE 2014, 9, e90397. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, N.; Ulbricht, J.; Lorson, T.; Schlaad, H.; Luxenhofer, R. Peptoids and polypeptoids at the frontier of supra- and macromolecular engineering. Chem. Rev. 2016, 116, 1753–1802. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Zuckermann, R.N. Protein side-chain translocation mutagenesis via incorporation of peptoid residues. ACS Chem. Biol. 2011, 6, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Unciti-Broceta, A.; Diezmann, F.; Ou-Yang, C.Y.; Fara, M.A.; Bradley, M. Synthesis, penetrability and intracellular targeting of fluorescein-tagged peptoids and peptide-peptoid hybrids. Bioorg. Med. Chem. 2009, 17, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, J.; Luo, Y.; Liang, X.; Supnet, C.; Kim, M.W.; Lotz, G.P.; Yang, G.; Muchowski, P.J.; Kodadek, T.; et al. Expanded polyglutamine-binding peptoid as a novel therapeutic agent for treatment of Huntington’s disease. Chem. Biol. 2011, 18, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Ren, G.; Liu, H.G.; Miao, Z.; Park, M.; Wang, Y.H.; Miller, T.M.; Barron, A.E.; Cheng, Z. In vivo biodistribution and small animal PET of 64Cu-labeled antimicrobial peptoids. Bioconj. Chem. 2012, 23, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Szekely, T.; Roy, O.; Faure, S.; Taillefumier, C. From glycopeptides to glycopeptoids. Eur. J. Org. Chem. 2014, 2014, 5641–5657. [Google Scholar] [CrossRef]
- Mao, J.; Bong, D. Synthesis of DNA-binding peptoids. Synlett 2015, 26, 1581–1585. [Google Scholar] [CrossRef]
- De Cola, C.; Manicardi, A.; Corradini, R.; Izzo, I.; De Riccardis, F. Carboxyalkyl peptoid PNAs: Synthesis and hybridization properties. Tetrahedron 2012, 68, 499–506. [Google Scholar] [CrossRef]
- Bolt, H.L.; Cobb, S.L. A practical method for the synthesis of peptoids containing both lysine-type and arginine-type monomers. Org. Biomol. Chem. 2016, 14, 1211–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojsoska, B.; Zuckermann, R.N.; Jenssen, H. Structure-activity relationship study of novel peptoids that mimic the structure of antimicrobial peptides. Antimicrob. Agents Chemother. 2015, 59, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids (oligo(N-substituted glycines)) by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
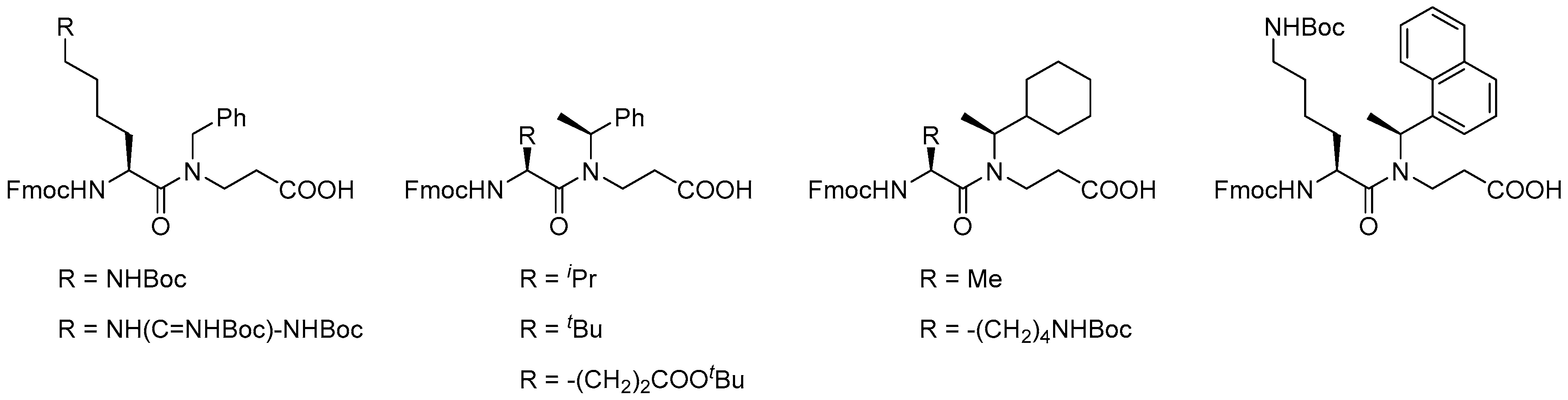
- Tal-Gan, Y.; Freeman, N.S.; Klein, S.; Levitzki, A.; Gilon, C. Synthesis and structure–activity relationship studies of peptidomimetic PKB/Akt inhibitors: The significance of backbone interactions. Bioorg. Med. Chem. 2010, 18, 2976–2985. [Google Scholar] [CrossRef] [PubMed]
- Vézina-Dawod, S.; Derson, A.; Biron, E. N-substituted arylsulfonamide building blocks as alternative submonomers for peptoid synthesis. Tetrahedron Lett. 2015, 56, 382–385. [Google Scholar] [CrossRef]
- Li, S.; Bowerman, D.; Marthandan, N.; Klyza, S.; Luebke, K.J.; Garner, H.R.; Kodadek, T. Photolithographic synthesis of peptoids. J. Am. Chem. Soc. 2004, 126, 4088–4089. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.-P.; Davis, P.; Lee, K.B.; Porreca, F.; Yamamura, H.I.; Hruby, V.J. Synthesis using a Fmoc-based strategy and biological activities of some reduced peptide bond pseudopeptide analogs of dynorphin A1. J. Med. Chem. 1995, 38, 3462–3468. [Google Scholar] [CrossRef] [PubMed]
- Horn, T.; Lee, B.-C.; Dill, K.A.; Zuckermann, R.N. Incorporation of chemoselective functionalities into peptoids via solid-phase submonomer synthesis. Bioconjug. Chem. 2004, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Rene, A.; Martinez, J.; Cavelier, F. N-substituted glycines with functional side-chains for peptoid synthesis. Eur. J. Org. Chem. 2014, 8142–8147. [Google Scholar] [CrossRef]
- Caumes, C.; Hjelmgaard, T.; Remuson, R.; Faure, S.; Taillefumier, C. Highly convenient gram-scale solution-phase peptoid synthesis and orthogonal side-chain post-modification. Synthesis 2011, 257–264. [Google Scholar] [CrossRef]
- Oddo, A.; Münzker, L.; Hansen, P.R. Peptide macrocycles featuring a backbone secondary amine: A convenient strategy for the synthesis of lipidated cyclic and bicyclic peptides on solid support. Org. Lett. 2015, 17, 2502–2505. [Google Scholar] [CrossRef] [PubMed]
- Figliozzi, G.; Goldsmith, R.; Ng, S.; Banville, S.; Zuckermann, R. Synthesis of N-substituted glycine peptoid libraries. Methods Enzymol. 1996, 267, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Biswas, G.; Park, S.; Kim, A.; Park, H.; Park, E.; Kim, J.; Kwon, Y.U. Unusual truncation of N-acylated peptoids under acidic conditions. Org. Biomol. Chem. 2014, 12, 5222–5226. [Google Scholar] [CrossRef] [PubMed]
- Goodson, B.; Ehrhardt, A.; Ng, S.; Nuss, J.; Johnson, K.; Giedlin, M.; Yamamoto, R.; Moos, W.H.; Krebber, A.; Ladner, M.; et al. Characterization of novel antimicrobial peptoids. Antimicrob. Agents Chemother. 1999, 43, 1429–1434. [Google Scholar] [PubMed]
- Ng, S.; Goodson, B.; Ehrhardt, A.; Moos, W.H.; Siani, M.; Winter, J. Combinatorial discovery process yields antimicrobial peptoids. Bioorg. Med. Chem. 1999, 7, 1781–1785. [Google Scholar] [CrossRef]
