Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers



1
Institute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
2
Research Group of Stereochemistry of the Hungarian Academy of Sciences, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
*
Authors to whom correspondence should be addressed.
Molecules 2017, 22(12), 2211; https://doi.org/10.3390/molecules22122211
Submission received: 3 November 2017
/
Revised: 6 December 2017
/
Accepted: 8 December 2017
/
Published: 13 December 2017
(This article belongs to the Section Organic Chemistry)
Abstract
:Efficient enzymatic resolutions are reported for the preparation of new eight-membered ring-fused enantiomeric β-amino acids [(1R,2S)-9 and (1S,2R)-9] and β-lactams [(1S,8R)-3, (1R,8S)-3 (1S,8R)-4 and (1R,8S)-7], through asymmetric acylation of (±)-4 (E > 100) or enantioselective hydrolysis (E > 200) of the corresponding inactivated (±)-3 or activated (±)-4 β-lactams, catalyzed by PSIM or CAL-B in an organic solvent. CAL-B-catalyzed ring cleavage of (±)-6 (E > 200) resulted in the unreacted (1S,8R)-6, potential intermediate for the synthesis of enantiomeric anatoxin-a. The best strategies, in view of E, reaction rate and product yields, which underline the importance of substrate engineering, are highlighted.
1. Introduction
Chiral β-amino acids and β-lactams are important compounds because of their pharmacological properties and their use in diverse syntheses. For example, (1R,2S)-2-aminocyclopentanecarboxylic acid (cispentacin) [1], the simplest, naturally occurring carbocyclic β-amino acid, is an antifungal antibiotic, but can also be found in the structures of some natural products, e.g., amipurimycin [2]. Its methylene derivative, (1R,2S)-2-amino-4-methylenecyclopentanecarboxylic acid (icofungipen, PLD-118) [3], in turn, is active in vitro against Candida species. Enantiomeric eight-membered ring-fused β-lactams (1R,8S)- and (1S,8R)-9-azabicyclo[6.2.0]dec-4-en-10-one are potential key intermediates [4] in the syntheses of anatoxin-a [5]. It is a neurotoxic alkaloid, one of the most toxic of the cyanobacterial toxins, but a potent and stereospecific agonist at nicotinic acetylcholine receptors [5,6,7]. A relatively large number of publications deal with its isolation from strains of Anabaena flos aqua, a freshwater blue-green alga, but synthetic methods [8,9,10], also for the preparation of new anatoxin-a homologues [11] have also been described. Cyclic β-amino acids can serve as building blocks for the synthesis of modified peptides with increased activity and stability [12] and with well-defined three-dimensional structures (foldamers) (e.g., β-peptides with possible antibiotic activity) similar to those of natural peptides [13,14]. Additionally, the alkene functionality in molecules is amenable to a range of transformations. Cyclic β-amino acids can also be used in heterocyclic [15,16] and combinatorial [17] chemistry.
In addition to conventional resolution methods for the preparation of enantiomeric β-amino acids and β-lactams, enzymatic strategies have also been described [18,19,20]. Our research group has also devised a number of efficient enzymatic kinetic and sequential kinetic resolution processes (acylations, deacylations and hydrolyses). Most of these methods have been reviewed [21,22,23].
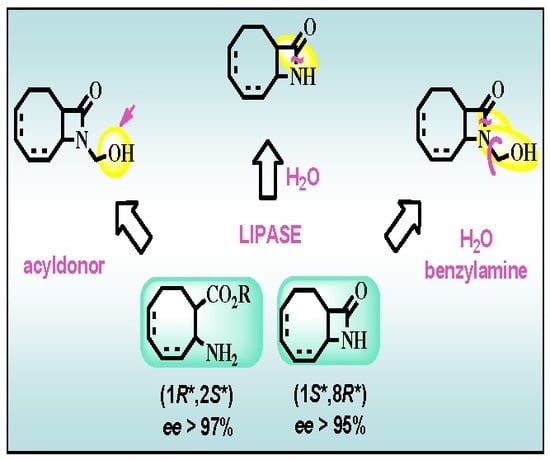
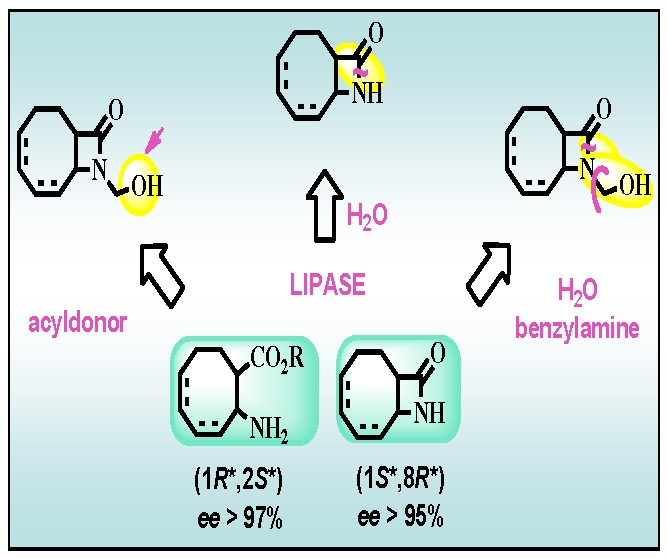
A primary aim of this work was to devise adequate enzymatic strategies for the preparation of valuable new enantiomeric eight-membered carbocyclic β-lactam and β-amino acid derivatives. In view of earlier results on enzymatic acylation of N-hydroxymethyl eight-membered carbocyclic β-lactams [4,24], we first planned to carry out enzymatic acylation of N-hydroymethyl-9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4] (Figure 1). The extensive investigations of the lipase-catalyzed ring cleavage of unactivated [25,26,27] and activated [28] β-lactams then suggested the possibility of the lipase-catalyzed enantioselective ring cleavage of racemic unactivated 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-3] and activated (±)-4 and N-hydroxymethyl 9-azabicyclo[6.2.0]dec-4-en-10-one [(±)-6]. A systematic comparison of the efficiency of these methods, with regard to E, reaction rate and yield for the products, was also intended.
2. Results and Discussion
2.1. Synthesis of β-lactams (±)-3–(±)-6
Lactams (±)-3 and (±)-5 were synthesised from cyclooctadiene 1 or 2 by the addition of chlorosulfonyl isocyanate (CSI), according to a slightly modified literature procedure (Scheme 1) [29,30]. Then product lactams were reacted with paraformaldehyde under sonication to form N-hydroxymethyl β-lactams (±)-4 and (±)-6 [4].
2.2. Lipase-Catalyzed O-acylation of (±)-4
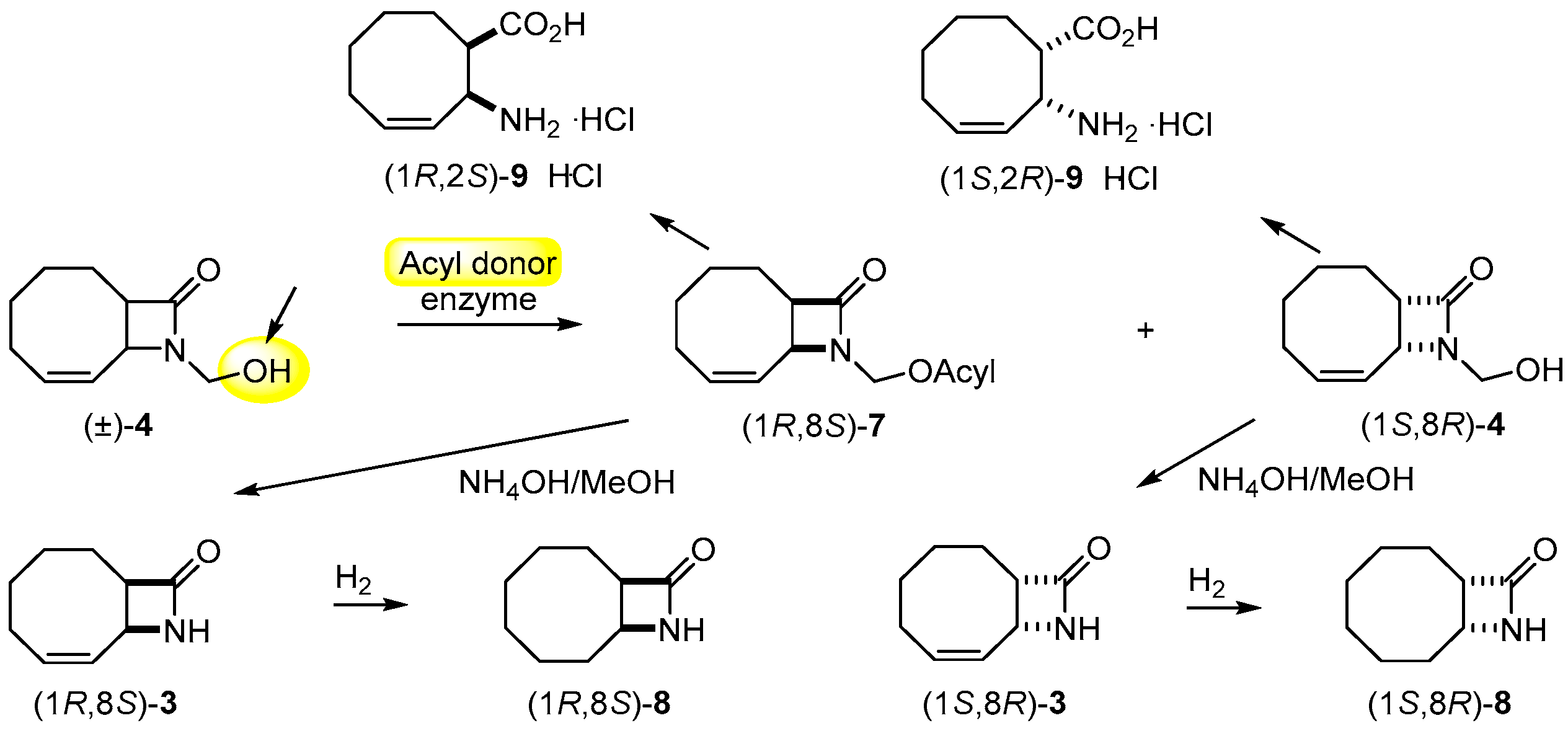
On the basis of the earlier results on the enzymatic resolution of N-hydroxymethyl 9-azabicyclo[6.2.0]dec-4-en-10-one [4] and N-hydroxymethyl 9-azabicyclo[6.2.0]decane-4-en-10-one [24], first the acylation of (±)-4 (Scheme 2) was carried out with vinyl butyrate (VB) in the presence of PSIM (Burkholderia cepacia) in diisopropyl ether (iPr2O) at −15 °C (Table 1, entry 1).
