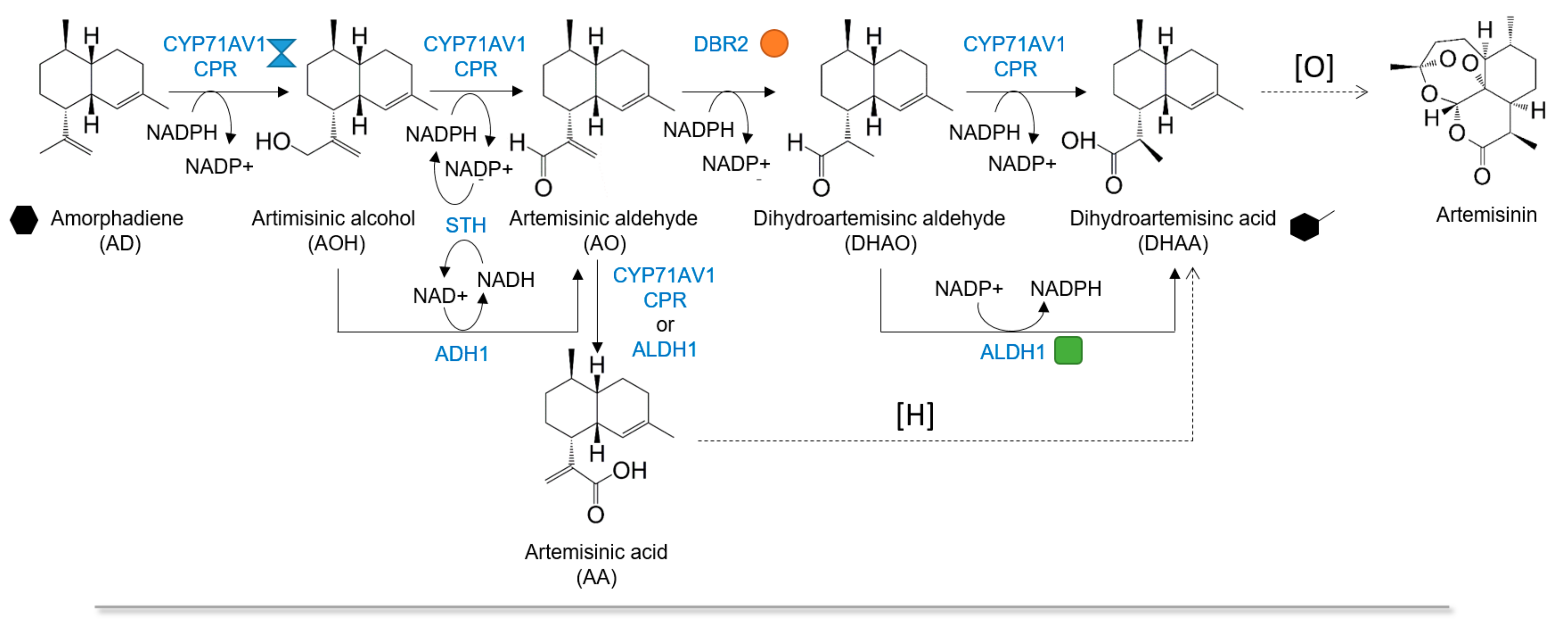
Multienzyme Biosynthesis of Dihydroartemisinic Acid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
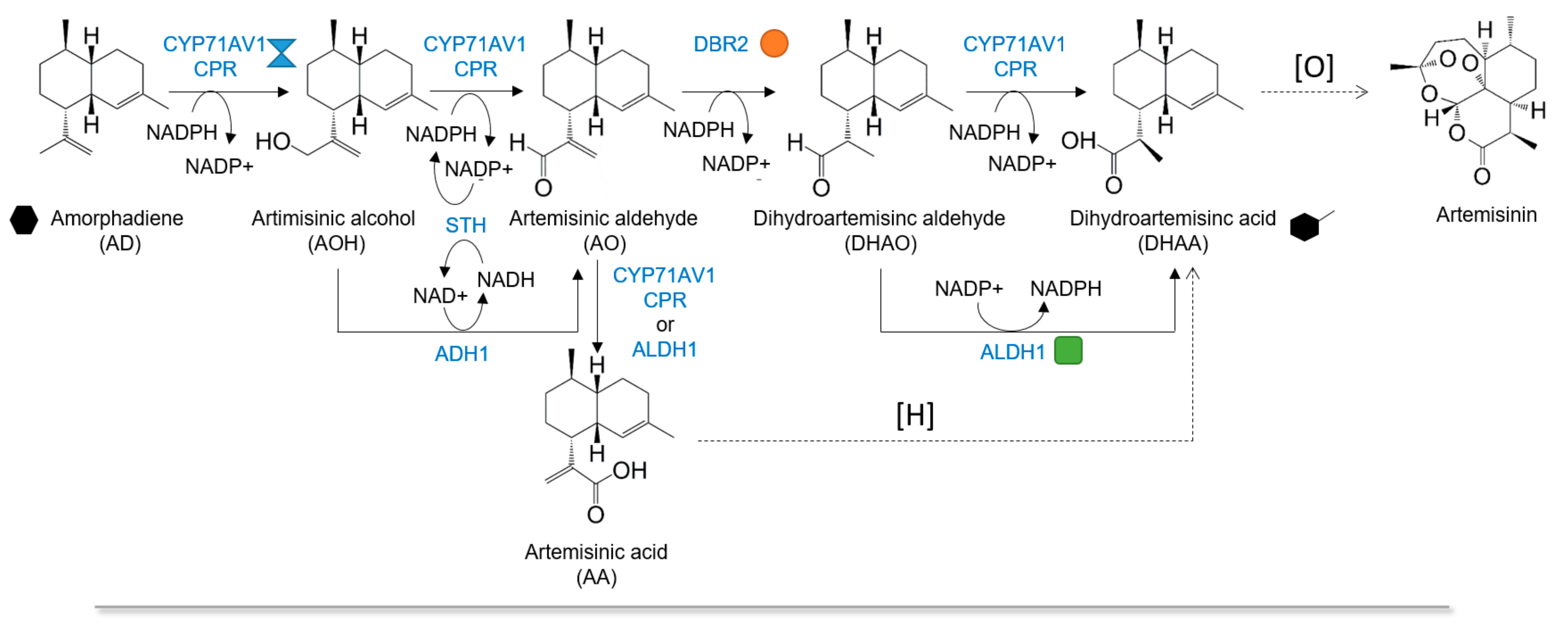
:1. Introduction
2. Results and Discussion
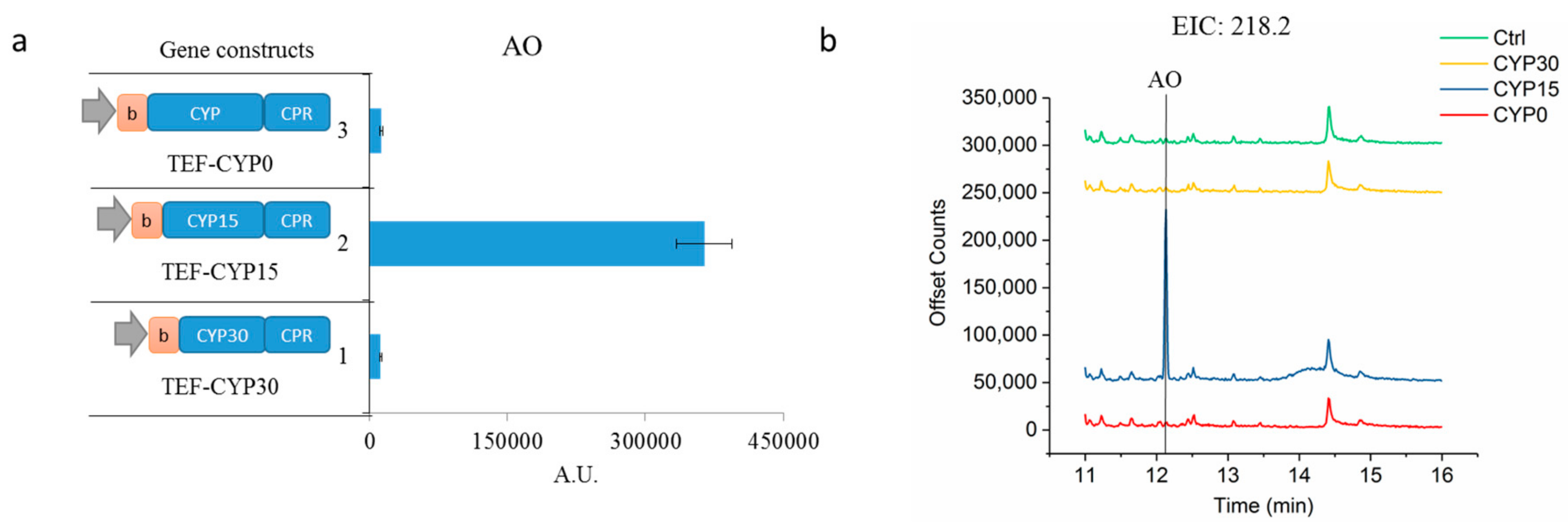
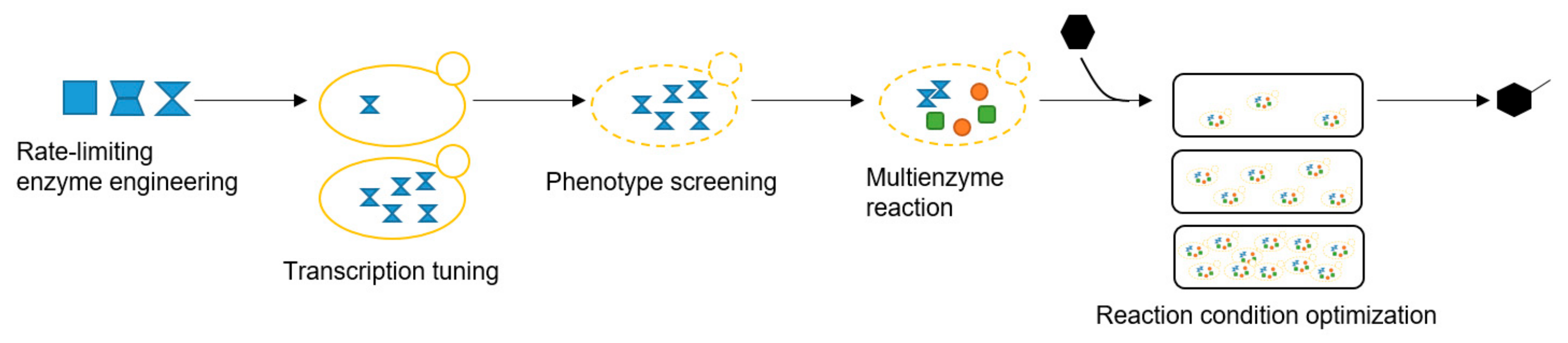
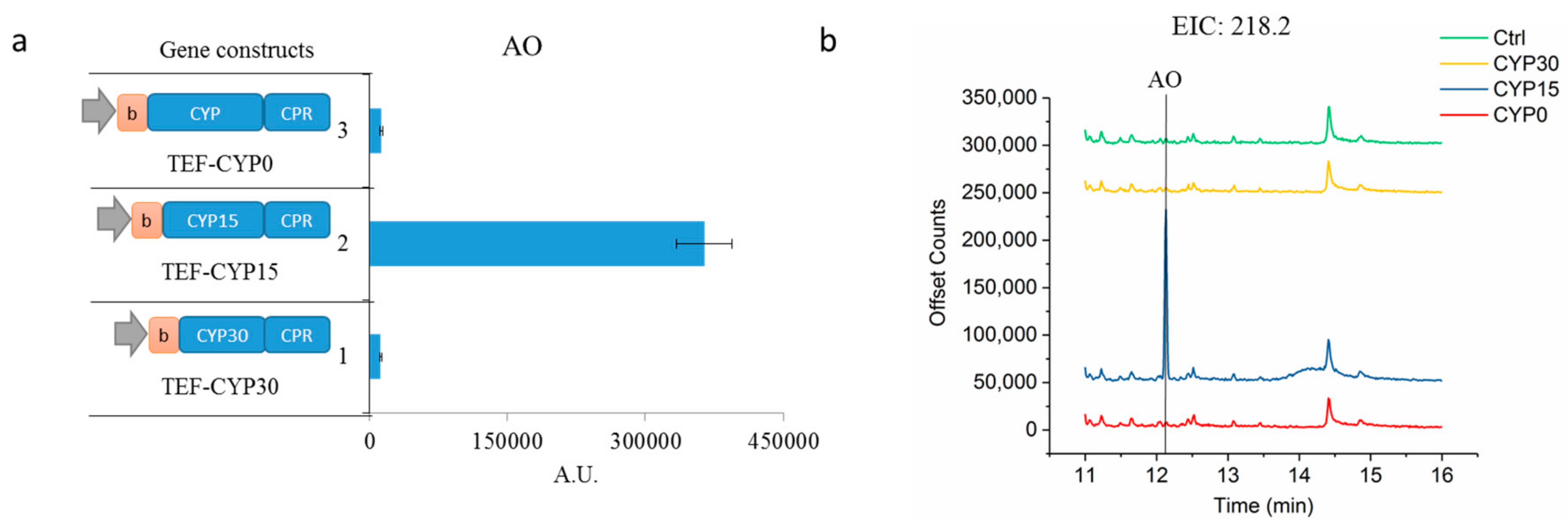
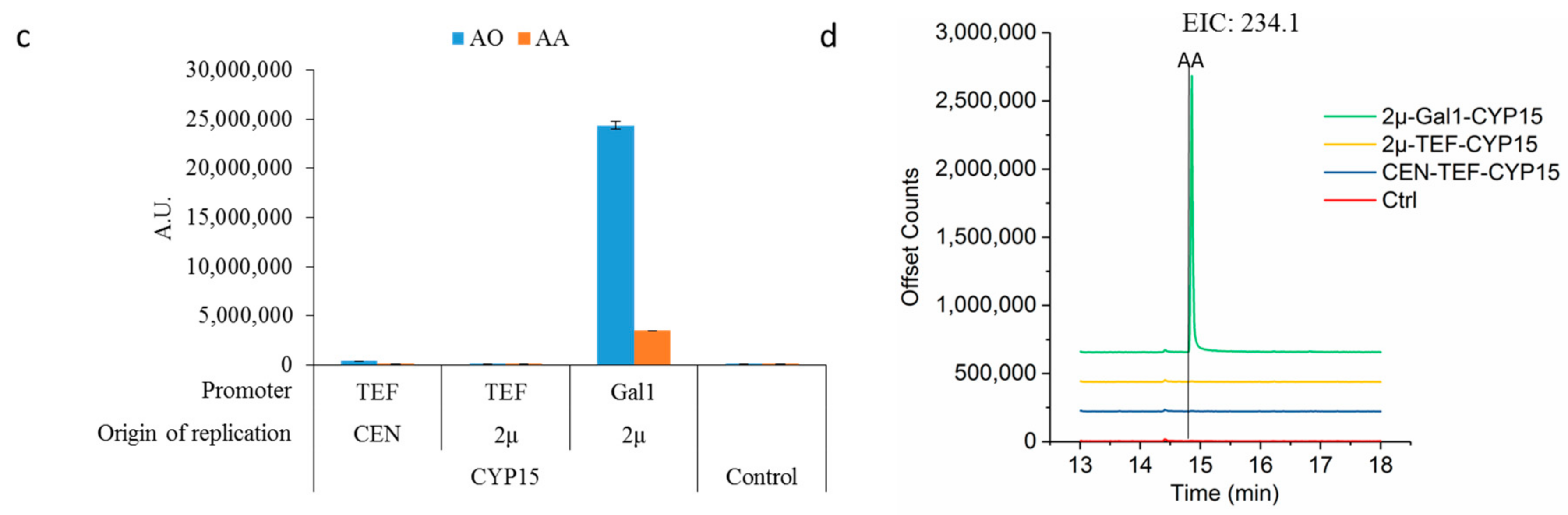
2.1. Enhancing CYP71AV1 Activity by Enzyme Engineering and Transcription Tuning
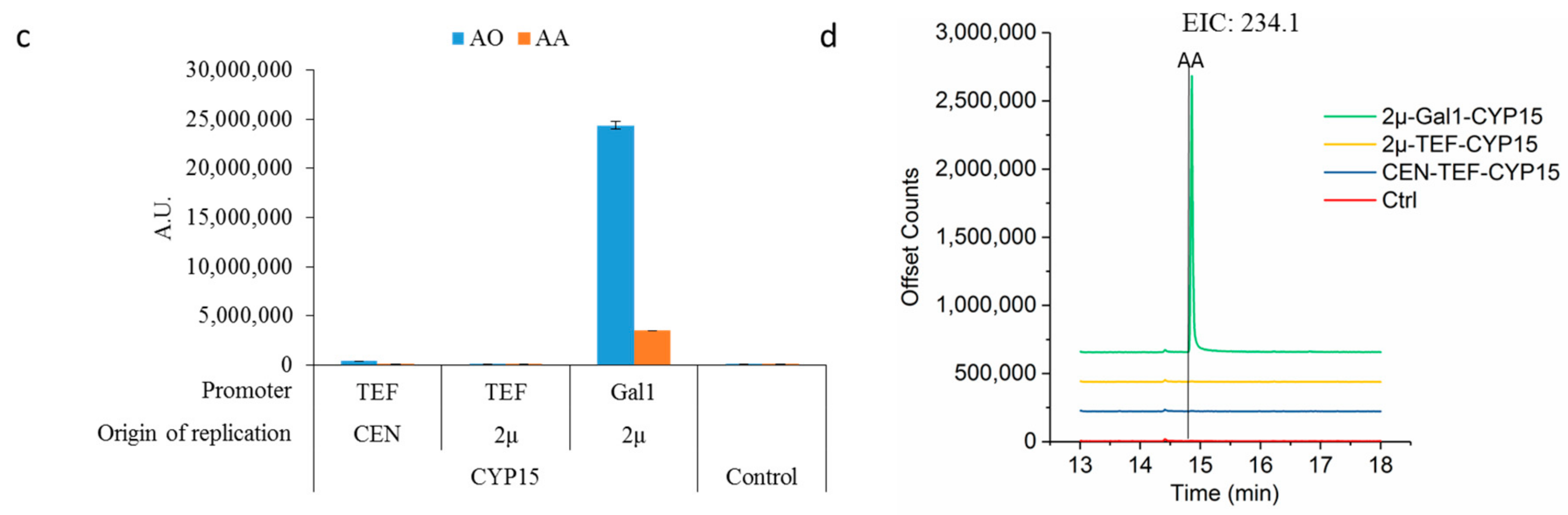
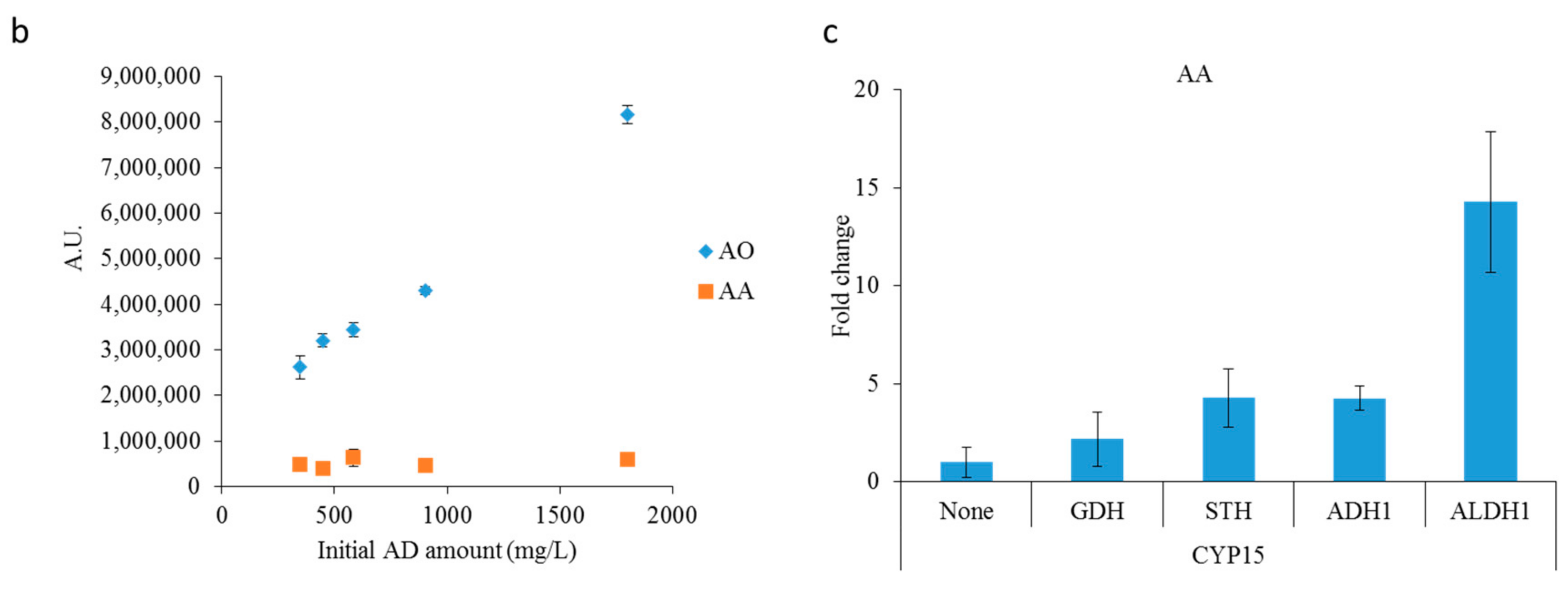
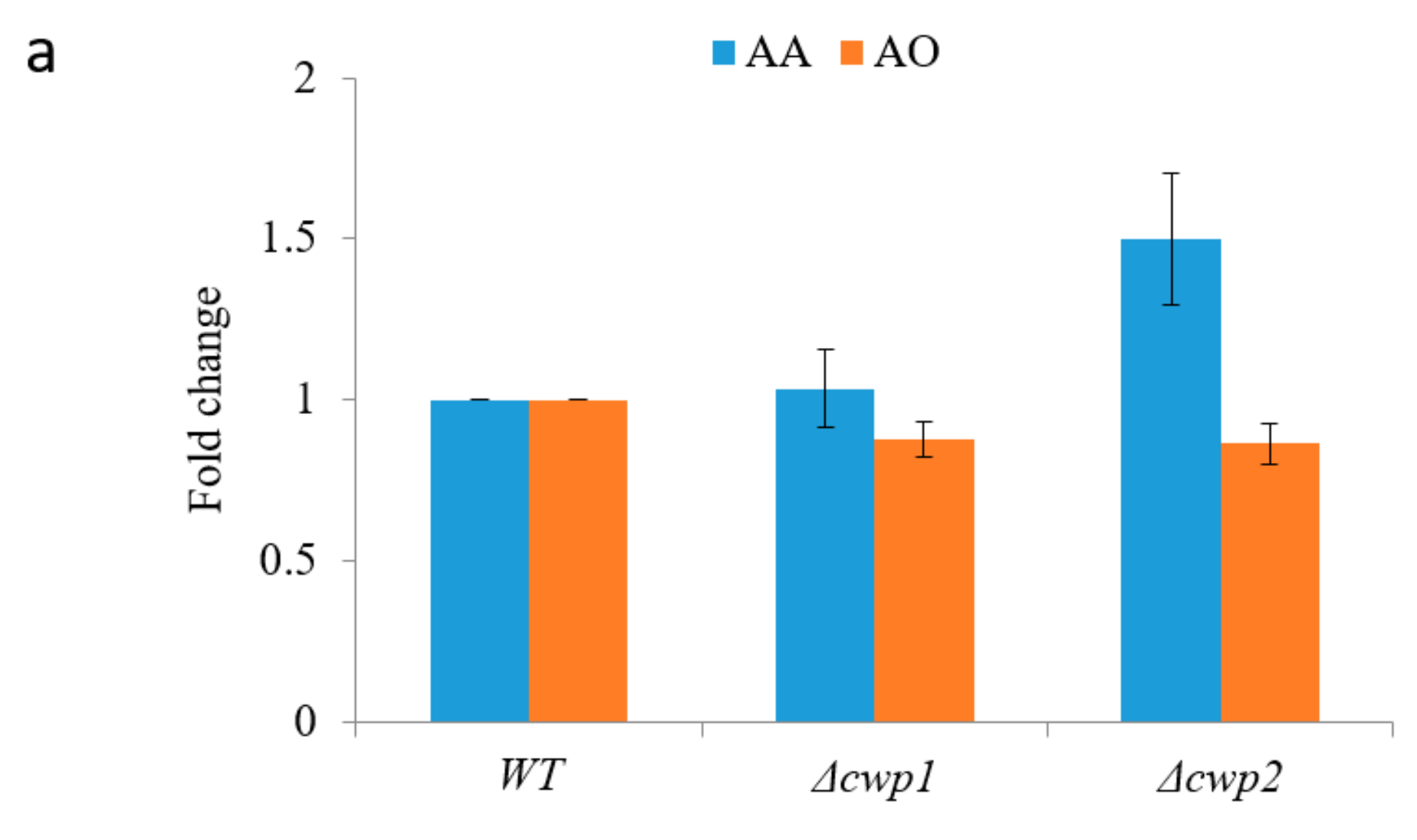
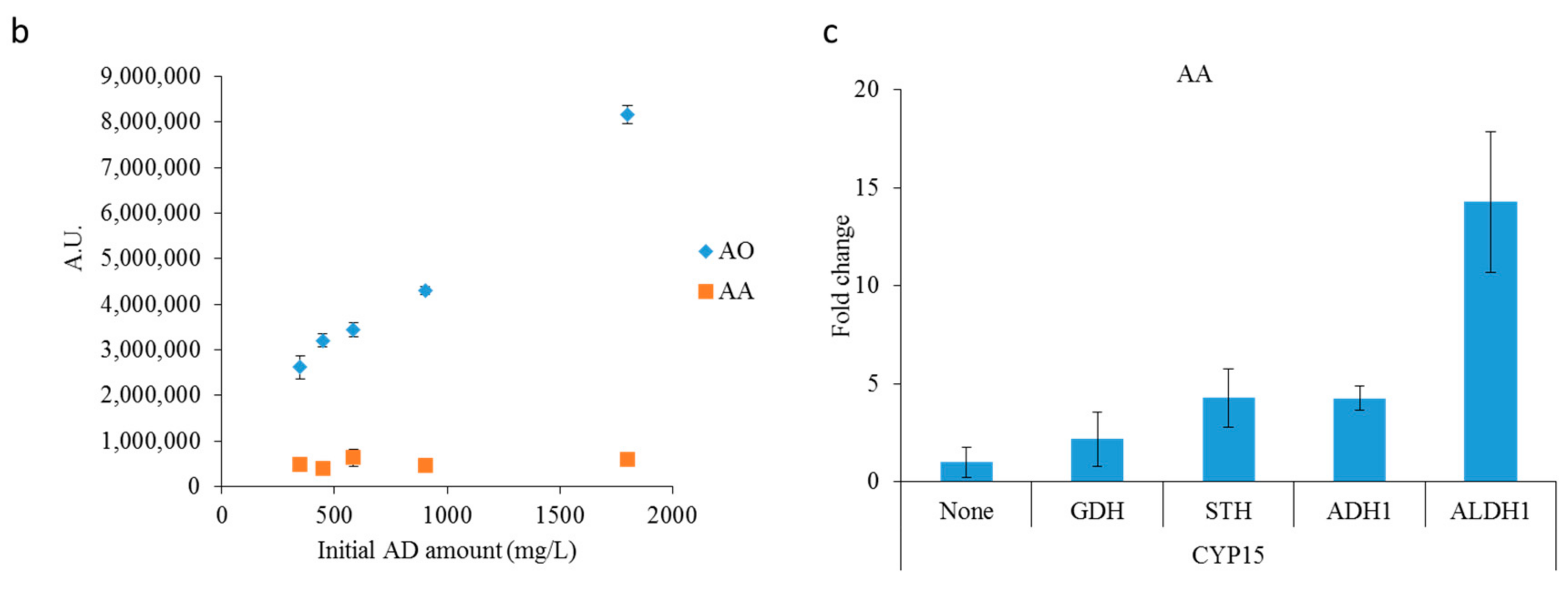
2.2. Optimizing Artemisinic Acid Production by Host Screening and Co-Factor Engineering
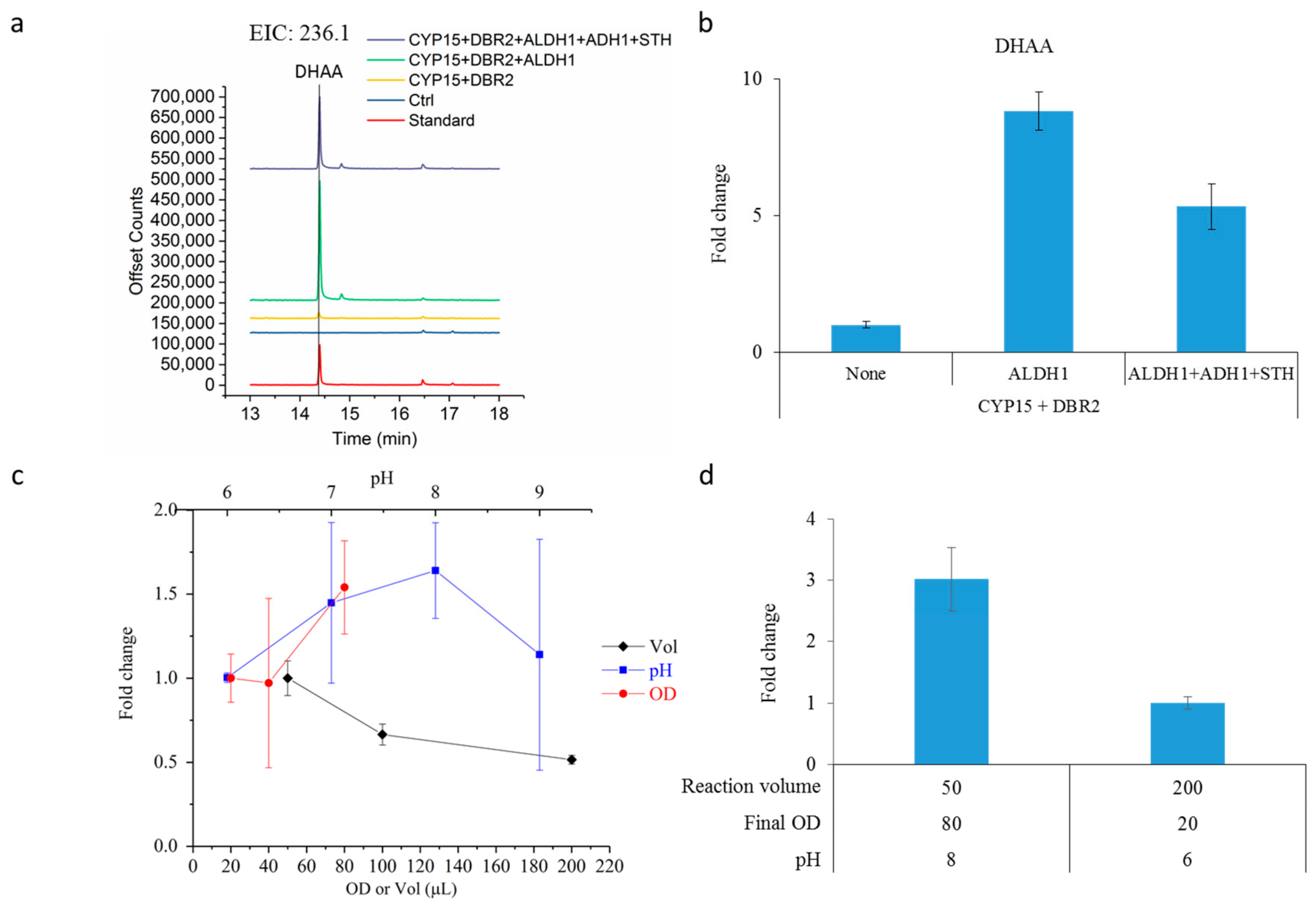
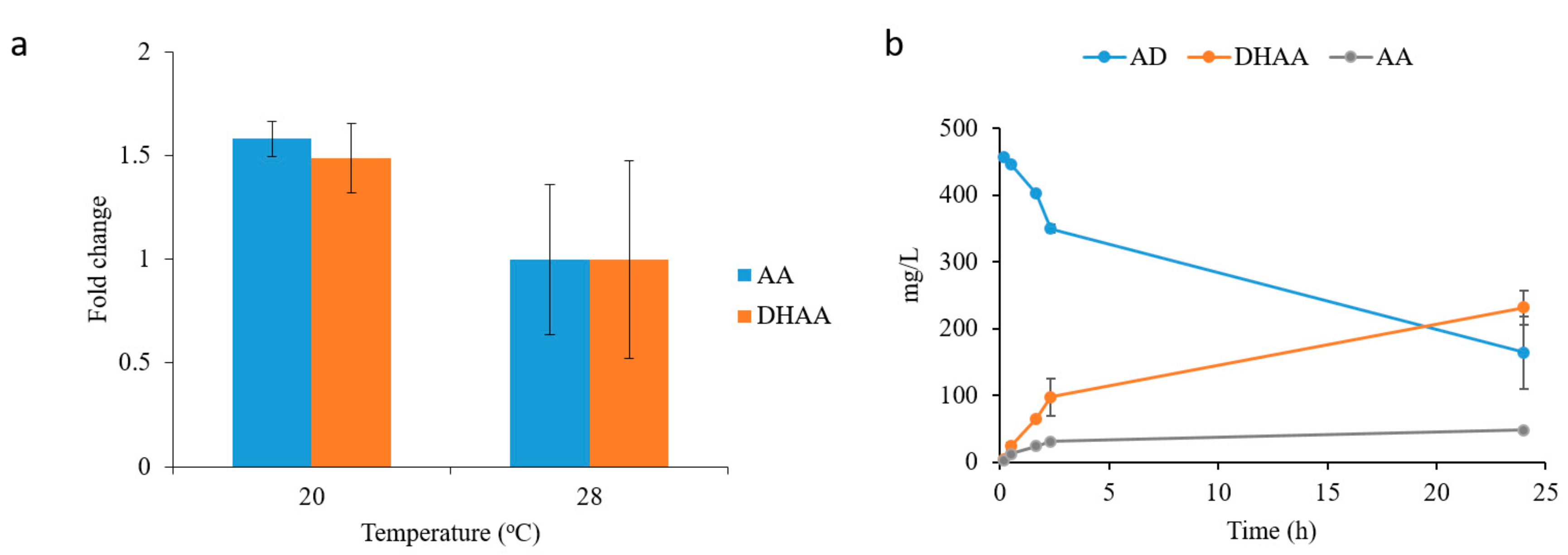
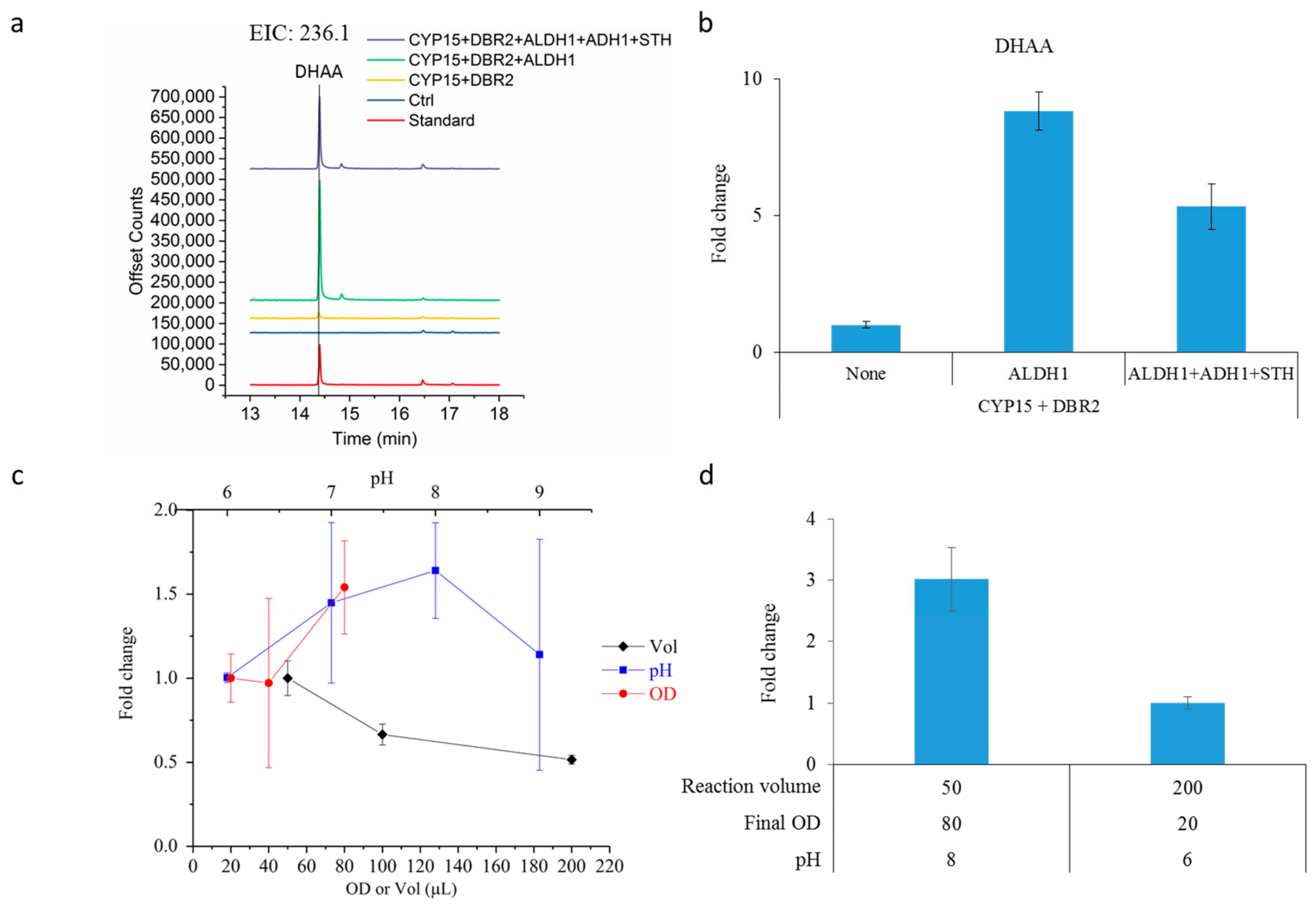
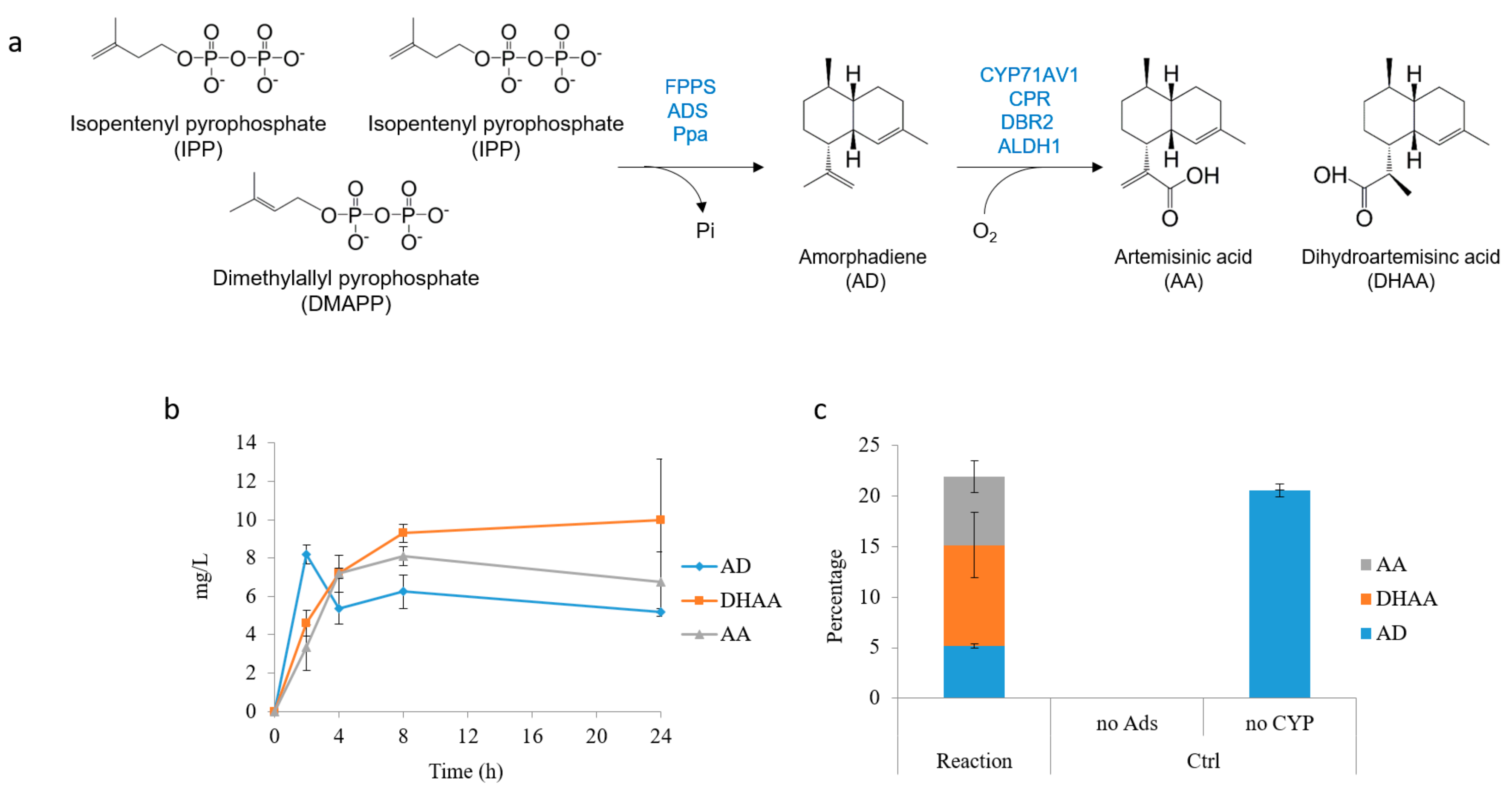
2.3. Dihydroartemisinic Acid Production and Reaction Condition Optimization
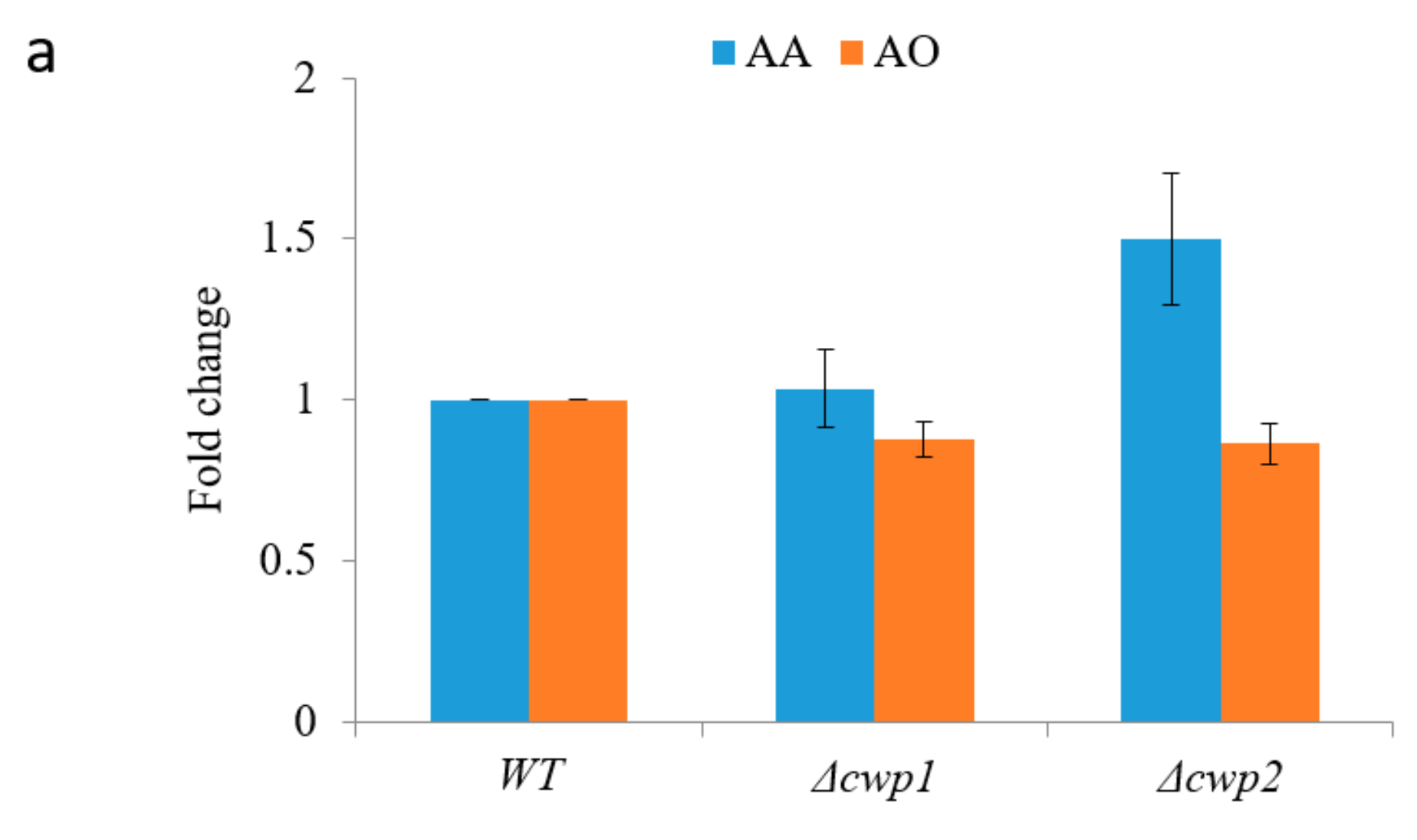
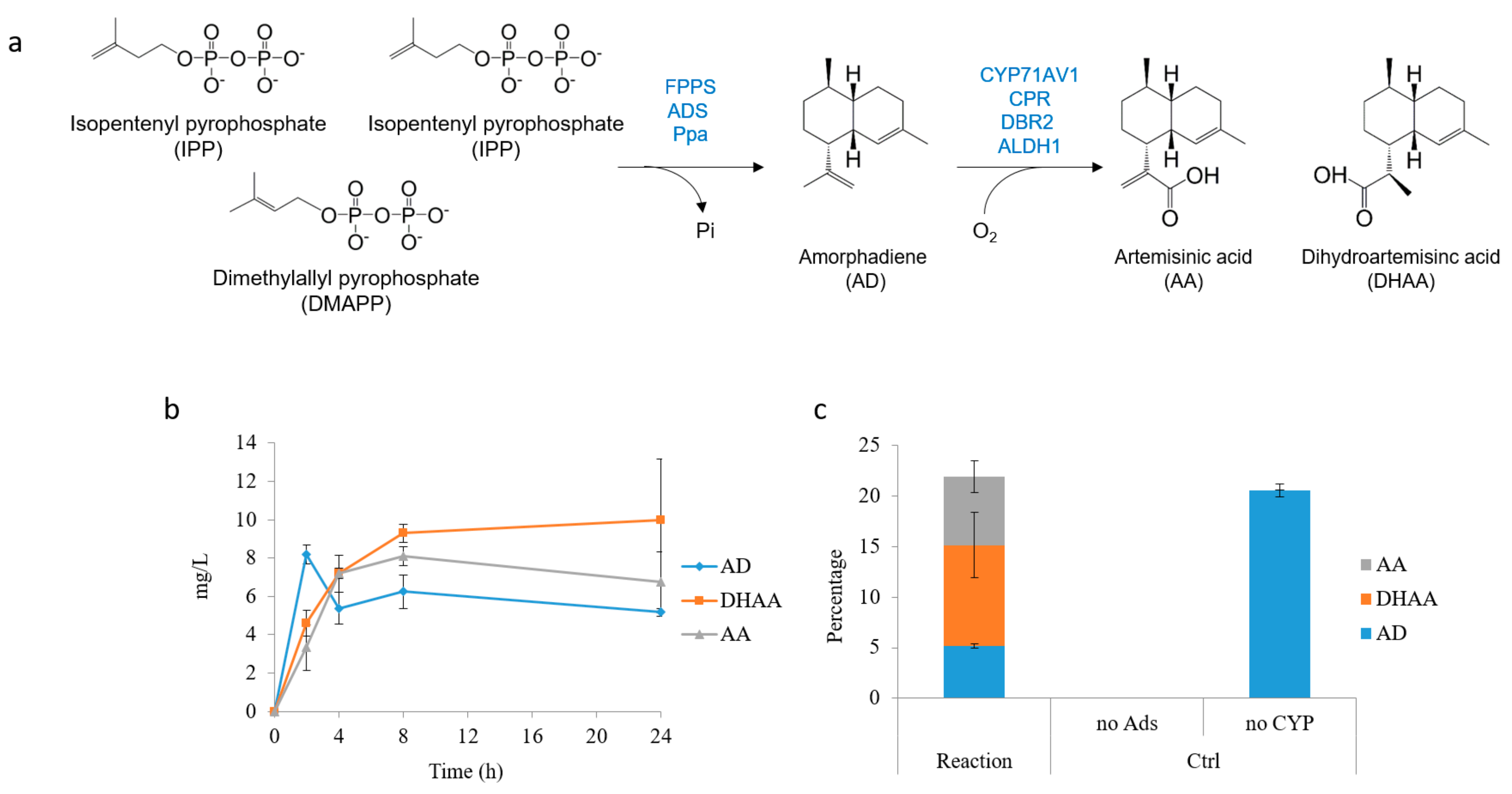
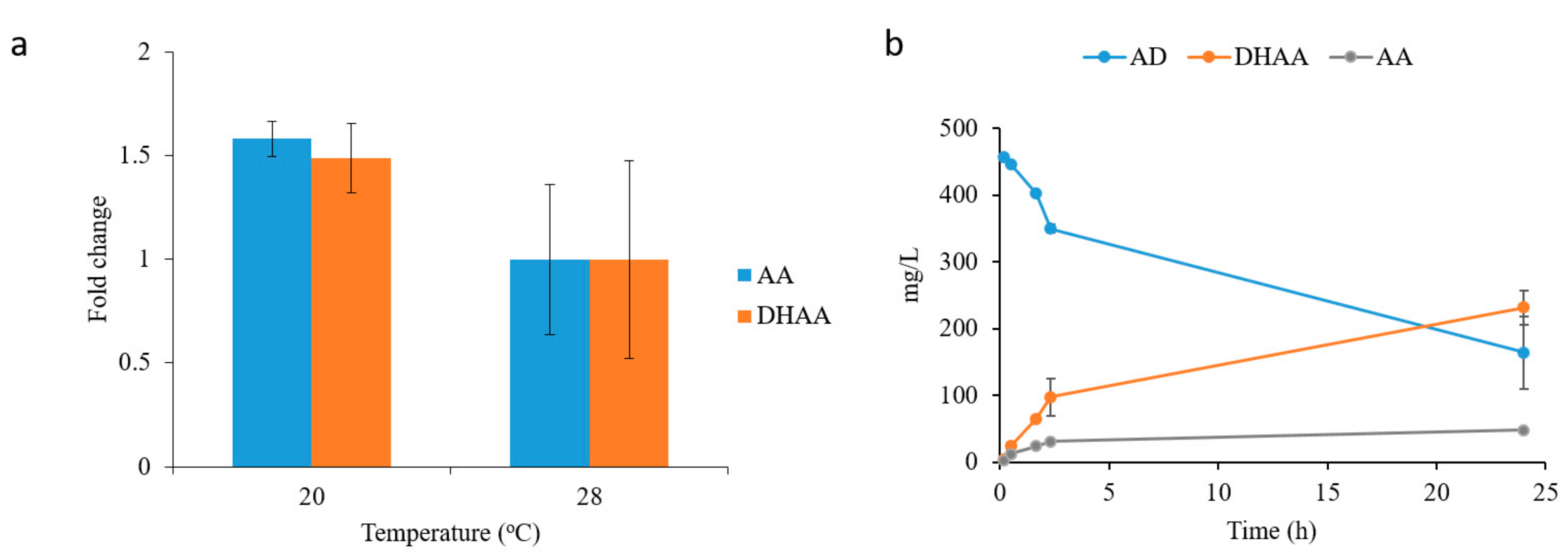
2.4. In Situ Production of Amorphadiene and Dihydroartemisinic Acid
3. Materials and Methods
3.1. Strains and Plasmids
