Multi-Objective Optimization Algorithm to Discover Condition-Specific Modules in Multiple Networks


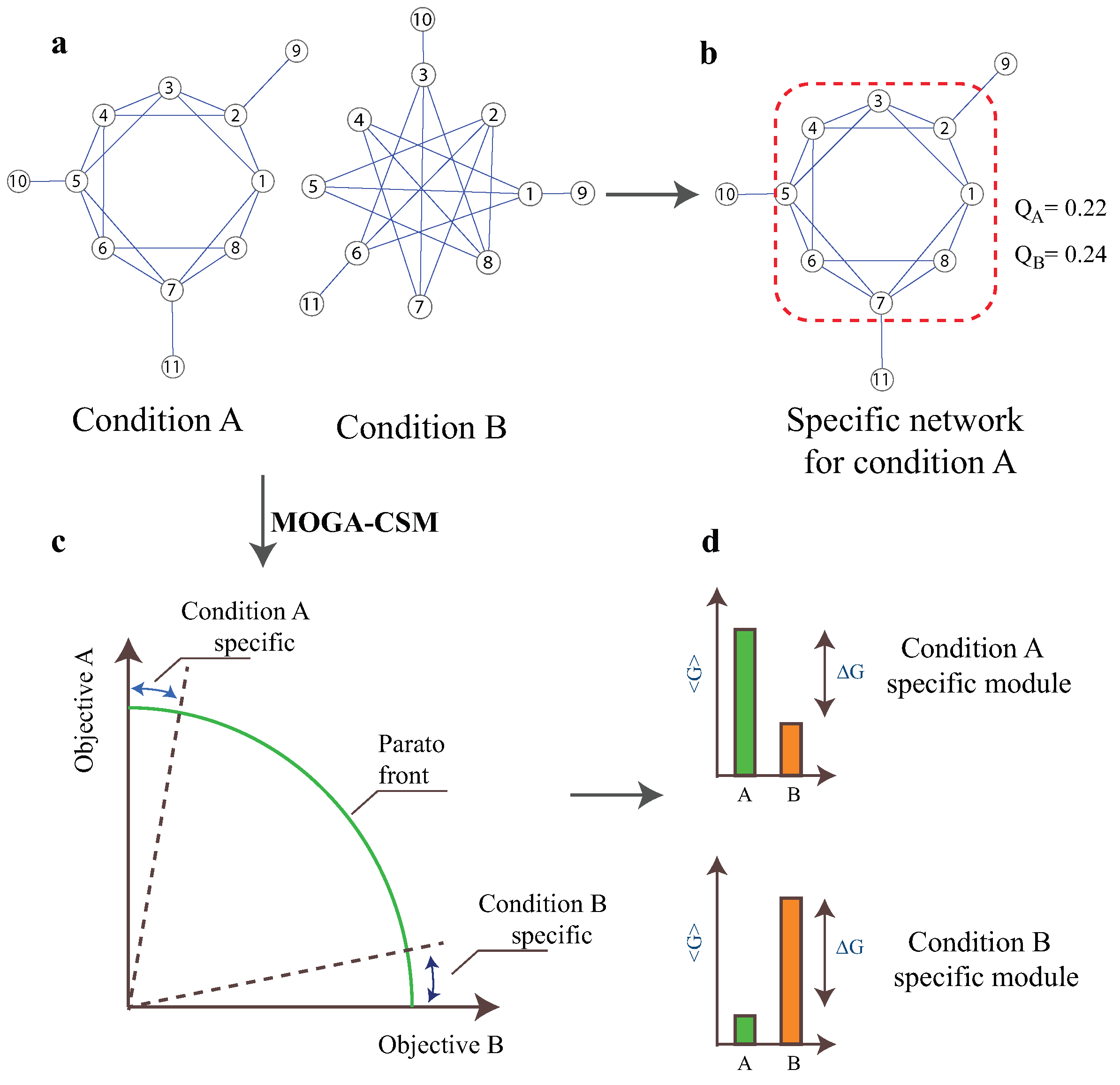{kind=link}
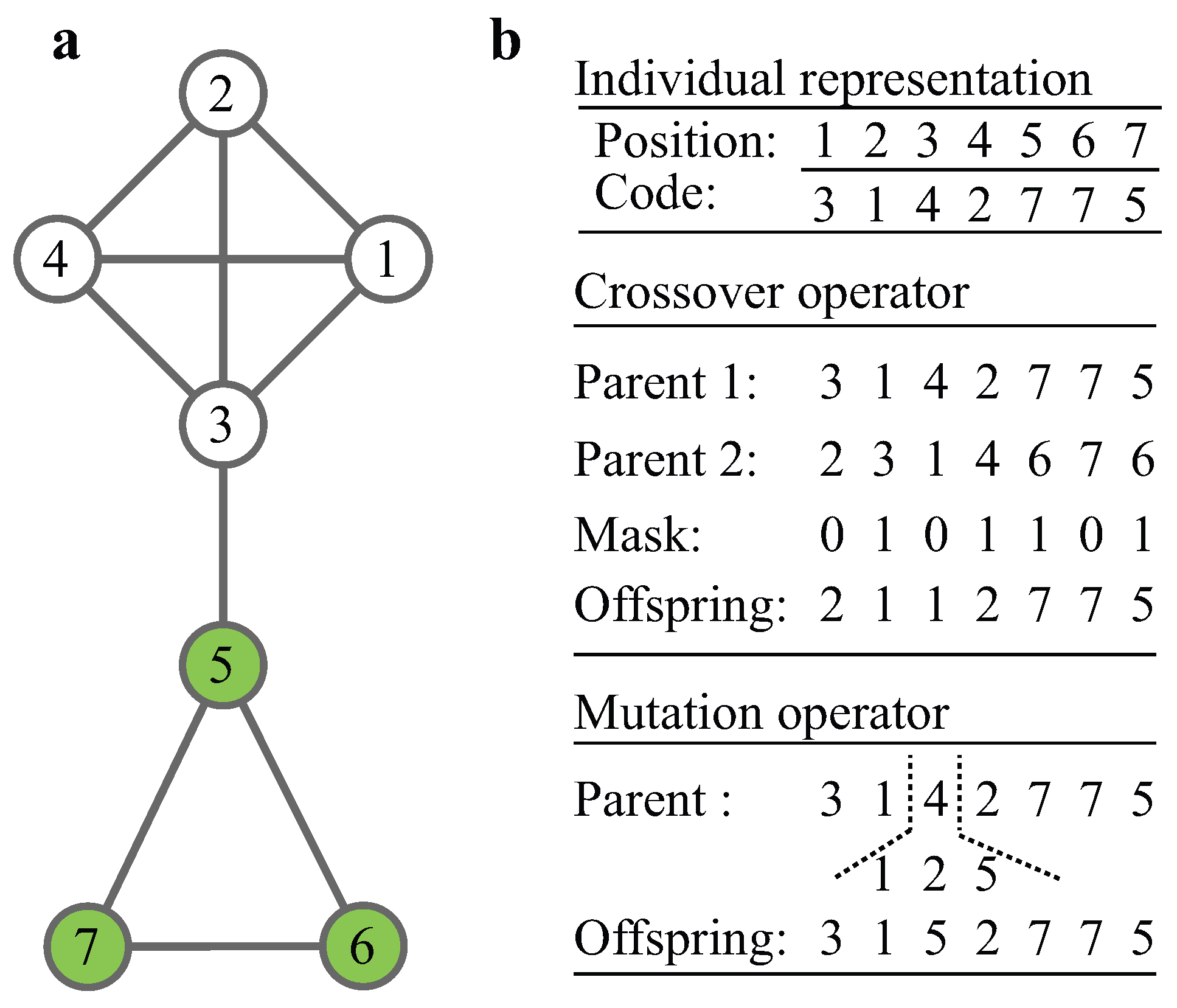{kind=link}
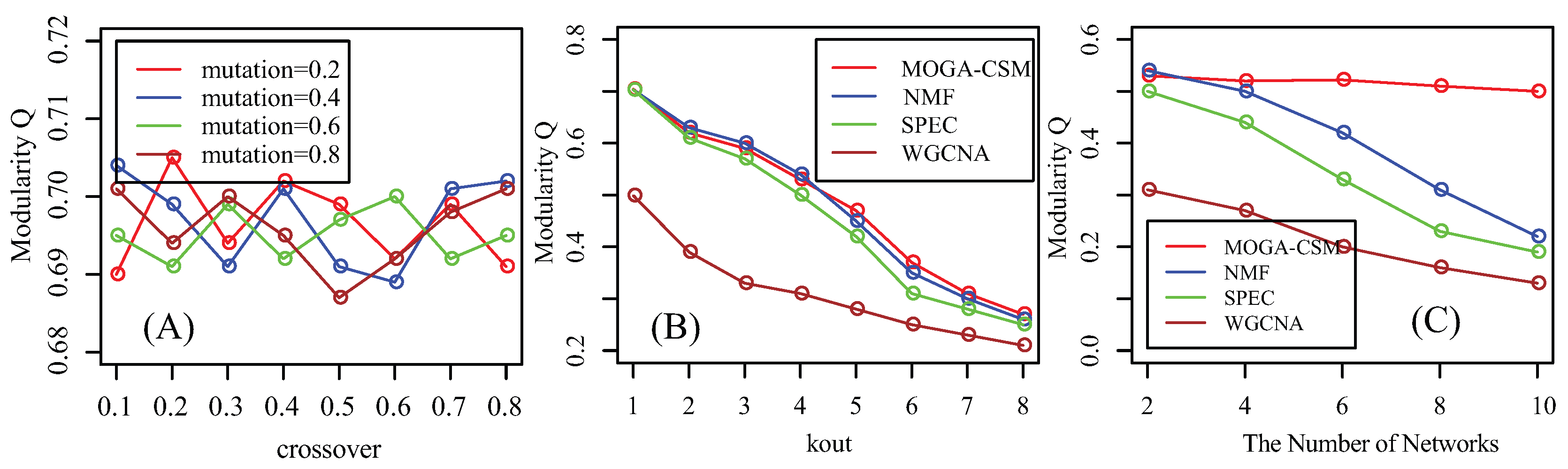{kind=link}
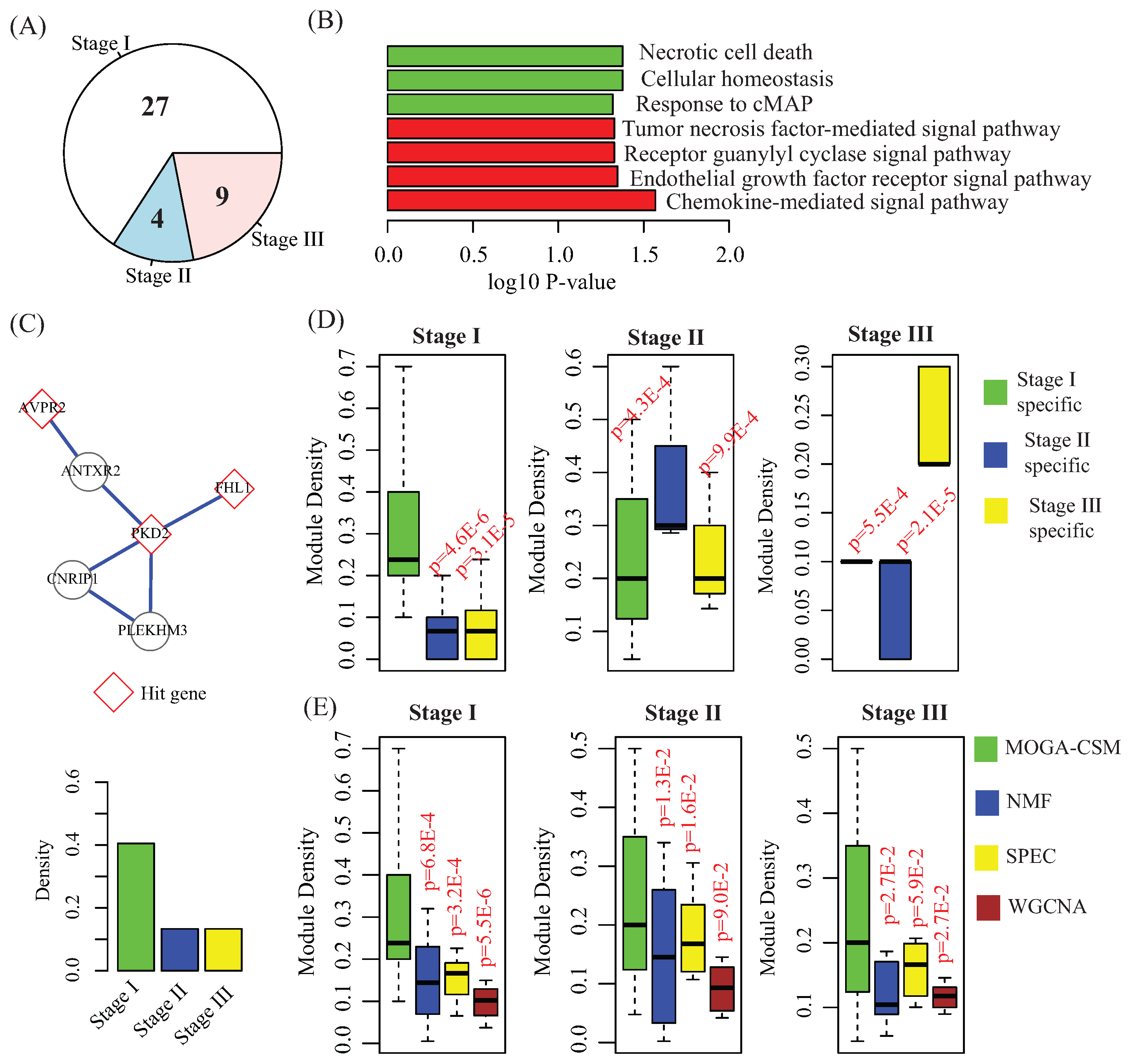{kind=link}
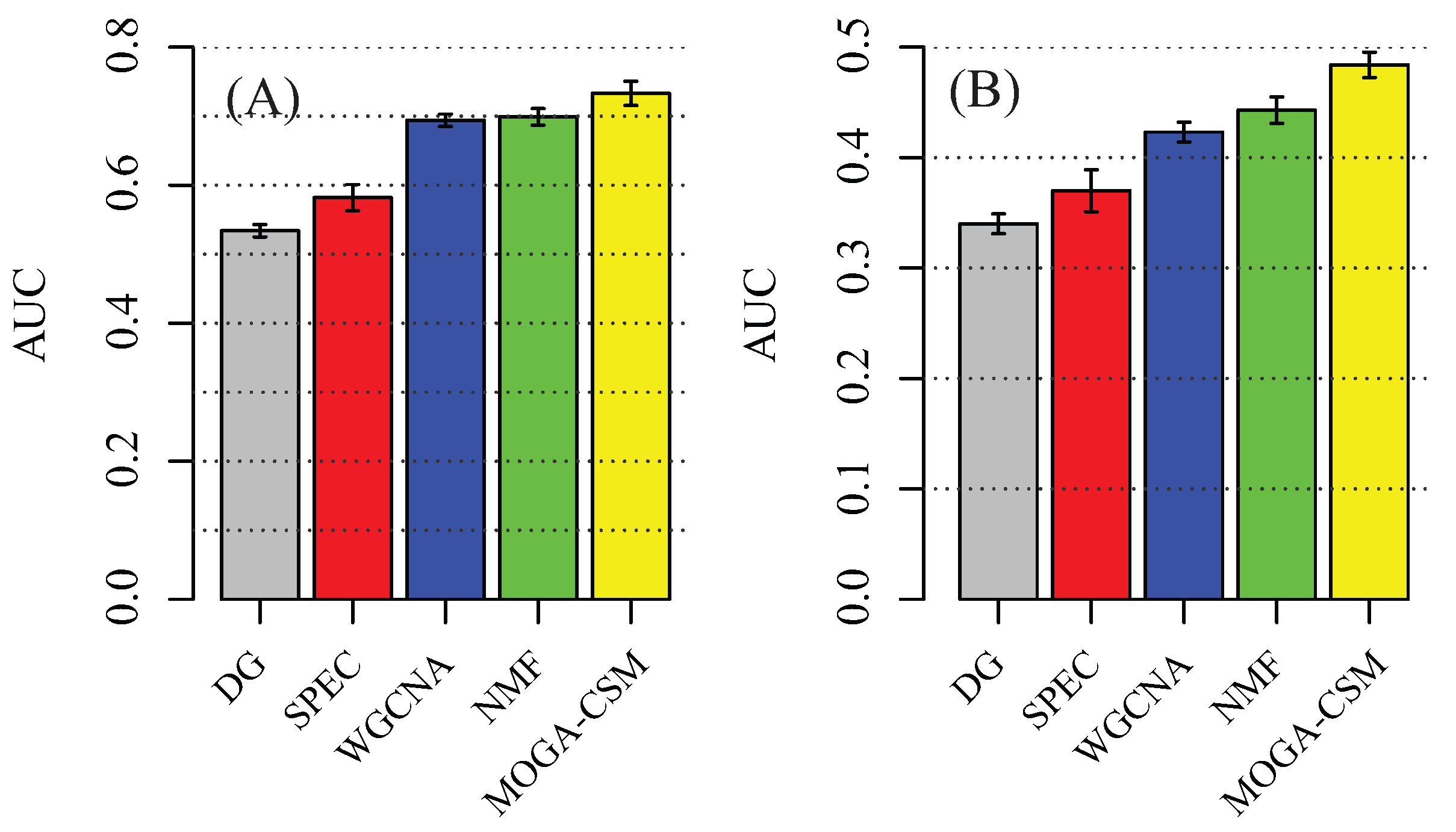{kind=link}
{kind=link}
Abstract
:1. Introduction
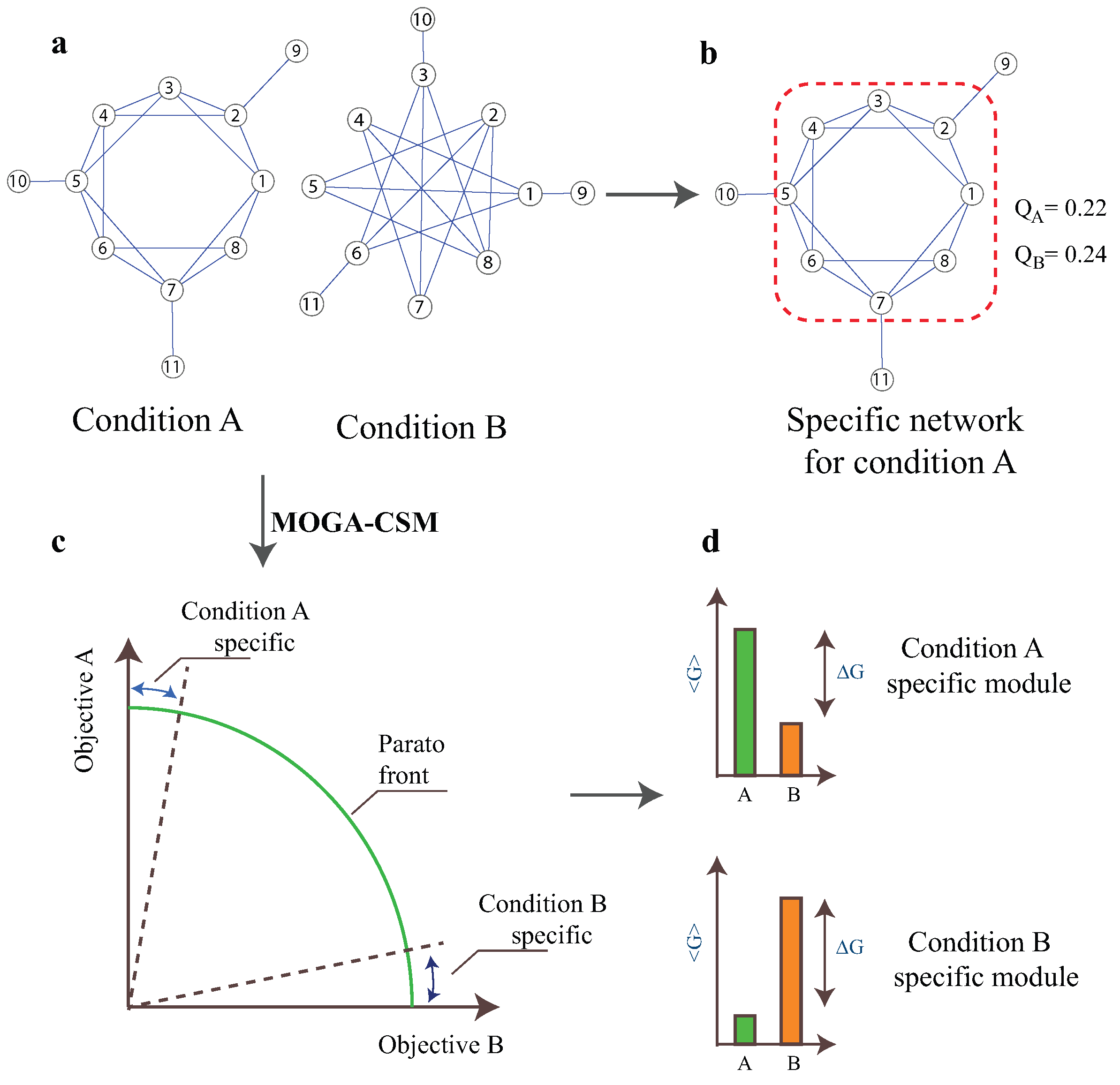
2. Methods
2.1. Multi-Objective Mathematical Model
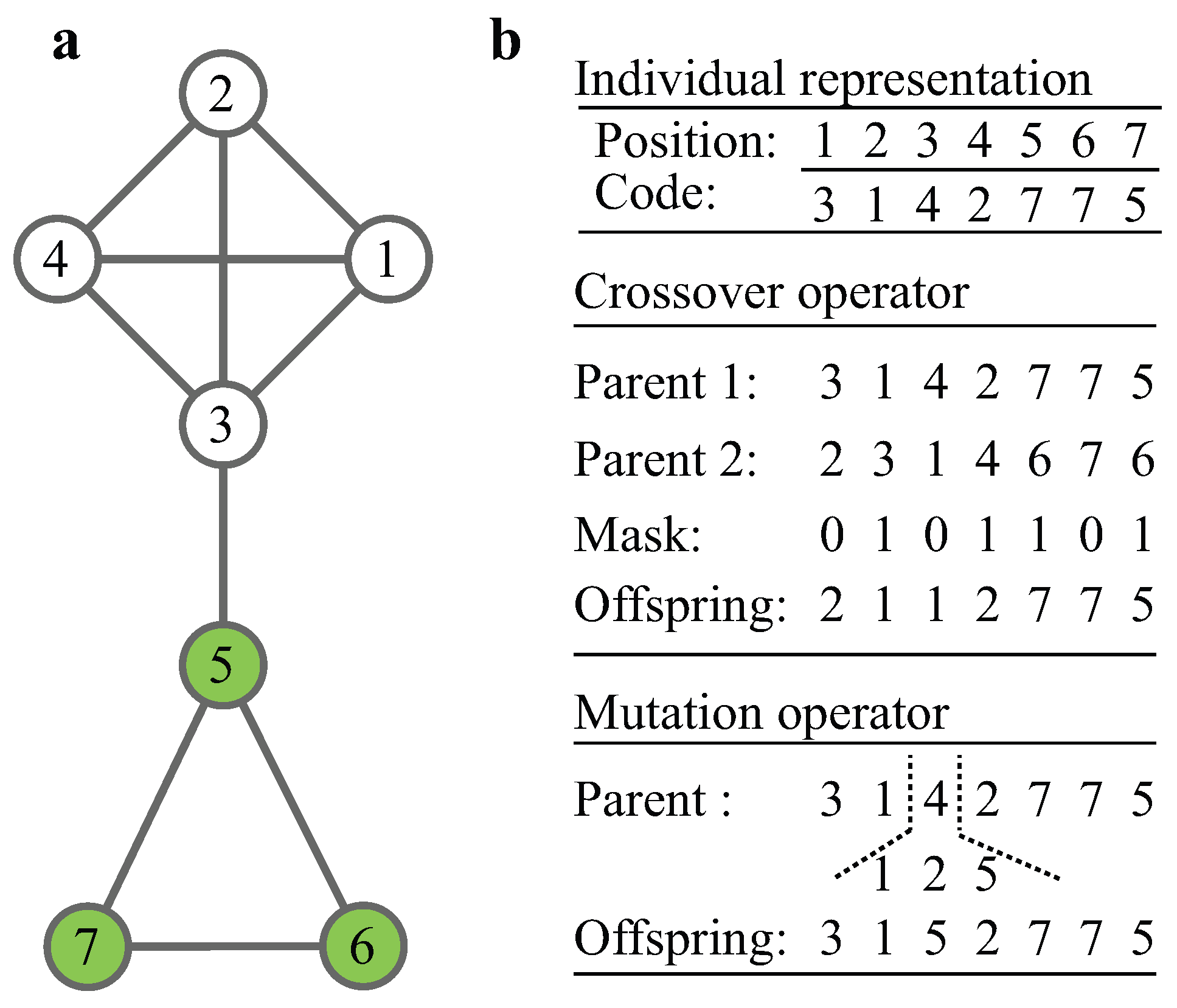
2.2. The MOGA-CSM
| Algorithm 1 Decoding Procedure |
| Input: P: an individual of the population. Output: : the module structure of P. 1: For each node , set and . 2: For each edge , find the roots of i and , denoted by and , respectively. 3: If , update ; else goto step 4. 4: If , update ; else update . 5: return Disjoint sets. |
| Algorithm 2 The MOGA-CSM |
| Input: : the involved multiple networks. m: the specific condition. Output: : the condition-specific modules. 1: Create a population of random individuals for . 2: Decode each individual P of the population using the decoding procedure. 