When the acylation was performed at higher temperatures, the reaction rate increased with the concomitant decrease in E (entries 2 and 3 vs. 1). As the amount of VB was increased from 2 to 10 equiv., the reaction rate increased while E apparently decreased (entry 4 vs. 3). The addition of a catalytic amount of Et3N and Na2SO4 resulted in a clear increase in E (entry 5 vs. 4).
To further increase the E values, acyl donors, such as 2,2,2-trifluoroethyl butyrate, vinyl acetate (VA), ethyl acetate (EtOAc) and acetic anhydride (Ac2O) were tested (entries 6–9). Unfortunately, none of the acyl donors tested exerted any beneficial influence on the reaction course. Several solvents have also been tested. When iPr2O was replaced by tBuOMe, practically no change was observed in the reaction course (entry 15 vs. 4). The same high E but somewhat lower reaction rate was observed in acetone (entry 17), while the best E and fastest reaction were detected in toluene (entry 16). Finally the environmentally less harmful iPr2O was selected as solvent.
In addition to lipase PSIM, several enzymes, such as lipase AK (Pseudomonas fluorescens), lipase AY (Candida rugosa), CAL-A (lipase A from Candida antarctica), CAL-B (lipase B from Candida antarctica) and PPL (porcine pancreatic lipase) have been investigated (entries 10–14). However, in terms of E and reaction rate, none of them proved to be better than PSIM.
2.3. Lipase-Catalyzed Ring Cleavage of (±)-3–(±)-6
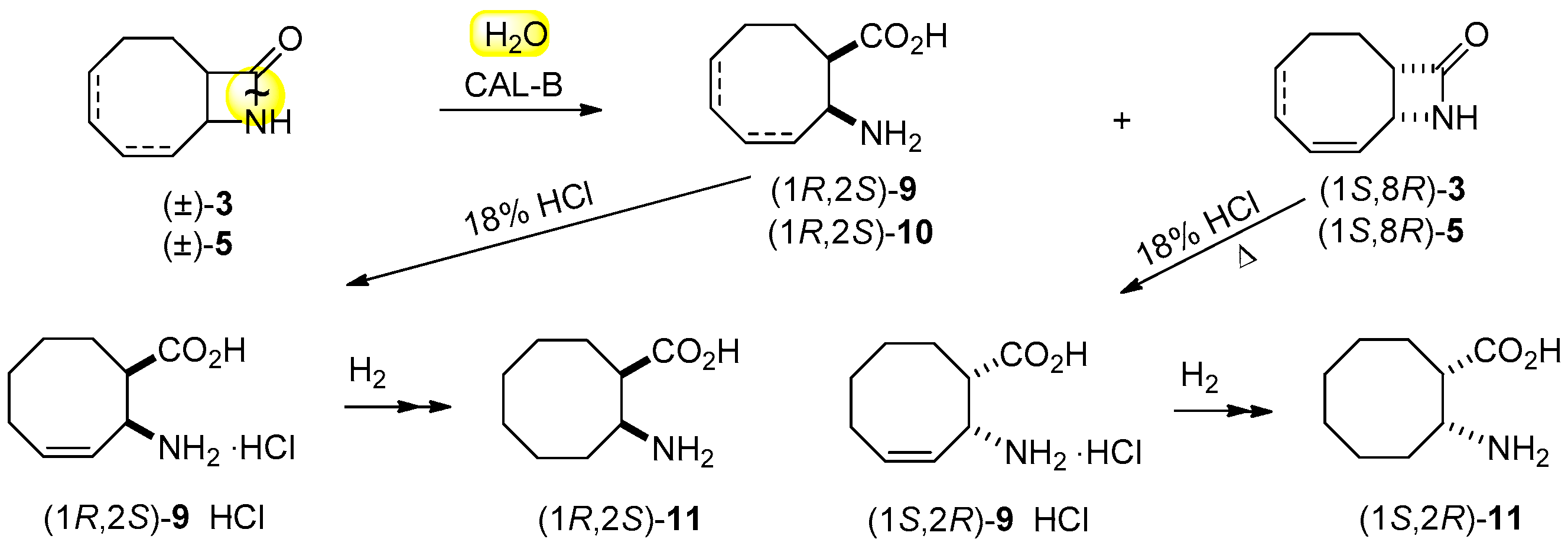
In view of the results on the lipase-catalyzed ring cleavage of carbocyclic β-lactams [25,26,27], the ring opening of (±)-3 was attempted with 1 equiv. of H2O in the presence of CAL-B (lipase B from Candida antarctica) in iPr2O at 60 °C (Scheme 3; Table 2, entry 1). When the ring cleavage of the regioisomeric 9-azabicyclo[6.2.0]dec-4-en-10-one [(±)-5] was performed under the same conditions, a similar fast reaction and the same high ee values (>98%) were observed (entry 2).