3.2. Yeast Growth and Protein Expression
3.3. Amorpha-4,11-Diene Purification
3.4. Overexpression and Purification of FPPS, ADS and Ppa
3.5. Yeast Whole Cell Biocatalysis and Product Extraction
3.6. GCMS Analysis of AD, AOH, AO and AA
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, B.; Sharma, R.A. Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. 3 Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef]
- Ajikumar, P.K.; Tyo, K.; Carlsen, S.; Mucha, O.; Phon, T.H.; Stephanopoulos, G. Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharm. 2008, 5, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Boutanaev, A.M.; Moses, T.; Zi, J.; Nelson, D.R.; Mugford, S.T.; Peters, R.J.; Osbourn, A. Investigation of terpene diversification across multiple sequenced plant genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E81–E88. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome p450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef] [PubMed]
- Pateraki, I.; Heskes, A.M.; Hamberger, B. Cytochromes p450 for terpene functionalisation and metabolic engineering. Adv. Biochem. Eng. Biotechnol. 2015, 148, 107–139. [Google Scholar] [PubMed]
- Chang, M.C.Y.; Eachus, R.A.; Trieu, W.; Ro, D.-K.; Keasling, J.D. Engineering Escherichia coli for production of functionalized terpenoids using plant p450s. Nat. Chem. Biol. 2007, 3, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Gavira, C.; Hofer, R.; Lesot, A.; Lambert, F.; Zucca, J.; Werck-Reichhart, D. Challenges and pitfalls of p450-dependent (+)-valencene bioconversion by Saccharomyces cerevisiae. Metab. Eng. 2013, 18, 25–35. [Google Scholar] [CrossRef]
- Biggs, B.W.; Lim, C.G.; Sagliani, K.; Shankar, S.; Stephanopoulos, G.; De Mey, M.; Ajikumar, P.K. Overcoming heterologous protein interdependency to optimize p450-mediated taxol precursor synthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, 3209–3214. [Google Scholar] [CrossRef]
- Zhang, C.; Zou, R.; Chen, X.; Stephanopoulos, G.; Too, H.P. Experimental design-aided systematic pathway optimization of glucose uptake and deoxyxylulose phosphate pathway for improved amorphadiene production. Appl. Microbiol. Biotechnol. 2015, 99, 3825–3837. [Google Scholar] [CrossRef]
- Ajikumar, P.K.; Xiao, W.H.; Tyo, K.E.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, M.; Jennewein, S.; Walker, K.; Croteau, R. Taxol biosynthesis. Chem. Biol. 2004, 11, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Qiao, K.; Edgar, S.; Stephanopoulos, G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotechnol. 