Looking Back, Looking Forward at Halogen Bonding in Drug Discovery


Abstract
: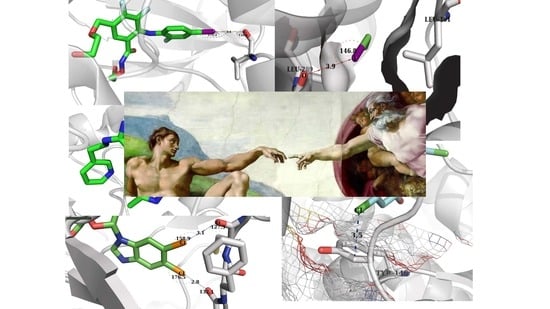
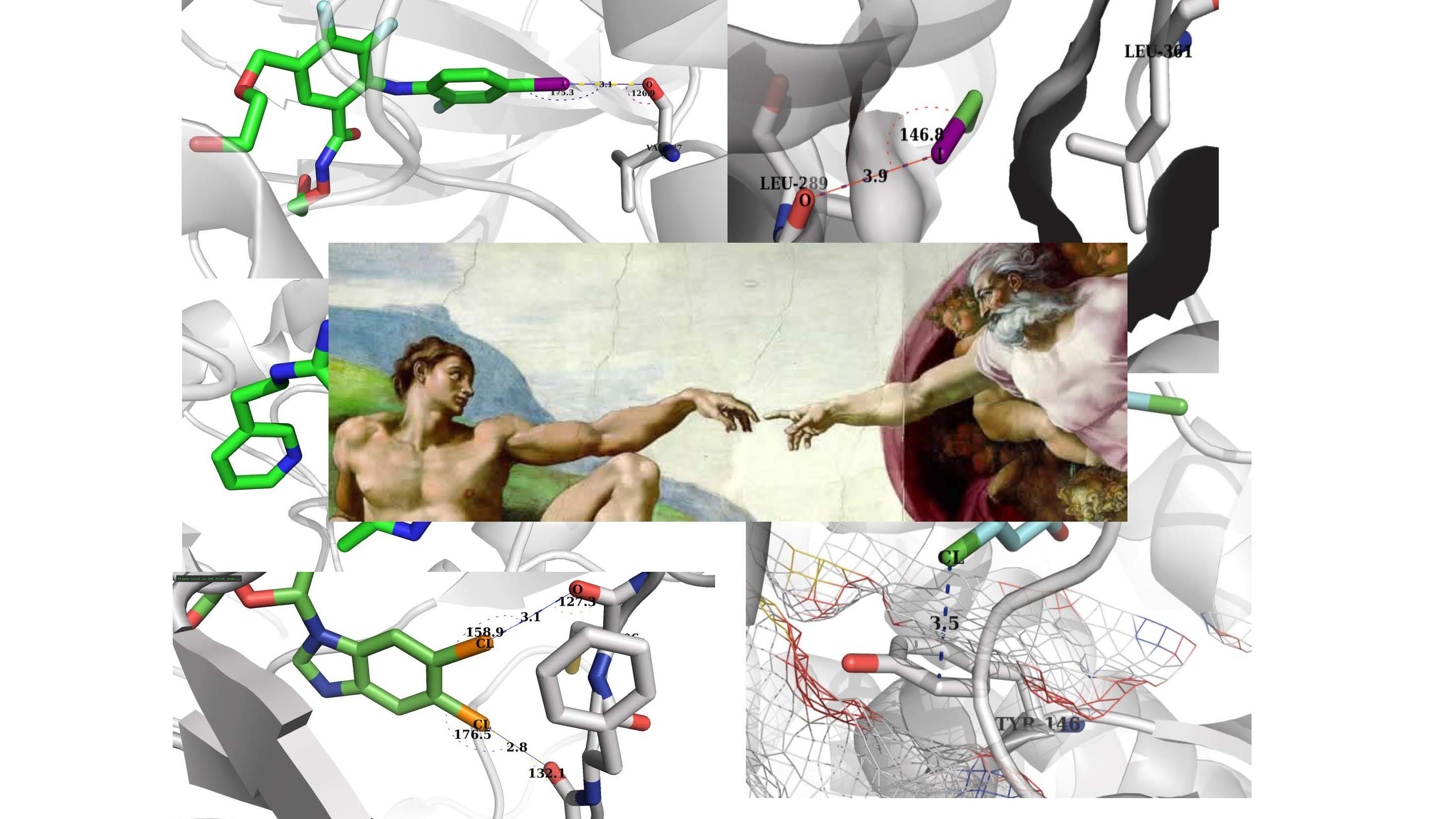{kind=link}
{kind=link}
{kind=link}
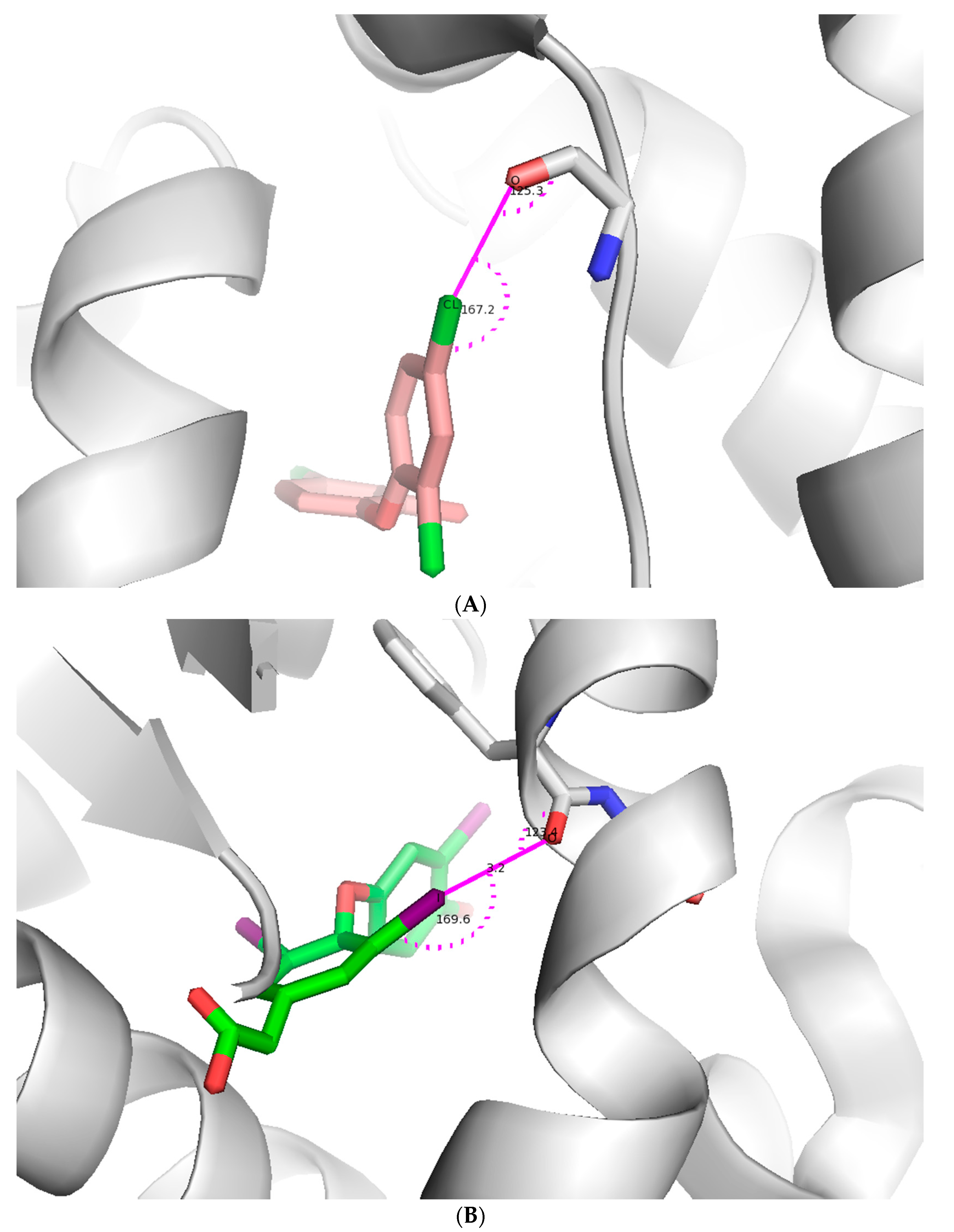{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
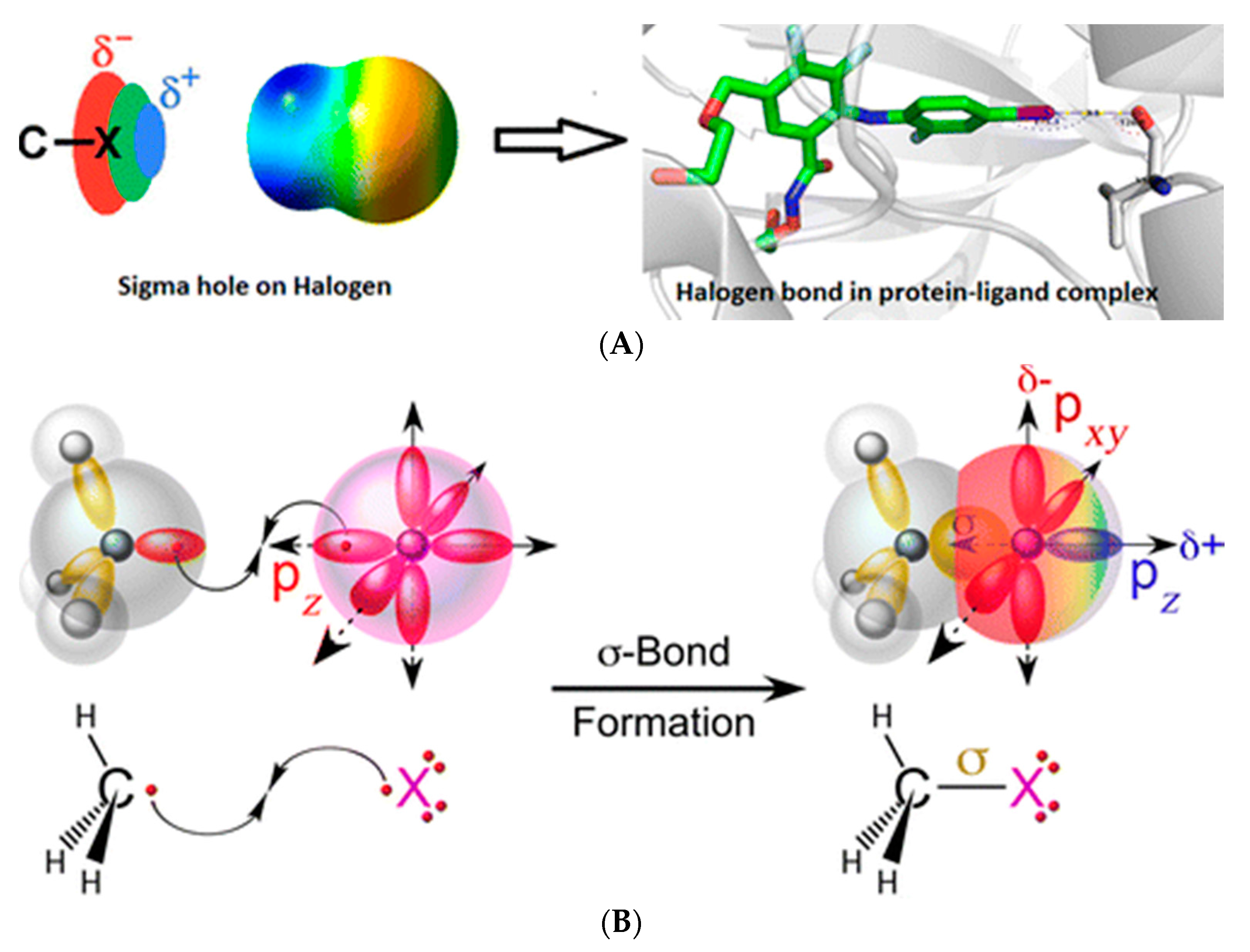
1. Introduction to Halogen Bonding
Origin of Halogen Bonding
2. Halogen Bonding in Biological Systems
2.1. Halogen Bonding between Small Molecules and Proteins
2.2. X-Bonds in Drug Discovery: Case Studies
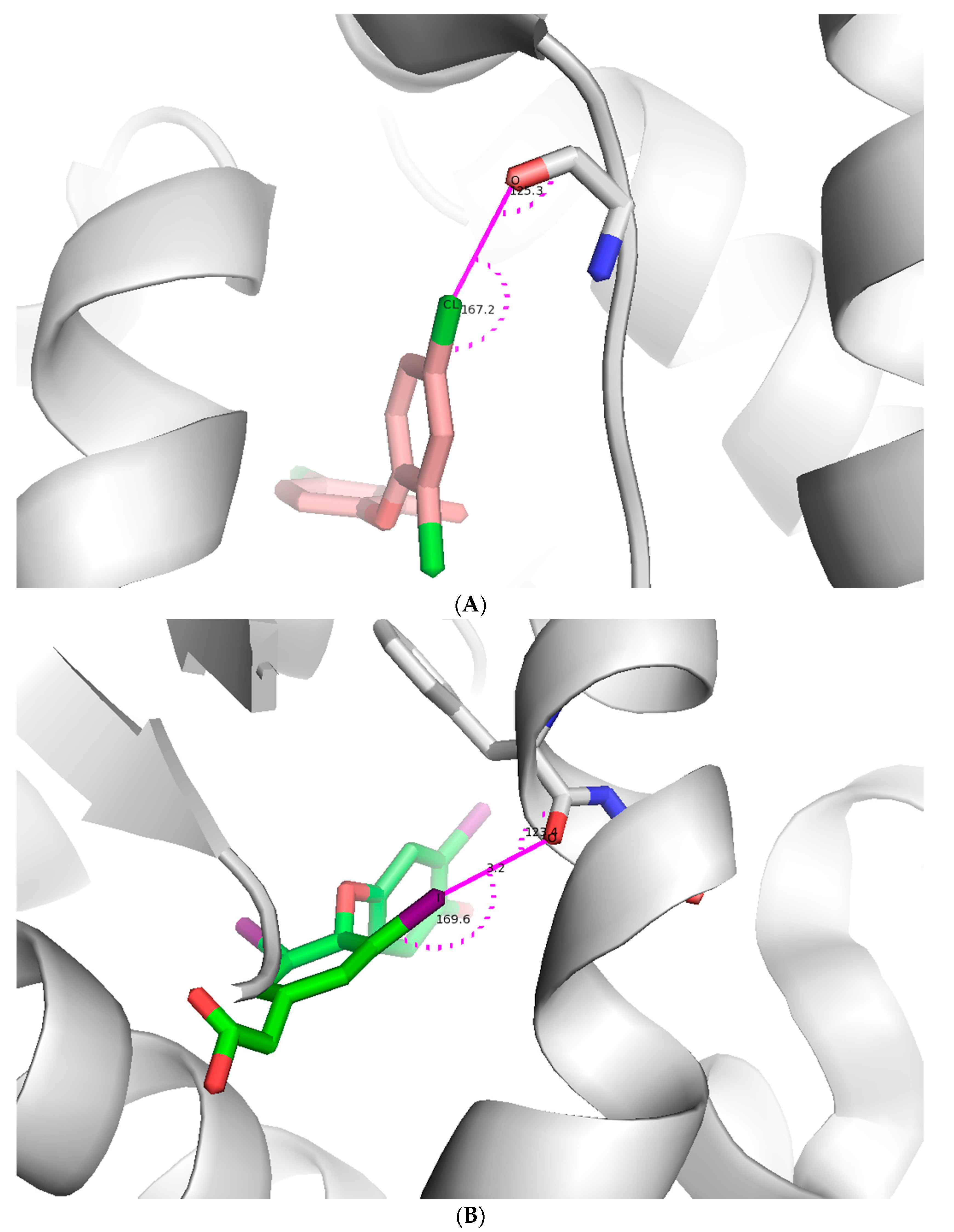
2.2.1. X-Bonding in Cathepsin
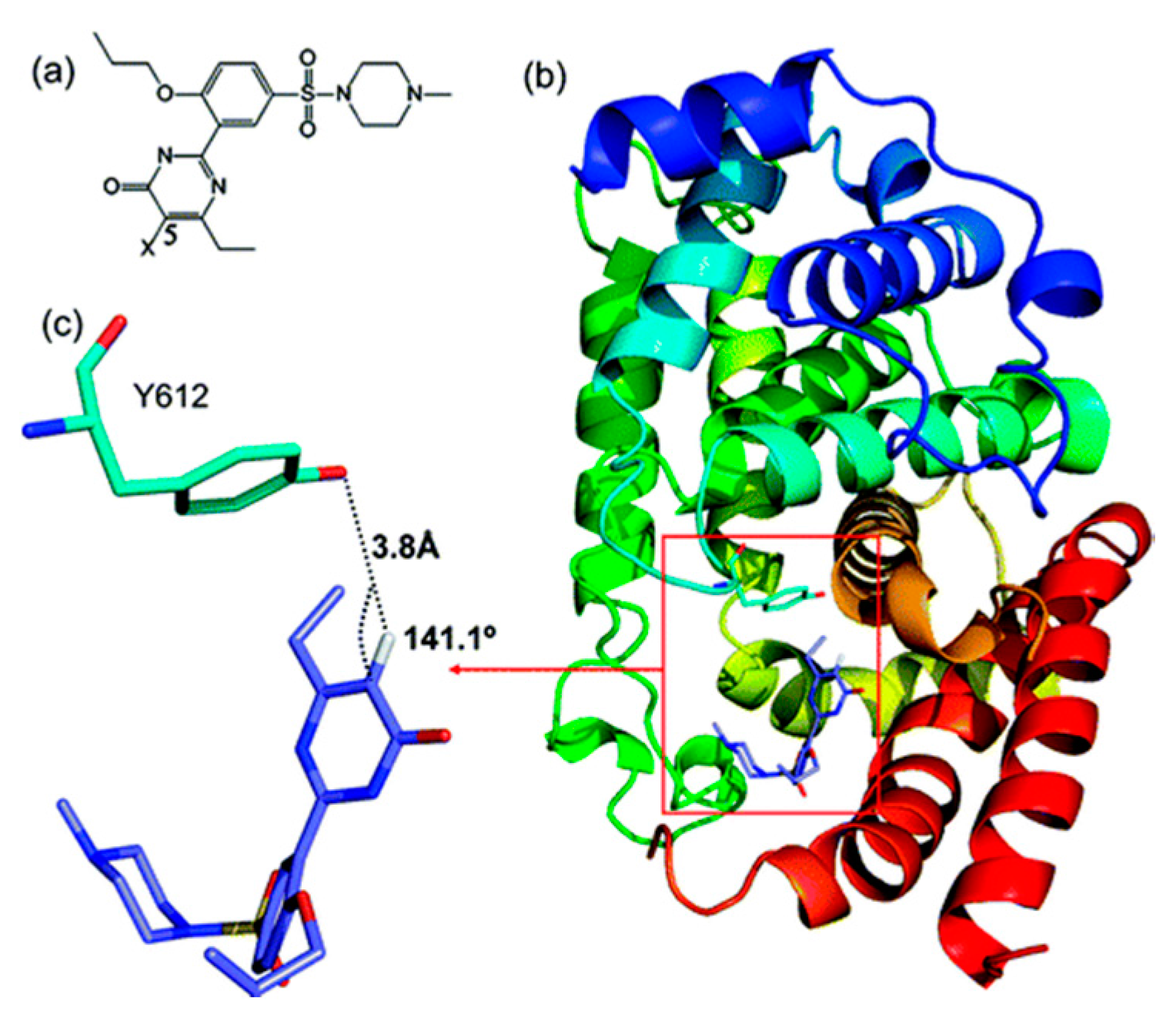
2.2.2. Sildenafil
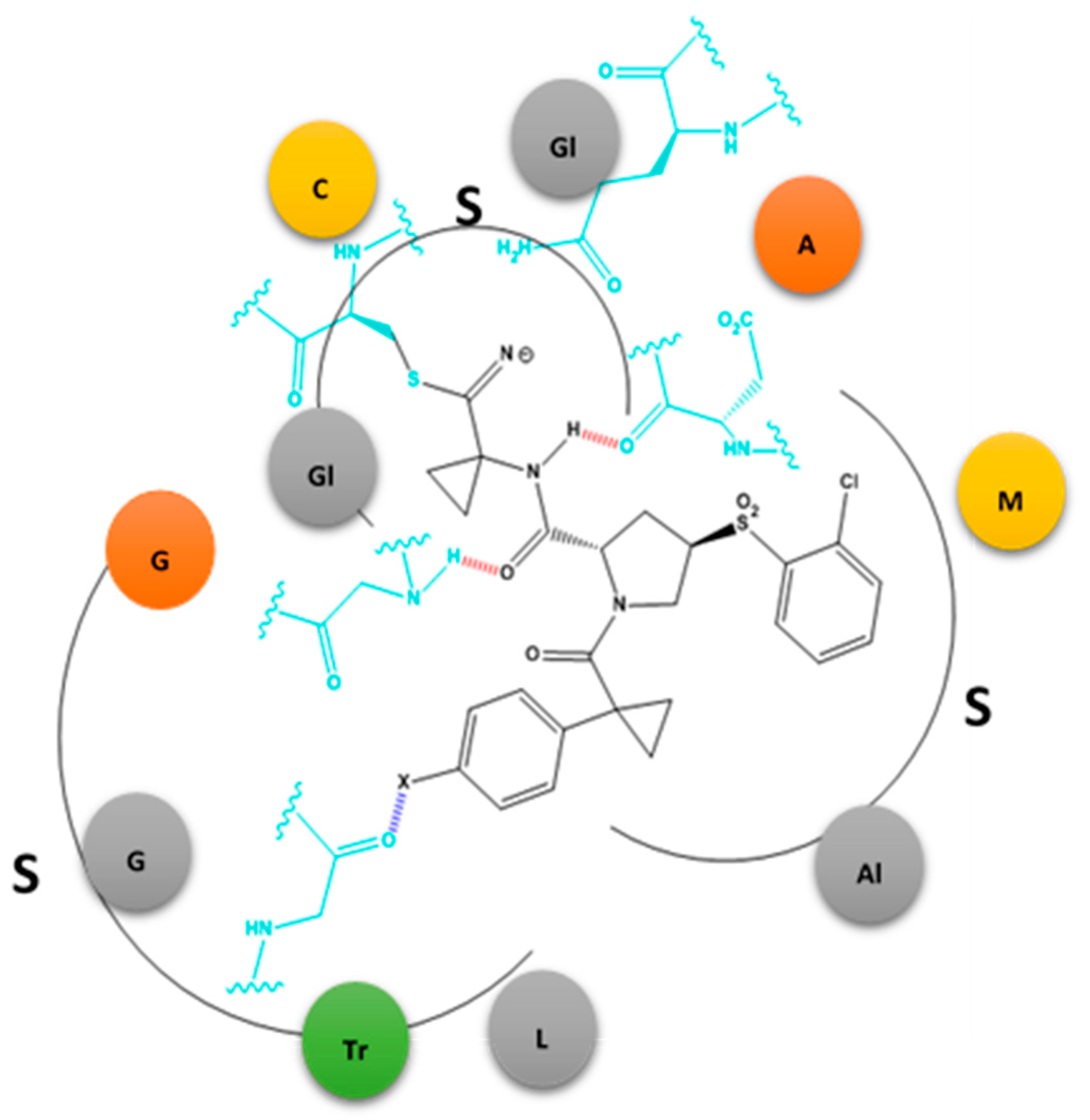
2.2.3. X-Bonding in Unnatural Amino Acid:Protein Complexes
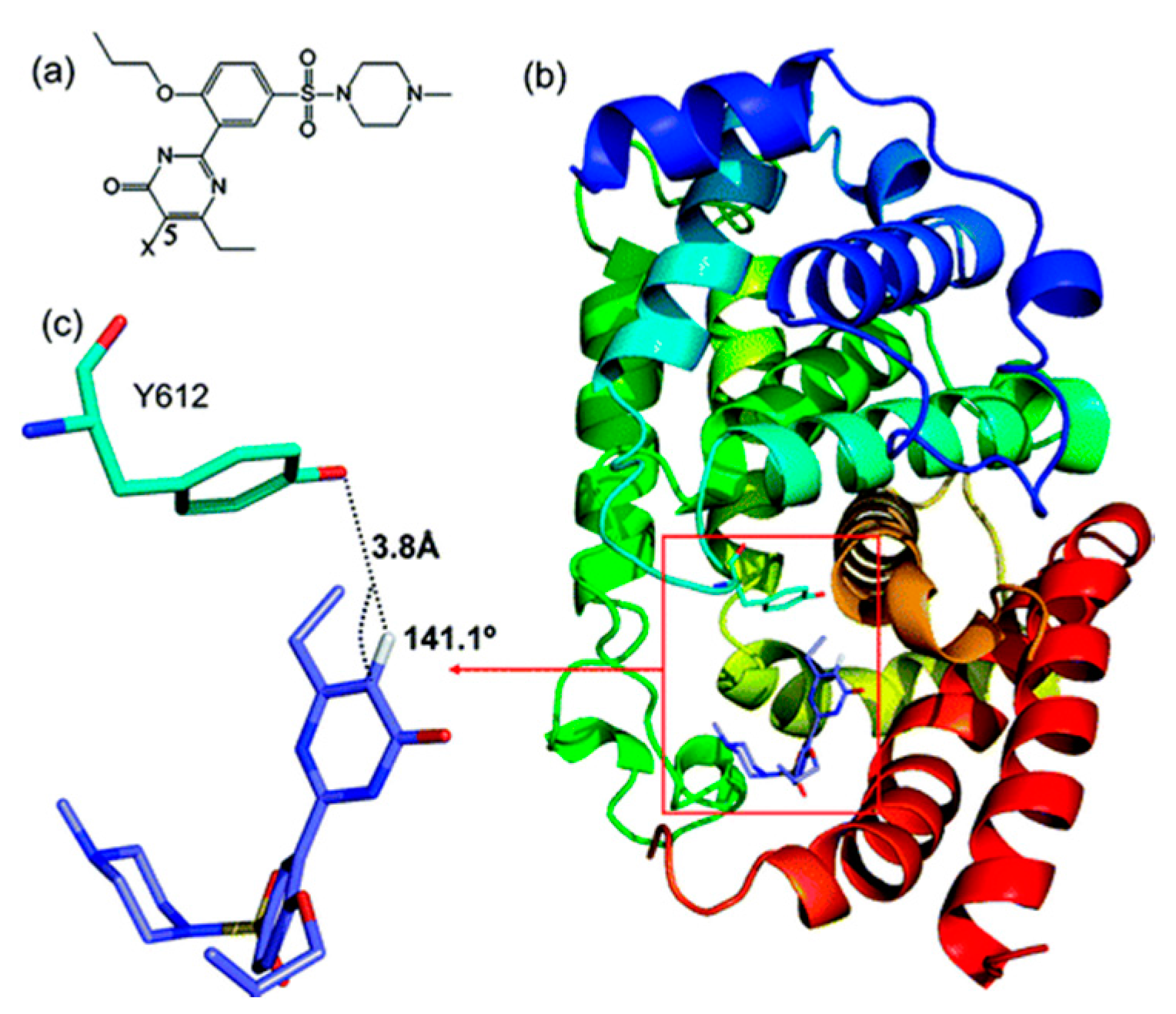
2.2.4. The Donepazil Study
2.3. Some Other Aspects and Applications of Halogen Bonding
2.3.1. Halogen Bonding in Nucleic Acids
2.3.2. Halogen Bonding and Anesthetics Specificity
2.3.3. Halogen Bonding and Drug Repositioning
2.3.4. Halogen Bonding in Directed Biosynthesis
3. Discussion
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Li, W.; Zeng, Y.; Zhang, X.; Zheng, S.; Meng, L. The enhancing effects of group V σ-hole interactions on the F-O halogen bond. Phys. Chem. Chem. Phys. 2014, 16, 19282–19289. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. The fluorine atom as a halogen bond donor, viz. a Positive Site. CrystEngComm 2011, 13, 6593–6596. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Ford, M.C.; Ho, P.S. Computational tools to model halogen bonds in medicinal chemistry. J. Med. Chem. 2016, 59, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, K.; Zariny, H. Halogen bonding: A lump-hole interaction. Chem. Phys. Lett. 2010, 492, 9–13. [Google Scholar] [CrossRef]