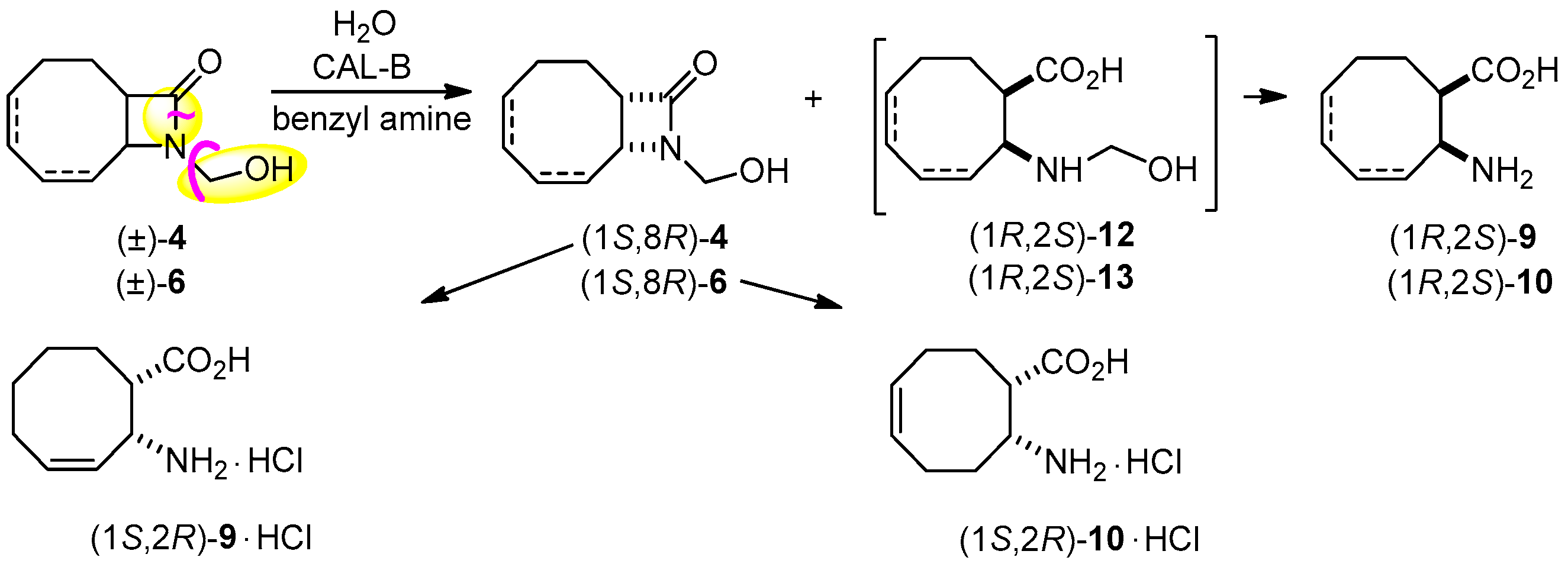
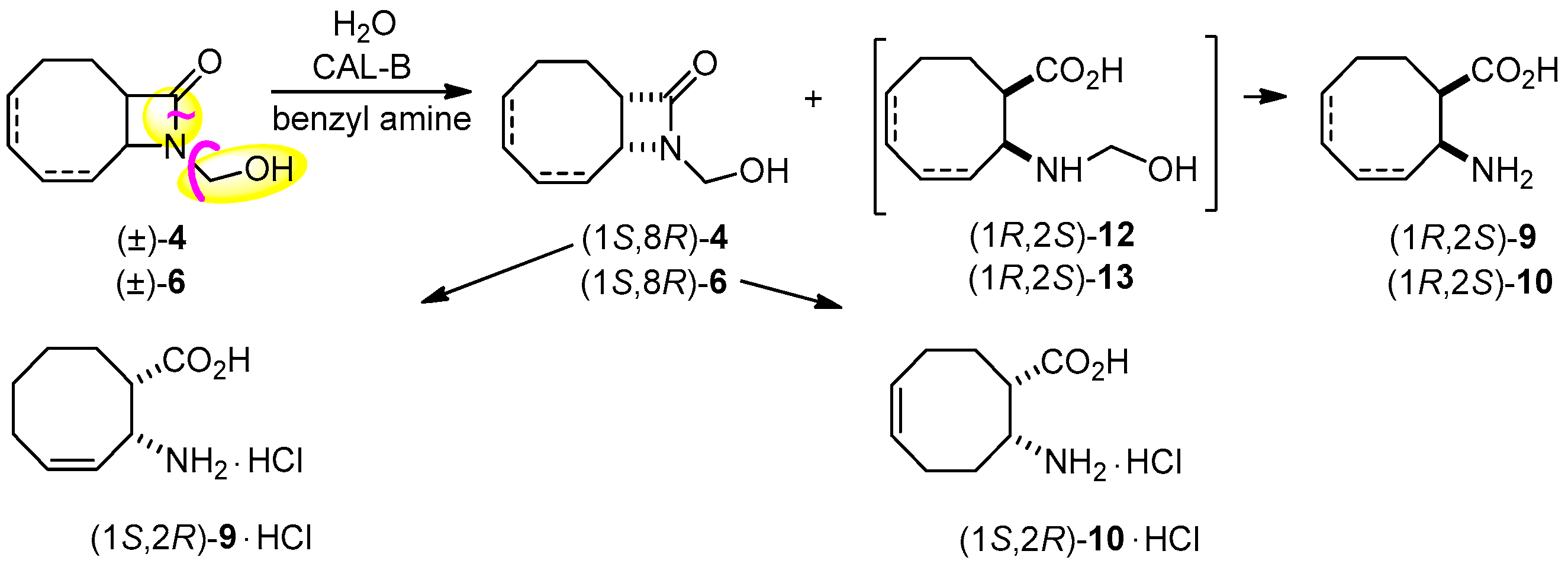
In further studies, we have probed our very recent results found about the ring cleavage of specially activated lactams [28], where the activating group underwent to a traceless, in situ degradation. Accordingly, the ring cleavage of (±)-4 was attempted with H2O in the presence of CAL-B and benzylamine to capture formaldehyde in iPr2O at 60 °C (Scheme 4, Table 2, entry 3). Excellent ee (>99%) characterized formed amino acid (1R,2S)-9 at a conversion close to 50%. The ring cleavage of regioisomeric (±)-6 was also carried out under the same conditions (entry 4), and the same high ee (>98%) for amino acid (1R,2S)-10 and unreacted lactam (1S,8R)-6, potential intermediate in the synthesis of enantiomeric anatoxin-a was observed. In good accordance with the earlier observation that the hydroxymethyl group activates the ring cleavage of lactams, racemic 4 and 6 underwent ring cleavage much faster than their corresponding inactivated counterparts (entry 3 vs. 1 and 4 vs. 2).
2.4. Further Transformations
Hydrolysis of enantiomeric 3, 4, 6 and 7 with 18% aqueous HCl (Scheme 2, Scheme 3 and Scheme 4) gave the corresponding hydrochloride salts 9·HCl and 10·HCl (ee > 97%), while treatment of enantiomeric 4 and 7 with NH4OH/MeOH (Scheme 2) resulted in β-lactams (1R,8S)-3 and (1S,8R)-3 (ee ≥ 95%). Catalytic reduction of enantiomeric lactams 3 and amino acids 9 with H2 or cyclohexene as a hydrogen donor and using palladium-on-carbon furnished saturated lactams (1R,8S)-8 and (1S,8R)-8, and amino acids (1R,2S)-11 and (1S,2R)-11, respectively (Scheme 2 and Scheme 3), without a drop in ee (>96%). Physical data on the enantiomers prepared are reported in Table 4 and Experimental Section.
2.5. Absolute Configurations
Absolute configurations were determined by comparing the [α] values of the enantiomeric compounds with literature data. Specifically, [α] values of enantiomeric compounds 8 (Table 4, entries 3 and 4), obtained from enantiomeric compounds 4 and 7 (Scheme 2), were compared with the literature data of (1S,8R)-9-azabicyclo[6.2.0]decan-10-one { = −18 (c = 0.5; CHCl3)} [25]. In a similar way, the [α] values of enantiomeric compounds 11 (Table 4, entries 9 and 10), obtained from enantiomeric compounds 9·HCl (Scheme 3), were compared with the literature data for (1R,2S)-2-aminocyclooctane-1-carboxylic acid { = +17.8 (c = 0.4; H2O)} [25]. Thus, the (1R,8S) configuration is determined for ester 9 and the corresponding lactam (3), and the (1S,8R) configuration is assigned to unreacted alcohol 4 and the corresponding lactam (3). The CAL-B-catalyzed ring cleavage of either inactivated lactams 3 and 5 or activated lactams 4 and 6 afforded the corresponding amino acids with (1R,2S) absolute configuration I.
3. Experimental Section
3.1. Materials and Methods
CAL-B (lipase B from Candida antarctica), produced by the submerged fermentation of a genetically modified Aspergillus oryzae microorganism and adsorbed on a macroporous resin (Catalogue no. L4777), and porcine pancreatic lipase (PPL) were from Sigma-Aldrich (St. Louis, MO, USA). CAL-A (lipase A from Candida antarctica) was purchased from Roche Diagnostics Corporation. Lipase PSIM (Burkholderia cepacia, immobilized on diatomaceous earth) was a kind gift from Amano Enzyme Europe Ltd. (Suqian, China) Lipase AK (Pseudomonas fluorescens) was from Amano Pharmaceuticals, and lipase AY (Candida rugosa) was from Fluka. The solvents were of the highest analytical grade. Melting points were determined with a Kofler apparatus. NMR spectra were recorded on a Bruker DRX 400 spectrometer. Optical rotations were measured with a Perkin-Elmer 341 polarimeter. Elemental analyses were performed with a Perkin-Elmer CHNS-2400 Ser II Elemental Analyzer. The syntheses of racemic N-hydroxymethyl 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-5] and N-hydroxymethyl 9-azabicyclo[6.2.0]dec-4-en-10-one [(±)-6] were performed according to our earlier method [4].