- Mojsoska, B.; Carretero, G.; Larsen, S.; Mateiu, R.V.; Jenssen, H. Peptoids successfully inhibit the growth of Gram-negative E. coli causing substantial membrane damage. Sci. Rep. 2017, 7, 42332. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.L.; Eggimann, G.A.; Jahoda, C.A.B.; Zuckermann, R.N.; Sharples, G.J.; Cobb, S.L. Exploring the links between peptoid antibacterial activity and toxicity. MedChemComm 2017, 8, 886–896. [Google Scholar] [CrossRef]
- Czyzewski, A.M.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Chongsiriwatana, N.P.; Yuen, E.; Hancock, R.E.W.; Barron, A.E. In vivo, in vitro, and in silico characterization of peptoids as antimicrobial agents. PLoS ONE 2016, 11, e0135961. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.K.; Nan, Y.H.; Lee, E.K.; Shin, S.Y. A novel Trp-rich model antimicrobial peptoid with increased protease stability. Bull. Korean Chem. Soc. 2010, 31, 2509–2513. [Google Scholar] [CrossRef]
- Chongsiriwatana, N.P.; Wetzler, M.; Barron, A.E. Functional synergy between antimicrobial peptoids and peptides against Gram-negative bacteria. Antimicrob. Agents Chemother. 2011, 55, 5399–5402. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Miller, T.M.; Wetzler, M.; Vakulenko, S.; Karlsson, A.J.; Palecek, S.P.; Mobashery, S.; Barron, A.E. Short alkylated peptoid mimics of antimicrobial lipopeptides. Antimicrob. Agents Chemother. 2011, 55, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Wadman, M.W.; Dohm, M.T.; Czyzewski, A.M.; Spormann, A.M.; Barron, A.E. Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2011, 55, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Findlay, B.; Szelemej, P.; Zhanel, G.G.; Schweizer, F. Guanidylation and tail effects in cationic antimicrobial lipopeptoids. PLoS ONE 2012, 7, e41141. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Turkett, J.A.; Corson, A.E.; Bicker, K.L. Peptoid library agar diffusion (PLAD) assay for the high-throughput identification of antimicrobial peptoids. ACS Comb. Sci. 2016, 18, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Turkett, J.A.; Bicker, K.L. Evaluating the effect of peptoid lipophilicity on antimicrobial potency, cytotoxicity, and combinatorial library design. ACS Comb. Sci. 2017, 19, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Shin, S.B.Y.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Benson, M.A.; Shin, S.B.Y.; Torres, V.J.; Kirshenbaum, K. Amphiphilic cyclic peptoids that exhibit antimicrobial activity by disrupting Staphylococcus aureus membranes. Eur. J. Org. Chem. 2013, 2013, 3560–3566. [Google Scholar] [CrossRef]
- Andreev, K.; Martynowycz, M.W.; Ivankin, A.; Huang, M.L.; Kuzmenko, I.; Meron, M.; Lin, B.H.; Kirshenbaum, K.; Gidalevitz, D. Cyclization improves membrane permeation by antimicrobial peptoids. Langmuir 2016, 32, 12905–12913. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.L.; Park, Y.; Park, I.S.; Park, Y.S.; Kim, Y.; Hahm, K.S.; Shin, S.Y. Improvement of bacterial cell selectivity of melittin by a single Trp mutation with a peptoid residue. Protein Pept. Lett. 2006, 13, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.H.; Park, K.H.; Jeon, Y.J.; Park, Y.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Antimicrobial and anti-inflammatory activities of a Leu/Lys-rich antimicrobial peptide with phe-peptoid residues. Protein Pept. Lett. 2007, 14, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-K.; Lee, S.-A.; Shin, S.; Lee, J.-Y.; Jeong, K.-W.; Nan, Y.H.; Park, Y.S.; Shin, S.Y.; Kim, Y. Structural flexibility and the positive charges are the key factors in bacterial cell selectivity and membrane penetration of peptoid-substituted analog of piscidin 1. Biochim. Biophys. Acta 2010, 1798, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y. Effect of double replacement of L-pro, D-pro, D-leu or Nleu in hydrophobic face of amphipathic alpha-helical model antimicrobial peptide on structure, cell selectivity and mechanism of action. Bull. Korean Chem. Soc. 2014, 35, 3267–3274. [Google Scholar] [CrossRef]
- Zhu, W.L.; Song, Y.M.; Park, Y.; Park, K.H.; Yang, S.T.; Kim, J.I.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Substitution of the leucine zipper sequence in melittin with peptoid residues affects self-association, cell selectivity, and mode of action. Biochim. Biophys. Acta 2007, 1768, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.L.; Hahm, K.S.; Shin, S.Y. Cathelicidin-derived Trp/Pro-rich antimicrobial peptides with lysine peptoid residue (Nlys): Therapeutic index and plausible mode of action. J. Pept. Sci. 2007, 13, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.C.; Jeon, D.; Shin, A.; Jin, S.; Shin, S.Y.; Park, Y.S.; Kim, Y. Effects of hydrophobic peptoid substitutions on the bacterial cell selectivity and antimicrobial activity of piscidin 1. Bull. Korean Chem. Soc. 2016, 37, 1545–1551. [Google Scholar] [CrossRef]