- Scholfield, M.R.; Zanden, C.M.V.; Carter, M.; Ho, P.S. Halogen bonding (X-bonding): A biological perspective. Protein Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Voth, A.R.; Khuu, P.; Oishi, K.; Ho, P.S. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 2009, 1, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Lv, J.; Zou, J.; Tian, F.; Shang, Z. Halogen-water-hydrogen bridges in biomolecules. J. Struct. Biol. 2010, 169, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Vasylyeva, V.; Nayak, S.K.; Terraneo, G.; Cavallo, G.; Metrangolo, P.; Resnati, G. Orthogonal halogen and hydrogen bonds involving a peptide bond model. CrystEngComm 2014, 16, 8102–8105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejos, M.; Auffinger, P.; Ho, P.S. Halogen interactions in biomolecular crystal structures. Int. Tables Crystallogr. 2012, F, 821–826. [Google Scholar]
- Hardegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Systematic investigation of halogen bonding in protein-ligand interactions. Angew. Chem. Int. Ed. 2011, 50, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Maillard, M.C.; Hom, R.K.; Benson, T.E.; Moon, J.B.; Mamo, S.; Bienkowski, M.; Tomasselli, A.G.; Woods, D.D.; Prince, D.B.; Paddock, D.J.; et al. Design, synthesis, and crystal structure of hydroxyethyl secondary amine-based. J. Med. Chem. 2007, 50, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Lafrance, L.V.; Calvo, R.R.; Milkiewicz, K.L.; Gupta, V.; Lattanze, J.; Ramachandren, K.; Carver, T.E.; Petrella, E.C.; Cummings, M.D.; et al. 1,4-Benzodiazepine-2,5-diones as small molecule antagonists of the HDM2-p53 interaction: Discovery and SAR. Bioorg. Med. Chem. Lett. 2005, 15, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Iltzsch, M.; Uber, S.S.; Tankersley, K.; Kouni, M.H. Structure-activity relationship for the binding of nucleoside ligands to adenosine kinase from Toxoplasma gondii. Biochem. Pharmacol. 