2015, 33, 377–383. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.A.; Yoshikuni, Y.; Fisher, K.J.; Woolard, F.X.; Ockey, D.; McPhee, D.J.; Renninger, N.S.; Chang, M.C.Y.; Baker, D.; Keasling, J.D. A novel semi-biosynthetic route for artemisinin production using engineered substrate-promiscuous p450bm3. ACS Chem. Biol. 2009, 4, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Teoh, K.H.; Reed, D.W.; Maes, L.; Goossens, A.; Olson, D.J.H.; Ross, A.R.S.; Covello, P.S. The molecular cloning of artemisinic aldehyde δ11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J. Biol. Chem. 2008, 283, 21501–21508. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.H.; Polichuk, D.R.; Reed, D.W.; Nowak, G.; Covello, P.S. Artemisia annua L. (asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome p450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett. 2006, 580, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Komori, A.; Suzuki, M.; Seki, H.; Nishizawa, T.; Meyer, J.J.; Shimizu, H.; Yokoyama, S.; Muranaka, T. Comparative functional analysis of CYP71AV1 natural variants reveals an important residue for the successive oxidation of amorpha-4,11-diene. FEBS Lett. 2013, 587, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; Leys, D.G.; McLean, K.J.; Marshall, K.R.; Ost, T.W.; Daff, S.; Miles, C.S.; Chapman, S.K.; Lysek, D.A.; Moser, C.C.; et al. P450 BM3: The very model of a modern flavocytochrome. Trends Biochem. Sci. 2002, 27, 250–257. [Google Scholar] [CrossRef]
- Haudenschild, C.; Schalk, M.; Karp, F.; Croteau, R. Functional expression of regiospecific cytochrome p450 limonene hydroxylases from mint (Mentha spp.) in Escherichia coli and Saccharomyces cerevisiae. Arch. Biochem. Biophys. 2000, 379, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Barnes, H.J.; Arlotto, M.P.; Waterman, M.R. Expression and enzymatic activity of recombinant cytochrome p450 17 alpha-hydroxylase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1991, 88, 5597–5601. [Google Scholar] [CrossRef] [PubMed]
- Van der Vaart, J.M.; Caro, L.H.; Chapman, J.W.; Klis, F.M.; Verrips, C.T. Identification of three mannoproteins in the cell wall of Saccharomyces cerevisiae. J. Bacteriol. 