- Shin, A.; Lee, E.; Jeon, D.; Park, Y.G.; Bang, J.K.; Park, Y.S.; Shin, S.Y.; Kim, Y. Peptoid-substituted hybrid antimicrobial peptide derived from papiliocin and magainin 2 with enhanced bacterial selectivity and anti-inflammatory activity. Biochemistry 2015, 54, 3921–3931. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Bang, J.K.; Kim, H.J.; Kim, J.K.; Kim, Y.; Shin, S.Y. Antimicrobial specificity and mechanism of action of disulfide-removed linear analogs of the plant-derived Cys-rich antimicrobial peptide Ib-AMP1. Peptides 2009, 30, 2144–2149. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, M.; Benincasa, M.; Bertoloni, G.; Biondi, B.; Dosselli, R.; Papini, E.; Reddi, E.; Rocchi, R.; Tavano, R.; Gennaro, R. Substitution of the arginine/leucine residues in apidaecin Ib with peptoid residues: Effect on antimicrobial activity, cellular uptake, and proteolytic degradation. J. Med. Chem. 2009, 52, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Bonke, G.; Larsen, C.J.; Yavari, N.; Nielsen, P.E.; Franzyk, H. Antibacterial peptide nucleic acid–antimicrobial peptide (PNA–AMP) conjugates: Antisense targeting of fatty acid biosynthesis. Bioconjug. Chem. 2016, 27, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Yan, T.T.; Wang, L.; Guo, F.F.; Ning, G.S.; Xiong, M. Statistical design, structural analysis, and in vitro susceptibility assay of antimicrobial peptoids to combat bacterial infections. J. Chemom. 2016, 30, 369–376. [Google Scholar] [CrossRef]
- Ryge, T.; Hansen, P. Novel lysine-peptoid hybrids with antibacterial properties. J. Pept. Sci. 2005, 11, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Frimodt-Moller, N.; Hansen, P.R. Antimicrobial activities of twenty lysine-peptoid hybrids against clinically relevant bacteria and fungi. Chemotherapy 2008, 54, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Ingmer, H.; Thomsen, L.E. The lysine-peptoid hybrid LP5 maintain activity under physiological conditions and affects virulence gene expression in Staphylococcus aureus. Peptides 2016, 78, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A. β-peptoid “foldamers”-why the additional methylene unit? Biopolymers 2011, 96, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.S.; Engel-Andreasen, J.; Olsen, C.A. β-peptoid foldamers at last. Acc. Chem. Res. 2015, 48, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Hamper, B.C.; Kolodziej, S.A.; Scatez, A.M.; Smith, R.G.; Cortez, E. Solid phase synthesis of β-peptiods: N-substituted β-aminopropionic acid oligomers. J. Org. Chem. 1998, 63, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Norgren, A.S.; Zhang, S.D.; Arvidsson, P.I. Synthesis and circular dichroism spectroscopic investigations of oligomeric β-peptoids with α-chiral side chains. Org. Lett. 2006, 8, 4533–4536. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Lambert, M.; Witt, M.; Franzyk, H.; Jaroszewski, J.W. Solid-phase peptide synthesis and circular dichroism study of chiral β-peptoid homooligomers. Amino Acids 2008, 34, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, R.D.; Frimodt-Møller, N.; Franzyk, H. Antimicrobial activity of peptidomimetics against multidrug-resistant Escherichia coli: A comparative study of different backbones. J. Med. Chem. 2012, 55, 7253–7261. [Google Scholar] [CrossRef] [PubMed]
- Shuey, S.W.; Delaney, W.J.; Shah, M.C.; Scialdone, M.A. Antimicrobial β-peptoids by a block synthesis approach. Bioorg. Med. Chem. Lett. 2006, 16, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Roy, O.; Faure, S.; Thery, V.; Didierjean, C.; Taillefumier, C. Cyclic β-peptoids. Org. Lett. 2008, 10, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Bonke, G.; Vedel, L.; Witt, M.; Jaroszewski, J.W.; Olsen, C.A.; Franzyk, H. Dimeric building blocks for solid-phase synthesis of α-peptide-β-peptoid chimeras. Synthesis-Stuttgart 2008, 2008, 2381–2390. [Google Scholar] [CrossRef]
- Olsen, C.A.; Bonke, G.; Vedel, L.; Adsersen, A.; Witt, M.; Franzyk, H.; Jaroszewski, J.W. α-peptide/β-peptoid chimeras. Org. Lett. 2007, 9, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Ziegler, H.L.; Nielsen, H.M.; Frimodt-Møller, N.; Jaroszewski, J.W.; Franzyk, H. Antimicrobial, hemolytic, and cytotoxic activities of β-peptoid–peptide hybrid oligomers: Improved properties compared to natural amps. ChemBioChem 2010, 11, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Hein-Kristensen, L.; Knapp, K.M.; Franzyk, H.; Gram, L. Bacterial membrane activity of α-peptide/β-peptoid chimeras: Influence of amino acid composition and chain length on the activity against different bacterial strains. BMC Microbiol. 2011, 11, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahnsen, R.O.; Sandberg-Schaal, A.; Frimodt-Moller, N.; Nielsen, H.M.; Franzyk, H. End group modification: Efficient tool for improving activity of antimicrobial peptide analogues towards Gram-positive bacteria. Eur. J. Pharm. Biopharm. 2015, 95, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Hansen, P.R.; Damborg, P.; Nielsen, H.M.; Franzyk, H. Lysine-based α-peptide/β-peptoid peptidomimetics: Influence of hydrophobicity, fluorination and distribution of cationic charge on antimicrobial activity and cytotoxicity. ChemMedChem 2017, 20, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Hein-Kristensen, L.; Knapp, K.M.; Franzyk, H.; Gram, L. Selectivity in the potentiation of antibacterial activity of α-peptide/β-peptoid peptidomimetics and antimicrobial peptides by human blood plasma. Res. Microbiol. 2013, 164, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Vedel, L.; Bonke, G.; Foged, C.; Ziegler, H.; Franzyk, H.; Jaroszewski, J.W.; Olsen, C.A. Antiplasmodial and prehemolytic activities of α-peptide-β-peptoid chimeras. ChemBioChem 2007, 8, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Knapp, K.M.; Yang, L.; Molin, S.; Franzyk, H.; Folkesson, A. High in vitro antimicrobial activity of β-peptoid-peptide hybrid oligomers against planktonic and biofilm cultures of Staphylococcus epidermidis. Int. J. Antimicrob. Agents 2013, 41, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Skovbakke, S.L.; Larsen, C.J.; Heegaard, P.M.H.; Moesby, L.; Franzyk, H. Lipidated α-peptide/β-peptoid hybrids with potent antiinflammatory activity. J. Med. Chem. 2015, 58, 801–813. [Google Scholar] [CrossRef] [PubMed]