1995, 49, 1501–1512. [Google Scholar] [CrossRef]
- Benjahad, A.; Guillemont, J.; Andries, K.; Nguyen, C.H.; Grierson, D.S. 3-Iodo-4-phenoxypyridinones (IOPY’s), a new family of highly potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2003, 13, 4309–4312. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase-inhibitor complex at 0.66 Å. Proteins Struct. Funct. Genet. 2004, 55, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Hardegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Halogen bonding at the active sites of human cathepsin L and MEK1 kinase: Efficient interactions in different environments. ChemMedChem 2011, 6, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, Z.; Chen, T.; Chen, T.; Wang, Z.; Tian, G.; Shi, J.; Wang, X.; Lu, Y.; Yan, X.; et al. Utilization of halogen bond in lead optimization: A case study of rational design of potent phosphodiesterase type 5 (PDE5) inhibitors. J. Med. Chem. 2011, 54, 5607–5611. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Wilkinson, J.; Gatzoulis, M.A. Sildenafil in primary pulmonary hypertension. N. Engl. J. Med. 2000, 343, 1342. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Hwang, K.; Jeon, Y.; Lee, J.; Heo, Y.; Kim, J.; Moon, J.; Yoon, J.; Hyun, Y.; Kim, E.; et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature 2003, 425, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Raha, K.; Peters, M.B.; Wang, B.; Yu, N.; Wollacott, A.M.; Westerhoff, L.M.; Merz, K.M. The role of quantum mechanics in structure-based drug design. Drug Discov. Today 2007, 12, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1 and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Application of the PM6 method to modeling proteins. J. Mol. Model. 