3.2. Typical Small-Scale Enzymatic Experiments
Racemic β-lactam in an organic solvent (0.05 M solution, 1 mL) was added to the lipase tested (30 mg mL−1). The tested acyl donor (2 or 10 equiv.) in acylations or H2O (1 equiv.) in hydrolyses was added. The mixture was shaken at −15 °C, 2–3 °C, 30 °C or 60 °C. The progress of the reaction was followed by taking samples from the reaction mixture at intervals and analysing them by using a gas chromatograph equipped with a chiral column. The ee values for the unreacted β-lactam enantiomers were determined directly on a Chromopack Chiralsil-Dex CB column [retention times are given in min; 140 °C for 25 min → 190 °C (temperature rise 20 °C∙min−1; 140 kPa, (1S,8R)-3: 27.74 (antipode: 27.56); 140 °C for 7 min → 190 °C (temperature rise 10 °C min−1; 140 kPa, (1S,8R)-5: 12.55 (antipode: 12.12)] or on a Chirasil-L-Val column, [140 °C for 7 min → 190 °C (temperature rise 10 °C/min; 140 kPa), (1S,8R)-4: 14.39 (antipode: 14.61); (1S,8R)-6: 14.40 (antipode: 14.87); (1R,8S)-7: 14.67 (antipode: 14.36)]. The ee values for the β-amino acids produced were determined on a Chirasil-L-Val column, after double derivatization with (i) CH2N2 [31] (caution! derivatization with diazomethane should be performed under a well-ventilated hood); (ii) Ac2O in the presence of 4-dimethylaminopyridine and pyridine [120 °C for 7 min → 190 °C (temperature rise 10 °C min−1; 140 kPa, (1R,2S)-9: 12,41 (antipode: 12.95); (1R,2S)-10: 12.88 (antipode: 14.06)] and after double derivatization with (i) CH2N2 and (ii) (PropCO)2O in the presence of 4-dimethylaminopyridine and pyridine [140 °C for 10 min → 190 °C (temperature rise 10 °C min−1; 140 kPa; (1R,2S)-11: 17.58 (antipode: 17.78)].
3.3. Synthesis of Racemic 9-Azabicyclo[6.2.0]dec-6-en-10-one [(±)-3]
A solution of CSI (2.28 mL, 26.2 mmol) in CH2Cl2 (25 mL) was added dropwise to a stirred solution of 1,3-cyclooctadiene (3 mL, 26.2 mmol) in CH2Cl2 (25 mL) at 0 °C. The reaction mixture was stirred at room temperature for 21 h, and the resulting liquid was then added dropwise to a vigorously stirred solution of Na2SO3 (0.33 g) and K2CO3 (7.12 g) in H2O (50 mL). After stirring the mixture for 3.5 h, the combined organic layers were dried (Na2SO4) and, after filtration, concentrated. The resulting white solid, racemic 3 (3.41 g, 86%), was recrystallized from iPr2O (m.p. 103–106 °C).
The 1H-NMR (400 MHz, CDCl3, 25 °C, TMS) δ (ppm) data for (±)-3: 1.46–2.11 (8H, m, 4 × CH2), 3.34–3.38 (1H, dd, J = 5.34, 12.26 Hz, H-1), 4.57–4.58 (1H, m, H-8), 5.41–5.79 (2H, m, CHCH), 6.01 (1H, bs, NH). Anal. calcd for C9H14NO: C, 71.49; H, 8.67; N, 9.26; Anal. found: C, 71. 30; H, 8.52; N, 9.23.
3.4. Synthesis of Racemic N-Hydroymethyl-9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4]
Racemic 3 (3 g, 19.84 mmol) was dissolved in THF (35 mL) followed by adding paraformaldehyde (0.6 g, 19.84 mmol), K2CO3 (0.17g, 1.19 mmol) and H2O (1.1 mL). The solution was sonicated for 4 h, the solvent was evaporated off and the residue was dissolved in ethyl acetate (50 mL). The solution was dried (Na2SO4) and then concentrated. The residue was recrystallised from iPr2O to afford (±)-4 as a white crystalline product (3.2 g, 89%; m.p. 81–84 °C).
The 1H-NMR (400 MHz, CD3OD, 25 °C, TMS) δ (ppm) data for (±)-4: 1.45–2.09 (8H, m, 4 × CH2), 3.30–3.32 (1H, dd, J = 1.89. 5.52 Hz, H-1), 3.33-3.39 (1H, s, OH), 4.60–4.64 (1H, dd, J = 7.78, 11,58 Hz, CH2OH), 4.69–4.71 (1H, m, H-8), 4.85–4.89 (1H, dd, J = 6.47, 11,66 Hz, CH2OH), 5.57–5.60 (2H, m, CHCH). Anal. calcd. for C10H15NO2: C, 66.27; H, 8.34; N, 7.73; found: C, 66.19; H, 8.44; N, 7.76.
3.5. Gram-Scale Resolution of N-hydroxymethyl-9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4] through Acylation
Racemic 4 (1 g, 5.51 mmol) was dissolved in iPr2O (40 mL). Lipase PSIM (1.2 g, 30 mg mL−1) then VB (6.29 mL, 55.1 mmol), Et3N (a few drops) and Na2SO4 (0.2 g) were added and the mixture was shaken at 30 °C. After 10 min, the enzyme was filtered off at 51% conversion and iPr2O was evaporated. The residue was chromatographed on silica (EtOAc:n-hexane 7:3) affording unreacted (1S,8R)-4 [0.44 g, 44%; [α]D25 = –142.4 (c = 0.45, EtOH); m.p. 79–83 °C (recrystallized from iPr2O); ee = 96%] and ester (1R,8S)-7 (0.63 g, 46%; = +39.2 (c = 35, EtOH); ee = 94%) as a pale-yellow oil.
The 1H-NMR (400 MHz, CDCl3, 25 °C, TMS) δ (ppm) data for (1R,8S)-7: 0.93–0.97 (3H, t, J = 7.4 Hz, CH3), 1.41–1.44 (2H, m, CH2CH2CH3), 1.57–2.33 (10H, m, CH2CH2CH3 and 4 × CH2 from ring), 3.31–3.32 (1H, m, H-1), 4.59–4.60 (1H, dd, J = 1.58, 3.88 Hz, H-8), 5.15–5.18 (1H, d, J = 11.36 Hz, CH2OCOPr), 5.20–5.23 (1H, d, J = 11.36 CH2OCOPr), 5.49–5.81 (2H, m, CHCH). Anal. calcd. for C14H21NO3: C, 66.91; H, 8.42; N, 5.57; found: C, 66.83; H, 8.60; N, 5.42.
The 1H-NMR (400 MHz, CDCl3, 25 °C, TMS) δ (ppm) data for (1S,8R)-4 are similar to those for (±)-4. Anal. found: C, 66.39; H, 8.24; N, 7.73.
3.6. Gram-Scale Resolution of 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-3] through Hydrolysis
Racemic 3 (0.5 g, 3.3 mmol) was dissolved in iPr2O (20 mL). CAL-B (0.6 g, 30 mg mL−1) and H2O (29.7 μL, 1.65 mmol) were added, and the mixture was shaken in an incubator shaker at 60 °C for 5.5 h. The reaction was stopped by filtering off the enzyme at 50% conversion. The solvent was evaporated off and the residue crystallized to give (1S,8R)-3 [240 mg, 48%; = −140.6 (c = 0.5; EtOH); m.p. 151–153 °C recrystallized from iPr2O; ee = 99%]. The filtered-off enzyme was washed with distilled water (3 × 15 mL), and the water was evaporated off, yielding crystalline β-amino acid (1R,2S)-9 [268 mg, 48%; = −17 (c = 0.35; H2O); 266–288 °C with sublimation (recrystallized from H2O/acetone); ee = 99%].
The 1H-NMR (400 MHz, CD3OD, 25 °C, TMS) δ (ppm) data for (1S,8R)-3 were similar to those for (±)-3. Anal. found: C, 71.33; H, 8.57; N, 9.10.
The 1H-NMR (400 MHz, D2O) δ (ppm) data for (1R,2S)-9: 1.45–2.24 (8H, m, 4 × CH2) 2.86–2.90 (1H, m, H-1) 4.42–4.45 (1H, m, H-2) 5.71–6.03 (2H, m, CHCH). Anal. calcd. for C9H15NO2: C, 63.88; H, 8.93; N, 8.28; found: C, 63.99; H, 8.84; N, 8.28.
3.7. Gram-Scale Resolution of N-hydroxymethyl 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4] through Hydrolysis
(±)-4 (250 mg, 1.37 mmol) was dissolved in iPr2O (10 mL) followed by the addition of CAL-B (300 mg, 30 mg mL−1), benzylamine (150 μL, 1.37 mmol) and H2O (12.3 μL, 0.68 mmol) and by shaking the mixture in an incubator shaker at 60 °C for 3 h. The reaction was stopped by filtering off the enzyme at 49% conversion. After solvent evaporation the residue (1S,8R)-4 crystallized out [120 mg, 48%; = −140.4 (c = 0.5; EtOH), m.p. 80–83 °C (recrystallized from iPr2O), ee = 98%]. The filtered enzyme was washed with distilled H2O (3 × 15 mL), and crystalline β-amino acid (1R,2S)-9 was isolated after evaporation of H2O. [109 mg, 47%; = −17.1 (c = 0.35; H2O), m.p. 264–265 °C (recrystallised from H2O/acetone), ee > 99%].
The 1H-NMR (400 MHz, CD3OD, 25 °C, TMS) δ (ppm) data for (1S,8R)-4 were similar to those for (±)-4. Anal. found: C, 66.18; H, 8.35; N, 7.73.