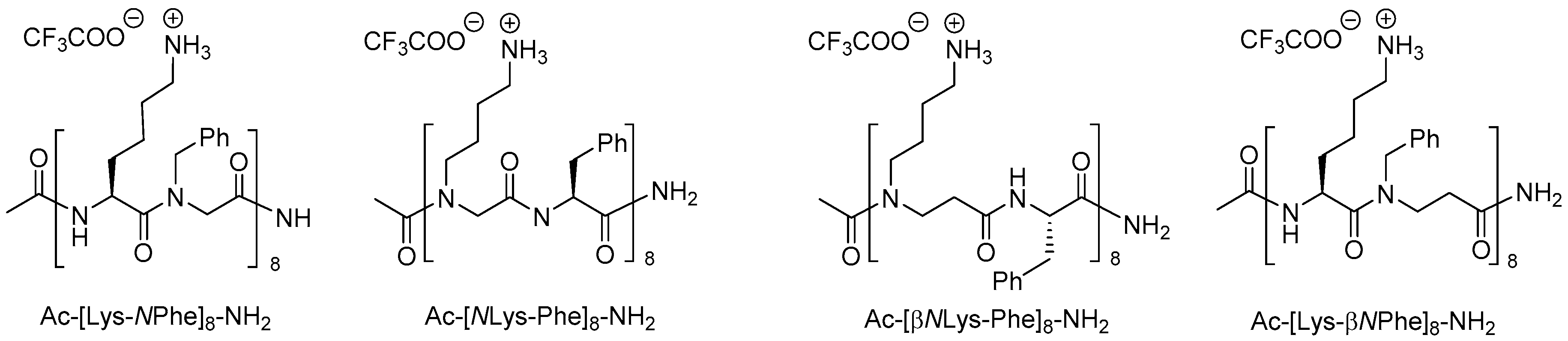
- Holdfeldt, A.; Skovbakke, S.L.; Winther, M.; Gabl, M.; Nielsen, C.; Perez-Gassol, I.; Larsen, C.J.; Wang, J.M.; Karlsson, A.; Dahlgren, C.; et al. The lipidated peptidomimetic Lau-((S)-Aoc)-(Lys-βNphe)6-NH2 is a novel formyl peptide receptor 2 agonist that activates both human and mouse neutrophil nadph oxidase. J. Biol. Chem. 2016, 291, 19888–19899. [Google Scholar] [CrossRef] [PubMed]
- Skovbakke, S.L.; Heegaard, P.M.H.; Larsen, C.J.; Franzyk, H.; Forsman, H.; Dahlgren, C. The proteolytically stable peptidomimetic Pam-(Lys-βNSpe)6-NH2 selectively inhibits human neutrophil activation via formyl peptide receptor 2. Biochem. Pharmacol. 2015, 93, 182–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreev, K.; Bianchi, C.; Laursen, J.S.; Citterio, L.; Hein-Kristensen, L.; Gram, L.; Kuzmenko, I.; Olsen, C.A.; Gidalevitz, D. Guanidino groups greatly enhance the action of antimicrobial peptidomimetics against bacterial cytoplasmic membranes. Biochim. Biophys. Acta 2014, 1838, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; de la Fuente-Nunez, C.; Hancock, R.E. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Skovbakke, S.L.; Winther, M.; Gabl, M.; Holdfeldt, A.; Linden, S.; Wang, J.M.; Dahlgren, C.; Franzyk, H.; Forsman, H. The peptidomimetic Lau-(Lys-βNSpe)6-NH2 antagonizes formyl peptide receptor 2 expressed in mouse neutrophils. Biochem. Pharmacol. 2016, 119, 56–65. [Google Scholar] [CrossRef] [PubMed]
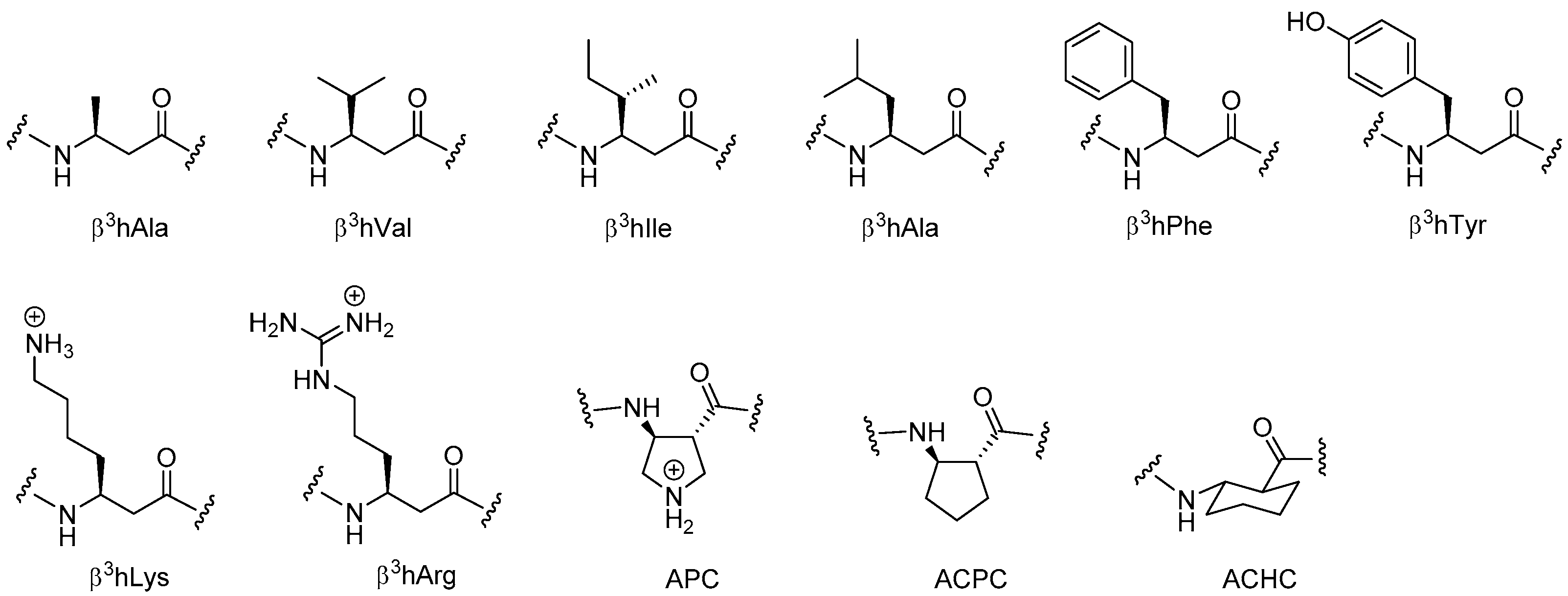
- Seebach, D.; Matthews, J.L. β-Peptides: A surprise at every turn. Chem. Commun. 1997, 2015–2022. [Google Scholar] [CrossRef]
- Arvidsson, P.I.; Frackenpohl, J.; Ryder, N.S.; Liechty, B.; Petersen, F.; Zimmermann, H.; Camenisch, G.P.; Woessner, R.; Seebach, D. On the antimicrobial and hemolytic activities of amphiphilic β-peptides. ChemBioChem 2001, 2, 771–773. [Google Scholar] [CrossRef]
- DeGrado, W.F.; Schneider, J.P.; Hamuro, Y. The twists and turns of β-peptides. J. Pept. Res. 1999, 54, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, Y.S.; Joel, P.; DeGrado, W.F. De novo design of antibacterial β-peptides. J. Am. Chem. Soc. 1999, 121, 12200–12201. [Google Scholar] [CrossRef]
- Porter, E.A.; Wang, X.; Lee, H.-S.; Weisblum, B.; Gellman, S.H. Antibiotics: Non-haemolytic β-amino-acid oligomers. Nature 2000, 404, 565. [Google Scholar] [CrossRef] [PubMed]
- Suresh Babu, V.V.; Gopi, H.N.; Anada, K. Homologation of α-amino acids to β-amino acids using Fmoc-amino acid pentafluorophenyl esters. J. Pept. Res. 1999, 53, 308–313. [Google Scholar] [CrossRef]
- Lee, H.S.; LePlae, P.R.; Porter, E.A.; Gellman, S.H. An efficient route to either enantiomer of orthogonally protected trans-3-aminopyrrolidine-4-carboxylic acid. J. Org. Chem. 2001, 66, 3597–3599. [Google Scholar] [CrossRef] [PubMed]
- LePlae, P.R.; Umezawa, N.; Lee, H.S.; Gellman, S.H. An efficient route to either enantiomer of trans-2-aminocyclopentanecarboxylic acid. J. Org. Chem. 2001, 66, 5629–5632. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; DeGrado, W. De novo design, synthesis, and characterization of antimicrobial β-peptides. J. Am. Chem. Soc. 2001, 123, 7553–7559. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [Google Scholar] [CrossRef] [PubMed]
- Geueke, B.; Namoto, K.; Agarkova, I.; Perriard, J.C.; Kohler, H.P.E.; Seebach, D. Bacterial cell penetration by β3-oligohomoarginines: Indications for passive transfer through the lipid bilayer. ChemBioChem 2005, 6, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Serrano, G.N.; Zhanel, G.G.; Schweizer, F. Antibacterial activity of ultrashort cationic lipo-β-peptides. Antimicrob. Agents Chemother. 2009, 53, 2215–2217. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.J.; Pomerantz, W.C.; Weisblum, B.; Gellman, S.H.; Palecek, S.P. Antifungal activity from 14-helical β-peptides. J. Am. Chem. Soc. 2006, 128, 12630–12631. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.J.; Pomerantz, W.C.; Neilsen, K.J.; Gellman, S.H.; Palecek, S.P. Effect of sequence and structural properties on 14-helical β-peptide activity against candida albicans planktonic cells and biofilms. ACS Chem. Biol. 2009, 4, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-R.; Raman, N.; Gellman, S.H.; Lynn, D.M.; Palecek, S.P. Hydrophobicity and helicity regulate the antifungal activity of 14-helical β-peptides. ACS Chem. Biol. 2014, 9, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Raman, N.; Lee, M.