2009, 15, 765–805. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Pitonak, M.; Jurecka, P.; Hobza, P. Stabilization and structure calculations for noncovalent interactions in extended molecular systems based on wave function and density functional theories. Chem. Rev. 2010, 110, 5023–5063. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; Fanfrlík, J.; Salahub, D.; Hobza, P. Semiempirical quantum chemical PM6 method augmented by dispersion and H-bonding correction terms reliably describes various types of noncovalent complexes. J. Chem. Theory Comput. 2009, 5, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, P.; Sponer, J.; Cerný, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Dobeš, P.; Fanfrlík, J.; Rezáč, J.; Otyepka, M.; Hobza, P. Transferable scoring function based on semiempirical quantum mechanical PM6-DH2 method: CDK2 with 15 structurally diverse inhibitors. J. Comput. Aided Mol. Des. 2011, 25, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Kortagere, S.; Ekins, S.; Welsh, W.J. Halogenated ligands and their interactions with amino acids: Implications for structure—Activity and structure—Toxicity relationships. J. Mol. Graph. Model. 2008, 27, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen interactions in protein-ligand complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Muzet, N.; Guillot, B.; Jelsch, C.; Howard, E.; Lecomte, C. Electrostatic complementarity in an aldose reductase complex from ultra-high-resolution crystallography and first-principles calculations. Proc. Natl. Acad. Sci. USA 2003, 100, 8742–8747. [Google Scholar] [CrossRef] [PubMed]