The 1H-NMR (400 MHz, D2O) data δ (ppm) for (1R,2S)-9 from (±)-4 were similar to those for (1R,2S)-9 from (±)-3 (4.4.). Anal. found: C, 63.95; H, 9.01; N, 8.25.
3.8. Gram-Scale Resolution of N-hydroxymethyl 9-azabicyclo[6.2.0]dec-4-en-10-one [(±)-6] through Hydrolysis
Via the procedure described above, the reaction of racemic 6 (250 mg, 1.37 mmol), benzylamine (150 μL, 1.37 mmol) and H2O (12.3 μL, 0.68 mmol) in iPr2O (10 mL) in the presence of CAL-B (300 mg, 30 mg mL−1) at 60 °C afforded amino acid (1R,2S)-10 [108 mg, 47%; = +24.9 (c = 0.3; H2O), lit. [26] = +23.9 (c = 0.3; H2O), ee = 95%; m.p.: 264–265 °C (recrystallized from H2O/acetone), lit. [26] 218–220 °C; ee = 99%] and unreacted (1S,8R)-6, [115 mg, 46%; = −28.7 (c = 0.5; EtOH), = −28.5 (c = 1; MeOH), lit. [4] = −27.0 (c = 1; MeOH), ee = 97%; m.p. 48–49 °C (recrystallized from n-hexane), lit. [4] 48–49 °C; ee = 99%] after 3 h.
The 1H-NMR (400 MHz, D2O) δ (ppm) data for (1R,2S)-10: 1.81–2.58 (8H, m, 4 × CH2) 2.79 (1H, m, H-1) 3.70–3.73 (1H, m, H-2) 5.72–5.73 (2H, m, CHCH). Anal. calcd. for C9H15NO2: C, 63.88; H, 8.93; N, 8.28; found: C, 63.59; H, 8.88; N, 8.18.
The 1H-NMR (400 MHz, CDCl3, 25 °C, TMS) data δ (ppm) for (1S,8R)-6 were similar to those for (±)-6: 1.86–2.21 and 2.37–2.44 (8H, m, 4 × CH2), 3.28–3.30 (1H, m, H-1), 3.71 (1H, bs, OH), 3.93–3.96 (1H, m, H-8), 4.45–4.48 (1H, d, J = 11.6 Hz, CH2OH), 4.73–4.79 (1H, d, J = 11.6 Hz, CH2OH), 5.67–5.71 (2H, m, CHCH). Anal. calcd. for C10H15NO2: C, 66.27; H, 8.34; N, 7.73; found: C, 66.10; H, 8.28; N, 7.66.
3.9. Synthesis of (1R,8S)-8 and (1S,8R)-8 through (1R,8S)-3 and (1S,8R)-3 from β-lactams (1S,8R)-4 and (1R,8S)-7
Ester (1R,8S)-7 (100 mg, 0.66 mmol) was dissolved in MeOH (10 mL), NH4OH (1 mL) was added and the mixture was stirred at room temperature for 6 h. The solvent was evaporated off, the residue chromatographed on silica (EtOAc:n-hexane 7:3) providing white crystals of (1R,8S)-3 [48 mg, 80%; = +147 (c = 0.5; EtOH); m.p. 152–153 °C (recrystallised from iPr2O); ee = 95%].
Similarly, (1S,8R)-4 (100 mg, 0.55 mmol) afforded white crystals of (1S,8R)-3 [59 mg, 71%; = −148.7 (c = 0.4; EtOH); m.p. 150–153 °C (recrystallised from iPr2O); ee = 96%].
The 1H-NMR (400 MHz, CD3OD, 25 °C, TMS) δ (ppm) data for (1S,8R)-3 and (1R,8S)-3 were similar to those for (±)-3. Anal. found for (1S,8R)-3: C, 71.45; H, 8.57; N, 9.22. Anal. found for (1R,8S)-3: C, 71.55; H, 8.67; N, 9.12.
Palladium-on-carbon (100 mg) was added to (1R,8S)-3 or (1S,8R)-3 (100 mg, 0.66 mmol) dissolved in a mixture of MeOH (10 mL) and cyclohexene (1 mL). The mixture was treated at reflux temperature for 3 h, and then the catalyst was filtered off. After evaporation, (1R,8S)-8 [51 mg, 51%; = +17.7 (c = 0.5; CHCl3); m.p. 104–106 °C (recrystallized from iPr2O); ee = 98%] or (1S,8R)-8 [47 mg, 47%; = −17.1 (c = 0.5; CHCl3), lit. [25] = −18 (c = 0.5; CHCl3)]; m.p. 105–107 °C, lit. [25] m.p. 108–112 °C; ee = 96%] was obtained as white crystalline product.
The 1H-NMR (400 MHz, CD3OD, 25 °C, TMS) δ (ppm) data for (1S,8R)-8 were similar to those for (1R,8S)-8: 1.30–2.09 (12H, m, 6xCH2), 3.03–3.06 (1H, m, H-1), 3.65–3.69 (1H, m, H-8), 5.89 (1H, bs, NH). Anal. found for C9H15NO, (1S,8R)-8: C, 70.48; H, 9.76; N, 9.14 and for (1R,8S)-8: C, 70.51; H, 9.82; N, 9.08.
3.10. Preparation of (1R,2S)-11 and (1S,2R)-11
Palladium-on-carbon (60 mg) was added to enantiomer (1R,2S)-9 or (1S,2R)-9 (100 mg) dissolved in MeOH (20 mL) and H2 was bubbled through the system at RT for 6 h. The catalyst was then filtered off and after evaporation white crystalline enantiomers of 2-aminocyclooctane-1-carboxylic acid (1R,2S)-11 [69.1 mg, 68%; = +19.2 (c = 0.4; H2O), lit. [25] = +17.8 (c = 0.4; H2O); m.p. 250–252 °C (recrystallized from H2O/acetone), lit. [25] m.p. 245–248 °C; ee = 99%] and (1S,2R)-11 [76.2 mg, 75%; = −19 (c = 0.33; H2O); m.p. 241–245 °C (recrystallized from H2O/acetone); ee = 99%] were isolated.
The 1H-NMR (400 MHz, D2O) data δ (ppm) for (1R,2S)-11 are similar to those for (1S,2R)-11: 1.50–1.92 (12H, m, 6xCH2) 2.78–2.79 (1H, m, H-1) 3.59–3.63 (1H, m, H-2). Anal. calcd. for C9H17NO2: C, 63.14; H, 10.01; N, 8.18; found for (1R,2S)-11: C, 63.20; H, 10.18; N, 8.02; found for (1S,2R)-11: C, 63.14; H, 9.91; N, 8.29.
3.11. Acidic Hydrolyses to β-amino Acid Hydrochlorides
When the enantiomeric lactam at issue (0.2 mmol) was treated with 18% aqueous HCl (5 mL) at reflux temperature, the desired amino acid hydrochloride was obtained, as follows:
(1R,2S)-9·HCl [37 mg, 92%; = −17.3 (c = 0.35; H2O); m.p. 218–220 °C (recrystallised from EtOH/Et2O); ee = 98%] from 50 mg (1R,8S)-7.
(1S,2R)-9·HCl [35 mg, 86%; = +19.6 (c = 0.5; H2O); m.p. 214–218 °C (recrystallised from EtOH/Et2O); ee = 99%] from 30 mg (1S,8R) 3.
(1S,2R)-9·HCl [31 mg, 76%; = +19.6 (c = 0.6; H2O); m.p. 219–220 °C (recrystallised from EtOH/Et2O); ee = 99%] from 36 mg (1S,8R) 4.
(1S,2R)-10·HCl [38 mg, 93%; = −15.0 (c = 0.5; H2O), lit. [25] = −15.9 (c = 0.3; H2O; m.p. 200–204 °C (recrystallised from EtOH/Et2O ee = 97%] from 36 mg (1S,8R)-6.
The 1H-NMR (400 MHz, D2O) data δ (ppm) for (1R,2S)-9·HCl are similar to those for (1S,2R)-9·HCl: 1.52–2.31 (8H, m, 4 × CH2) 3.21–3.24 (1H, m, H-1) 4.55–4.58 (1H, m, H-2) 5.76–6.14 (2H, m, CHCH).
The 1H-NMR (400 MHz, D2O) data δ (ppm) for (1S,2R)-10·HCl: 1.64–2.33 (8H, m, 4 × CH2) 2.91–2.98 (1H, m, H-1) 3.66–3.69 (1H, m, H-2) 5.56–5.63 (2H, m, CHCH).
Anal. calcd. for C9H16ClNO2: C, 52.56; H, 7.84; N, 6.81; found for (1R,2S)-9·HCl: C, 52.50; H, 7.68; N, 6.80; found for (1S,2R)-9·HCl from (1S,8R) 3: C, 52.68; H, 7.64; N, 6.78; found for (1S,2R)-9·HCl from (1S,8R)-4: C, 52.56; H, 7.66; N, 6.95; found for (1S,2R)-10·HCl: C, 52.47; H, 7.77; N, 6.62.