-R.; Lynn, D.; Palecek, S. Antifungal activity of 14-helical β-peptides against planktonic cells and biofilms of candida species. Pharmaceuticals 2015, 8, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Mora-Navarro, C.; Caraballo-Leon, J.; Torres-Lugo, M.; Ortiz-Bermudez, P. Synthetic antimicrobial β-peptide in dual-treatment with fluconazole or ketoconazole enhances the in vitro inhibition of planktonic and biofilm Candida albicans. J. Pept. Sci. 2015, 21, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Mora-Navarro, C.; Mendez-Vega, J.; Caraballo-Leon, J.; Lee, M.R.; Palecek, S.; Torres-Lugo, M.; Ortiz-Bermudez, P. Hydrophobicity of antifungal β-peptides is associated with their cytotoxic effect on in vitro human colon Caco-2 and liver HepG2 cells. PLoS ONE 2016, 11, e0149271. [Google Scholar] [CrossRef]
- Raman, N.; Lee, M.R.; Lopez, A.D.R.; Palecek, S.P.; Lynn, D.M. Antifungal activity of a β-peptide in synthetic urine media: Toward materials-based approaches to reducing catheter-associated urinary tract fungal infections. Acta Biomater. 2016, 43, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Raman, N.; Marchillo, K.; Lee, M.R.; Lopez, A.D.R.; Andes, D.R.; Palecek, S.P.; Lynn, D.M. Intraluminal release of an antifungal β-peptide enhances the antifungal and anti-biofilm activities of multilayer-coated catheters in a rat model of venous catheter infection. ACS Biomater. Sci. Eng. 2016, 2, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.A.; Weisblum, B.; Gellman, S.H. Unexpected relationships between structure and function in α,β-peptides: Antimicrobial foldamers with heterogeneous backbones. J. Am. Chem. Soc. 2004, 126, 6848–6849. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.A.; Weisblum, B.; Gellman, S.H. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of α/β-peptides. J. Am. Chem. Soc. 2007, 129, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, X.; Sebti, S.M.; Chen, J.; Cai, J. Design and synthesis of AApeptides: A new class of peptide mimics. Bioorg. Med. Chem. Lett. 2011, 21, 1469–1471. [Google Scholar] [CrossRef] [PubMed]
- Padhee, S.; Hu, Y.; Niu, Y.; Bai, G.; Wu, H.; Costanza, F.; West, L.; Harrington, L.; Shaw, L.N.; Cao, C.; et al. Non-hemolytic α-AApeptides as antimicrobial peptidomimetics. Chem. Commun.(Camb.) 2011, 47, 9729–9731. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Padhee, S.; Wu, H.; Bai, G.; Harrington, L.; Burda, W.N.; Shaw, L.N.; Cao, C.; Cai, J. Identification of γ-AApeptides with potent and broad-spectrum antimicrobial activity. Chem. Commun. 2011, 47, 12197–12199. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.G.; Amin, M.N.; Padhee, S.; Wang, R.S.E.; Qiao, Q.; Bai, G.; Li, Y.Q.; Mathew, A.; Cao, C.H.; Cai, J.F. Lipidated peptidomimetics with improved antimicrobial activity. ACS Med. Chem. Lett. 2012, 3, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Padhee, S.; Smith, C.; Wu, H.; Li, Y.; Manoj, N.; Qiao, Q.; Khan, Z.; Cao, C.; Yin, H.; Cai, J. The development of antimicrobial α-AApeptides that suppress proinflammatory immune responses. ChemBioChem 2014, 15, 688–694. [Google Scholar] [CrossRef] [PubMed]
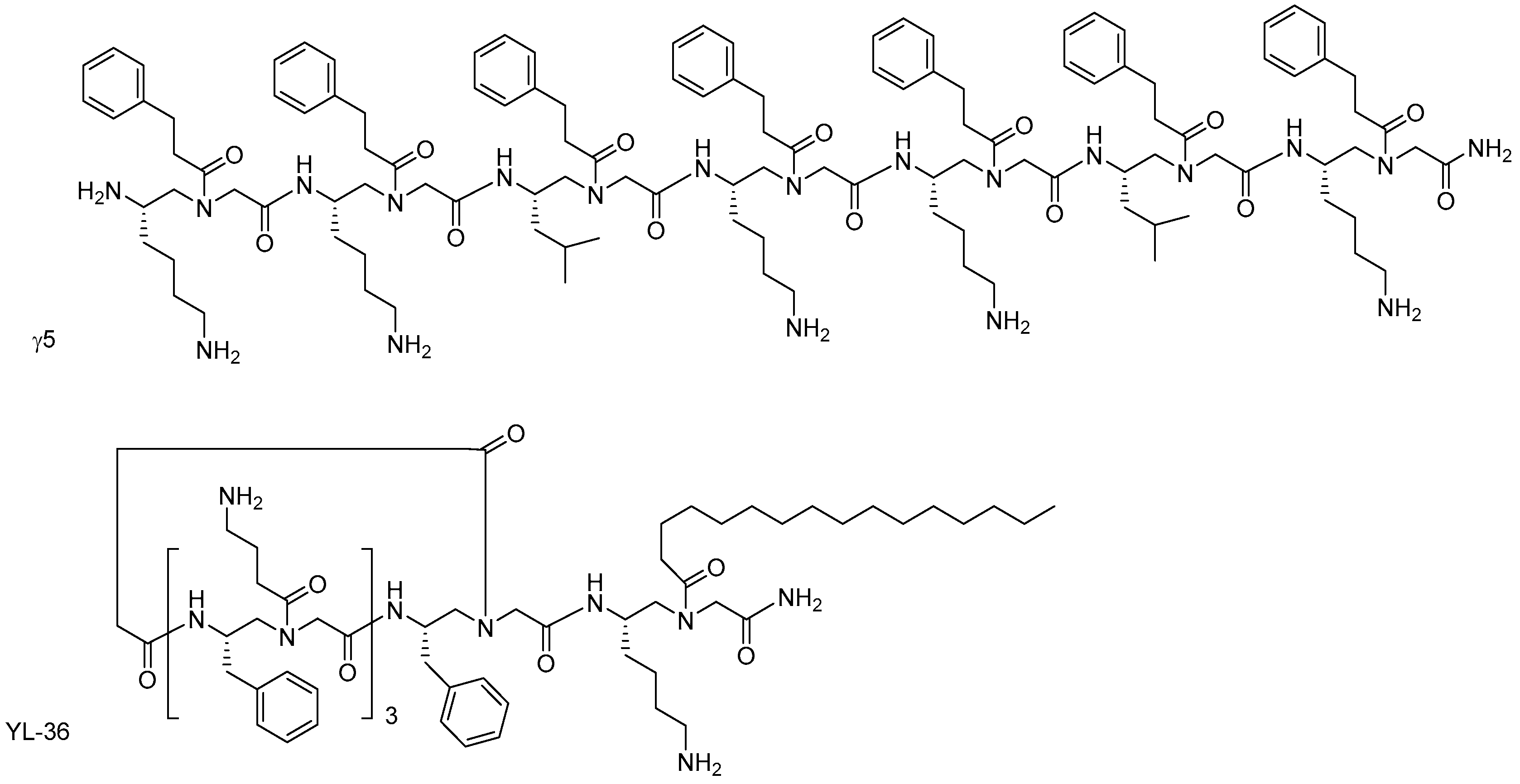
- Niu, Y.; Padhee, S.; Wu, H.; Bai, G.; Qiao, Q.; Hu, Y.; Harrington, L.; Burda, W.N.; Shaw, L.N.; Cao, C.; et al. Lipo-γ-AApeptides as a new class of potent and broad-spectrum antimicrobial agents. J. Med. Chem. 2012, 55, 4003–4009. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Niu, Y.; Padhee, S.; Wang, R.E.; Li, Y.; Qiao, Q.; Bai, G.; Cao, C.; Cai, J. Design and synthesis of unprecedented cyclic γ-AApeptides for antimicrobial development. Chem. Sci. 2012, 3, 2570–2575. [Google Scholar] [CrossRef]
- Li, Y.Q.; Smith, C.; Wu, H.F.; Padhee, S.; Manoj, N.; Cardiello, J.; Qiao, Q.; Cao, C.H.; Yin, H.; Cai, J.F. Lipidated cyclic γ-AApeptides display both antimicrobial and anti-inflammatory activity. ACS Chem. Biol. 2014, 9, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Wu, H.F.; Teng, P.; Bai, G.; Lin, X.Y.; Zuo, X.B.; Cao, C.H.; Cai, J.F. Helical antimicrobial sulfono-γ-AApeptides. J. Med. Chem. 2015, 58, 4802–4811. [Google Scholar] [CrossRef] [PubMed]
- She, F.; Nimmagadda, A.; Teng, P.; Su, M.; Zuo, X.; Cai, J. Helical 1:1 α/sulfono-γ-AAheterogeneous peptides with antibacterial activity. Biomacromolecules 2016, 17, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
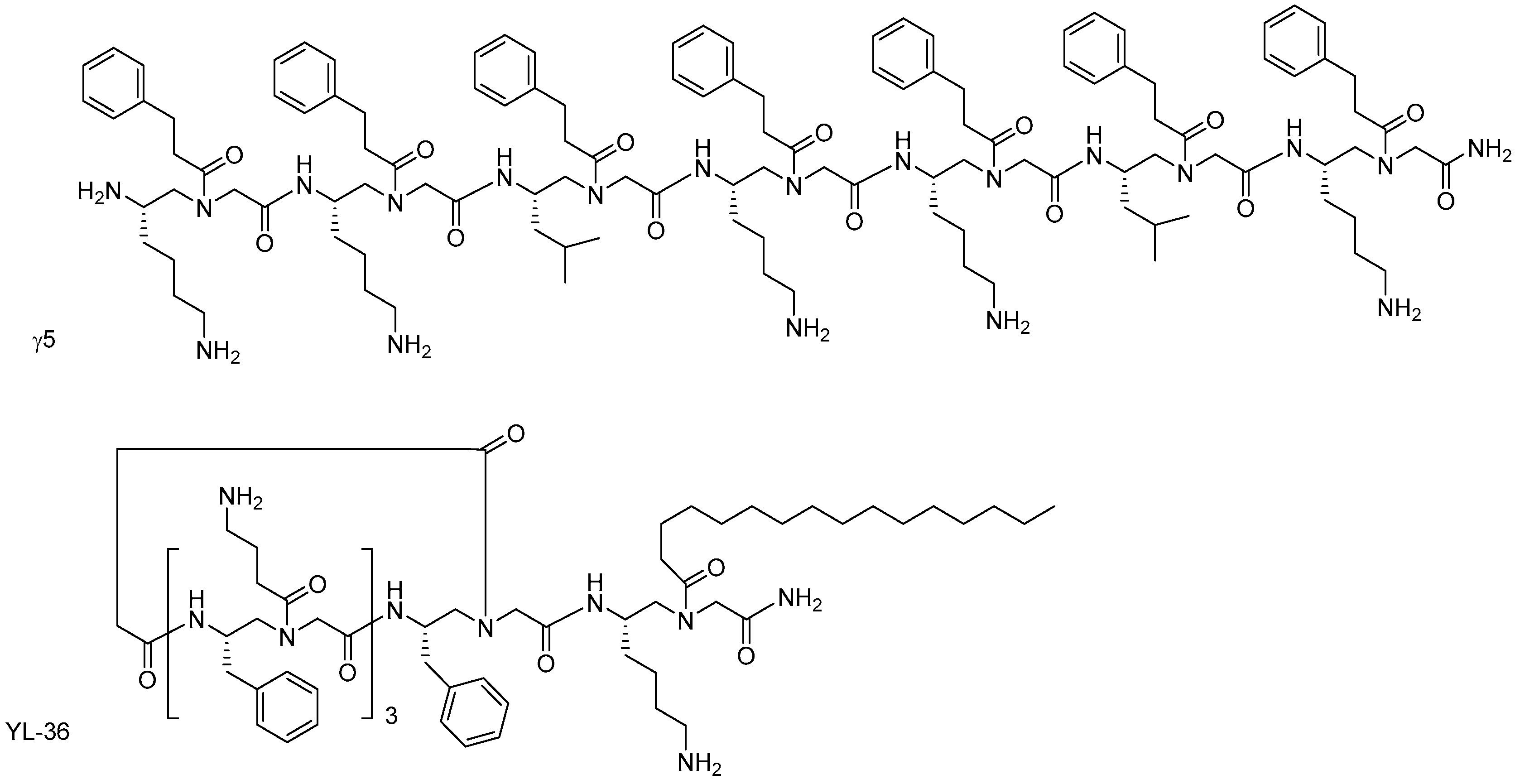
- Teng, P.; Shi, Y.; Sang, P.; Cai, J. γ-AApeptides as a new class of peptidomimetics. Chem. Eur. J. 2016, 22, 5458–5466. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Teng, P.; Sang, P.; She, F.; Wei, L.; Cai, J. γ-AApeptides: Design, structure, and applications. Acc. Chem. Res. 2016, 49, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Bolarinwa, O.; Nimmagadda, A.; Su, M.; Cai, J. Structure and function of AApeptides. Biochemistry 2017, 56, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Hu, Y.; Wu, H.; Cai, J. Synthesis of AApeptides. In Peptide Modifications to Increase Metabolic Stability and Activity; Cudic, P., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 35–46. [Google Scholar]
