Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study

 ,
, 
Abstract
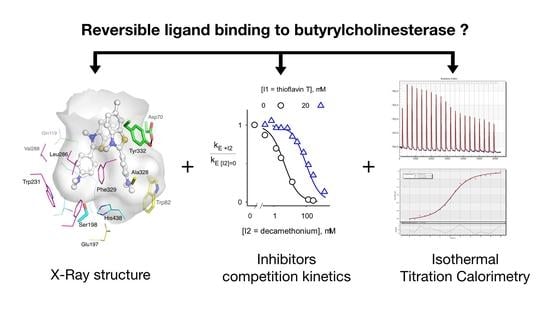
:
1. Introduction
2. Results
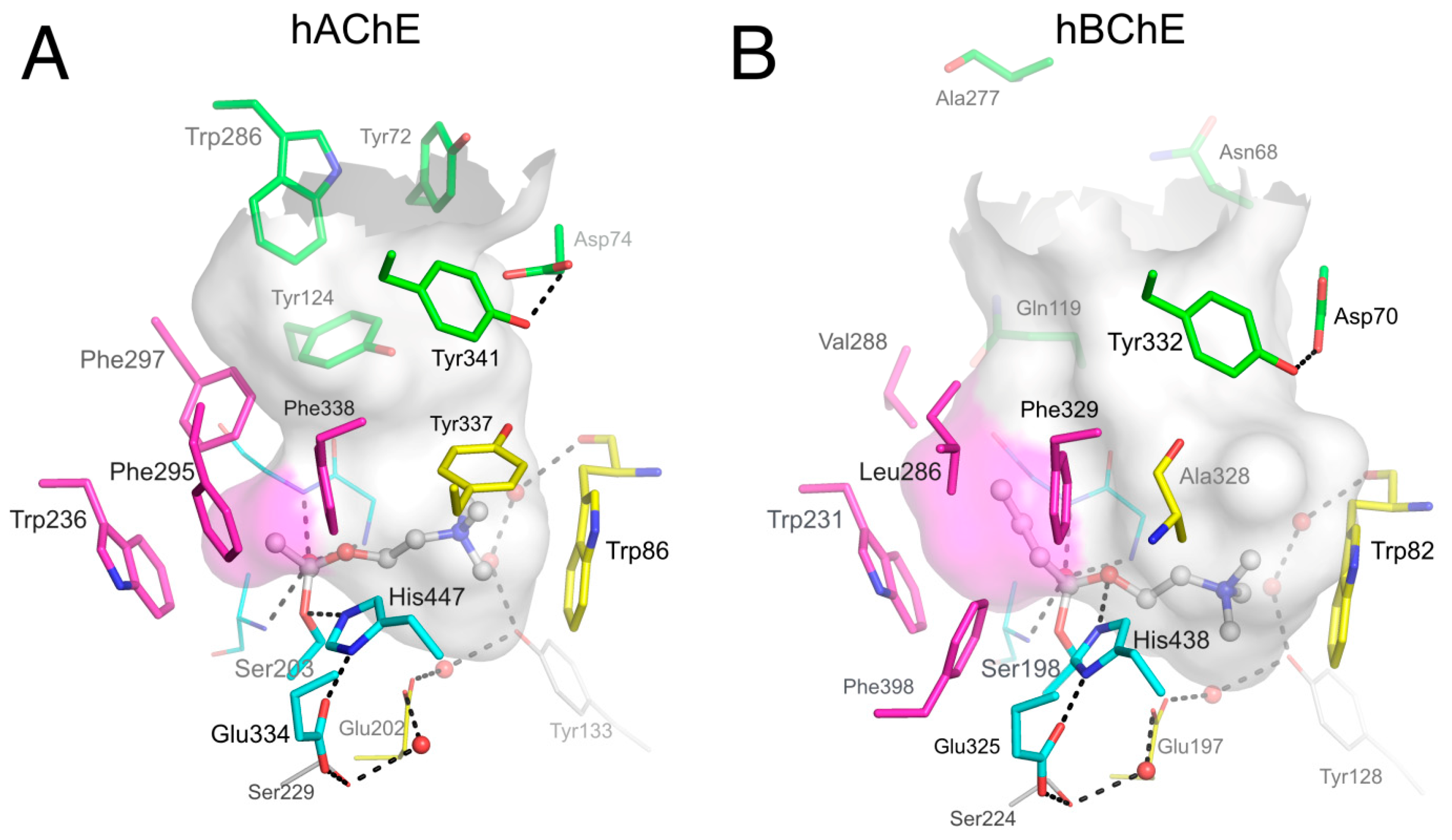
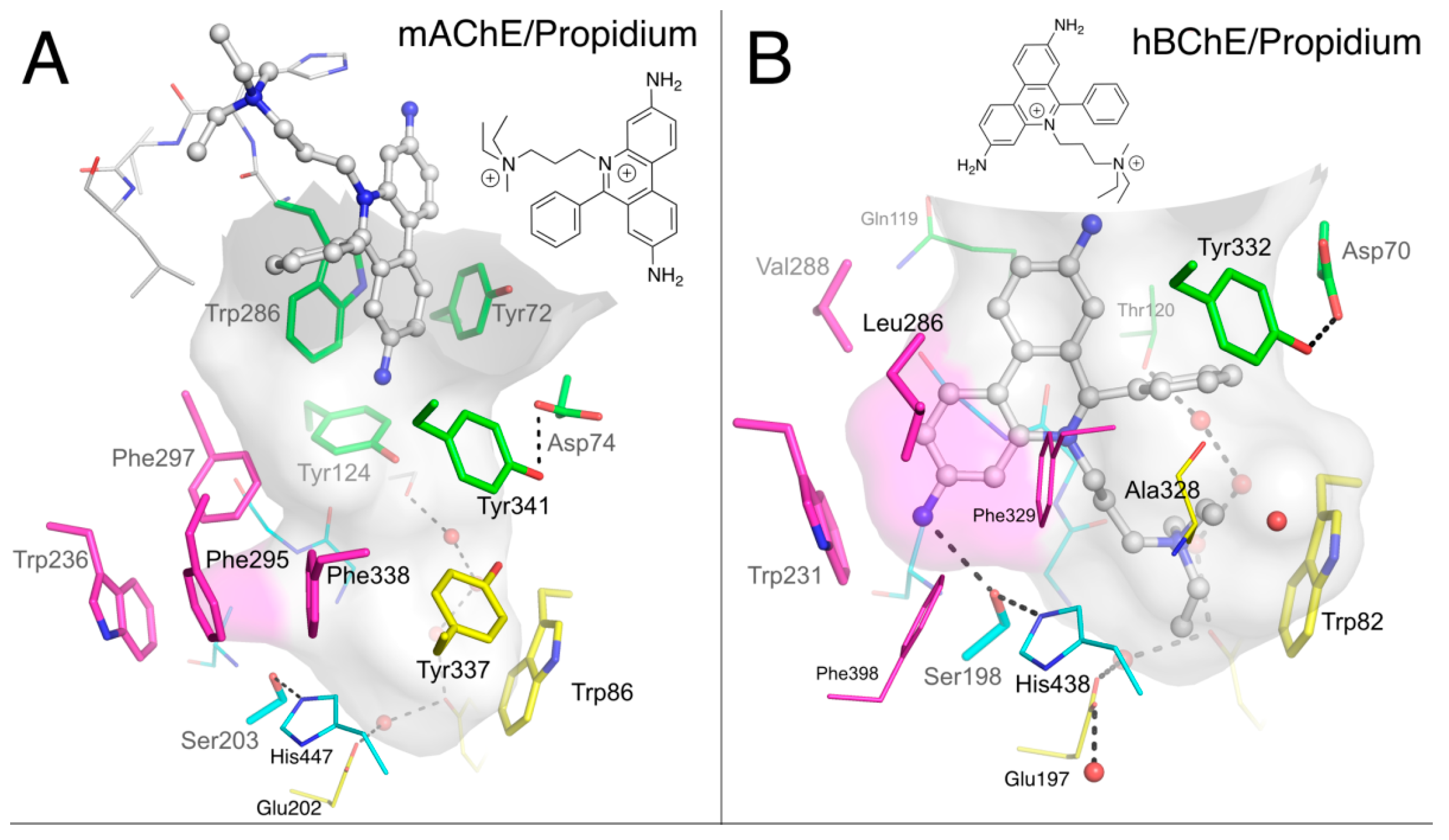
2.1. X-ray Structures of Human BChE-Ligand Complexes
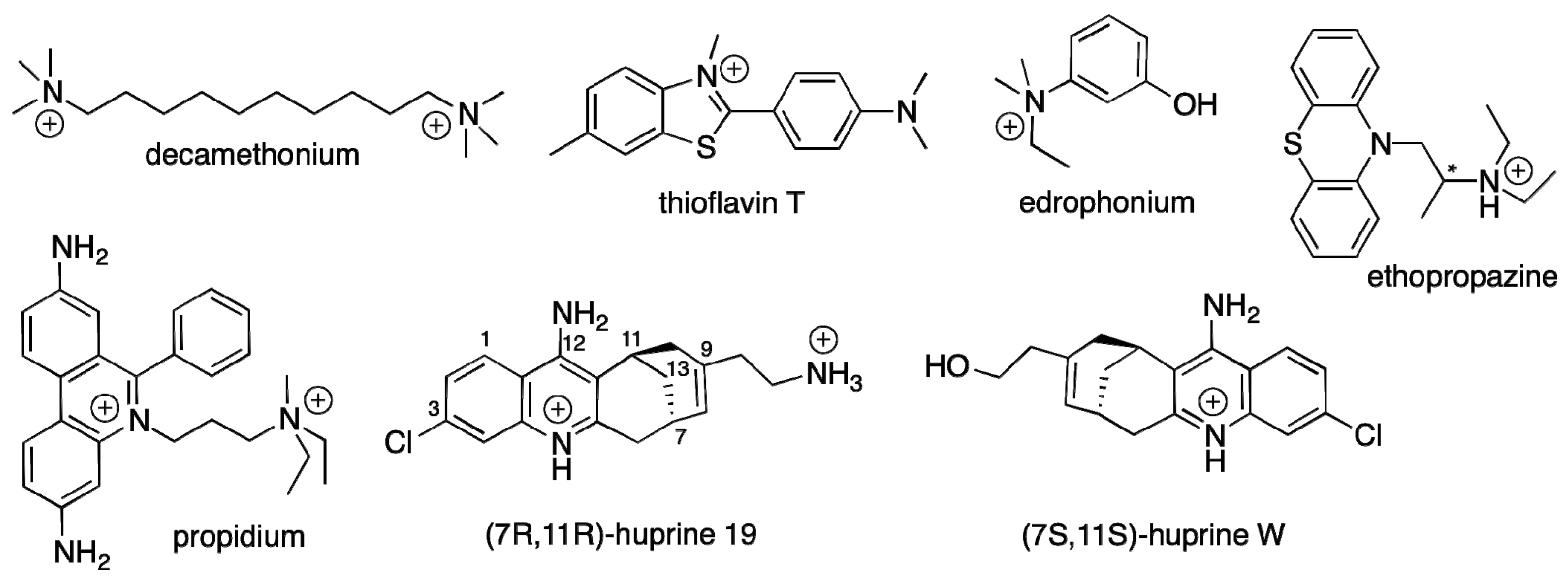
2.1.1. Decamethonium
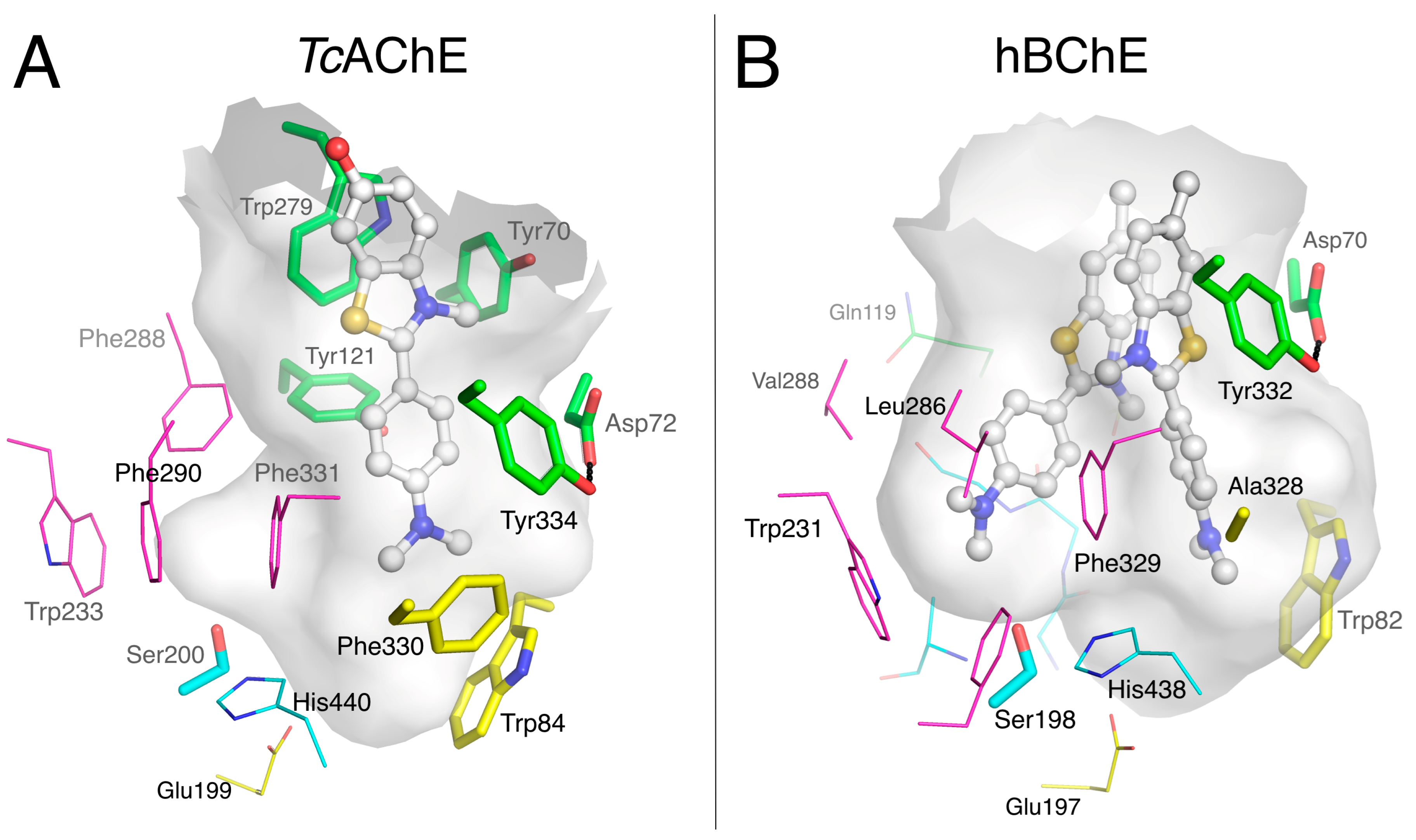
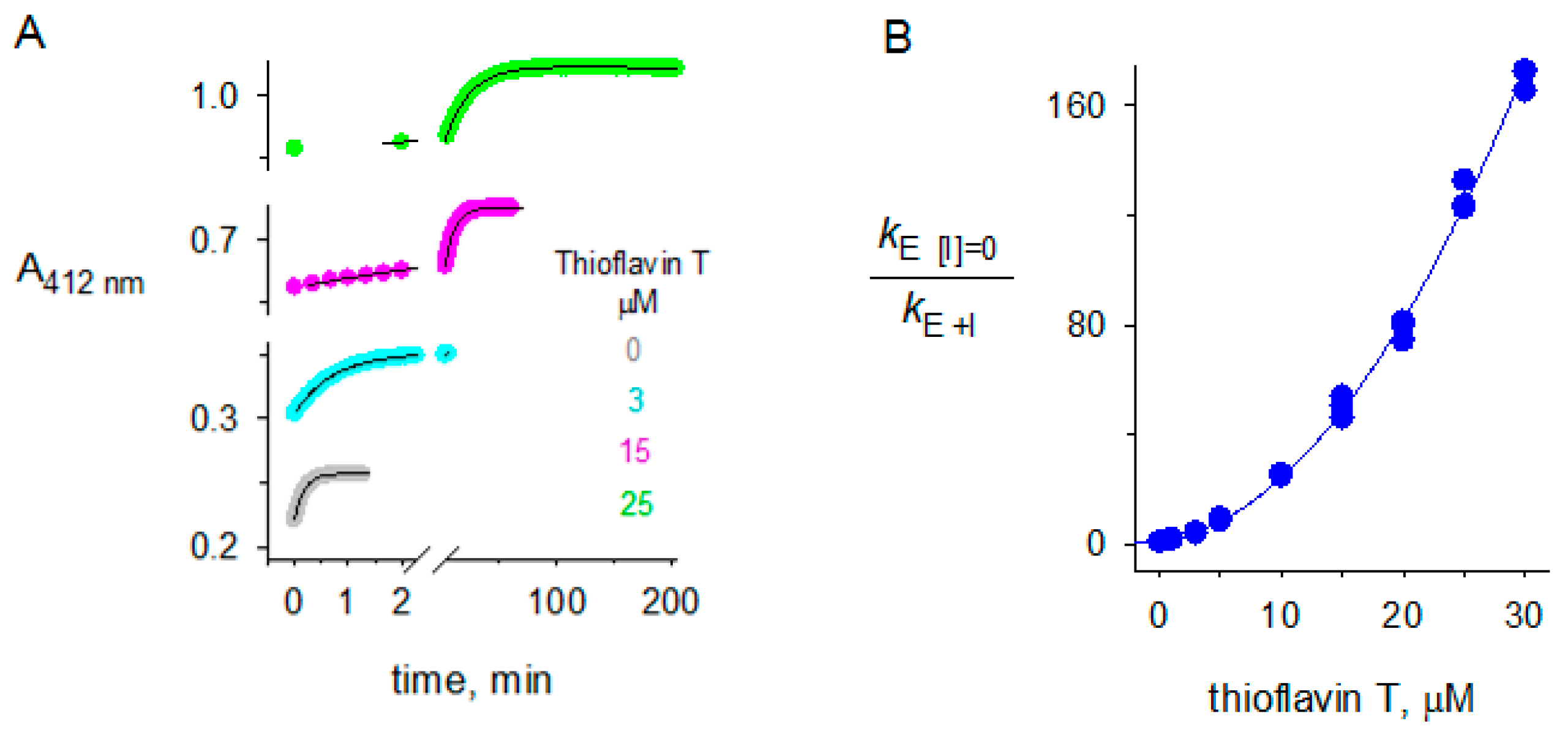
2.1.2. Thioflavin T
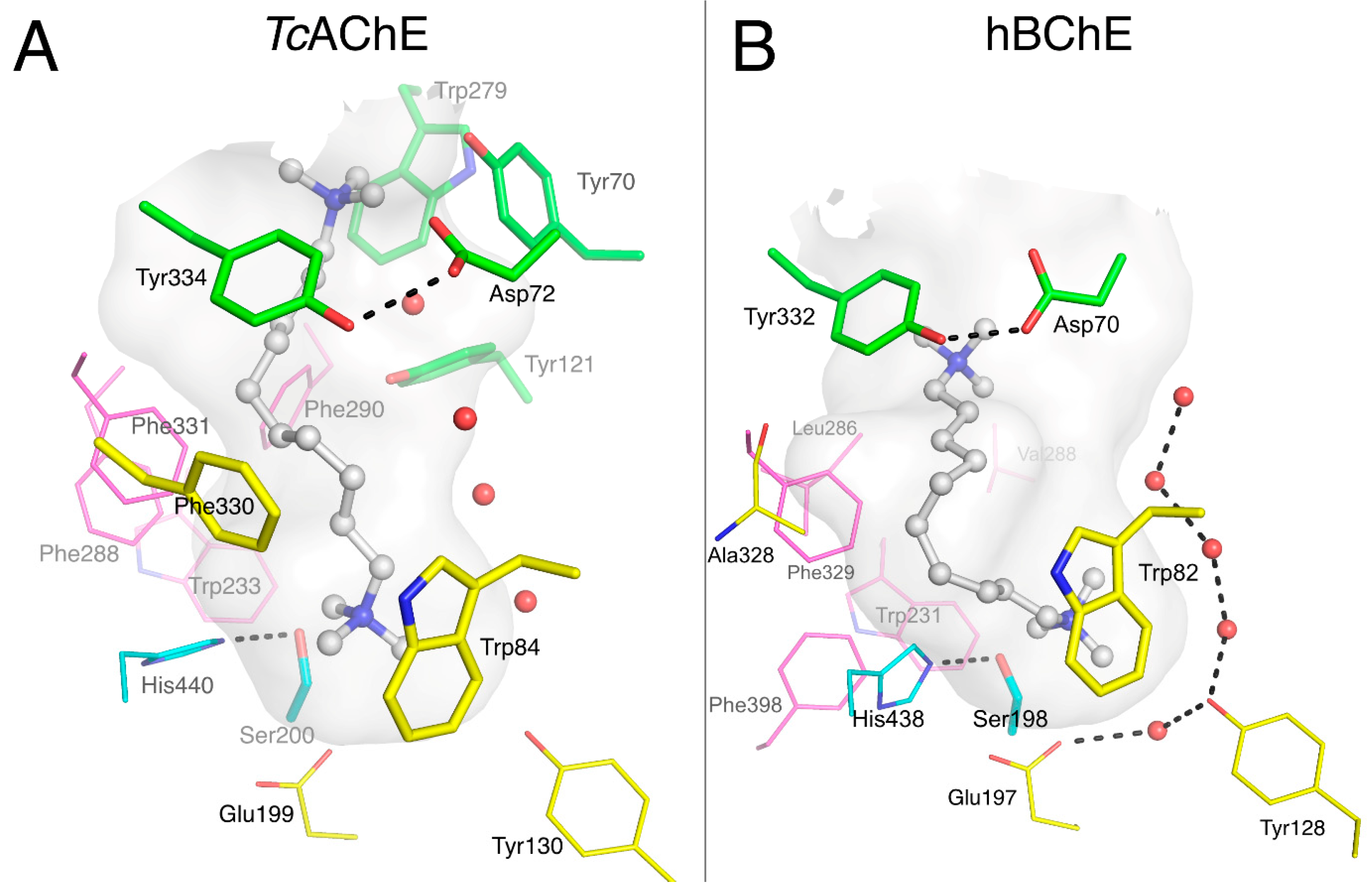
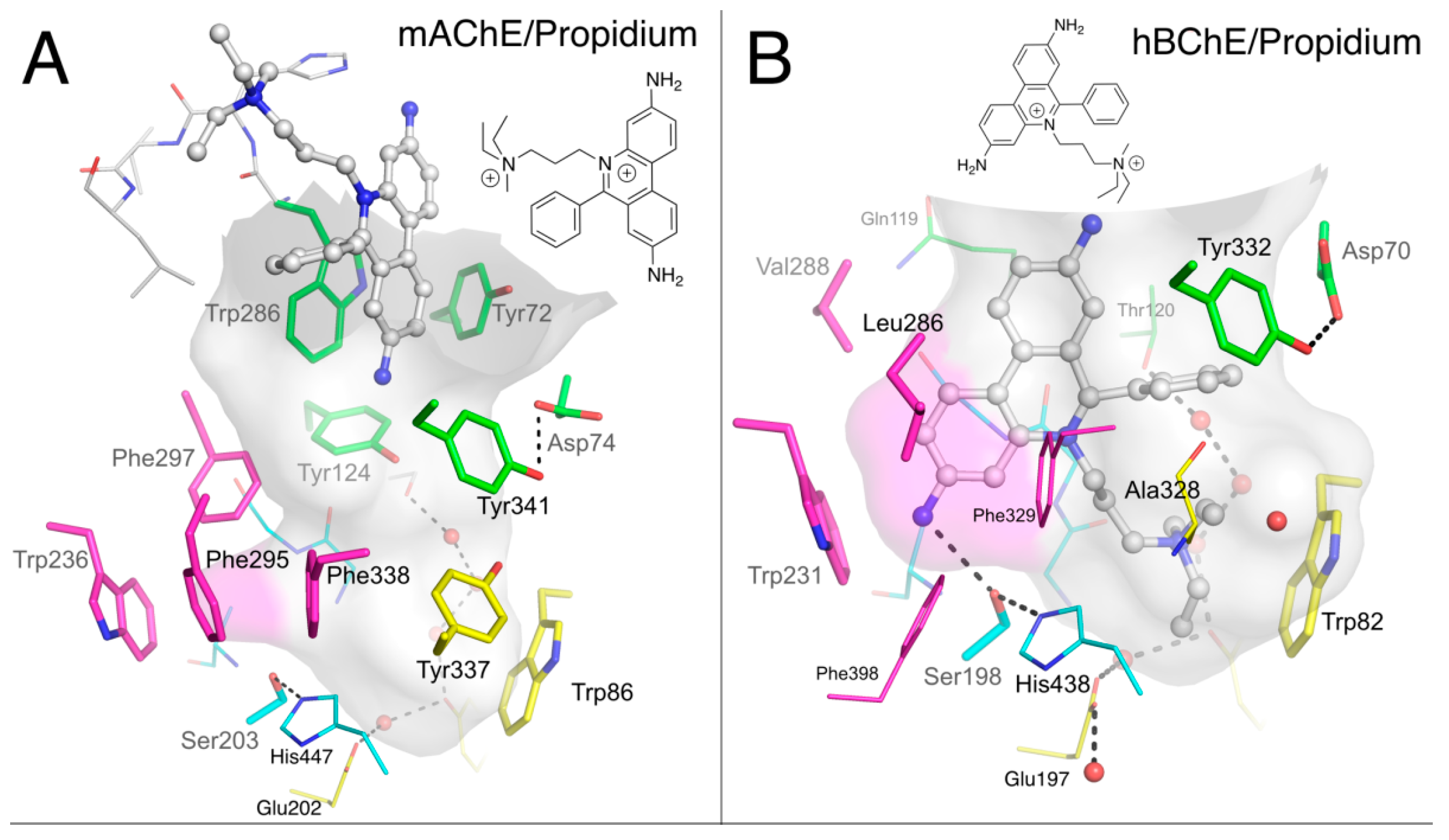
2.1.3. Propidium
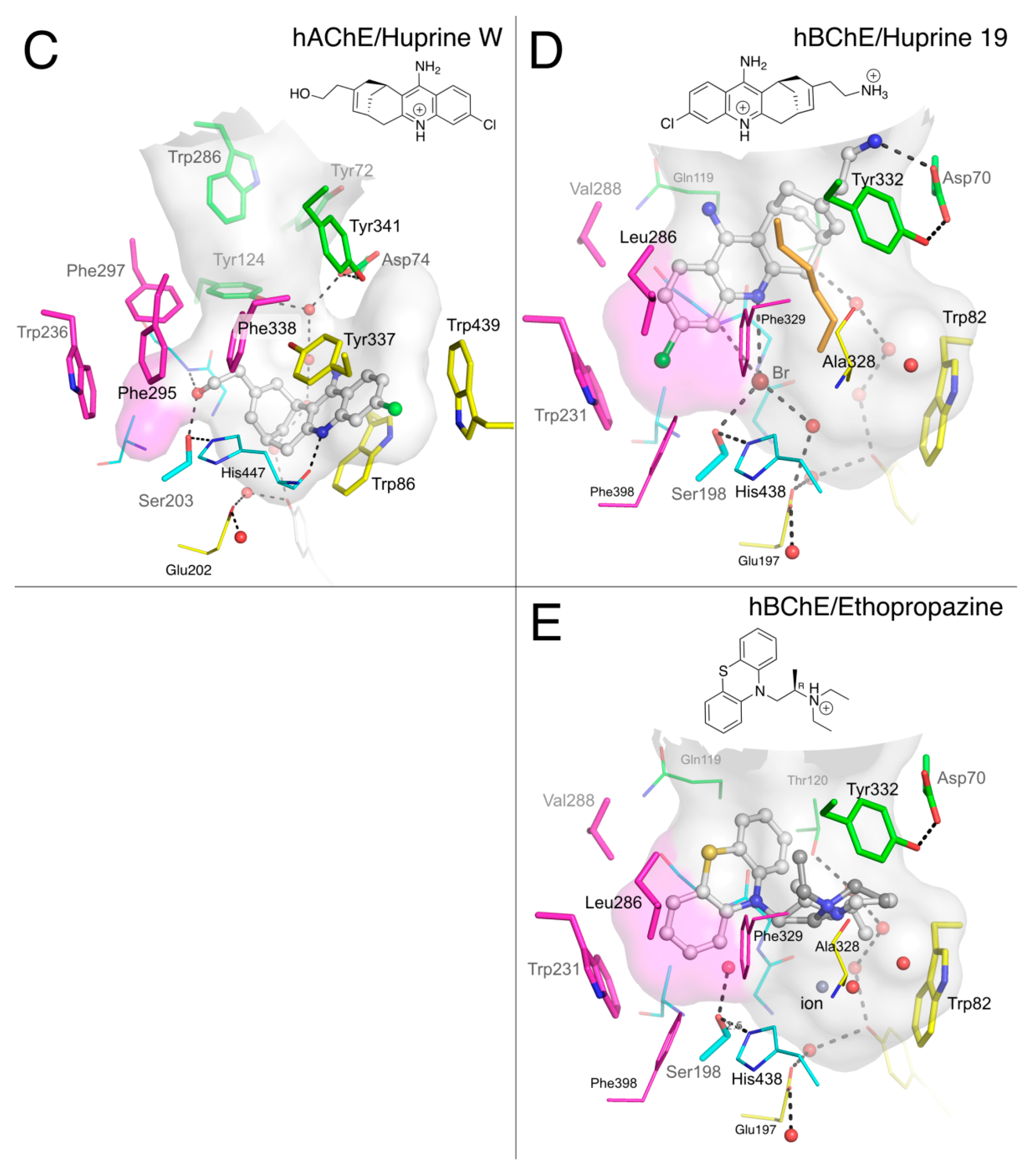
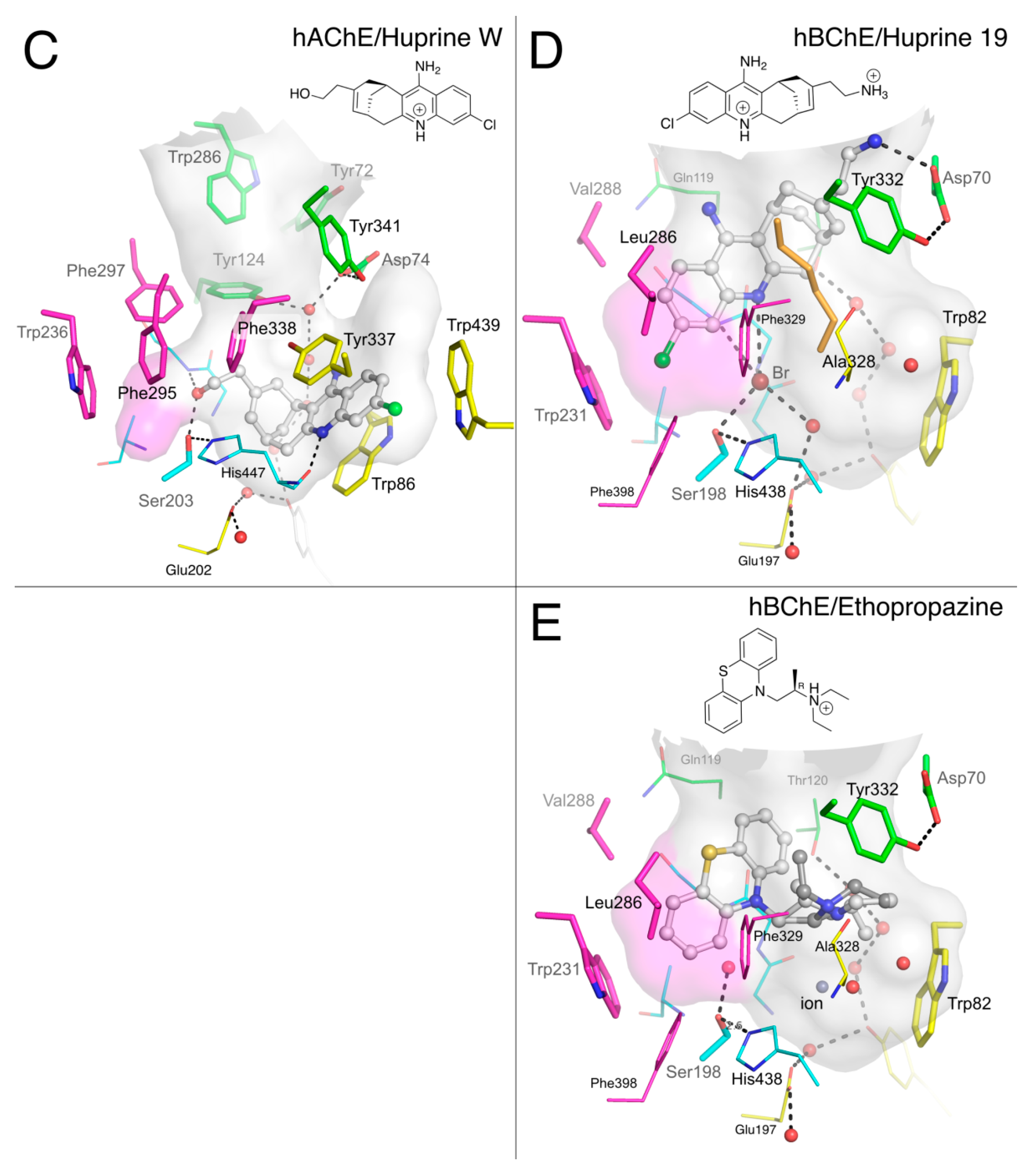
2.1.4. Huprines
2.1.5. Ethopropazine
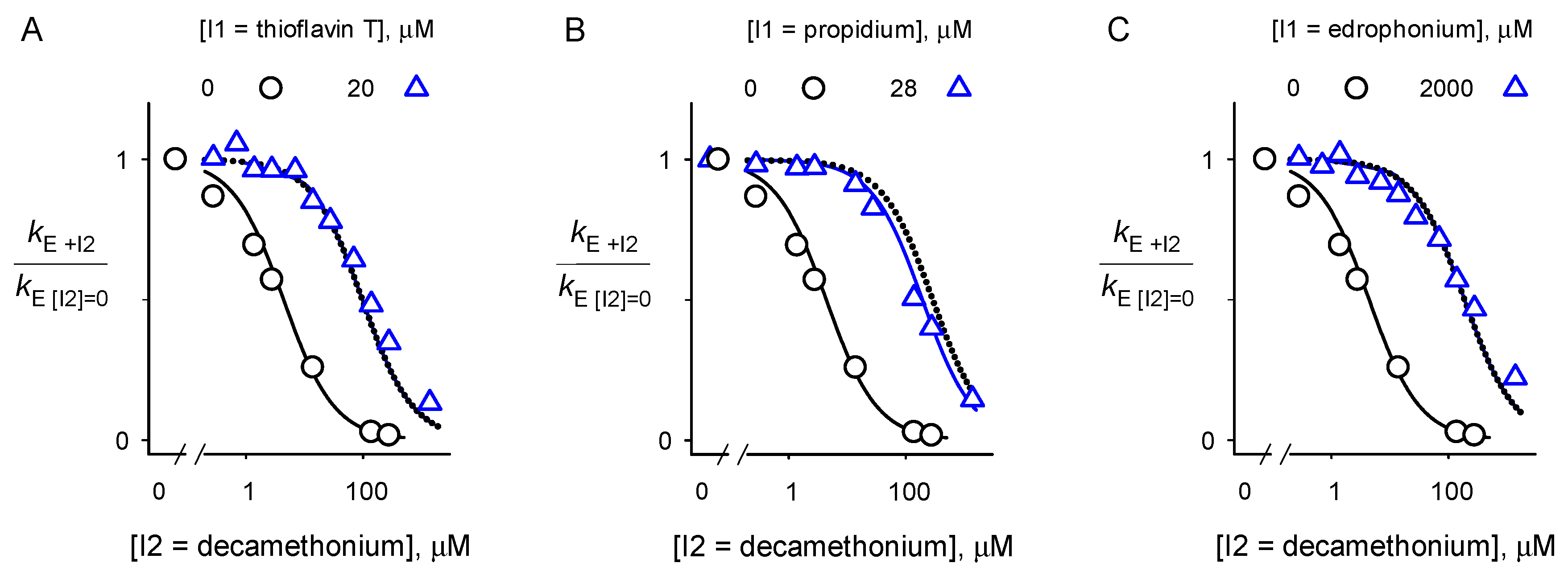
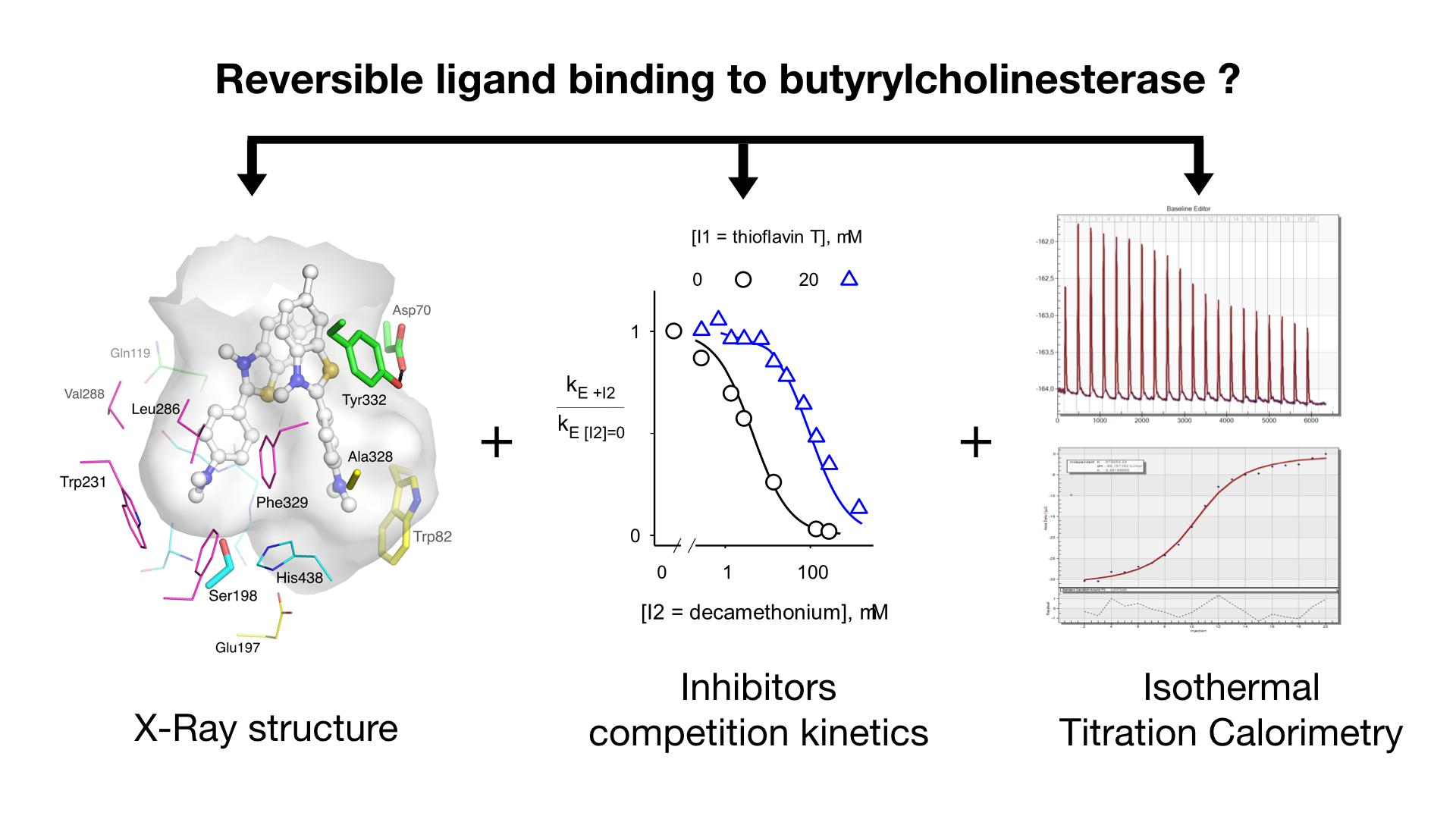
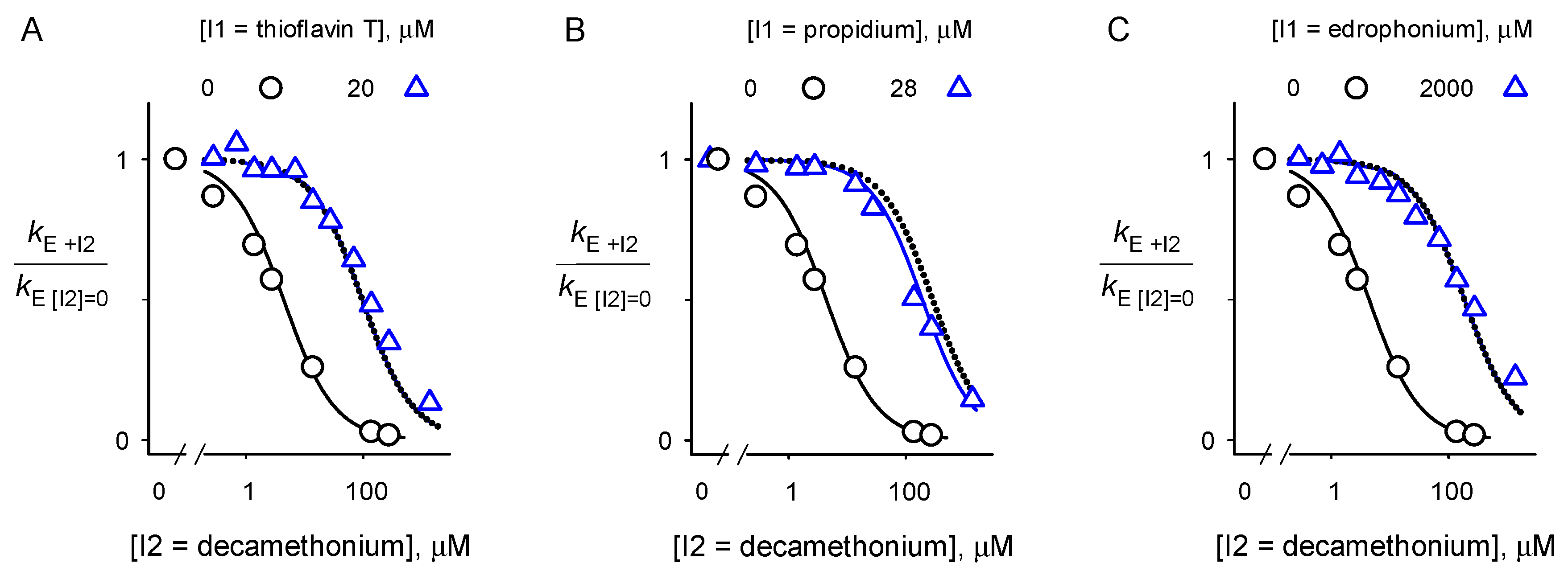
2.2. Inhibitor Competition
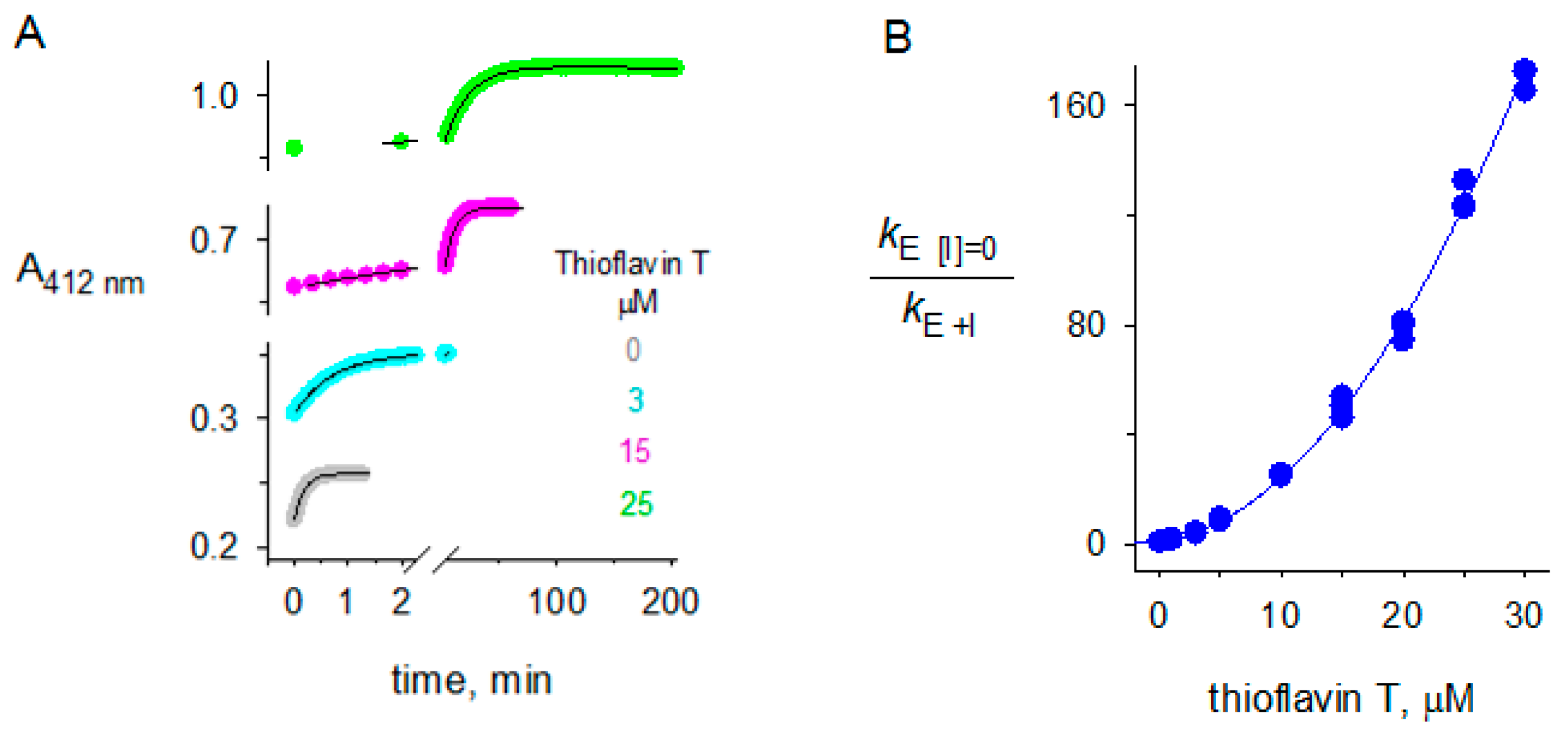
2.3. Thioflavin T Inhibition of BChE
2.4. Isothermal Titration Calorimetry
3. Discussion
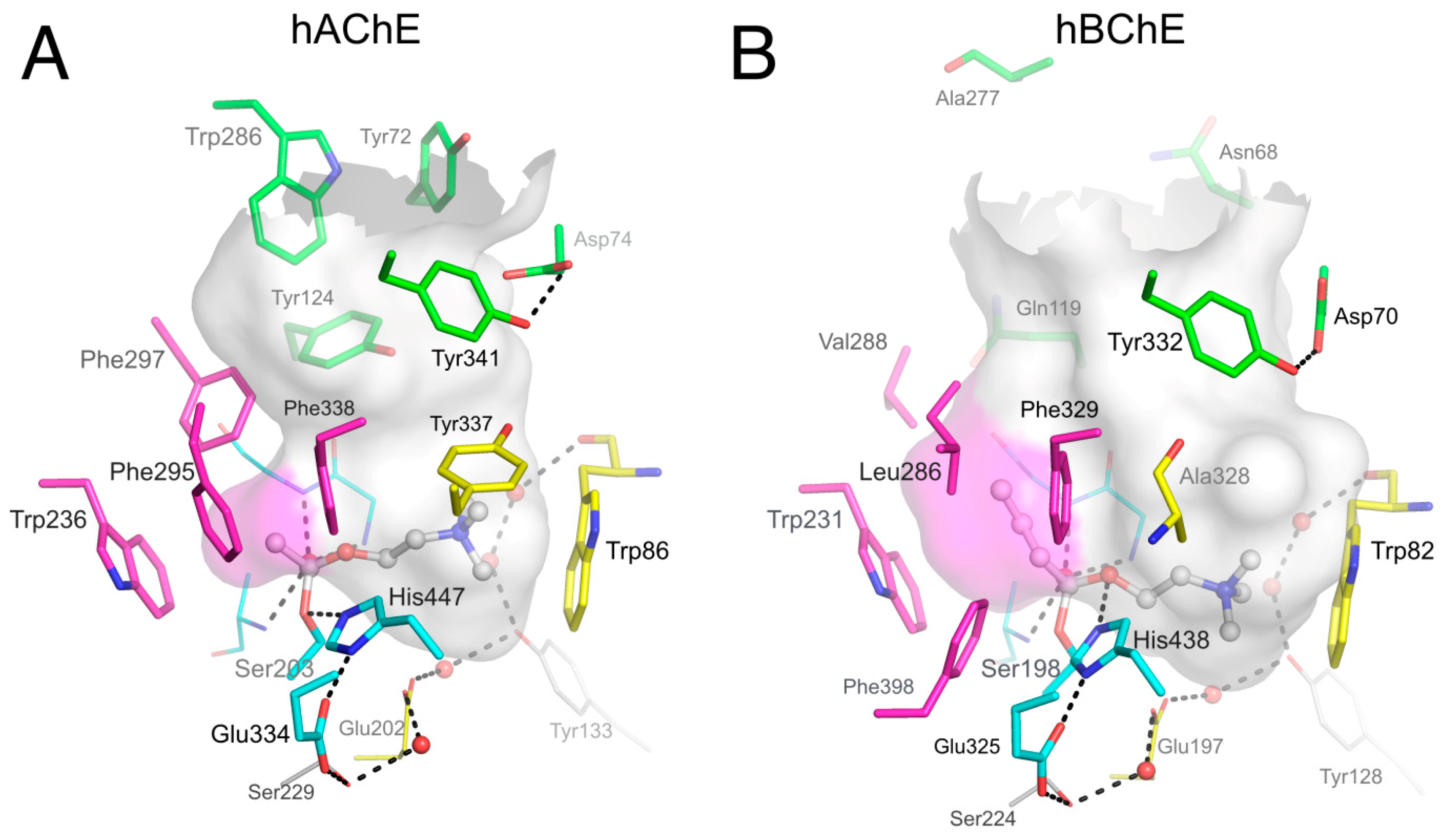
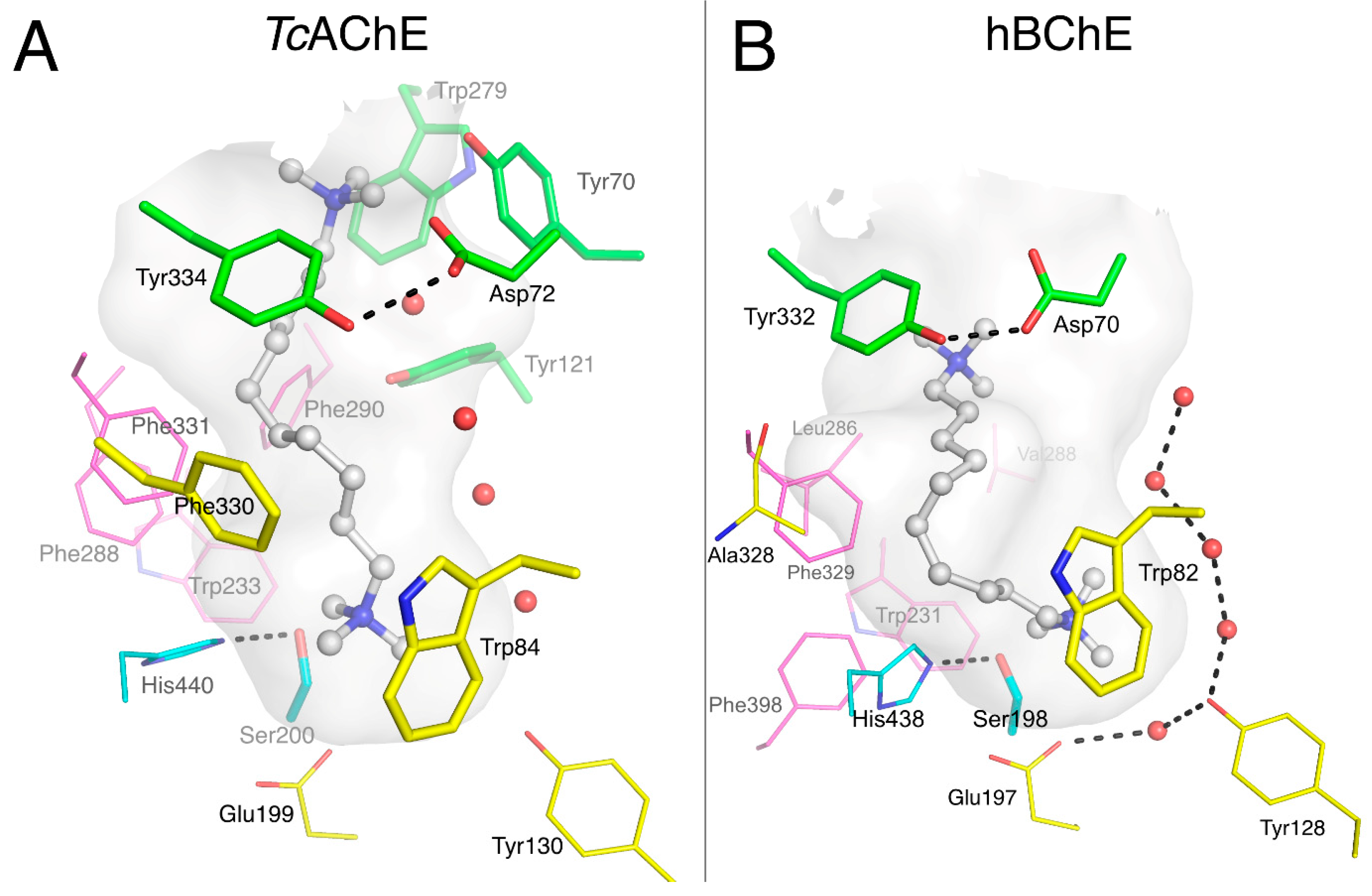
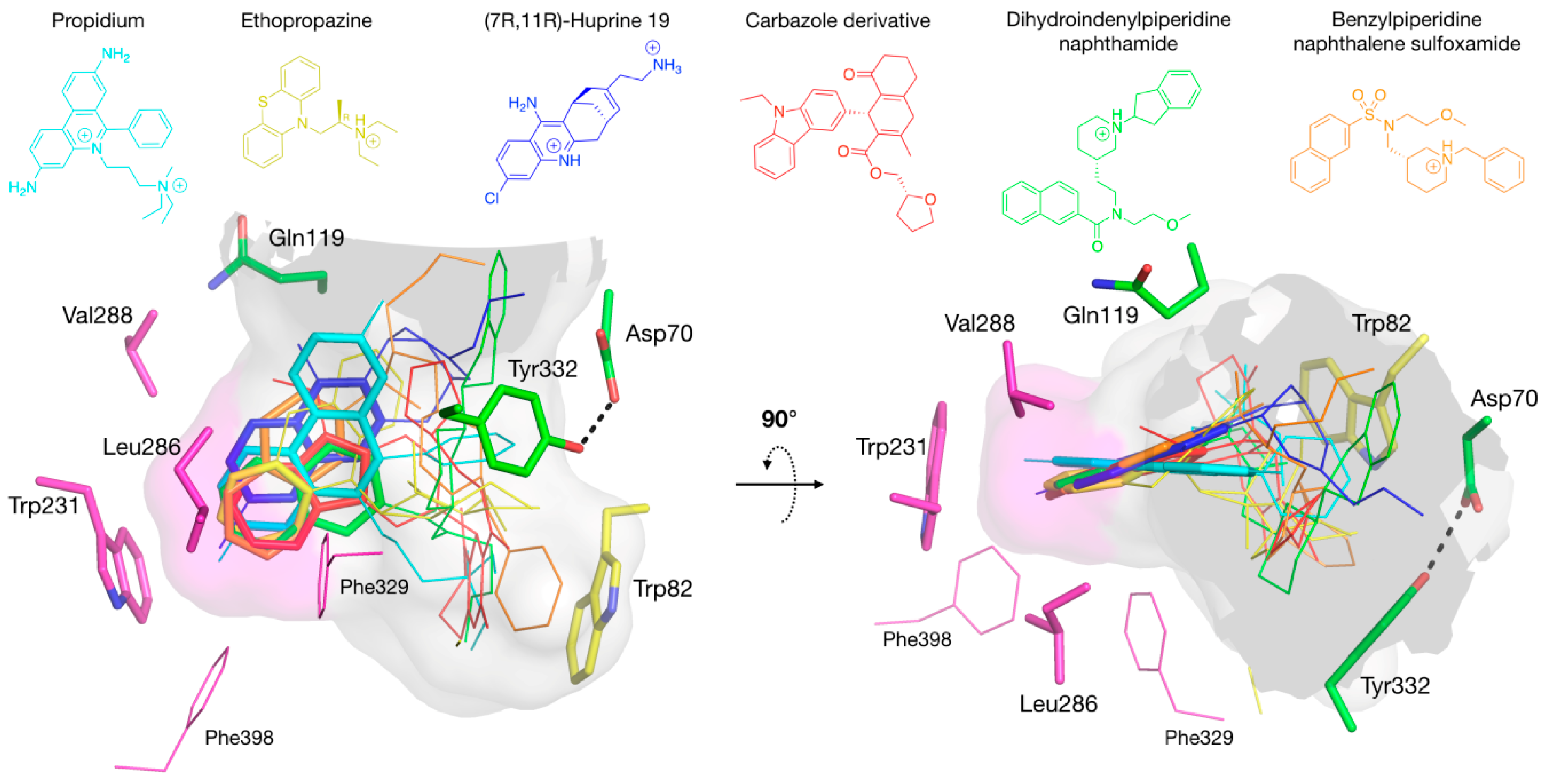
3.1. Is There a True BChE P-Site?
3.2. Dual Binding of Thioflavin T to BChE
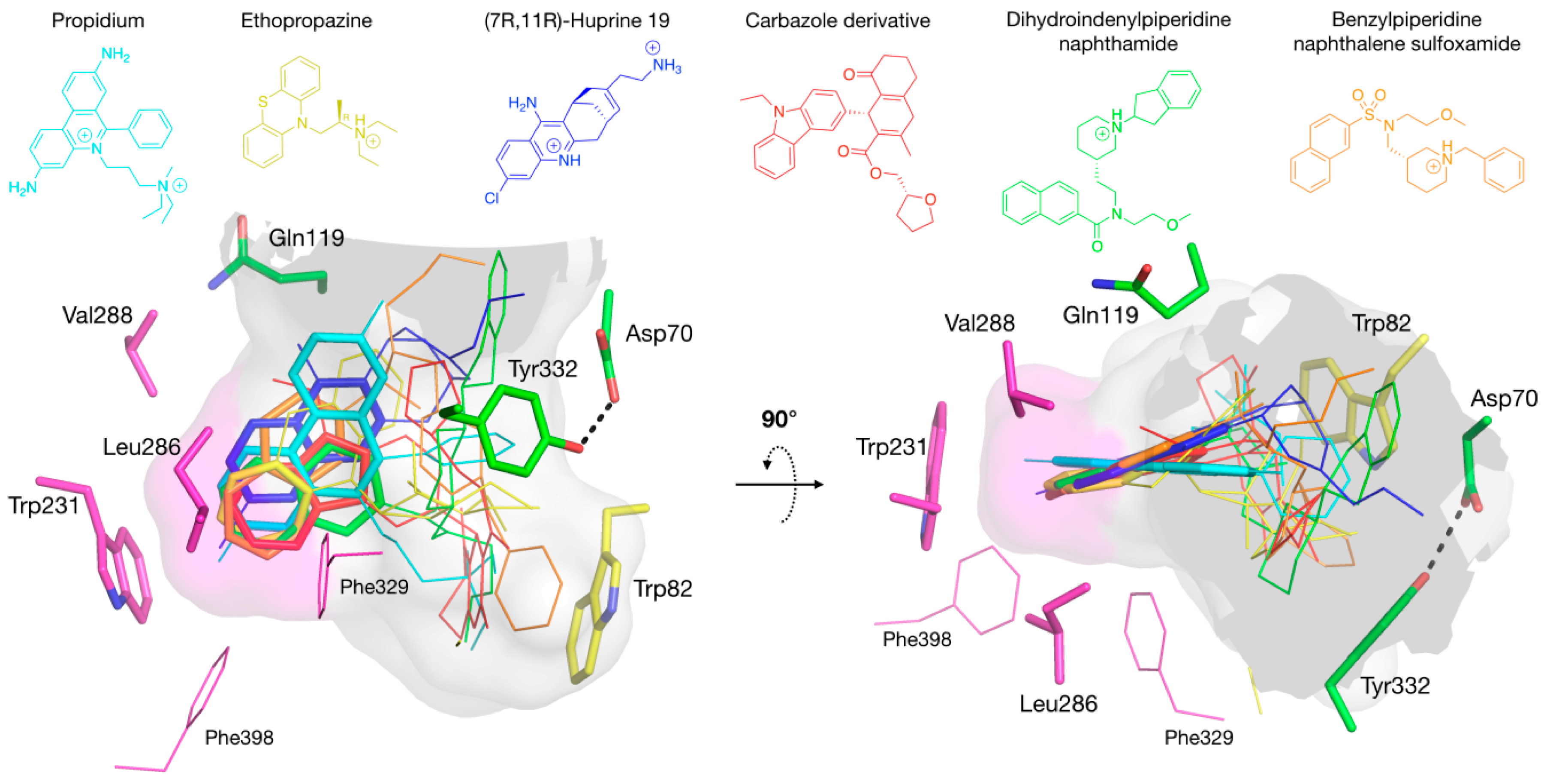
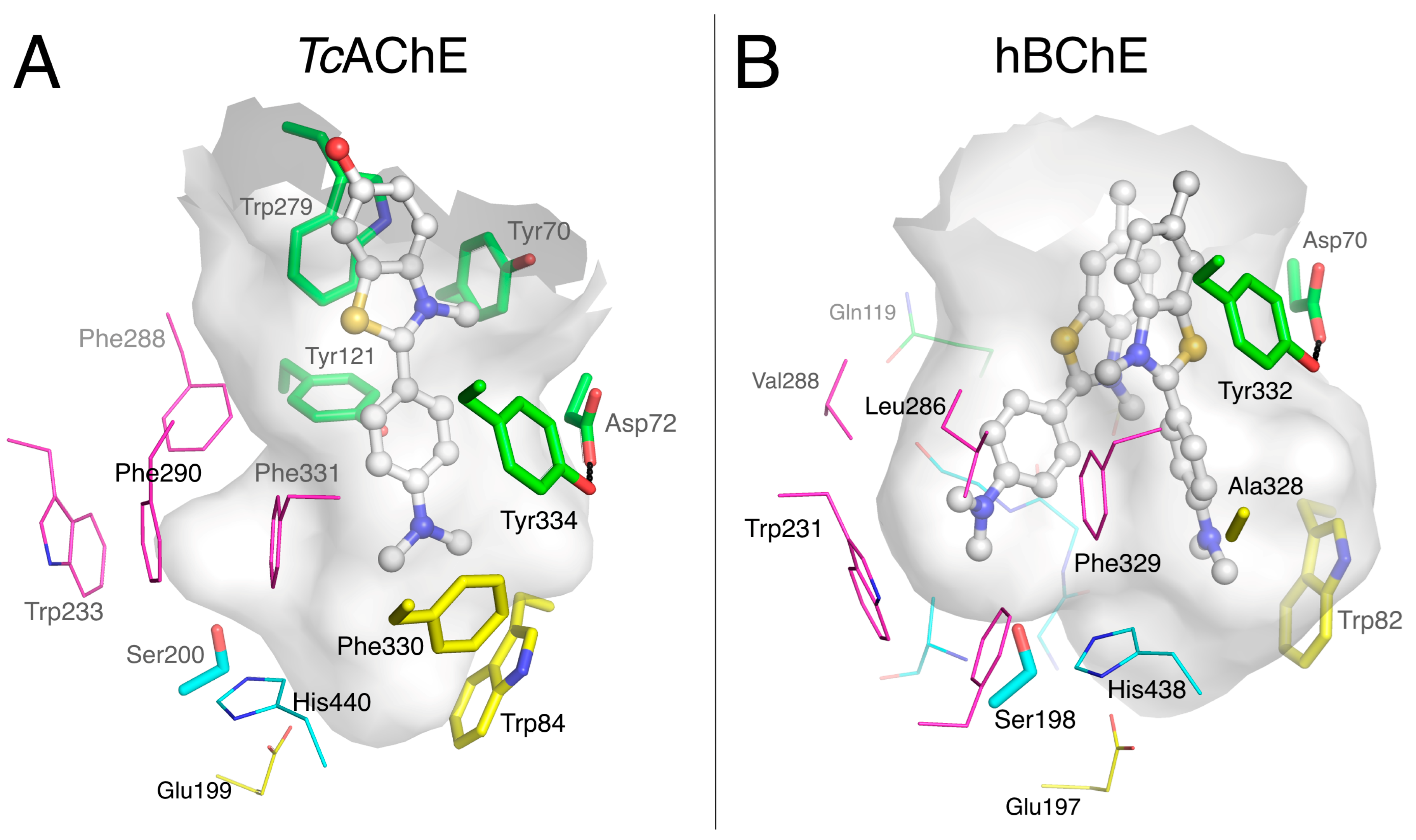
3.3. Specific Binding of Aromatic Compounds to the BChE Acyl-Binding Pocket
3.4. Molecular Docking to the AChE and BChE Active Site Gorges
4. Materials and Methods
4.1. Enzymes and Chemicals
4.2. Crystals of the hBChE Complexes
4.2.1. Decamethonium and Ethopropazine
4.2.2. Huprine 19
4.2.3. Thioflavin T and Propidium
4.3. X-ray Data Collection and Structure of BChE-Ligand Complexes
4.4. Inhibition of Second-Order Substrate Hydrolysis
4.5. Inhibitor Competition
4.6. Isothermal Titration Calorimetry (ITC) Measurements
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Silver, A. The Biology of Cholinesterases; Elsevier: Amsterdam, The Netherlands, 1974; p. 596. [Google Scholar]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.P.; Fournier, D.; Greenblatt, H.M.; Stojan, J.; Sussman, J.L.; Zaccai, G.; Silman, I.; Weik, M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006, 25, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Radic, Z.; Sulzenbacher, G.; Kim, E.; Taylor, P.; Marchot, P. Substrate and product trafficking through the active center gorge of acetylcholinesterase analyzed by crystallography and equilibrium binding. J. Biol. Chem. 2006, 281, 29256–29267. [Google Scholar] [CrossRef] [PubMed]