- Borozan, S.Z.; Stojanović, S.Đ. Halogen bonding in complexes of proteins and non-natural amino acids. Comput. Boil. Chem. 2013, 47, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Dorronsoro, I.; Rubio, L.; Munoz, P.; García-Palomero, E.; Del Monte, M.; Medina, M. Donepezil-tacrine hybrid related derivatives as new dual binding site inhibitors of AChE. Bioorg. Med. Chem. 2005, 13, 6588–6597. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. Perspective: The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zou, J.; Tian, F.; Shang, Z. Fluorine bonding—How does it work in protein—Ligand interactions? J. Chem. Inf. Model. 2009, 49, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Bronowska, A.K.; Řezáč, J.; Přenosil, O.; Konvalinka, J.; Hobza, P. A reliable docking/scoring scheme based on the semiempirical quantum mechanical PM6-DH2 method accurately covering dispersion and H-bonding: HIV-1 protease with 22 ligands. J. Phys. Chem. B 2010, 114, 12666–12678. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Fotouhi, N.; Wovkulich, P.; Roberts, J. Cell cycle inhibitors for the treatment of cancer. Drugs Future 2003, 28, 881. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. A halogen-bonding correction for the semiempirical PM6 method. Chem. Phys. Lett. 2011, 506, 286–289. [Google Scholar] [CrossRef]
- Dobes, P.; Rezac, J.; Fanfrlik, J.; Otyepka, M.; Hobza, P. Semiempirical quantum mechanical method PM6-DH2X describes the geometry and energetics of CK2-inhibitor complexes involving halogen bonds well, while the empirical potential fails. J. Phys. Chem. 2011, 115, 8581–8589. [Google Scholar] [CrossRef] [PubMed]