- Wu, H.; Teng, P.; Cai, J. Rapid access to multiple classes of peptidomimetics from common γ-AApeptide building blocks. Eur. J. Org. Chem. 2014, 2014, 1760–1765. [Google Scholar] [CrossRef]
- Wu, H.; Amin, M.N.; Niu, Y.; Qiao, Q.; Harfouch, N.; Nimer, A.; Cai, J. Solid-phase synthesis of γ-AApeptides using a submonomeric approach. Org. Lett. 2012, 14, 3446–3449. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.H.; Jones, A.; Wu, H.F.; Varani, G.; Cai, J.F. γ-AApeptides bind to RNA by mimicking RNA-binding proteins. Org. Biomol. Chem. 2011, 9, 6604–6609. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niu, Y.; Hong, H.; Wu, H.; Zhang, Y.; Engle, J.W.; Barnhart, T.E.; Cai, J.; Cai, W. Radiolabeled γ-AApeptides: A new class of tracers for positron emission tomography. Chem. Commun. 2012, 48, 7850–7852. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.G.; Cheng, N.; Wu, H.F.; Kang, S.; Ye, R.D.; Cai, J.F. Design, synthesis and characterization of fMLF-mimicking aapeptides. ChemBioChem 2014, 15, 2420–2426. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.F.; Li, Y.Q.; Bai, G.; Niu, Y.H.; Qiao, Q.; Tipton, J.D.; Cao, C.H.; Cai, J.F. γ-AApeptide-based small-molecule ligands that inhibit Aβ aggregation. Chem. Commun. 2014, 50, 5206–5208. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Wu, H.; Huang, R.; Qiao, Q.; Costanza, F.; Wang, X.-S.; Hu, Y.; Amin, M.N.; Nguyen, A.-M.; Zhang, J.; et al. Nanorods formed from a new class of peptidomimetics. Macromolecules 2012, 45, 7350–7355. [Google Scholar] [CrossRef]
- Padhee, S.; Li, Y.; Cai, J. Activity of lipo-cyclic γ-aapeptides against biofilms of Staphylococcus epidermidis and Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2015, 25, 2565–2569. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Li, Y.; Cai, J.; Song, L. Selective membrane disruption mechanism of an antibacterial γ-AApeptide defined by epr spectroscopy. Biophys. J. 2016, 110, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Wu, H.; Li, Y.; Hu, Y.; Padhee, S.; Li, Q.; Cao, C.; Cai, J. AApeptides as a new class of antimicrobial agents. Org. Biomol. Chem. 2013, 11, 4283–4290. [Google Scholar] [CrossRef] [PubMed]
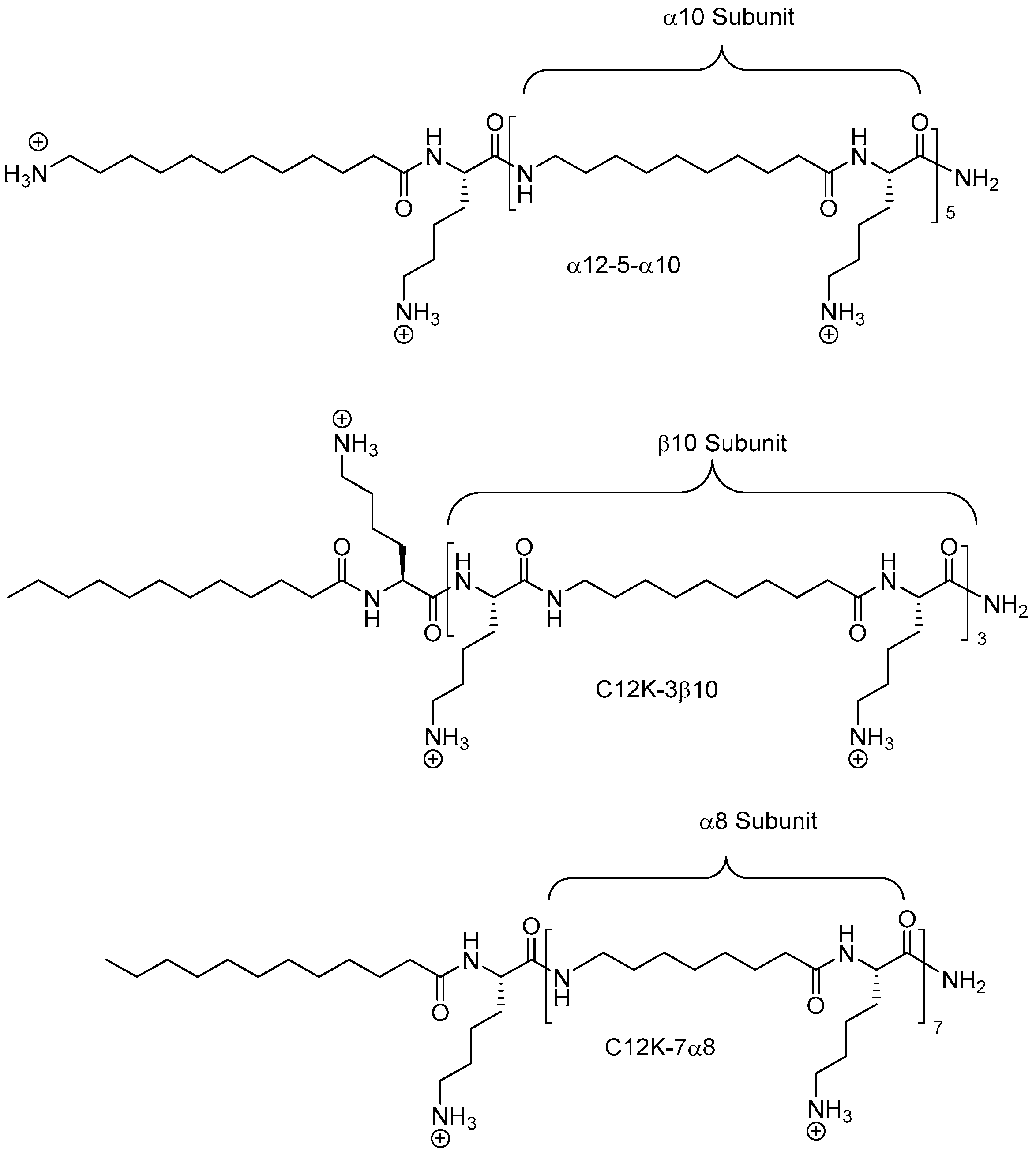
- Radzishevsky, I.S.; Rotem, S.; Bourdetsky, D.; Navon-Venezia, S.; Carmeli, Y.; Mor, A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 2007, 25, 657–659. [Google Scholar] [CrossRef] [PubMed]
- Rotem, S.; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 2009, 1788, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Mor, A. Engineered oaks against antibiotic resistance and for bacterial detection. In Host Defense Peptides and Their Potential as Therapeutic Agents; Epand, R.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 205–226. [Google Scholar]
- Livne, L.; Kovachi, T.; Sarig, H.; Epand, R.F.; Zaknoon, F.; Epand, R.M.; Mor, A. Design and characterization of a broad-spectrum bactericidal acyl-lysyl oligomer. Chem. Biol. 2009, 16, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Livne, L.; Epand, R.F.; Papahadjopoulos-Sternberg, B.; Epand, R.M.; Mor, A. OAK-based cochleates as a novel approach to overcome multidrug resistance in bacteria. FASEB J. 2010, 24, 5092–5101. [Google Scholar] [CrossRef] [PubMed]
- Zaknoon, F.; Goldberg, K.; Sarig, H.; Epand, R.F.; Epand, R.M.; Mor, A. Antibacterial properties of an oligo-acyl-lysyl hexamer targeting Gram-negative species. Antimicrob. Agents Chemother. 2012, 56, 4827–4832. [Google Scholar] [CrossRef] [PubMed]
- Kaneti, G.; Sarig, H.; Marjieh, I.; Fadia, Z.; Mor, A. Simultaneous breakdown of multiple antibiotic resistance mechanisms in S. aureus. FASEB J. 2013, 27, 4834–4843. [Google Scholar] [CrossRef] [PubMed]
- Makobongo, M.O.; Kovachi, T.; Gancz, H.; Mor, A.; Merrell, D.S. In vitro antibacterial activity of acyl-lysyl oligomers against Helicobacter pylori. Antimicrob. Agents Chemother. 2009, 53, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Makobongo, M.O.; Gancz, H.; Carpenter, B.M.; McDaniel, D.P.; Merrell, D.S. The oligo-acyl lysyl antimicrobial peptide C12k-2β12 exhibits a dual mechanism of action and demonstrates strong in vivo efficacy against Helicobacter pylori. Antimicrob. Agents Chemother. 2012, 56, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Zaknoon, F.; Wein, S.; Krugliak, M.; Meir, O.; Rotem, S.; Ginsburg, H.; Vial, H.; Mor, A. Antiplasmodial properties of acyl-lysyl oligomers in culture and animal models of malaria. Antimicrob. Agents Chemother. 2011, 55, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- Radzishevsky, I.; Krugliak, M.; Ginsburg, H.; Mor, A. Antiplasmodial activity of lauryl-lysine oligomers. Antimicrob. Agents Chemother. 2007, 51, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Jammal, J.; Zaknoon, F.; Kaneti, G.; Goldberg, K.; Mor, A. Sensitization of Gram-negative bacteria to rifampin and OAK combinations. Sci. Rep. 2015, 5, 9216. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, Y.; Zaknoon, F.; Mor, A. Experimental conditions that enhance potency of an antibacterial oligo-acyl-lysyl. Antimicrob. Agents Chemother. 2010, 54, 2590–2595. [Google Scholar] [CrossRef] [PubMed]