- Nachmansohn, D.; Wilson, I.B. The enzymic hydrolysis and synthesis of acetylcholine. Adv. Enzymol. Relat. Subj. Biochem. 1951, 12, 259–339. [Google Scholar] [PubMed]
- Mallender, W.D.; Szegletes, T.; Rosenberry, T.L. Acetylthiocholine binds to asp74 at the peripheral site of human acetylcholinesterase as the first step in the catalytic pathway. Biochemistry 2000, 39, 7753–7763. [Google Scholar] [CrossRef] [PubMed]
- Szegletes, T.; Mallender, W.D.; Rosenberry, T.L. Nonequilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands. Biochemistry 1998, 37, 4206–4216. [Google Scholar] [CrossRef] [PubMed]
- Radic, Z.; Pickering, N.A.; Vellom, D.C.; Camp, S.; Taylor, P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993, 32, 12074–12084. [Google Scholar] [CrossRef] [PubMed]
- Barak, D.; Kronman, C.; Ordentlich, A.; Ariel, N.; Bromberg, A.; Marcus, D.; Lazar, A.; Velan, B.; Shafferman, A. Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core. J. Biol. Chem. 1994, 269, 6296–6305. [Google Scholar] [PubMed]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Sonoda, L.K.; Silman, I.; Sussman, J.L.; Rosenberry, T.L. Crystal structure of thioflavin T bound to the peripheral site of Torpedo californica acetylcholinesterase reveals how thioflavin T acts as a sensitive fluorescent reporter of ligand binding to the acylation site. J. Am. Chem. Soc. 2008, 130, 7856–7861. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Quinn, D.; Nair, H.; Silman, I.; Sussman, J. The X-ray structure of a transition state analog complex reveals the molecular origins of the catalytic power and substrate specificity of acetylcholinesterase. J. Am. Chem. Soc. 1996, 118, 2340–2346. [Google Scholar] [CrossRef]
- Bourne, Y.; Taylor, P.; Radic, Z.; Marchot, P. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 2003, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Auletta, J.T.; Johnson, J.L.; Rosenberry, T.L. Molecular basis of inhibition of substrate hydrolysis by a ligand bound to the peripheral site of acetylcholinesterase. Chem. Biol. Interact. 2010, 187, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Sussman, J.L.; Krejci, E.; Bon, S.; Chanal, P.; Massoulie, J.; Silman, I. Conversion of acetylcholinesterase to butyrylcholinesterase: Modeling and mutagenesis. Proc. Natl. Acad. Sci. USA 1992, 89, 10827–10831. [Google Scholar] [CrossRef] [PubMed]