- Voth, A.R.; Ho, P.S. The role of halogen bonding in inhibitor recognition and binding by protein kinases. Curr. Top. Med. Chem. 2007, 7, 1336–1348. [Google Scholar] [PubMed]
- Gianoncelli, A.; Cozza, G.; Orzeszko, A.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Tetraiodobenzimidazoles are potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. 2009, 17, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP site-directed inhibitors of protein kinase 2: An update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Pinna, L.A. Inspecting the structure-activity relationship of protein kinase CK2 inhibitors derived from tetrabromo-Benzimidazole. Chem. Biol. 2005, 12, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Cendron, L.; Bortolato, A.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Moro, S.; Pinna, L.A. The ATP-binding site of protein kinase CK2 holds a positive electrostatic area and conserved water molecules. ChemBioChem 2007, 8, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; De Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001, 10, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A. AMBER empirical potential describes the geometry and energy of noncovalent halogen interactions better than advanced semiempirical quantum mechanical method PM6-DH2X. J. Phys. Chem. B 2012, 116, 3659–3669. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen bonding—A novel interaction for rational drug design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A. Molecular mechanical study of halogen bonding in drug discovery. J. Comput. Chem. 2011, 32, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ali, M.T.; Shawan, M.M.A.K.; Sarwar, M.G.; Khan, M.A.K.; Halim, M.A. Halogen-directed drug design for Alzheimer’s disease: A combined density functional and molecular docking study. SpringerPlus 2016, 5, 1346. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.W.; Kollman, P.A. Advancing beyond the atom-centered model in additive and nonadditive molecular mechanics. J. Comput. Chem. 1997, 18, 1632–1646. [Google Scholar] [CrossRef]
- Cieplak, P.; Caldwell, J.; Kollman, P. Organic and biological systems going beyond the atom centered two body additive approximation: Aqueous solution free energies of methanol and amide hydrogen bonding and chloroform/water partition coefficients of the nucleic acid bases. J. Comput. Chem. 2001, 22, 1048–1057. [Google Scholar] [CrossRef]
- Halgren, T.A.; Damm, W. Polarizable force fields. Curr. Opin. Struct. Biol. 2001, 11, 236–242. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A. Performance assessment of semiempirical molecular orbital methods in describing halogen bonding: quantum mechanical and quantum mechanical/molecular mechanical-molecular dynamics study. J. Chem. Inf. Model. 2011, 51, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Kolar, M.; Hobza, P. On extension of the current biomolecular empirical force field for the description of halogen bonds. J. Chem. Theory Comput. 2012, 8, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OLPS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.; Tirado-Rives, J. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, Z.; Yang, Z.; Liu, Y.; Wang, J.A.; Shao, Q.; Zhu, W. The stabilization effect of dielectric constant and acidic amino acids on arginine-arginine (Arg-Arg) pairings: Database survey and computational studies. J. Phys. Chem. B 2013, 117, 4827–4835. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xu, Z.; Liu, Y.; Wang, J.; Shi, J.; Chen, K.; Zhu, W. Unstable, metastable, or stable halogen bonding interaction involving negatively charged donors? A statistical and computational chemistry study. J. Phys. Chem. B 2014, 118, 14223–14233. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.H.; Tabarrini, O. Halogen bonding in nucleic acid complexes. J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Lin, J.; Liu, Q.; Wang, S.; Lv, G.; Li, K.; Shi, H.; Huang, Z.; Bertaccini, E.J. The role of the hydroxyl group in propofol-protein target recognition: Insights from ONIOM studies. J. Phys. Chem. B 2017, 121, 5883–5896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, B.; Xu, Z.; Li, B.; Cai, T.; Zhang, X.; Yu, Y.; Wang, H.; Shi, J.; Zhu, W. Repositioning organohalogen drugs: A case study for identification of potent B-Raf V600E inhibitors via docking and bioassay. Sci. Rep. 2016, 6, 31074. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, L.; Cui, D.; Yao, Z.; Gao, B.; Lin, J.; Wei, D. The important role of halogen bond in substrate selectivity of enzymatic catalysis. Sci. Rep. 2016, 6, 34750. [Google Scholar] [CrossRef] [PubMed]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Chemin. 2016, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking Back, Looking Forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. https://doi.org/10.3390/molecules22091397
Mendez L, Henriquez G, Sirimulla S, Narayan M. Looking Back, Looking Forward at Halogen Bonding in Drug Discovery. Molecules. 2017; 22(9):1397. https://doi.org/10.3390/molecules22091397
Chicago/Turabian StyleMendez, Lois, Gabriela Henriquez, Suman Sirimulla, and Mahesh Narayan. 2017. "Looking Back, Looking Forward at Halogen Bonding in Drug Discovery" Molecules 22, no. 9: 1397. https://doi.org/10.3390/molecules22091397