1995, 177, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Spaans, S.; Weusthuis, R.; Van Der Oost, J.; Kengen, S. Nadph-generating systems in bacteria and archaea. Front Microbiol. 2015, 6, 742. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.H.; Polichuk, D.R.; Reed, D.W.; Covello, P.S. Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. Botany 2009, 87, 635–642. [Google Scholar] [CrossRef]
- Bertea, C.M.; Freije, J.R.; van der Woude, H.; Verstappen, F.W.; Perk, L.; Marquez, V.; De Kraker, J.W.; Posthumus, M.A.; Jansen, B.J.; de Groot, A.; et al. Identification of intermediates and enzymes involved in the early steps of artemisinin biosynthesis in Artemisia annua. Planta Med. 2005, 71, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Wriessnegger, T.; Augustin, P.; Engleder, M.; Leitner, E.; Müller, M.; Kaluzna, I.; Schürmann, M.; Mink, D.; Zellnig, G.; Schwab, H.; et al. Production of the sesquiterpenoid (+)-nootkatone by metabolic engineering of Pichia pastoris. Metab. Eng. 2014, 24, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Carter, O.A.; Peters, R.J.; Croteau, R. Monoterpene biosynthesis pathway construction in Escherichia coli. Phytochemistry 2003, 64, 425–433. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, C.; Zou, R.; Zhou, K.; Stephanopoulos, G.; Too, H.P. Statistical experimental design guided optimization of a one-pot biphasic multienzyme total synthesis of amorpha-4,11-diene. PLoS ONE 2013, 8, e79650. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, C.; Zou, R.; Stephanopoulos, G.; Too, H.-P. In vitro metabolic engineering of amorpha-4,11-diene biosynthesis at enhanced rate and specific yield of production. ACS Synth. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Korman, T.P.; Opgenorth, P.H.; Bowie, J.U. A synthetic biochemistry platform for cell free production of monoterpenes from glucose. Nat. Commun. 2017, 8, 15526. [Google Scholar] [CrossRef]
- Zhu, F.; Zhong, X.; Hu, M.; Lu, L.; Deng, Z.; Liu, T. In vitro reconstitution of mevalonate pathway and targeted engineering of farnesene overproduction in Escherichia coli. Biotechnol. Bioeng. 2014, 111, 1396–1405. [Google Scholar] [CrossRef]
- Cheng, T.; Liu, H.; Zou, H.; Chen, N.; Shi, M.; Xie, C.; Zhao, G.; Xian, M. Enzymatic process optimization for the in vitro production of isoprene from mevalonate. Microb. Cell Fact. 2017, 16, 8. [Google Scholar] [CrossRef]