- Tormos, J.R.; Wiley, K.L.; Seravalli, J.; Nachon, F.; Masson, P.; Nicolet, Y.; Quinn, D.M. The reactant state for substrate-activated turnover of acetylthiocholine by butyrylcholinesterase is a tetrahedral intermediate. J. Am. Chem. Soc. 2005, 127, 14538–14539. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Cusack, B.; Davies, M.P.; Fauq, A.; Rosenberry, T.L. Unmasking tandem site interaction in human acetylcholinesterase. Substrate activation with a cationic acetanilide substrate. Biochemistry 2003, 42, 5438–5452. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Legrand, P.; Bartels, C.F.; Froment, M.T.; Schopfer, L.M.; Lockridge, O. Role of aspartate 70 and tryptophan 82 in binding of succinyldithiocholine to human butyrylcholinesterase. Biochemistry 1997, 36, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Ehret-Sabatier, L.; Loew, D.; Colas, C.; van Dorsselaer, A.; Goeldner, M. Trp82 and Tyr332 are involved in two quaternary ammonium binding domains of human butyrylcholinesterase as revealed by photoaffinity labeling with [3H]DDF. Biochemistry 1998, 37, 10507–10513. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Xie, W.; Froment, M.T.; Levitsky, V.; Fortier, P.L.; Albaret, C.; Lockridge, O. Interaction between the peripheral site residues of human butyrylcholinesterase, D70 and Y332, in binding and hydrolysis of substrates. Biochim. Biophys. Acta 1999, 1433, 281–293. [Google Scholar] [CrossRef]
- Masson, P.; Froment, M.T.; Bartels, C.F.; Lockridge, O. Asp7O in the peripheral anionic site of human butyrylcholinesterase. Eur. J. Biochem. 1996, 235, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.R.; Martin, E.; Rosenberry, T.L.; Darvesh, S. Probing the peripheral site of human butyrylcholinesterase. Biochemistry 2012, 51, 7046–7053. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Wandhammer, M.; de Koning, M.; van Grol, M.; Loiodice, M.; Saurel, L.; Noort, D.; Goeldner, M.; Nachon, F. A step toward the reactivation of aged cholinesterases—Crystal structure of ligands binding to aged human butyrylcholinesterase. Chem. Biol. Interact. 2013, 203, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Brus, B.; Coquelle, N.; Sosic, I.; Sink, R.; Brazzolotto, X.; Mravljak, J.; Colletier, J.P.; Gobec, S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2015, 23, 4442–4452. [Google Scholar] [CrossRef] [PubMed]