- Sarig, H.; Ohana, D.; Epand, R.F.; Mor, A.; Epand, R.M. Functional studies of cochleate assemblies of an oligo-acyl-lysyl with lipid mixtures for combating bacterial multidrug resistance. FASEB J. 2011, 25, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Sarig, H.; Ohana, D.; Papahadjopoulos-Sternberg, B.; Mor, A.; Epand, R.M. Physical properties affecting cochleate formation and morphology using antimicrobial oligo-acyl-lysyl peptide mimetics and mixtures mimicking the composition of bacterial membranes in the absence of divalent cations. J. Phys. Chem. B. 2011, 115, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Radzishevsky, I.S.; Kovachi, T.; Porat, Y.; Ziserman, L.; Zaknoon, F.; Danino, D.; Mor, A. Structure-activity relationships of antibacterial acyl-lysine oligomers. Chem. Biol. 2008, 15, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Sarig, H.; Mor, A.; Epand, R.M. Cell-wall interactions and the selective bacteriostatic activity of a miniature oligo-acyl-lysyl. Biophys. J. 2009, 97, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, K.; Sarig, H.; Zaknoon, F.; Epand, R.F.; Epand, R.M.; Mor, A. Sensitization of Gram-negative bacteria by targeting the membrane potential. FASEB J. 2013, 27, 3818–3826. [Google Scholar] [CrossRef] [PubMed]
- Sarig, H.; Goldfeder, Y.; Rotem, S.; Mor, A. Mechanisms mediating bactericidal properties and conditions that enhance the potency of a broad-spectrum oligo-acyl-lysyl. Antimicrob. Agents Chemother. 2011, 55, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Forde, E.; Devocelle, M. Pro-moieties of antimicrobial peptide prodrugs. Molecules 2015, 20, 1210–1227. [Google Scholar] [CrossRef] [PubMed]
- Falciani, C.; Lozzi, L.; Scali, S.; Brunetti, J.; Bracci, L.; Pini, A. Site-specific pegylation of an antimicrobial peptide increases resistance to Pseudomonas aeruginosa elastase. Amino Acids 2014, 46, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Water, J.J.; Smart, S.; Franzyk, H.; Foged, C.; Nielsen, H.M. Nanoparticle-mediated delivery of the antimicrobial peptide plectasin against Staphylococcus aureus in infected epithelial cells. Eur. J. Pharm. Biopharm. 2015, 92, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Mor, A.; Epand, R.M. Lipid complexes with cationic peptides and oaks; their role in antimicrobial action and in the delivery of antimicrobial agents. Cell. Mol. Life Sci. 2011, 68, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, I.; Casciaro, B.; Miro, A.; Quaglia, F.; Mangoni, M.L.; Ungaro, F. Overcoming barriers in Pseudomonas aeruginosa lung infections: Engineered nanoparticles for local delivery of a cationic antimicrobial peptide. Colloids Surf. B Biointerfaces 2015, 135, 717–725. [Google Scholar] [CrossRef] [PubMed]
- De Mello, M.B.; da Silva Malheiros, P.; Brandelli, A.; Pesce da Silveira, N.; Jantzen, M.M.; de Souza da Motta, A. Characterization and antilisterial effect of phosphatidylcholine nanovesicles containing the antimicrobial peptide pediocin. Probiotics Antimicrob. Proteins 2013, 5, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Domalaon, R.; Findlay, B.; Ogunsina, M.; Arthur, G.; Schweizer, F. Ultrashort cationic lipopeptides and lipopeptoids: Evaluation and mechanistic insights against epithelial cancer cells. Peptides 2016, 84, 58–67. [Google Scholar] [CrossRef] [PubMed]
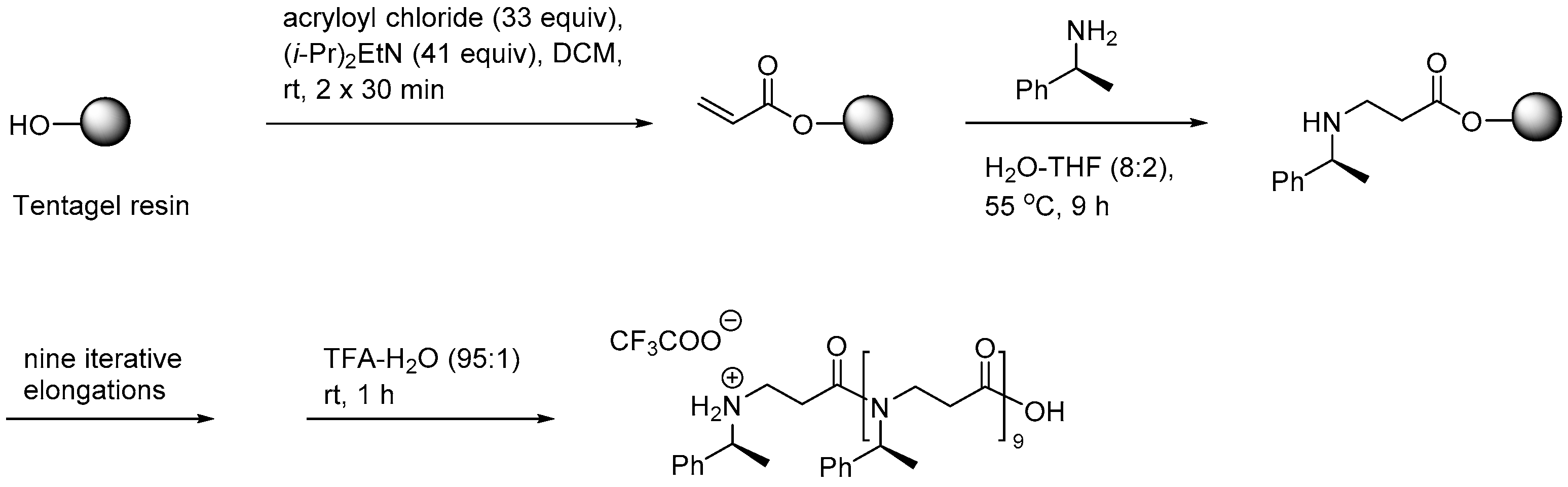
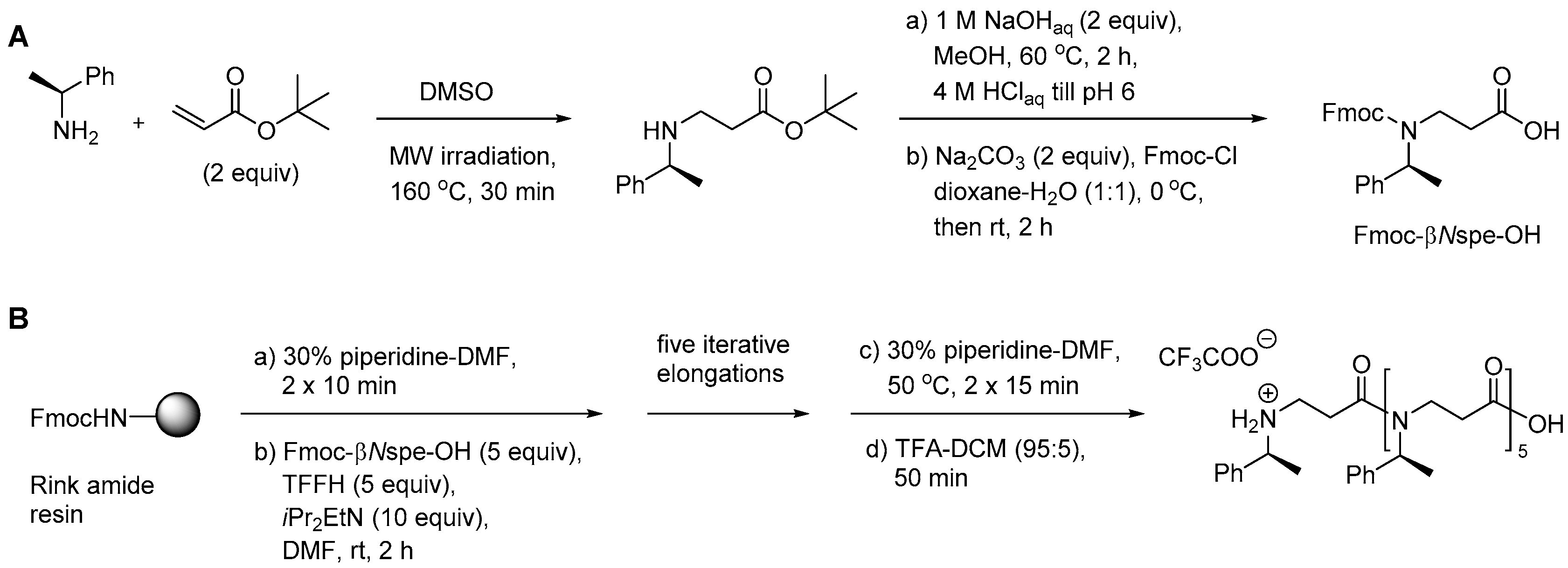
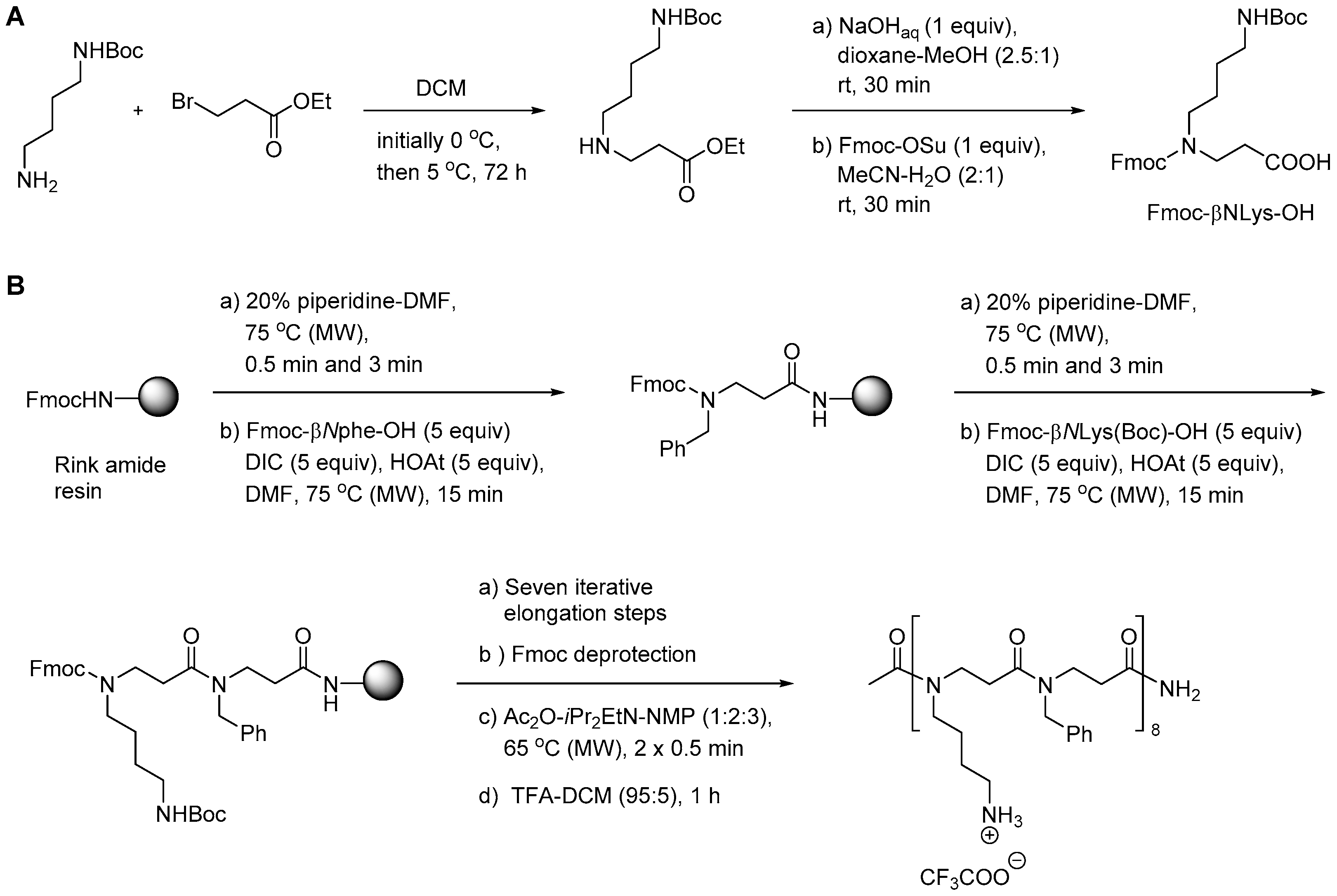
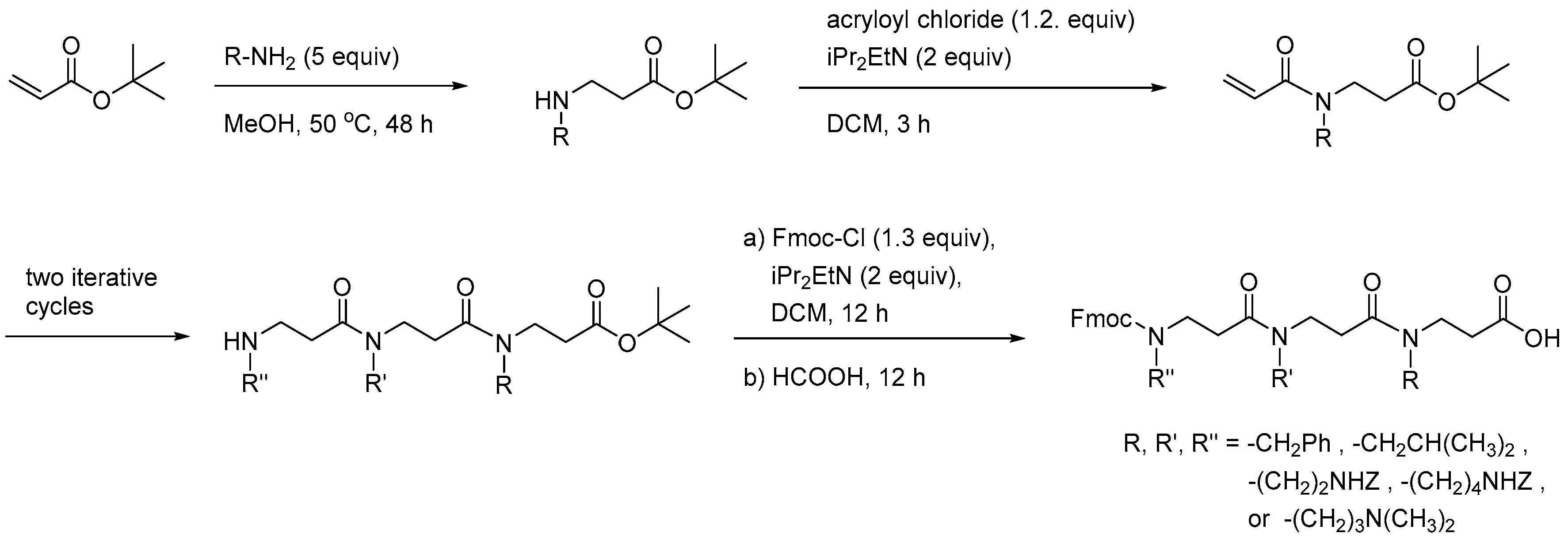
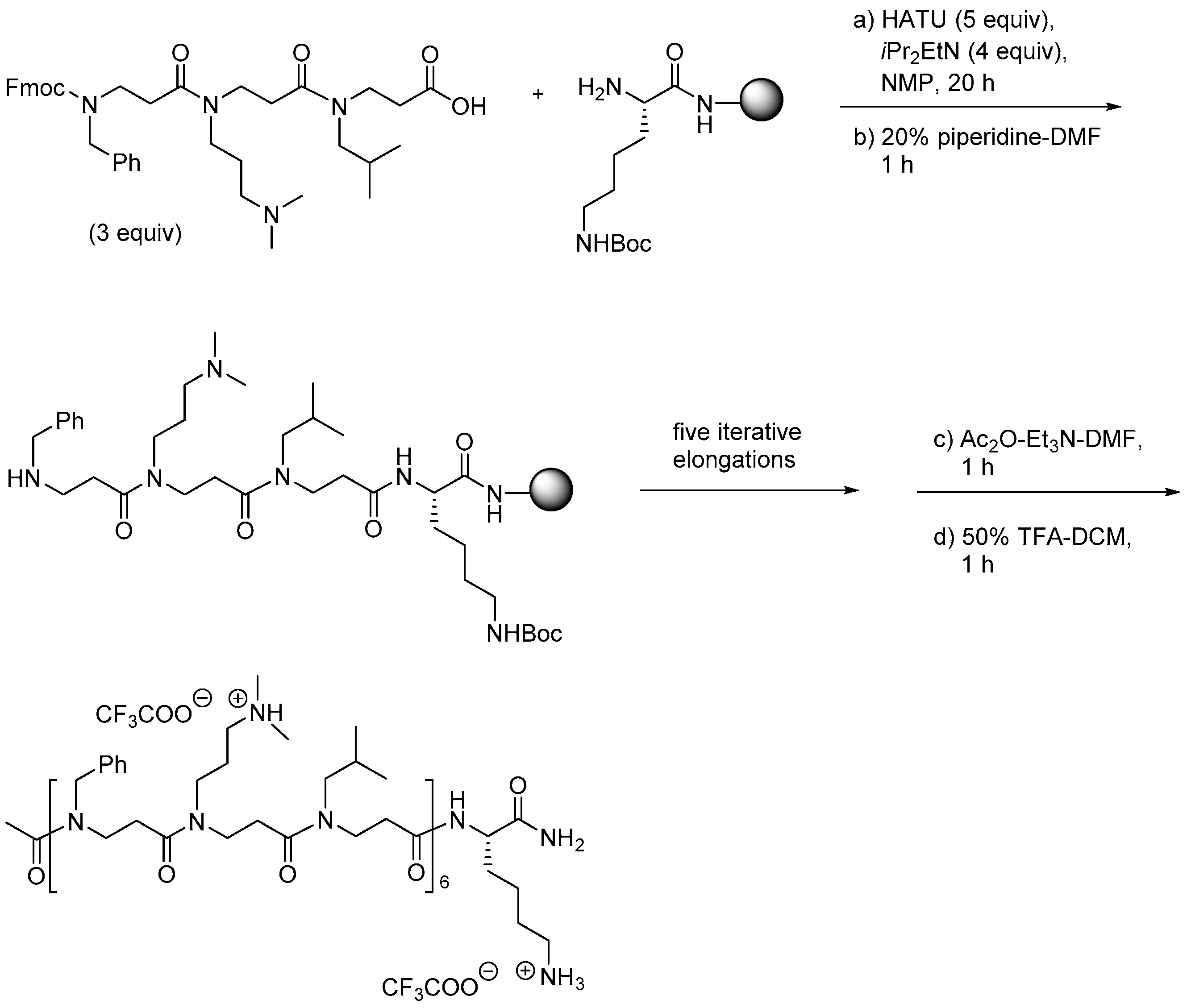
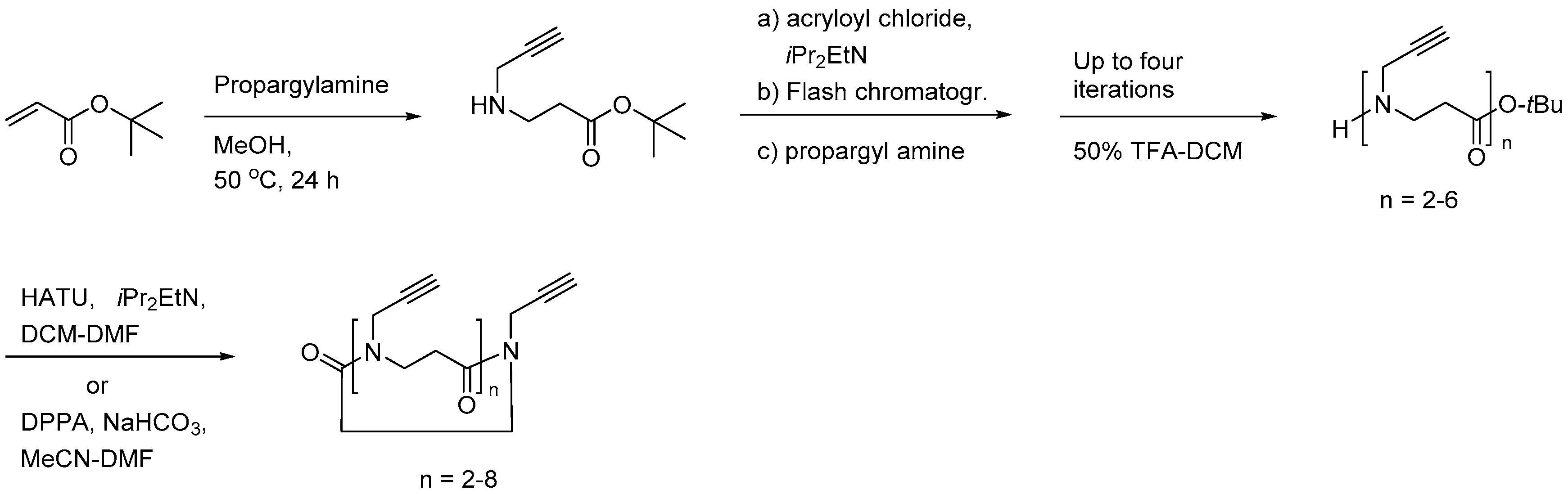
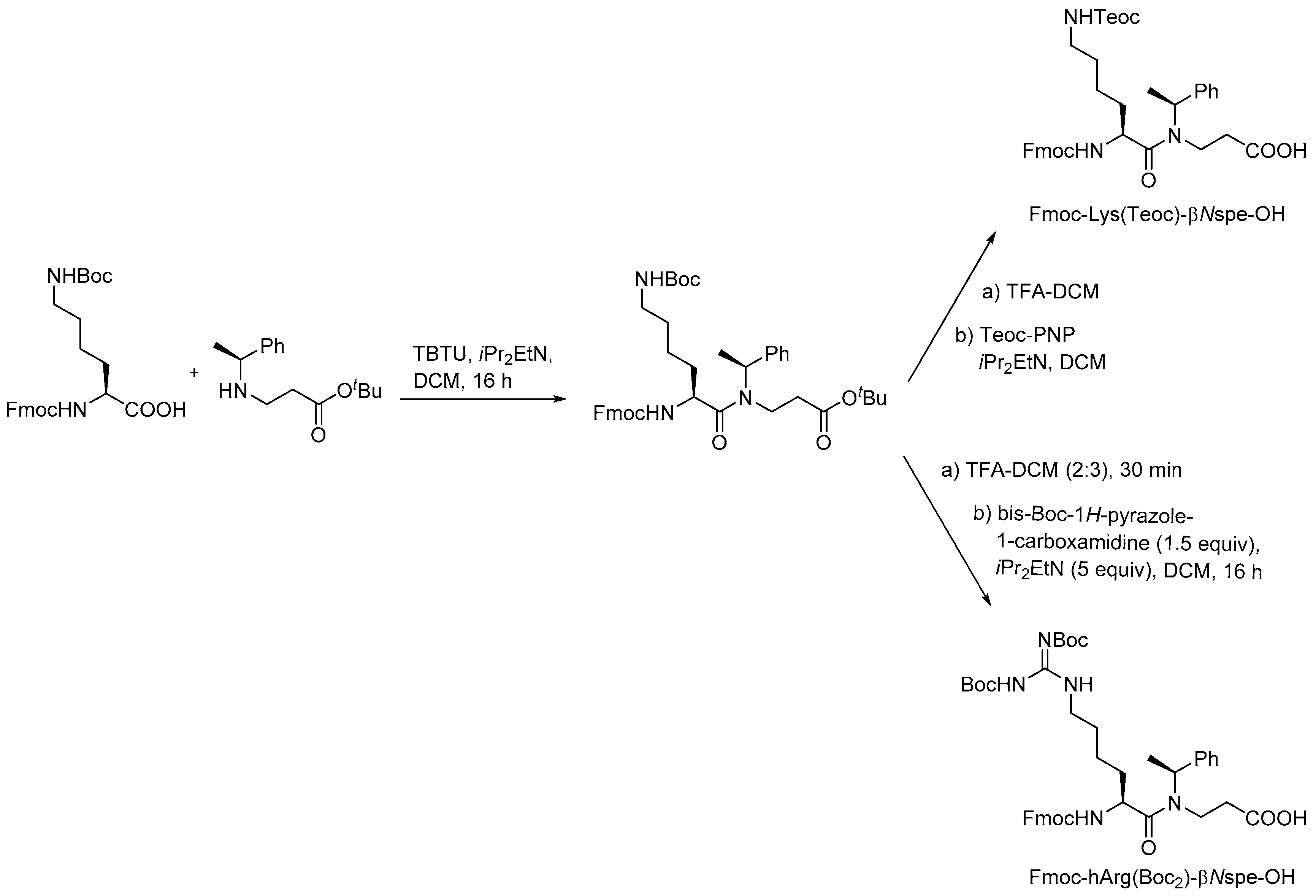
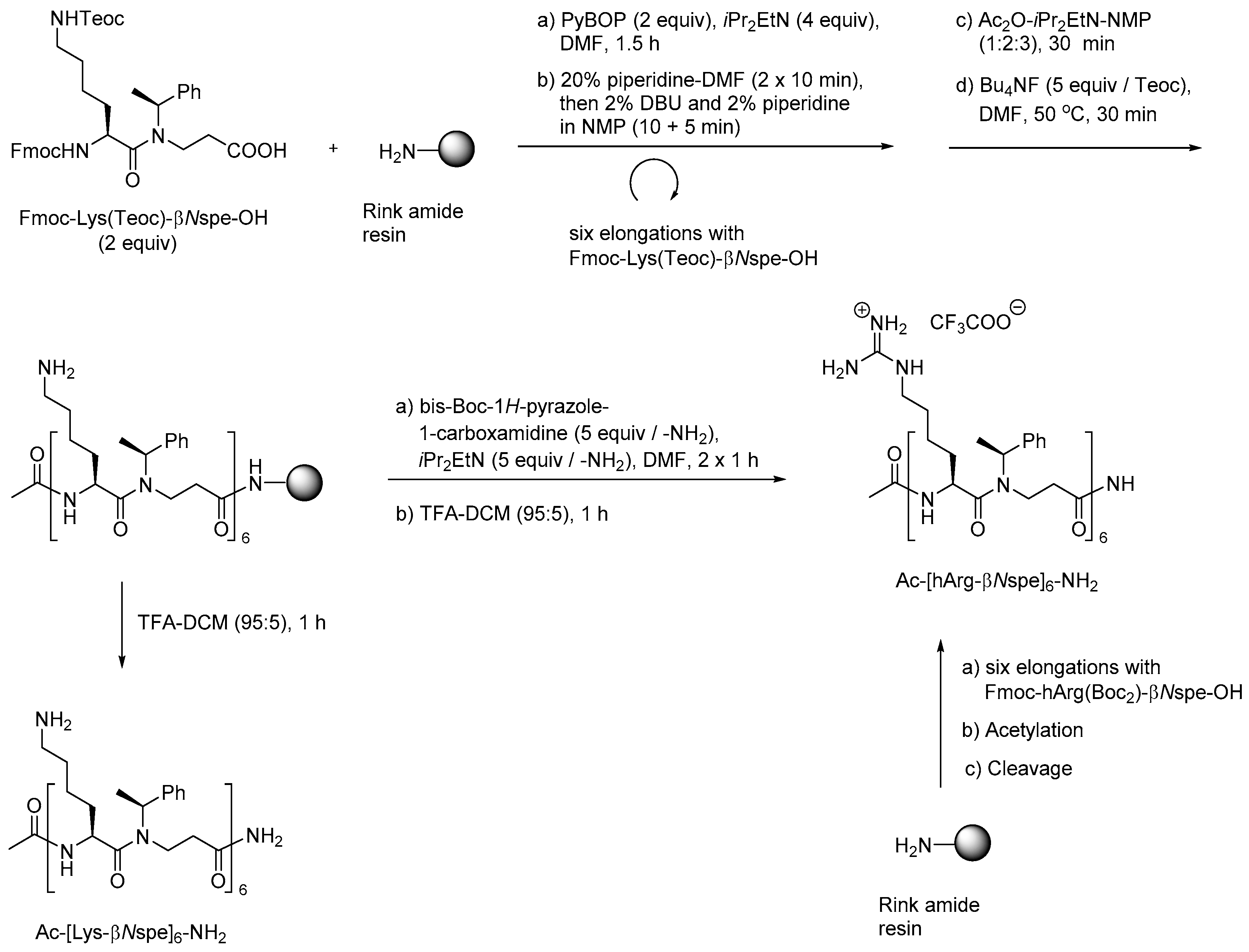

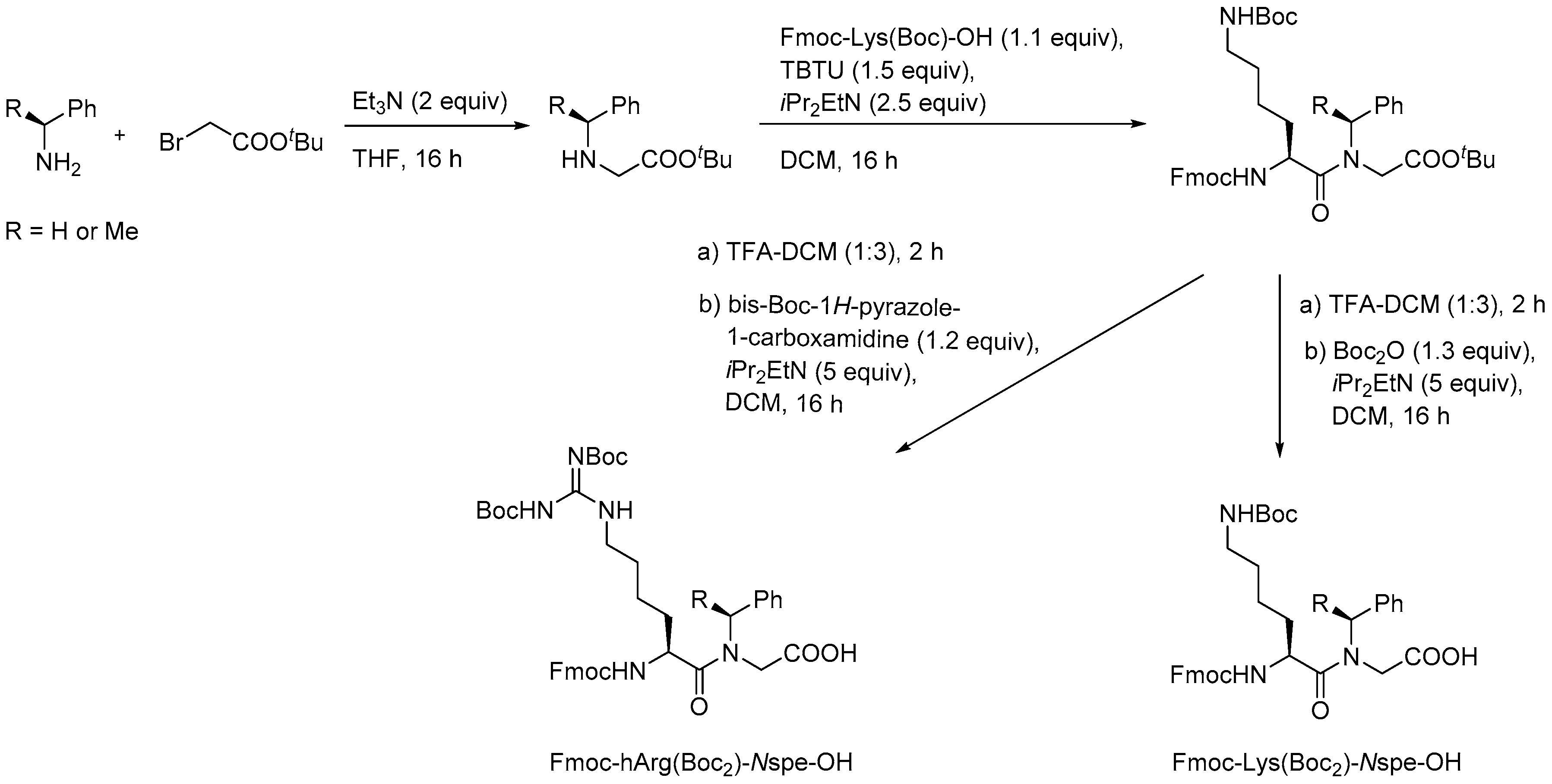

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
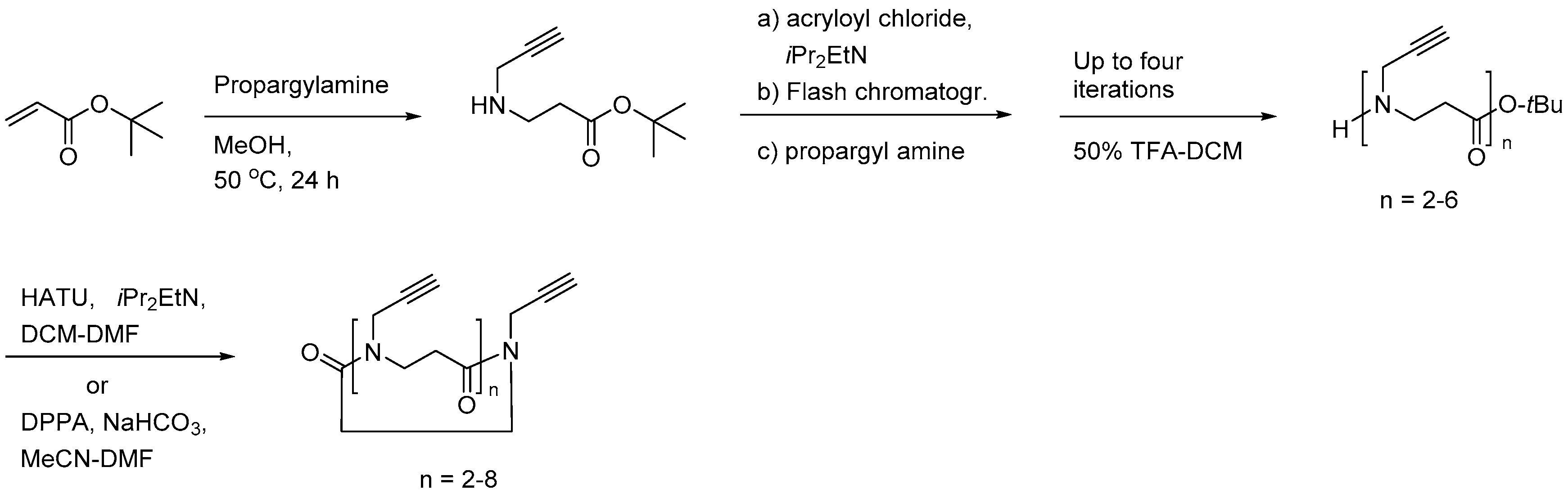{kind=link}
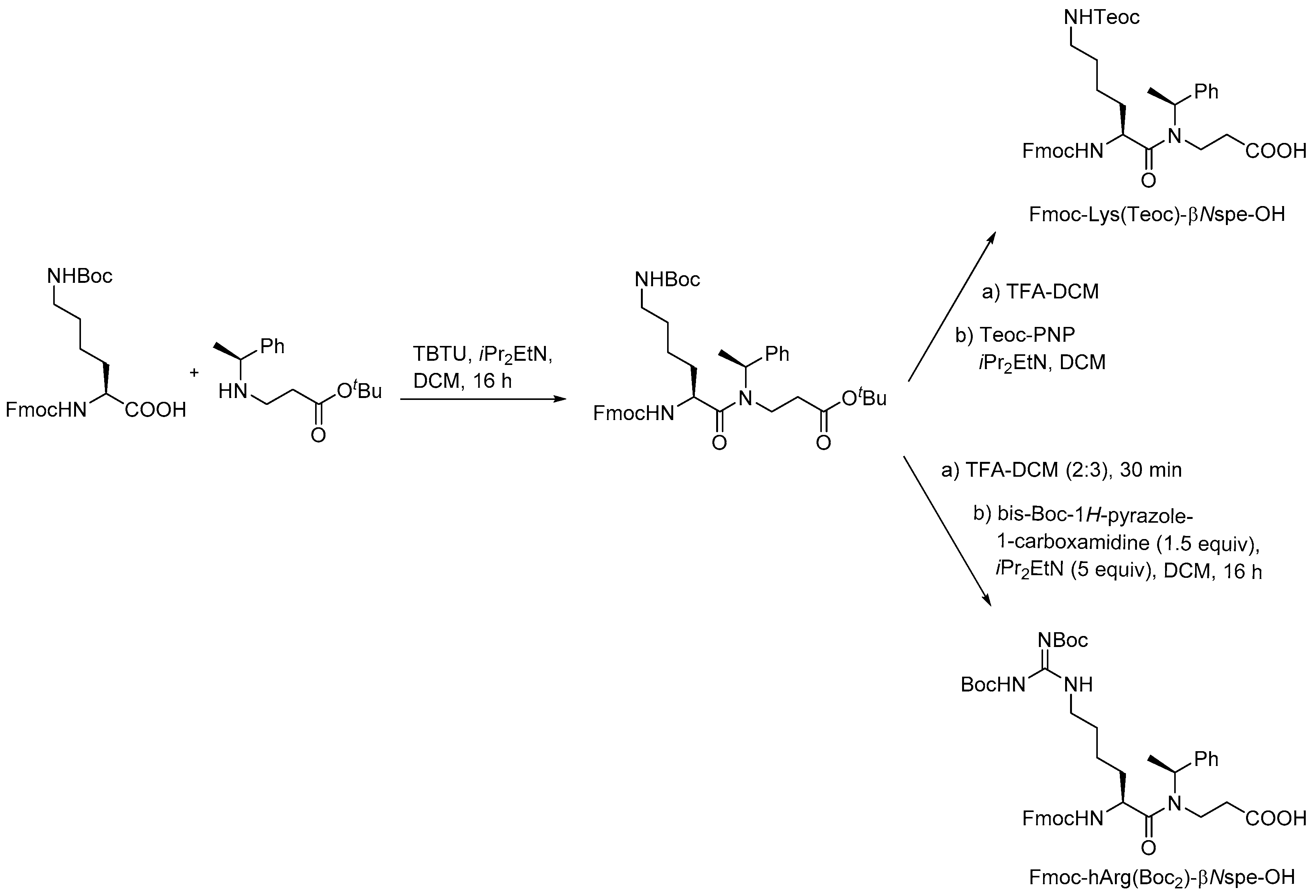{kind=link}
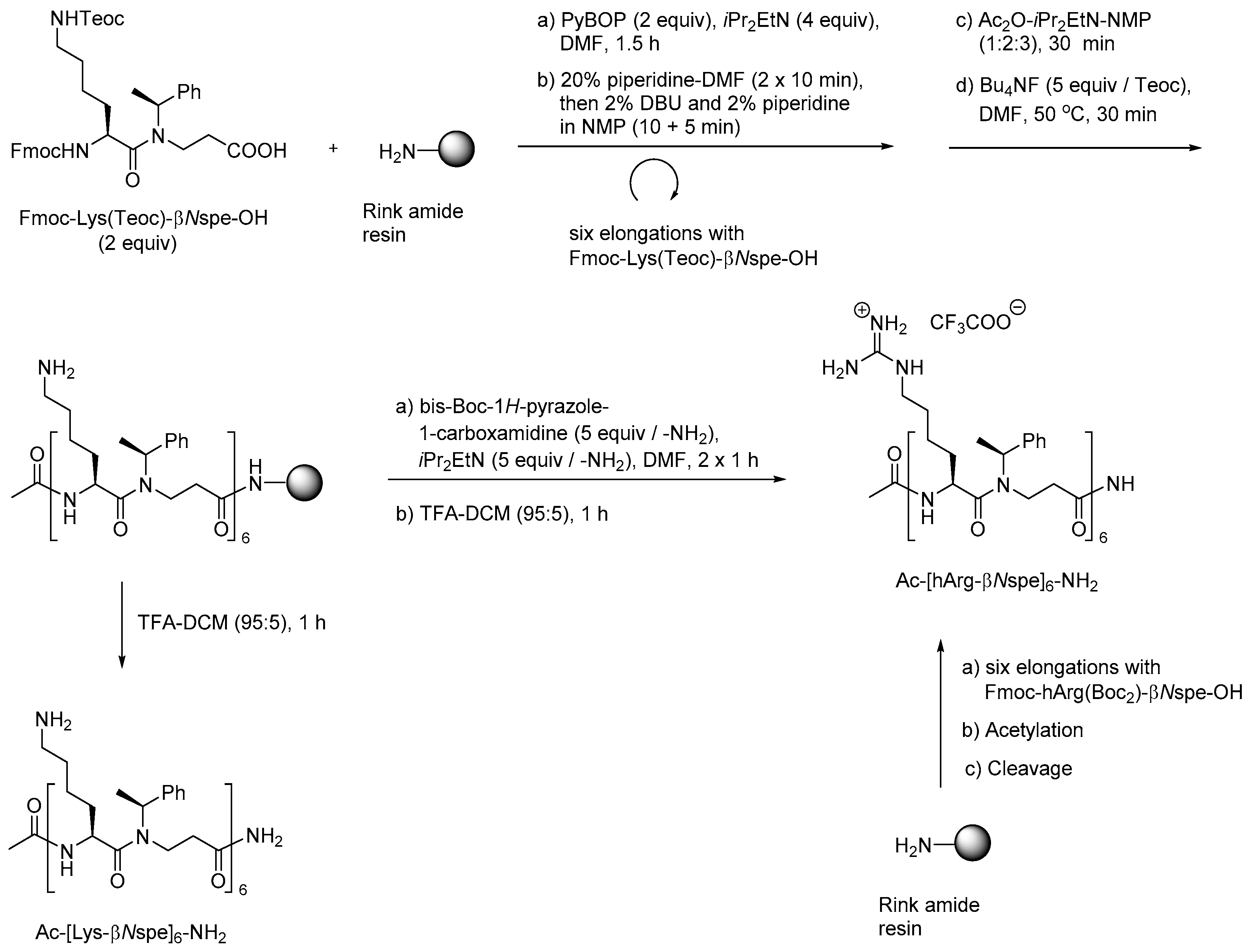{kind=link}
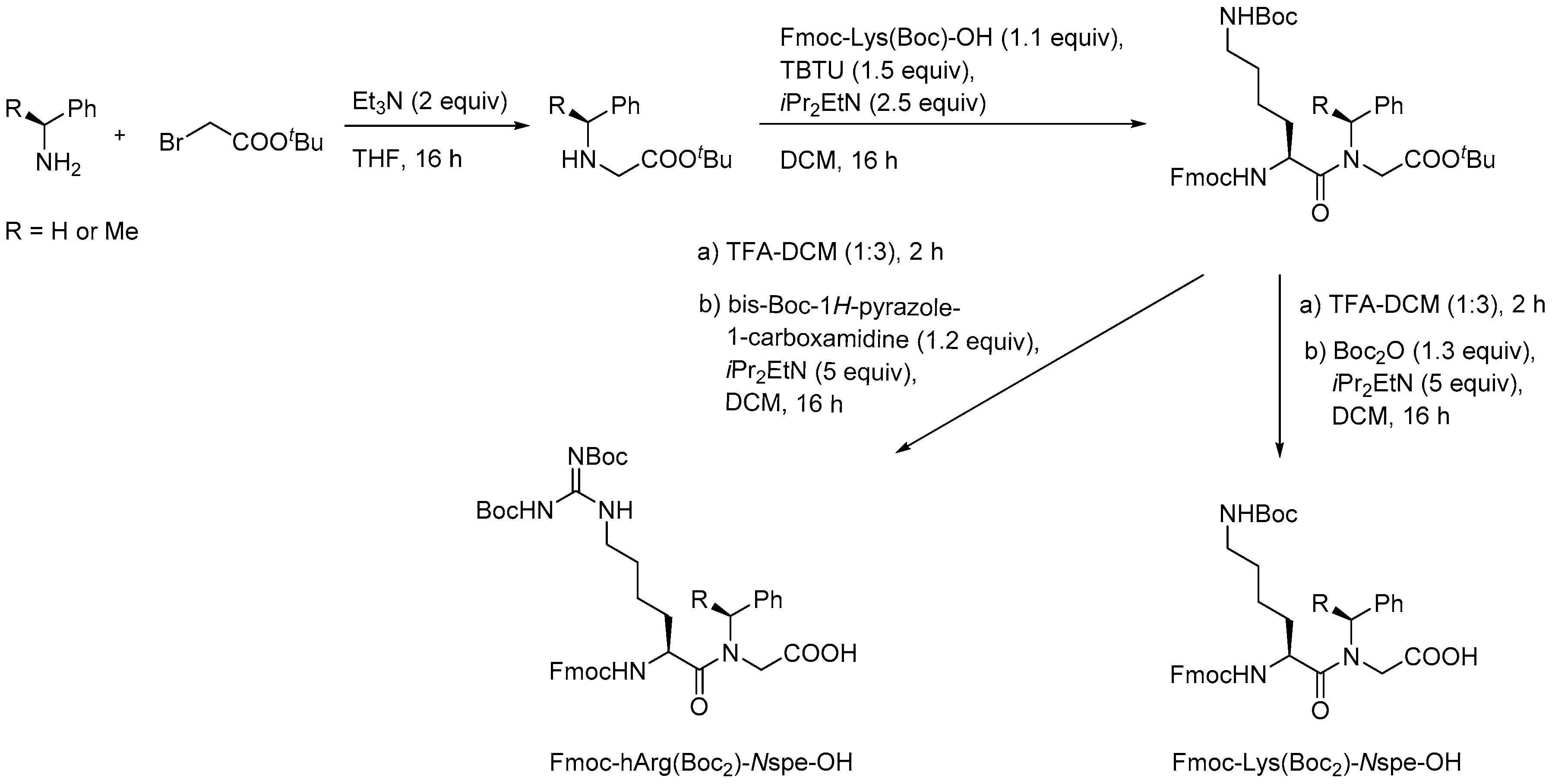{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMP | AMP source | Target | Phase | Company | Admin. | Refs. |
|---|---|---|---|---|---|---|
| NVB-302 | Lantibiotic | C. difficile | I * | Novacta | Oral | [62] |
| hlf1-11 | Lactoferricin analog | Infection following transplantation | I/II | AM-Pharma | I.v. | [63] |
| Wap-8294A2 | Lysobactor spp. | G+ bacteria (VRE and MRSA) | I/II | aRigen | Top. | [64] |
| C16G2 | Synthetic peptide | Prevention of tooth decay caused by Streptococcus mutans | II | C3 Jian Inc. | Mouth wash | [65,66] |
| IMX942 (SGX942) | Indolicidin analog | Immunomodulation during oral mucositis | II | Soligenix | Oral rinse | [67] |
| DPK-060 | Human protein kininogen | Acute external otitis | II | Pergamum AB | Ear drops | [68] |
| PXL01 | Human lactoferrin analog | Surgical adhesion prevention | II | Pergamum AB | Top. | [69] |
| Brilacidin | Defensin analog | Radiation-induced oral mucositis in patients with head and neck cancer | II | Cellceutix Corp | Oral rinse | [70] |
| PAC113 | Histatin analog | Oral candidiasis in HIV seropositive patients | II | Pacgen Biopharmaceuticals Corp. | Mouth rinse | [71] |
| POL7080 | Protegrin analog | Ventilator-associated P. aeruginosa pneumonia | II *** | Polyphor Ltd. | I.v. | [72,73] |