4. Conclusions
A highly efficient CAL-B (lipase B from Candida antarctica)-catalyzed ring-cleavage reaction (E > 200) of 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-3)] with 1 equiv. of H2O in iPr2O at 60 °C resulted new eight-membered ring-fused β-lactam (1S,8R)-3 and β-amino acid (1R,2S)-9 (ee ≥ 99%). Even more efficient two-step transformation was carried out for the ring cleavage of activated lactams (±)-4 with 1 equiv. of H2O, in the presence of CAL-B using 1 equiv. of benzylamine in iPr2O at 60 °C. The ring cleavage of regioisomeric (±)-6, carried out under the same conditions resulted unreacted (1S,8R)-6, potential intermediate for the synthesis of enantiomeric anatoxin-a. These results underline the importance of substrate engineering, since faster reactions were clearly detected when activated lactams vs. their inactivated counterparts were reacted. Advantages of these reactions are the spontaneous degradation of N-hydroxymethyl groups and easy product separation by organic solvent–H2O extraction.
An indirect enzymatic method, in view of the synthesis of enantiomeric β-amino acids, has also been devised, through a relatively fast acylation of N-hydroxymethyl 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4)] with 10 equiv. of VB mediated by lipase PSIM (Burkholderia cepacia) using a catalytic amount of Et3N and Na2SO4 in iPr2O at 30 °C (E = 114). This route is somewhat less efficient and, in fact, it is a longer procedure to prepare amino acid enantiomers. However, it ensures the simultaneous preparation of new β-lactam enantiomers [(1S,8R)-4 and (1R,8S)-7, and (1S,8R)-3 and (1R,8S)-3, respectively].
Acknowledgments
The authors are grateful to the Hungarian Research Foundation (OTKA No K 71938 and K115731) for financial support. This research was supported by EU-funded Hungarian grant GINOP-2.3.2-15-2016-00014.
Author Contributions
Enikő Forró and Ferenc Fülöp conceived and designed the experiments. Loránd Kiss and Judit Árva performed the experiments. Enikő Forró analyzed the data and wrote the paper. All authors read and approved the final manuscript. Ferenc Fülöp revised and edited the manuscript.
Conflicts of Interest
There are no conflict to declare.
References
- Fülöp, F. The chemistry of 2-aminocycloalkanecarboxylic acids. Chem. Rev. 2001, 101, 2181–2204. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, A.; Hahn, M.G.; Dumic, M.; Mittendorf, J. Alicyclic β-amino acids in medicinal chemistry. Amino Acids 2005, 29, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, J.; Kunisch, F.; Matzke, M.; Militzer, H.C.; Schmidt, A.; Schönfeld, W. Novel Antifungal β-Amino Acids: Synthesis and Activity against Candida albicans. Bioorg. Med. Chem. Lett. 2003, 14, 433–436. [Google Scholar] [CrossRef]
- Forró, E.; Árva, J.; Fülöp, F. Preparation of (1R,8S)- and (1S,8R)-9-azabicyclo[6.2.0]dec-4-en-10-one: Potential starting compounds for the synthesis of anatoxin-a. Tetrahedron Asymmetry 2001, 12, 643–649. [Google Scholar] [CrossRef]
- Parsons, P.J.; Camp, N.P.; Edwards, N.; Sumoreeah, L.R. Synthesis of (±)-anatoxin-a and analogues. Tetrahedron 2000, 56, 309–315. [Google Scholar] [CrossRef]
- Wonnacott, S.; Kaiser, S.; Mogg, A.; Soliakov, L.; Jones, I.W. Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. Eur. J. Pharmacol. 2000, 393, 51–58. [Google Scholar] [CrossRef]
- Sharples, C.G.V.; Kaiser, S.; Soliakov, L.; Marks, M.J.; Collins, A.C.; Washburn, M.; Wright, E.; Spencer, J.A.; Gallagher, T.; Whiteaker, P.; et al. UB-165: A Novel Nicotinic Agonist with Subtype Selectivity Implicates the α4β2* Subtype in the Modulation of Dopamine Release from Rat Striatal Synaptosomes. J. Neurosci. 2000, 20, 2783–2791. [Google Scholar] [PubMed]
- Rodriguez, V.; Moura, S.; Pinto, E.; Pereira, C.M.P.; Braga, R.C. Toxicological and chemical aspects of anatoxin-a and its analogs. Quim. Nova 2006, 29, 1465–1471. [Google Scholar]
- Mansell, H.L. Synthetic approaches to anatoxin-a. Tetrahedron 1996, 52, 6025–6061. [Google Scholar] [CrossRef]
- Masato, K.; Shingo, H.; Yasumasa, H.; Tetsuhiro, N. Formal amide insertion strategy for the synthesis of anatoxin-a using rhodium catalysis. Tetrahedron 2016, 72, 1495–1499. [Google Scholar]
- Marc, M.; Outurguin, F.; Renard, P.Y.; Créminon, C.; Franck, X. Synthesis of a (+)-anatoxin-a analogue for monoclonal antibodies production. Tetrahedron Lett. 2009, 50, 4554–4557. [Google Scholar] [CrossRef]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-Amino acids: Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Mándity, I.M.; Wéber, E.; Martinek, T.A.; Olajos, G.; Tóth, G.K.; Vass, E.; Fülöp, F. Design of peptidic foldamer helices: A stereochemical patterning approach. Angew. Chem. Int. Ed. 2009, 48, 2171–2175. [Google Scholar] [CrossRef] [PubMed]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Miklós, F.; Forró, E. Diexo-3-aminonorbornane-2-carboxylic acid as highly applicable chiral source for the enantioselective synthesis of heterocycles. Synlett 2008, 1687–1689. [Google Scholar] [CrossRef]
- Kazi, B.; Kiss, L.; Forró, E.; Fülöp, F. Synthesis of orthogonally protected azepane β-amino ester enantiomers. Tetrahedron Lett. 2010, 51, 82–85. [Google Scholar] [CrossRef]
- Kanizsai, I.; Gyónfalvi, S.; Szakonyi, Z.; Sillanpää, R.; Fülöp, F. Synthesis of bi- and tricyclic beta-lactam libraries in aqueous medium. Green Chem. 2007, 9, 357–360. [Google Scholar] [CrossRef]
- Liljeblad, A.; Kanerva, L.T. Biocatalysis as a profound tool in the preparation of highly enantiopure beta-amino acids. Tetrahedron 2006, 62, 5831–5854. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernande, V.; Gotor, V. Hydrolases in the stereoselective synthesis of N-heterocyclic amines and amino acid derivatives. Chem. Rev. 2011, 111, 3998–4035. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, M.; Tabassum, R.; Ahmad, M.M.; Hassan, N.A.; Oku, H.; Rivera, G. Enantioselective synthesis of β-amino acids: A Review. Med. Chem. 2015, 5, 295–309. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Direct and indirect enzymatic methods for the preparation of enantiopure cyclic β-amino acids and derivatives from β-lactams. Mini Rev. Org. Chem. 2004, 1, 93–102. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Recent lipase-catalyzed hydrolytic approaches to pharmacologically important β-and γ-amino acids. Curr. Med. Chem. 2012, 19, 6178–6187. [Google Scholar] [PubMed]
- Forró, E.; Fülöp, F. Cispentacin, enzymatic highlights of its 25-years history. Mini Rev. Org. Chem. 2016, 14, 219–226. [Google Scholar] [CrossRef]
- Gyarmati, Z.C.; Liljeblad, A.; Rintola, M.; Bernáth, G.; Kanerva, L.T. Lipase-catalyzed kinetic resolution of 7-, 8- and 12-membered alicyclic β-amino esters and N-hydroxymethyl-β-lactam enantiomers. Tetrahedron Asymmetry 2003, 14, 3805–3814. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Lipase-catalyzed enantioselective ring opening of unactivated alicyclic-fused β-lactams in an organic solvent. Org. Lett. 2003, 5, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Forró, E.; Fülöp, F. Advanced procedure for the enzymatic ring opening of unsaturated alicyclic β-lactams. Tetrahedron Asymmetry 2004, 15, 2875–2880. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. An efficient enzymatic synthesis of benzocispentacin and its new six- and seven-membered homologues. Chem. Eur. J. 2006, 12, 2587–2592. [Google Scholar] [CrossRef] [PubMed]
- Forró, E.; Galla, Z.; Fülöp, F. The N-hydroxymethyl group as a traceless activating group for the CAL-B-catalysed ring cleavage of β-lactams: A type of two-Step cascade reaction. Eur. J. Org. Chem. 2016, 2647–2652. [Google Scholar] [CrossRef]
- Nativ, E.; Rona, P. 2-Azabicyclo [3.2.0] heptane-3-one. Isr. J. Chem. 1972, 10, 55–58. [Google Scholar] [CrossRef]
- Singh, R.; Cooper, R.D.G. Synthesis and biological evaluation of 6-azabicyclo [3.2.0] hept-2-ene derivatives as potential anti-bacterial agents and β-lactamase inhibitors. Tetrahedron 1994, 50, 12049–12064. [Google Scholar] [CrossRef]
- Forró, E. New gas chromatographic method for the enantioseparation of β-amino acids by a quick double-derivatization technique. J. Chromatogr. A. 2009, 1216, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors in mg quantities. |
Figure 1.