- Brus, B.; Kosak, U.; Turk, S.; Pislar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.P.; Gobec, S. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef] [PubMed]
- Kosak, U.; Brus, B.; Knez, D.; Sink, R.; Zakelj, S.; Trontelj, J.; Pislar, A.; Slenc, J.; Gobec, M.; Zivin, M.; et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [PubMed]
- Kosak, U.; Knez, D.; Coquelle, N.; Brus, B.; Pislar, A.; Nachon, F.; Brazzolotto, X.; Kos, J.; Colletier, J.P.; Gobec, S. N-Propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: Potential multifunctional anti-Alzheimer’s agents. Bioorg. Med. Chem. 2017, 25, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Dighe, S.N.; Deora, G.S.; De la Mora, E.; Nachon, F.; Chan, S.; Parat, M.O.; Brazzolotto, X.; Ross, B.P. Discovery and Structure-Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening. J. Med. Chem. 2016, 59, 7683–7689. [Google Scholar] [CrossRef] [PubMed]
- Driant, T.; Nachon, F.; Ollivier, C.; Renard, P.Y.; Derat, E. On the Influence of the Protonation States of Active Site Residues on AChE Reactivation: A QM/MM Approach. ChemBioChem 2017, 18, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.; Mishra, B.K.; Andersson, C.D.; Ekstrom, F.; Linusson, A. The Nature of Activated Non-classical Hydrogen Bonds: A Case Study on Acetylcholinesterase-Ligand Complexes. Chemistry 2016, 22, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Lappi, S. Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding. Biochemistry 1975, 14, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; Cusack, B.; Mallender, W.D.; El Achab, R.E.; Morral, J.; Munoz-Torrero, D.; Rosenberry, T.L. Huprine X is a novel high-affinity inhibitor of acetylcholinesterase that is of interest for treatment of Alzheimer’s disease. Mol. Pharmacol. 2000, 57, 409–417. [Google Scholar] [PubMed]
- Dvir, H.; Wong, D.M.; Harel, M.; Barril, X.; Orozco, M.; Luque, F.J.; Munoz-Torrero, D.; Camps, P.; Rosenberry, T.L.; Silman, I.; et al. 3D structure of Torpedo californica acetylcholinesterase complexed with huprine X at 2.1 A resolution: Kinetic and molecular dynamic correlates. Biochemistry 2002, 41, 2970–2981. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Carletti, E.; Colletier, J.P.; Weik, M.; Nachon, F.; Jean, L.; Renard, P.Y. Huprine derivatives as sub-nanomolar human acetylcholinesterase inhibitors: From rational design to validation by X-ray crystallography. ChemMedChem 2012, 7, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Foucault, R.; Gillon, E.; Bohn, P.; Nachon, F.; Jean, L.; Renard, P.Y. New huprine derivatives functionalized at position 9 as highly potent acetylcholinesterase inhibitors. ChemMedChem 2011, 6, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Brazzolotto, X.; Wandhammer, M.; Ronco, C.; Trovaslet, M.; Jean, L.; Lockridge, O.; Renard, P.Y.; Nachon, F. Human butyrylcholinesterase produced in insect cells: Huprine-based affinity purification and crystal structure. FEBS J. 2012, 279, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Wandhammer, M.; Nicolet, Y.; Schopfer, L.M.; Masson, P.; Lockridge, O. X-ray crystallographic snapshots of reaction intermediates in the G117H mutant of human butyrylcholinesterase, a nerve agent target engineered into a catalytic bioscavenger. Biochem. J. 2011, 434, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Redman, A.M.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in active site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Biochemistry 1997, 36, 14642–14651. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Mallender, W.D.; Inestrosa, N.C.; Rosenberry, T.L. Thioflavin T is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J. Biol. Chem. 2001, 276, 23282–23287. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.A.; Zheng, X.; Sept, D.; Auletta, J.T.; Rosenberry, T.L.; Marshall, G.R. Drug-like leads for steric discrimination between substrate and inhibitors of human acetylcholinesterase. Chem. Biol. Drug Des. 2011, 78, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Beri, V.; Wildman, S.A.; Shiomi, K.; Al-Rashid, Z.F.; Cheung, J.; Rosenberry, T.L. The natural product dihydrotanshinone I provides a prototype for uncharged inhibitors that bind specifically to the acetylcholinesterase peripheral site with nanomolar affinity. Biochemistry 2013, 52, 7486–7499. [Google Scholar] [CrossRef] [PubMed]