| LTX-109 (Lytixar) | Synthetic peptidomimetic | Nasal decolonisation of MRSA G+ skin infections | IIa * | Lytix Biopharma | Nasal Top. | [74,75,76] |
| Diabetic foot ulcer | PC | |||||
| OP-145 | LL-37 | Chronic middle ear infection | II * | Dr. Reddy’s Research | Ear drops | [77,78] |
| LL-37 | Human cathelicidin | Leg ulcer | IIb | ProMore Pharma | Top. | [79,80] |
| Novexatin® (NP213) | Cyclic cationic peptide | Fungal nail infection | IIb | Novabiotics | Top. | [81] |
| p2TA (AB 103) | Synthetic peptide | Necrotizing soft tissue infections | III | Atox Bio Ltd | I.v. | [82] |
| Iseganan | Protegrin analog | Ventilator-associated pneumonia | III ** | IntraBiotics Pharmaceuticals | Top. | [83,84] |
| Locilex (MSI-78) | Magainin analog (Pexiganan) | Infections of diabetic foot ulcers | III ** | Dipexium Pharmaceuticals | Top. | [85] |
| Omiganan (CLS001) | Indolicidin derivative | CAUTI caused by S. aureus; | IIIb ** | Mallinckrodt | Top. | [86] |
| Topical skin antisepsis, rosacea; | III | Mallinckrodt | ||||
| Vulvaryl intraepithelial neoplasia, acne, atopic dermatitis | II | Cutanea Life Sciences | ||||
| Suroto- mycin | Cyclic lipopeptide | C. difficile (diarrhea) | III * | Cubist Pharmaceuticals/Merck | Oral | [87] |
| Ramoplanin (NTI-851) | Actinoplanes spp | VRE (G+) | III | Nano-therapeutics | Oral | [88,89] |
| C. difficile (G+) | II |
| AMP | AMP Source | Target | Phase | Company | Admin. | Refs. |
|---|---|---|---|---|---|---|
| MU1140 | Streptococcus mutans (lantibiotic) | G+ bacteria (MRSA, C. difficile) | PC | Oragenics | Not specified | [90,91] |
| HB1345 | Lipopeptide | Broad-spectrum antibiotic, acne | PC | Helix Biomedix | Top. | [92] |
| Novarifyn (NP432) | Synthetic antimicrobial | MRSA, P. aeruginosa, C. difficile, A. baumannii, E. coli | PC | Novabiotics | Top. | [93] |
| Arenicin (AP139) | Lugworm Arenicol marina | G− bacteria, UTI | PC | Adenium Biotech | Not specified | [94,95] |
| AP138 | Arenicin analog | MRSA implant infections | PC | Adenium | Not specified | [96] |
| AP114 | Arenicin analog | C. difficile | PC | Adenium | Not specified | [96] |
| Avidocin and purocin | Modified R-type bacteriocin | G+ and G− bacteria | PC | AvidBiotics | Oral | [97,98] |
| Compound | MIC Values Cytotoxicity | ||||
|---|---|---|---|---|---|
| S. aureus | E. coli | P. aeruginosa | EC50 a/HC50 b | Refs. | |
| H-(NhArg-Nspe-Nspe)4-NH2 | 1 µM | 6 µM | 13 µM | 12 µM a | [179] |
| H-(NLys)4(NhTrp)4-NIle-NH2 | 8 µg/mL | 8 µg/mL | 4 µg/mL | 128 µg/mL c | [162,177] |
| c(Nap-Ndpe)3 | 3.9 µg/mL | ND | 7.8 µg/mL | >250 µg/mL b | [190] |
| Compound | MIC (in µM) Hemolysis | ||||
|---|---|---|---|---|---|
| S. aureus | E. coli | P. aeruginosa | HC50 | Ref. | |
| Pis-1[NkG] a | 4 | 2 | 4 | >200 | [197] |
| KLW-f b | 1 | 2 | 1 | >100 | [192] |
| L8K9W1-NLeu c | 2 | 2 | 1 | >100 | [194] |
| STPk d | 1 | 1 | 2 | >200 | [196] |
| Compound | MIC (in µM or µg/mL) Hemolysis | |||
|---|---|---|---|---|
| S. aureus | P. aeruginosa | HA a | Ref. | |
| IK-Nssb-NLys-VRK-Nssb-NH2 | 3.1 µg/mL | 11.8 µg/mL | - | [202] |
| NLys-NLys-W-Nsmb-IKRW-NH2 | 9.2 µg/mL | 5.7 µg/mL | - | [202] |
| Nnal-Nmphe-Nnal-Nnle-KKKKK-NH2 | 1.6 µM | 12.5 µM | 3% | [204] |
| KKKK-Nnal-Nmphe-Nnal-Nnle-NH2 | 1.6 µM | 6.25 µM | 6% | [204] |
| Compound | MIC (in µM) Hemolysis (in µM) | ||||
|---|---|---|---|---|---|
| S. aureus | E. coli | P. aeruginosa | HC10 | Refs. | |
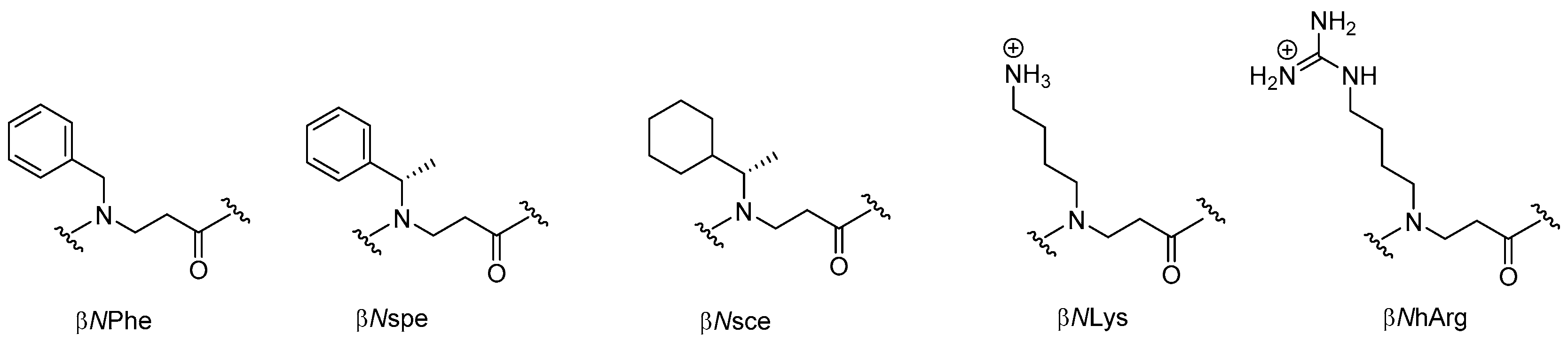
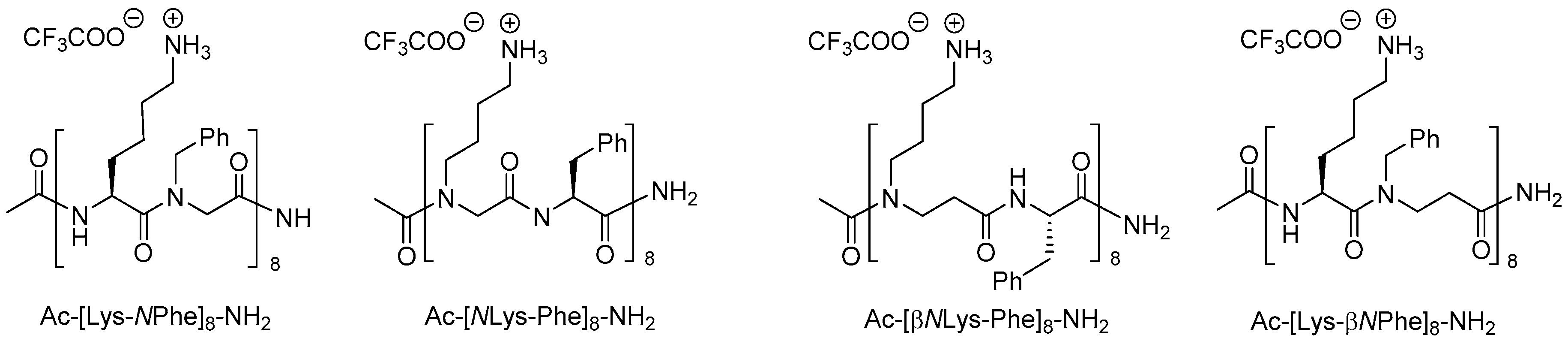
| Ac-[Lys-βNspe]8-NH2 | 37 | 9.1 | 4.7 | >500 | [216] |
| Ac-[hArg-βNPhe]6-NH2 | 5.9 | 1.5 | - | >500 | [216,217] |
| Ac-[hArg-βNspe]6-NH2 | 2.8 | 2.8 | - | >500 | [216,217] |
| Ac-[Lys-βNPhe-hArg-βNPhe]4-NH2 | 32 | 1 | - | - | [120] |
| cHex-[hArg-βNspe-Lys-βNspe]4-NH2 | 1 | 2 | - | 600 a | [218] |
| Ac-[Lys-βN(4-F-Phe)]8-NH2 | 0.8 | 3.2 | 1.6 | >150 | [219] |
| Compound | MIC (in µg/mL) Hemolysis (in µg/mL) | |||
|---|---|---|---|---|
| S. aureus | E. coli | HC10 | Ref. | |
| H-ACPC-Leu-APC-Lys-APC-Leu-ACPC-Lys-APC-Leu-ACPC-Leu-ACPC-Leu-ACPC-NH2 | 12.5 | 6.3 | 100 | [250] |
| H-APC-Leu-ACPC-Lys-ACPC-Ala-APC-Ala-ACPC-Lys-ACPC-Ala-APC-Leu-ACPC-NH2 | 12.5 | 6.3 | >400 | [250] |
| MIC Hemolysis | ||||||
|---|---|---|---|---|---|---|
| Compound type | ID | MRSA | P. aeruginosa | K. pneumonia | HC50 | Ref. |
| α-AApeptide | α4 | 4 | 8 | 8 | >250 | [274] |
| γ-AApeptide | γ-5 | 5 | ND | 5 | 300 | [253] |
| Cyclic γ-AApeptide | HW-B-13 | 1 | 8 | 8 | 100 | [257] |
| Lipo-γ-AA | 13 | 3 | 3 | 3 | >500 | [256] |
| Cyclic lipo-γ-AA | YL-36 | 2 | 5 | 3 | 100 | [258] |
| Sulfono-γ-AApeptide | 7 | 2 | 4 | ND | 100 | [259] |
| 1:1 α/sulfono-γ-AApeptide | 6 | 3 | 4 | 4 | >250 | [260] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules 2017, 22, 1430. https://doi.org/10.3390/molecules22091430
Molchanova N, Hansen PR, Franzyk H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules. 2017; 22(9):1430. https://doi.org/10.3390/molecules22091430
Chicago/Turabian StyleMolchanova, Natalia, Paul R. Hansen, and Henrik Franzyk. 2017. "Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs" Molecules 22, no. 9: 1430. https://doi.org/10.3390/molecules22091430