3: Obtain the rank of each individual according to nondomination rank. 4: Generate new offspring using the crossover and mutation operators. 5: Combine the parents and offspring into a new pool and rank them. 6: Select the individuals with lower rank for the next generation. 7: If the termination criterion is not satisfied, goto step 1; otherwise, goto step 8. 8: return with the maximum modularity. |
2.3. Algorithm Analysis
3. Materials
3.1. Statistical Significance of Specific Modules
3.2. Features for Support Vector Machine on Specific Modules
3.3. Artificial Networks
3.4. Breast Cancer Networks
4. Results
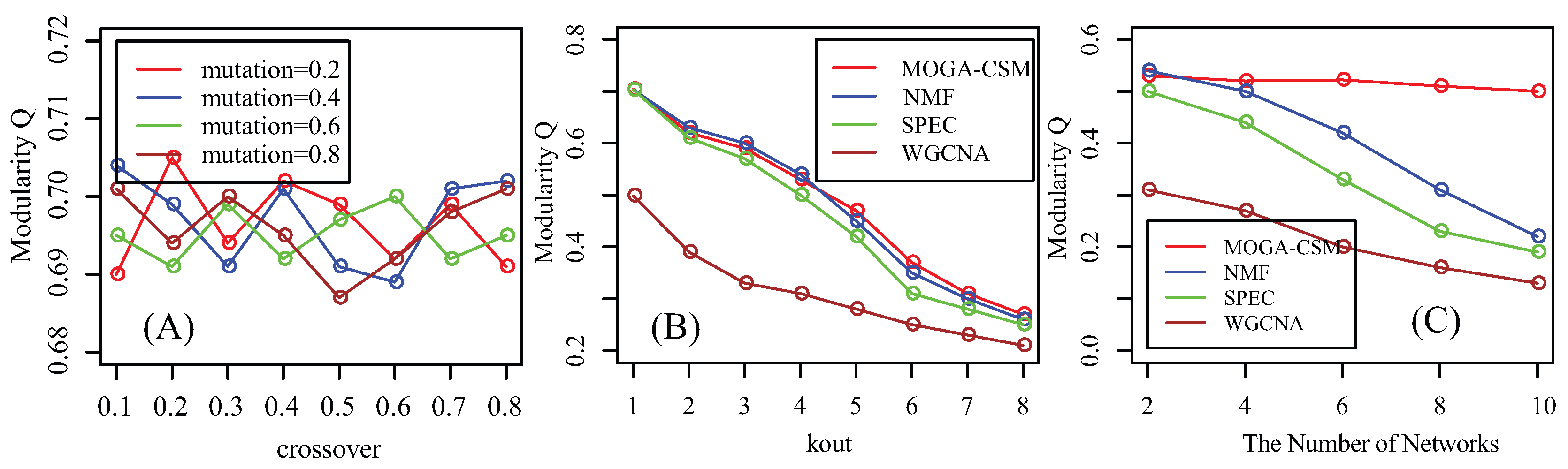
4.1. Benchmarking Performance of the Artificial Networks
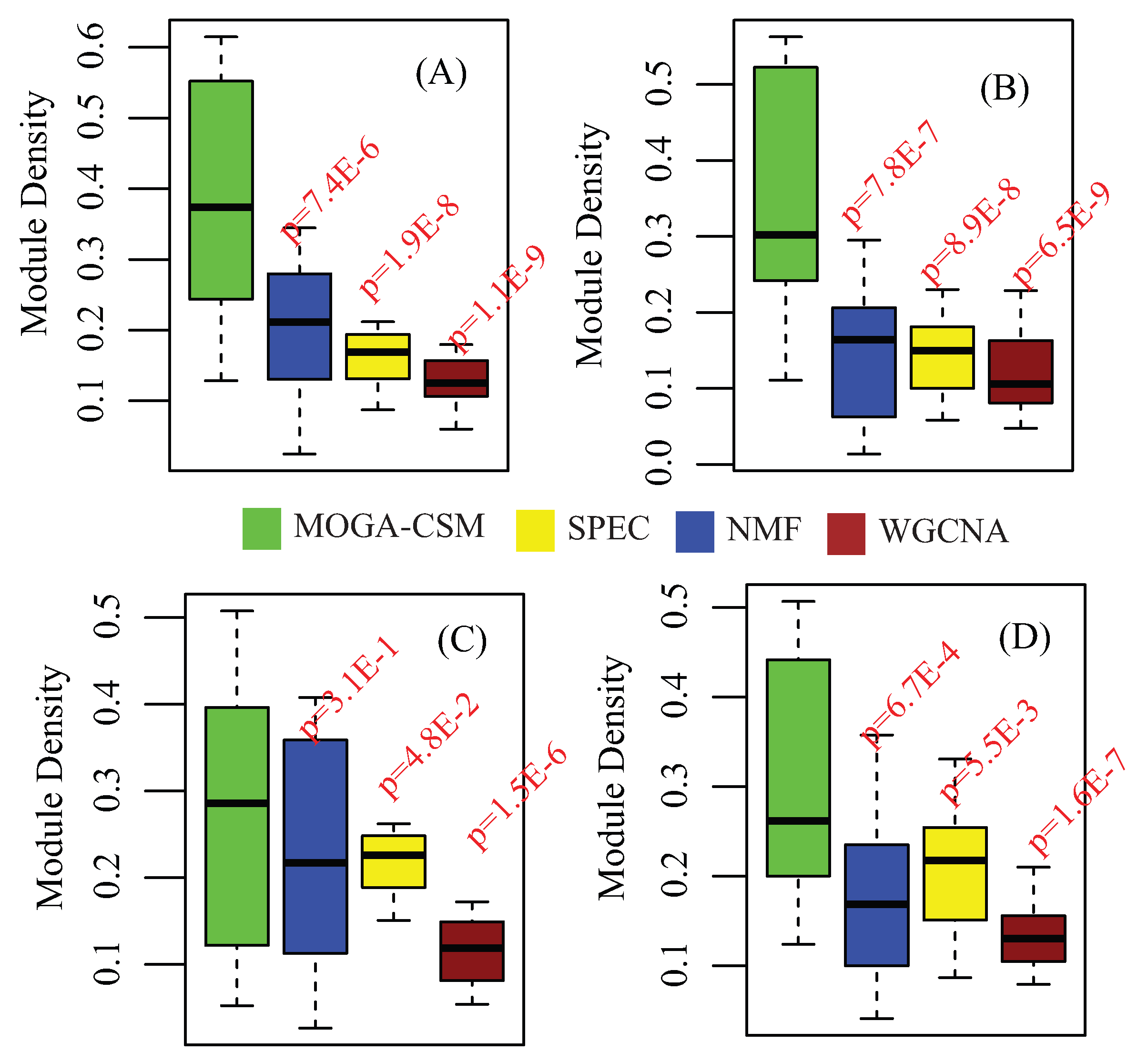
4.2. Benchmarking Performance of the Breast Cancer Networks
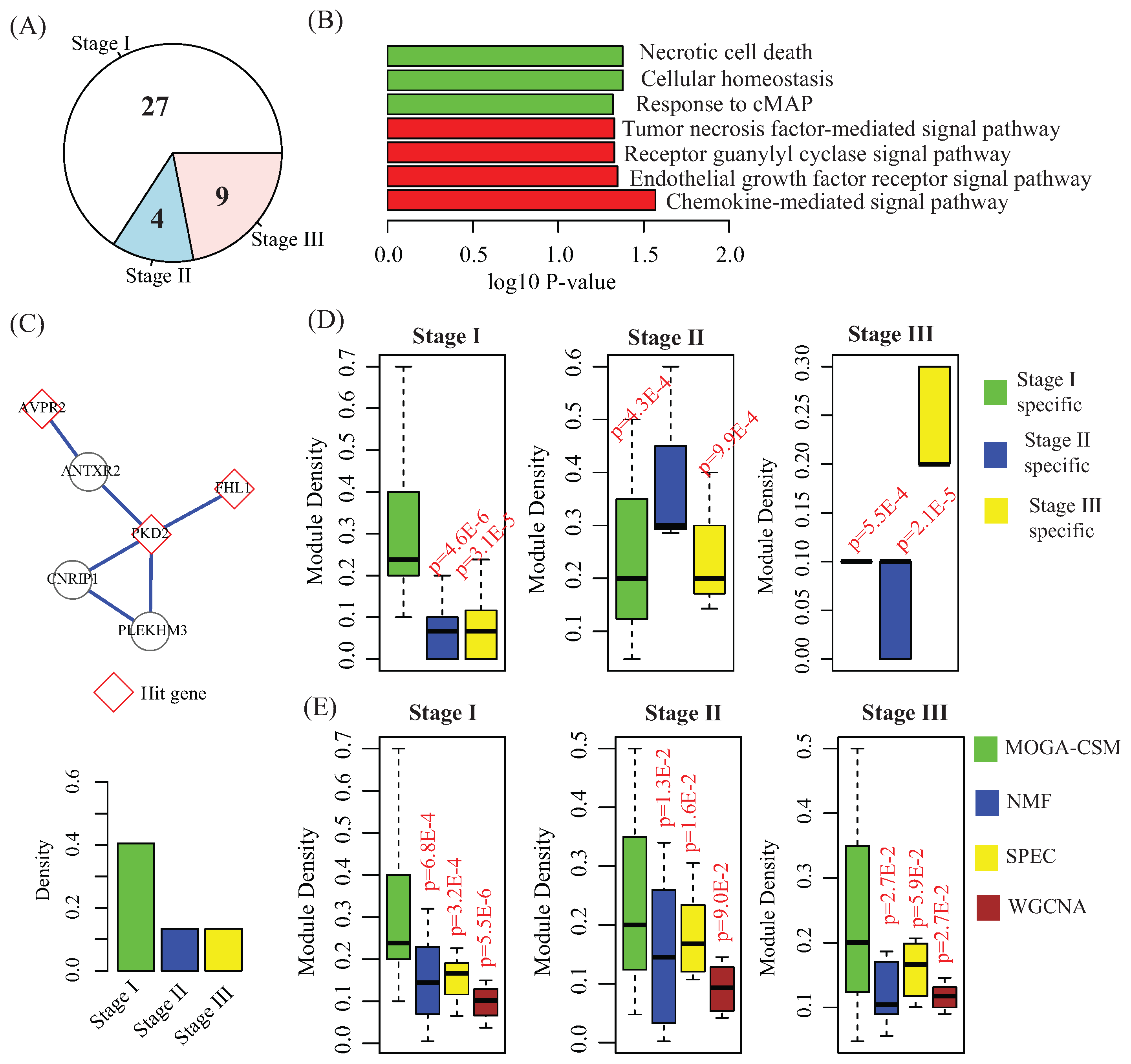
4.3. Stage-Specific Modules Serve as Biomarkers to Predict Breast Cancer Stages
4.4. Benchmarking Performance of Cancer Co-Methylation Networks
5. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Biotechnol. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signaling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Schwikowski, B.; Uetz, P.; Fields, S. A network of protein-protein interactions in yeast. Nat. Biotechnol. 2010, 18, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.-L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 841. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Guo, M.; Wang, C.; Liu, X.; Zou, Q. An overview of SNP interactions in genome-wide association studies. Brief. Funct. Genom. 2015, 14, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Taylor, I.W.; Linding, R.; Warde-Farley, D.; Liu, Y.; Pesquita, C.; Faria, D.; Bull, S.; Pawson, T.; Morris, Q.; Wrana, J.L. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009, 27, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Lee, E.; Liu, Y.; Lee, D.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center & Research Institute at Christiana Care Health Services; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed]
- De Lichtenberg, U.; Jensen, L.J.; Brunak, S.; Bork, P. Dynamic complex formation during the yeast cell cycle. Science 2005, 307, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Sharan, R.; Karp, R.M.; Sittler, T.; Root, D.E.; Stockwell, B.R.; Ideker, T. Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proc. Natl. Acad. Sci. USA 2003, 100, 11394–11399. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, U.; Miranda, M.; Suresh, S.; Davis, R.W.; St Onge, R.P. Multiplex assay for condition-dependent changes in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 9213–9218. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tang, W.; Wang, P.; Guo, X.; Gao, L. Extracting stage-specific and dynamic modules through analyzing multiple networks associated with cancer progression. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016. [Google Scholar] [CrossRef]
- Ma, X.; Gao, L.; Tan, K. Modeling disease progression using dynamics of module connectivity. Bioinformatics 2014, 30, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Gao, L.; Karamanlidis, G.; Gao, P.; Lee, C.F.; Garcia-Menendez, L.; Tian, R.; Tan, K. Revealing module dynamics in heart diseases by analyzing multiple differential networks. PLoS Comput. Biol. 2015, 11, e1004332. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Tibshirani, R.; Plevritis, S.K. Pancancer analysis of DNA methylation-driven genes using MethyMix. Genome Biol. 2015, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sun, P.; Qin, G. Identifying condition-specific modules by clustering multiple networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef]
- Kwok, N.W.; Shi, H.Y.; Ha, Q.; Jia, X. Simultaneous image color correction and enhancement using particle swarm optimization. Eng. Appl. Artif. Intell. 2013, 26, 2356–2371. [Google Scholar] [CrossRef]
- Mai, C.; Nguyen, M.T.T.; Kwok, N.W. A modified snsharp masking method using particle swarm optimization. In Proceedings of the 2011 4th International Conference on Image and Signal Processing, Shanghai, China, 15–17 October 2011; Volume 2, pp. 646–650. [Google Scholar]
- Fang, G.; Kwok, N.M.; Ha, Q. Automatic fuzzy membership function tuning using the particle swarm optimization. In Proceedings of the 2008 IEEE Pacific-Asia Workshop on Computational Intelligence and Industrial Application, Wuhan, China, 19–20 December 2008; Volume 2, pp. 324–328. [Google Scholar]
- Folino, F.; Pizzuti, C. An evolutionary multiobjective approach for community discovery in dynamic networks. IEEE Trans. Knowl. Data Eng. 2014, 26, 1838–1852. [Google Scholar] [CrossRef]
- Handl, J.; Kell, D.B.; Knowles, J. Multiobjective optimization in Bioinformatics and Computational Biology. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E 2004, 77, 026113. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.H. Adaption in Natural and Artificial Systems; University of Michigan Press: Ann Harbor, MI, USA, 1975. [Google Scholar]
- Sriniva, N.; Deb, K. Multi-objective optimization using nondominated sorting in genetic algorithms. Evol. Comput. 1994, 2, 221–248. [Google Scholar] [CrossRef]
- Gu, X.; Yu, Y.; Li, J.; Li, Y.; Alamdari, M.M. Semi-active storey isolation system employing MRE isolator with parameter identification based on NSGA-II with DCD. Earthq. Struct. 2016, 11, 1101–1121. [Google Scholar] [CrossRef]