- Wachtmeister, J.; Rother, D. Recent advances in whole cell biocatalysis techniques bridging from investigative to industrial scale. Curr. Opin. Biotechnol. 2016, 42, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Zhou, K.; Stephanopoulos, G.; Too, H.P. Combinatorial engineering of 1-deoxy-d-xylulose 5-phosphate pathway using cross-lapping in vitro assembly (cliva) method. PLoS ONE 2013, 8, e79557. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. Frozen competent yeast cells that can be transformed with high efficiency using the liac/ss carrier DNA/peg method. Nat. Protoc. 2007, 2, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Porath, J. Immobilized metal ion affinity chromatography. Protein Expr. Purif. 1992, 3, 263–281. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Zhang, C.; Too, H.-P. Multienzyme Biosynthesis of Dihydroartemisinic Acid. Molecules 2017, 22, 1422. https://doi.org/10.3390/molecules22091422
Chen X, Zhang C, Too H-P. Multienzyme Biosynthesis of Dihydroartemisinic Acid. Molecules. 2017; 22(9):1422. https://doi.org/10.3390/molecules22091422
Chicago/Turabian StyleChen, Xixian, Congqiang Zhang, and Heng-Phon Too. 2017. "Multienzyme Biosynthesis of Dihydroartemisinic Acid" Molecules 22, no. 9: 1422. https://doi.org/10.3390/molecules22091422