Substrates (±)-3, (±)-4 and (±)-6 in the enzymatic strategies planned.

Scheme 1.
Synthesis of (±)-3–(±)-6.
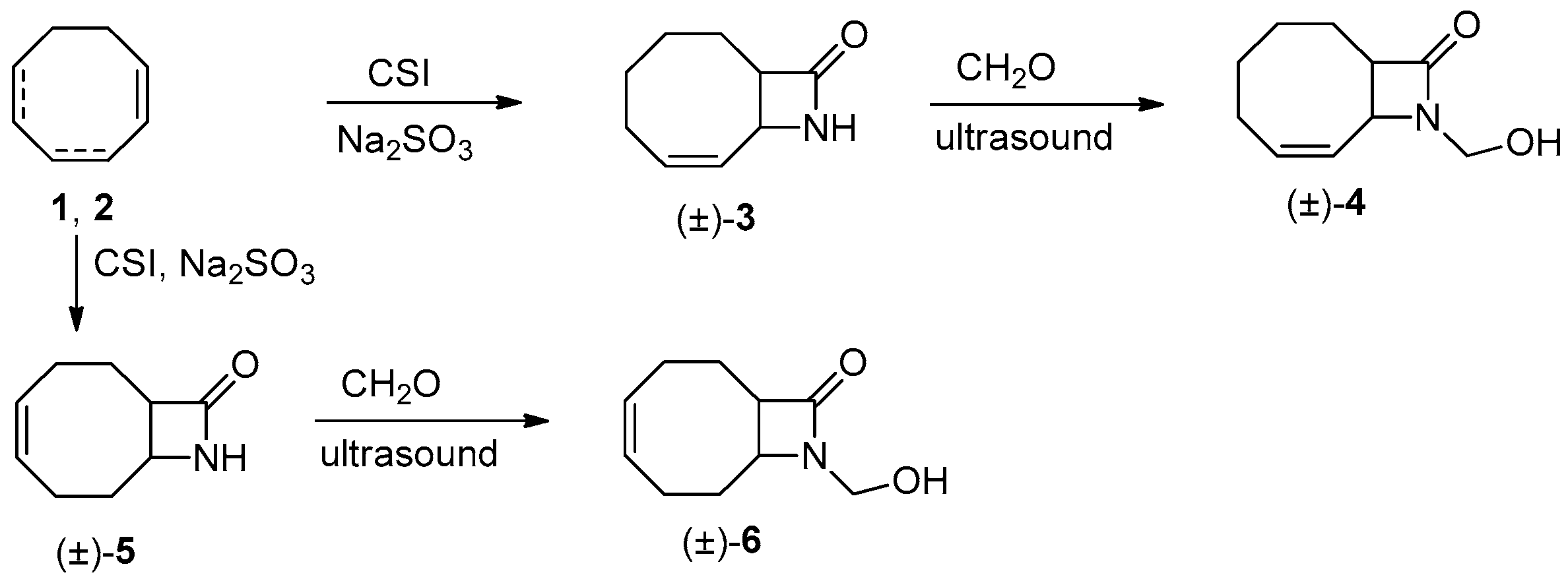
Scheme 2.
Lipase-catalyzed O-acylation of (±)-4.
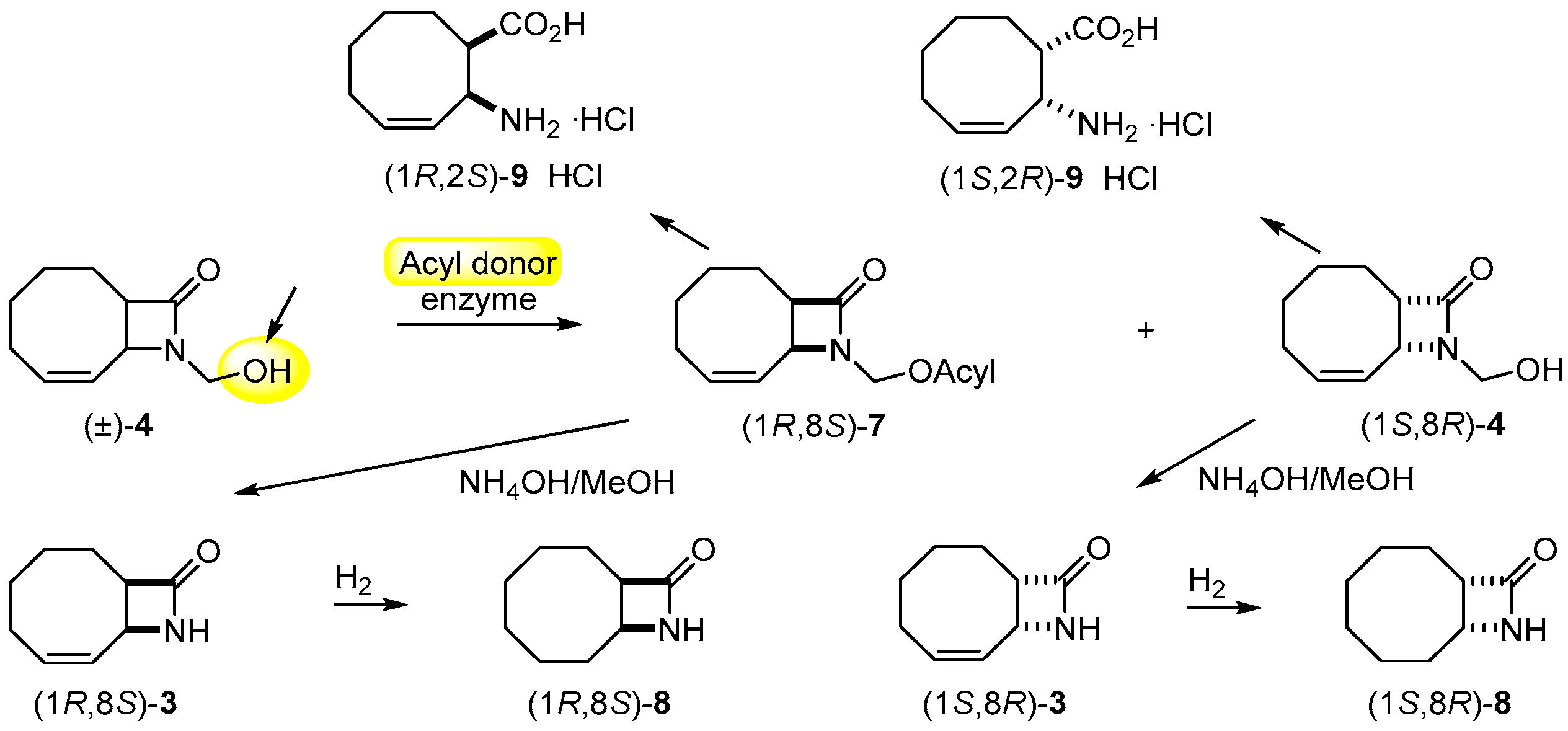
Scheme 3.
Lipase-catalyzed ring cleavage of (±)-3 and (±)-5.