- Falconer, R.J. Applications of isothermal titration calorimetry—The research and technical developments from 2011 to 2015. J. Mol. Recognit. 2016, 29, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Sinko, G.; Kovarik, Z.; Reiner, E.; Simeon-Rudolf, V.; Stojan, J. Mechanism of stereoselective interaction between butyrylcholinesterase and ethopropazine enantiomers. Biochimie 2011, 93, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Martin, P.K.; Nix, A.J.; Wildman, S.A.; Cheung, J.; Snyder, S.A.; Tan, R.X. Hopeahainol A binds reversibly at the acetylcholinesterase (AChE) peripheral site and inhibits enzyme activity with a novel higher order concentration dependence. Chem. Biol. Interact. 2016, 259, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Allgardsson, A.; David Andersson, C.; Akfur, C.; Worek, F.; Linusson, A.; Ekstrom, F. An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet. Molecules 2017, 22, 1433. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; McDonald, R.S.; Penwell, A.; Conrad, S.; Darvesh, K.V.; Mataija, D.; Gomez, G.; Caines, A.; Walsh, R.; Martin, E. Structure-activity relationships for inhibition of human cholinesterases by alkyl amide phenothiazine derivatives. Bioorg. Med. Chem. 2005, 13, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; McDonald, R.S.; Darvesh, K.V.; Mataija, D.; Conrad, S.; Gomez, G.; Walsh, R.; Martin, E. Selective reversible inhibition of human butyrylcholinesterase by aryl amide derivatives of phenothiazine. Bioorg. Med. Chem. 2007, 15, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Darvesh, K.V.; McDonald, R.S.; Mataija, D.; Walsh, R.; Mothana, S.; Lockridge, O.; Martin, E. Carbamates with differential mechanism of inhibition toward acetylcholinesterase and butyrylcholinesterase. J. Med. Chem. 2008, 51, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Pottie, I.R.; Darvesh, K.V.; McDonald, R.S.; Walsh, R.; Conrad, S.; Penwell, A.; Mataija, D.; Martin, E. Differential binding of phenothiazine urea derivatives to wild-type human cholinesterases and butyrylcholinesterase mutants. Bioorg. Med. Chem. 2010, 18, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Colletier, J.P.; Schopfer, L.M.; Santoni, G.; Masson, P.; Lockridge, O.; Nachon, F.; Weik, M. Inhibition pathways of the potent organophosphate CBDP with cholinesterases revealed by X-ray crystallographic snapshots and mass spectrometry. Chem. Res. Toxicol. 