- Deb, K. Multi-Objective Optimization Using Evolutionary Algorithms; Wiley: Chichester, UK, 2001. [Google Scholar]
- Pizzuti, C. A multi-objective genetic algorithm to find communities in complex networks. IEEE Trans. Evol. Comput. 2012, 3, 418–430. [Google Scholar] [CrossRef]
- Handle, J.; Knowles, J. An evolutionary genetic algorithm to multi-objective clustering. IEEE Trans. Evol. Comput. 2007, 11, 56–76. [Google Scholar] [CrossRef]
- Park, Y.; Song, M. A genetic algorithm for clustering problems. In Proceedings of the 3rd International Conference on Genetic Algorithms, Madison, WI, USA, 22–25 July 1998; pp. 2–9. [Google Scholar]
- Cormen, T.H.; Leiserson, C.E.; Rivest, R.L.; Stein, C. Introduction to Algorithms, 3rd ed.; MIT Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methadol. 1995, 57, 289–300. [Google Scholar]
- Reverter, A.; Chan, E.K. Combining partial correlation and an information theory approach to the reverse engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E 2006, 74, 036104. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yu, L.; Wang, P.; Yang, X. Discovering DNA methylation patterns for long non-coding RNAs associated with cancer subtypes. Comput. Biol. Chem. 2017, 69, 164–170. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
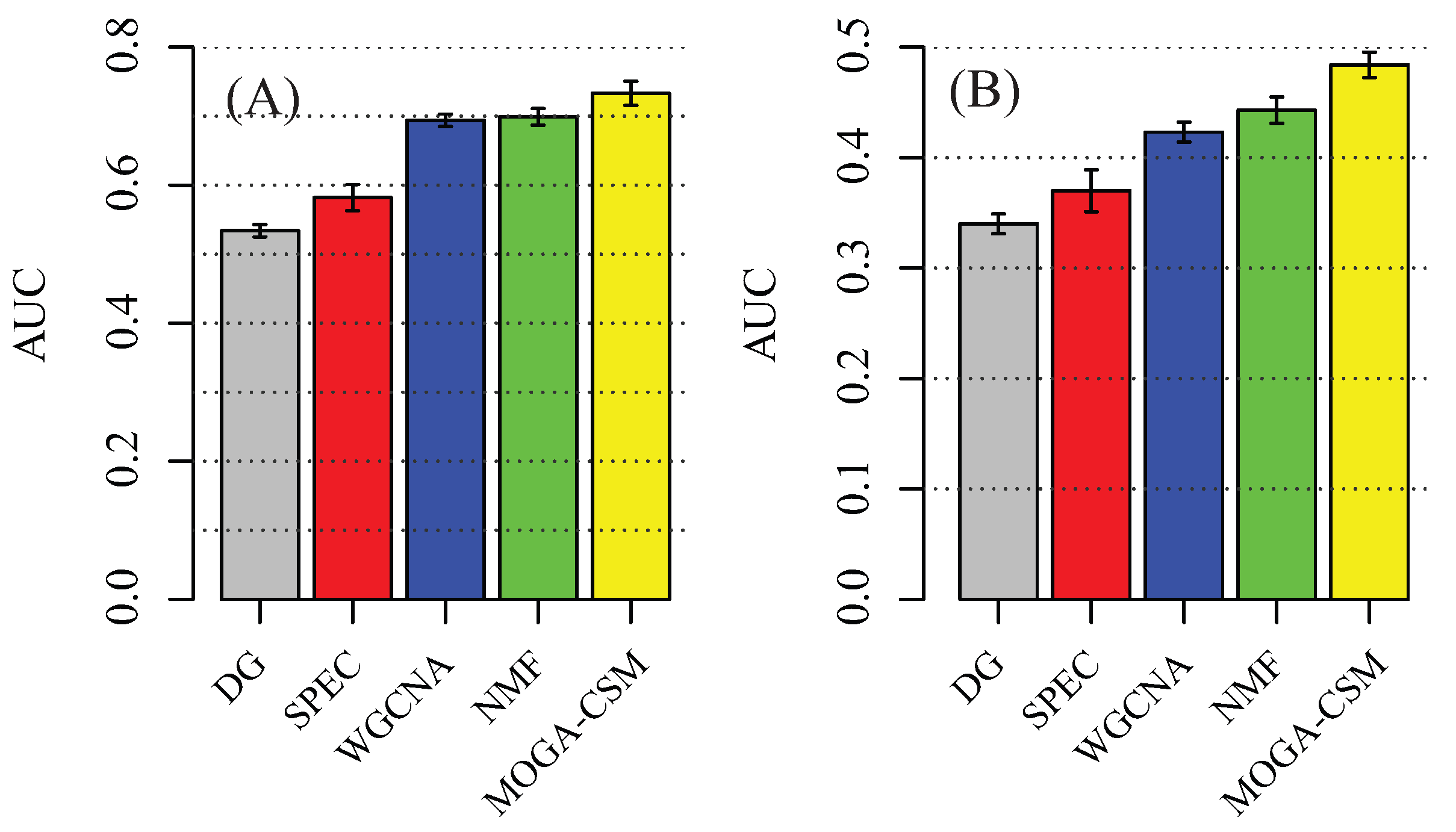

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Sun, P.; Zhao, J. Multi-Objective Optimization Algorithm to Discover Condition-Specific Modules in Multiple Networks. Molecules 2017, 22, 2228. https://doi.org/10.3390/molecules22122228
Ma X, Sun P, Zhao J. Multi-Objective Optimization Algorithm to Discover Condition-Specific Modules in Multiple Networks. Molecules. 2017; 22(12):2228. https://doi.org/10.3390/molecules22122228
Chicago/Turabian StyleMa, Xiaoke, Penggang Sun, and Jianbang Zhao. 2017. "Multi-Objective Optimization Algorithm to Discover Condition-Specific Modules in Multiple Networks" Molecules 22, no. 12: 2228. https://doi.org/10.3390/molecules22122228
APA StyleMa, X., Sun, P., & Zhao, J. (2017). Multi-Objective Optimization Algorithm to Discover Condition-Specific Modules in Multiple Networks. Molecules, 22(12), 2228. https://doi.org/10.3390/molecules22122228