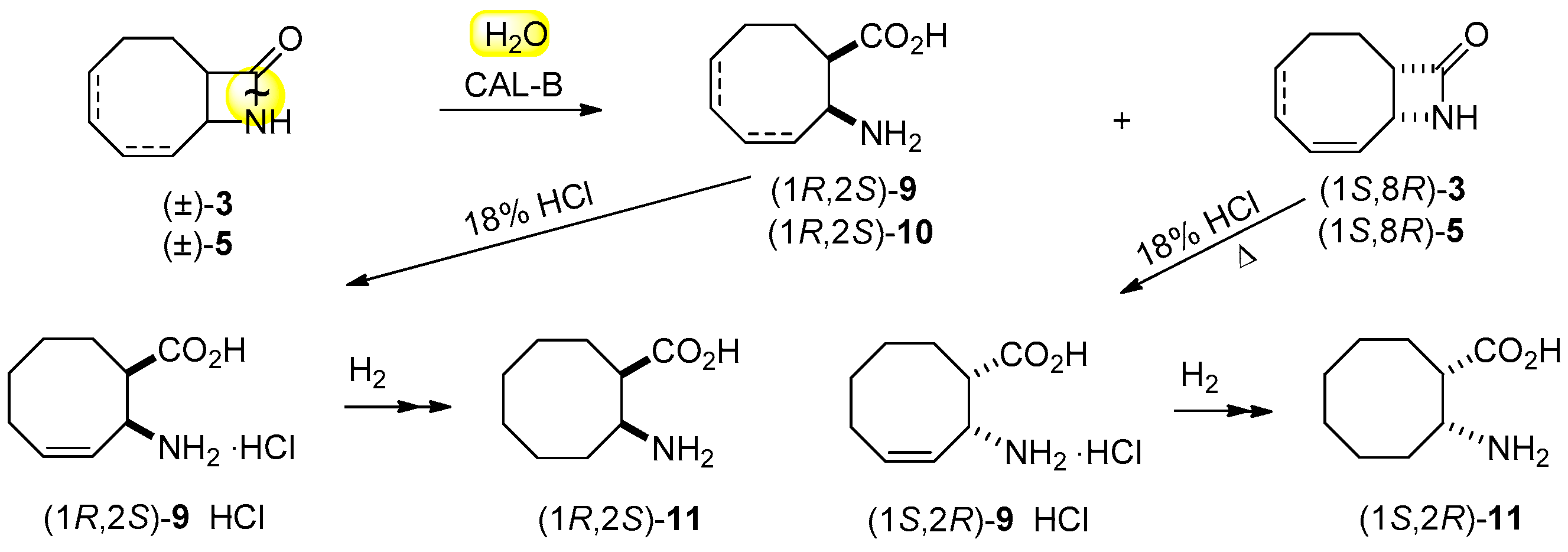
Scheme 4.
Lipase-catalyzed two-step transformation of (±)-4 and (±)-6.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Enzyme-catalyzed acylation of (±)-4 a.
| Entry | Enzyme (30 mg mL−1) | Acyl Donor (Equiv) | Solvent | Temp. (°C) | R. Time (Min) | Conv. (%) | ees b (%) | eep b (%) | E |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PSIM | VB (2) | iPr2O | -15 | 120 | 44 | 76 | 96 | 112 |
| 2 | PSIM | VB (2) | iPr2O | 2-3 | 60 | 43 | 70 | 94 | 67 |
| 3 | PSIM | VB (2) | iPr2O | 30 | 10 | 46 | 77 | 90 | 44 |
| 4 | PSIM | VB (10) | iPr2O | 30 | 10 | 51 | 87 | 84 | 32 |
| 5 | PSIM | VB (10) + Et3N + Na2SO4 | iPr2O | 30 | 10 25 | 45 50 | 77 91 | 96 92 | 114 76 |
| 6 | PSIM | 2,2,2-Trifluoroethyl-butyrate(10) | iPr2O | 30 | 20 | 49 | 83 | 86 | 34 |
| 7 | PSIM | VA (10) | iPr2O | 30 | 10 | 50 | 81 | 82 | 25 |
| 8 | PSIM | EtOAc(10) | iPr2O | 30 | 240 | 16 | 12 | 62 | 5 |
| 9 | PSIM | Ac2O (10) | iPr2O | 30 | 10 | 52 | 87 | 80 | 25 |
| 10 | AK c | VB (10) | iPr2O | 30 | 20 | 49 | 74 | 78 | 17 |
| 11 | AY c | VB (10) | iPr2O | 30 | 240 | 25 | 14 | 42 | 28 |
| 12 | CAL-A c | VB (10) | iPr2O | 30 | 10 | 29 | 12 | 29 | 2 |
| 14 | CAL-B | VB (10) | iPr2O | 30 | 50 | 67 | 2 | 1 | 1 |
| 14 | PPL | VB (10) | iPr2O | 30 | 120 | 20 | 23 | 91 | 26 |
| 15 | PSIM | VB (10) | tBuOMe | 30 | 10 | 51 | 87 | 85 | 34 |
| 16 | PSIM | VB (10) | Toluene | 30 | 5 | 49 | 89 | 93 | 82 |
| 17 | PSIM | VB (10) | Acetone | 30 | 60 | 49 | 86 | 89 | 47 |
a 0.015 M substrate; b According to GC (Experimental Section); c Contains 20% (w/w) of lipase adsorbed on Celite in the presence of sucrose.
Table 2.
CAL-B-catalyzed ring cleavage of racemic 3 a, 4 b, 5 a and 6 b.
| Entry | Substrate | R. Time (h) | Conv. c (%) | ees d (%) | eep e (%) |
|---|---|---|---|---|---|
| 1 | (±)-3 | 5 | 43 | 75 | >99 |
| 2 | (±)-5 | 5 | 46 | 84 | >99 |
| 3 | (±)-4 | 3 | 49 | 96 | >99 |
| 4 | (±)-6 | 3 | 50 | >99 | 98 |
a 0.015 M substrate, 30 mg mL−1 CAL-B, 0.5 equiv. of H2O, iPr2O, 60 °C; b 0.015 M substrate, 30 mg mL−1 CAL-B, 0.5 equiv. of H2O, 1 equiv. of benzylamine, iPr2O, 60 °C; c Calculated from eeS and eeP; d According to GC (Experimental Section); e Determined by using GC, after double derivatization (DD) [31].
Table 3.
Preparative-scale resolutions.
| Product | Unreacted substrate | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction Partner | R. Time (Min) | Conv. a (%) | Yield (%) | Isomer | eeP (%) | Yield (%) | Isomer | eeS (%) | (EtOH) | ||
| (±)-3 b | H2O | 330 | 50 | 48 | (1R,2S)-9 | 99 c | −17 d | 49 | (1S,8R)-3 | 99 e | −140.6 f |
| (±)-4 g | VB | 10 | 51 | 46 | (1R,8S)-7 | 94 e | +39.2 h | 44 | (1S,8R)-4 | 96 e | −142.4 i |
| (±)-4 j | H2O | 180 | 49 | 47 | (1R,2S)-9 | >99 c | −17.1 d | 48 | (1S,8R)-4 | 98 e | −140.4 f |
| (±)-6 j | H2O | 180 | 50 | 47 | (1R,2S)-10 | 99 c | +24.9 k | 46 | (1S,8R)-6 | 99 e | −28.7 l |
a Calculated from eeS and eeP; b 0.015 M substrate, 30 mg mL−1 CAL-B, 0.5 equiv. of H2O, iPr2O, 60 °C; c Determined by GC after DD (Experimental Section); d c = 0.35; H2O; e Determined by GC (Experimental Section); f c = 0.5; g 0.015 M substrate, 30 mg mL−1 PSIM, 10 equiv. of VB, catalytic amount of Et3N and Na2SO4, iPr2O 30 °C; h c = 0.35; EtOH; i c = 0.45; j 0.015 M substrate, 30 mg mL−1 CAL-B, 0.5 equiv. of H2O, 1 equiv. of benzylamine, iPr2O, 60 °C; k c = 0.3; H2O; l c = 0.5.
Table 4.
Physical data on enantiomers prepared.
| Entry | Enantiomers | ee (%) | |
|---|---|---|---|
| 1 | (1R,8S) 3 from (1R,8S)-7 | 95 | +147 (c = 0.5; EtOH) |
| 2 | (1S,8R) 3 from (1S,8R)-4 | 96 | −148.7 (c = 0.4; EtOH) |
| 3 | (1R,8S) 8 from (1R,8S)-3 | 98 | +17.7 (c = 0.5; CHCl3) |
| 4 | (1S,8R) 8 from (1S,8R)-3 | 96 | −17.1 (c = 0.5; CHCl3) |
| 5 | (1S,2R)-9·HCl from (1S,8R) 3 | 99 | +19.6 (c = 0.5; H2O) |
| 6 | (1S,2R)-9·HCl from (1S,8R) 4 | 99 | +19.6 (c = 0.6; H2O) |
| 7 | (1R,2S)-9·HCl from (1R,8S)-7 | 98 | −17.3 (c = 0.35; H2O) |
| 8 | (1S,2R)-10·HCl from (1S,8R)-6 | 97 | −15.0 (c = 0.5; H2O) |
| 9 | (1R,2S)-11 from (1R,2S)-9 | 99 | +19.2 (c = 0.4; H2O) |
| 10 | (1S,2R)-11 from (1S,2R)-9 | 99 | −19 (c = 0.33; H2O) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Forró, E.; Kiss, L.; Árva, J.; Fülöp, F. Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers. Molecules 2017, 22, 2211. https://doi.org/10.3390/molecules22122211
AMA Style
Forró E, Kiss L, Árva J, Fülöp F. Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers. Molecules. 2017; 22(12):2211. https://doi.org/10.3390/molecules22122211
Chicago/Turabian StyleForró, Enikő, Loránd Kiss, Judit Árva, and Ferenc Fülöp. 2017. "Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers" Molecules 22, no. 12: 2211. https://doi.org/10.3390/molecules22122211