2013, 26, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Koellner, G.; Kryger, G.; Millard, C.B.; Silman, I.; Sussman, J.L.; Steiner, T. Active-site gorge and buried water molecules in crystal structures of acetylcholinesterase from Torpedo californica. J. Mol. Biol. 2000, 296, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Aurbek, N.; Gillon, E.; Loiodice, M.; Nicolet, Y.; Fontecilla-Camps, J.C.; Masson, P.; Thiermann, H.; Nachon, F.; Worek, F. Structure-activity analysis of aging and reactivation of human butyrylcholinesterase inhibited by analogues of tabun. Biochem. J. 2009, 421, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Wandhammer, M.; Carletti, E.; Van der Schans, M.; Gillon, E.; Nicolet, Y.; Masson, P.; Goeldner, M.; Noort, D.; Nachon, F. Structural study of the complex stereoselectivity of human butyrylcholinesterase for the neurotoxic V-agents. J. Biol. Chem. 2011, 286, 16783–16789. [Google Scholar] [CrossRef] [PubMed]
- Golicnik, M.; Sinko, G.; Simeon-Rudolf, V.; Grubic, Z.; Stojan, J. Kinetic model of ethopropazine interaction with horse serum butyrylcholinesterase and its docking into the active site. Arch. Biochem. Biophys. 2002, 398, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Nicolet, Y.; Viguie, N.; Masson, P.; Fontecilla-Camps, J.C.; Lockridge, O. Engineering of a monomeric and low-glycosylated form of human butyrylcholinesterase: Expression, purification, characterization and crystallization. Eur. J. Biochem. 2002, 269, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; McDonald, R.S.; Darvesh, K.V.; Mataija, D.; Mothana, S.; Cook, H.; Carneiro, K.M.; Richard, N.; Walsh, R.; Martin, E. On the active site for hydrolysis of aryl amides and choline esters by human cholinesterases. Bioorg. Med. Chem. 2006, 14, 4586–4599. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Kumar, R.; Roberts, S.; Walsh, R.; Martin, E. Butyrylcholinesterase-Mediated enhancement of the enzymatic activity of trypsin. Cell. Mol. Neurobiol. 2001, 21, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Li, H.; Li, B.; Ekstrom, F.; Nicolet, Y.; Loiodice, M.; Gillon, E.; Froment, M.T.; Lockridge, O.; Schopfer, L.M.; et al. Aging of cholinesterases phosphylated by tabun proceeds through O-dealkylation. J. Am. Chem. Soc. 2008, 130, 16011–16020. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Collaborative-Computational-Project-4, The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763.
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.K.; Gruswitz, F. HOLLOW: Generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 2008, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Bernhard, S.A. Studies of catalysis by acetylcholinesterase. I. Fluorescent titration with a carbamoylating agent. Biochemistry 1971, 10, 4114–4120. [Google Scholar] [CrossRef] [PubMed]
- Eastman, J.; Wilson, E.J.; Cervenansky, C.; Rosenberry, T.L. Fasciculin 2 binds to the peripheral site on acetylcholinesterase and inhibits substrate hydrolysis by slowing a step involving proton transfer during enzyme acylation. J. Biol. Chem. 1995, 270, 19694–19701. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | KI (µM) | K12/K1 |
|---|---|---|
| Decamethonium | 4.5 ± 0.2 | - |
| Propidium | 0.43 ± 0.06 a | 118 |
| Thioflavin-T | 0.8 ± 0.1 a | >200 |
| Edrophonium | 49 ± 3 a | >200 |
| Enzyme | Compound | KD (±SD) (µM) | n (±SD) | ΔH (±SD) (kJ·mol−1) | ΔG (±SD) (kJ·mol−1) | TΔS (kJ·mol−1) | ∆S (J·mol−1·K−1) |
|---|---|---|---|---|---|---|---|
| hAChE | Edrophonium | 1.4 ± 0.4 | 0.8 ± 0.3 | −26.4 ± 7.9 | −33.6 ± 0.8 | −7.2 | −24.2 |
| Ethopropazine | 26.7 ± 14.7 | 1.5 ± 0.3 | −10.9 ± 1.1 | −26.4 ± 1.6 | −15.5 | −52.0 | |
| Propidium | 6.7 ± 0.7 | 0.8 ± 0.1 | −45.4 ± 5.0 | −29.6 ± 0.3 | 15.8 | 52.9 | |
| Thioflavin-T | 17.7 | 1 | −23.7 | −27.2 | −3.5 | −11.8 | |
| hBChE | Edrophonium | 27.2 ± 2.4 | 0.9 ± 0.1 | −12.6 ± 1.7 | −26.1 ± 0.3 | −13.5 | −45.3 |
| Ethopropazine | 1.1 ± 0.1 | 1.0 ± 0.1 | −43.6 ± 6.4 | −34.1 ± 0.2 | 9.5 | 31.8 | |
| Propidium | 2.1 ± 0.4 | 0.8 ± 0.1 | −43.1 ± 19.1 | −31.7 ± 0.4 | 11.4 | 38.2 | |
| Thioflavin-T | 2.6 ± 0.5 | 2.2 ± 0.1 | −11.4 ± 0.6 | −32.0 ± 0.6 | −20.6 | −69.1 |
| Compound | AChE | BChE | ||
|---|---|---|---|---|
| KI (µM) a | K12/K1 a | KI (µM) a | K12/K1 a | |
| Thioflavin T | 3.9 ± 0.4 | - | 0.8 ± 0.1 b | - |
| Propidium | 7.2 ± 0.6 | 93 (C) | 0.43 ± 0.06 | 23 (PC) |
| Edrophonium | 0.82 ± 0.05 | 2.9 ± 0.2 (NC) a | 49 ± 3 | 60 (PC) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098. https://doi.org/10.3390/molecules22122098
Rosenberry TL, Brazzolotto X, Macdonald IR, Wandhammer M, Trovaslet-Leroy M, Darvesh S, Nachon F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules. 2017; 22(12):2098. https://doi.org/10.3390/molecules22122098
Chicago/Turabian StyleRosenberry, Terrone L., Xavier Brazzolotto, Ian R. Macdonald, Marielle Wandhammer, Marie Trovaslet-Leroy, Sultan Darvesh, and Florian Nachon. 2017. "Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study" Molecules 22, no. 12: 2098. https://doi.org/10.3390/molecules22122098