Species-Specific Inactivation of Triosephosphate Isomerase from Trypanosoma brucei: Kinetic and Molecular Dynamics Studies

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
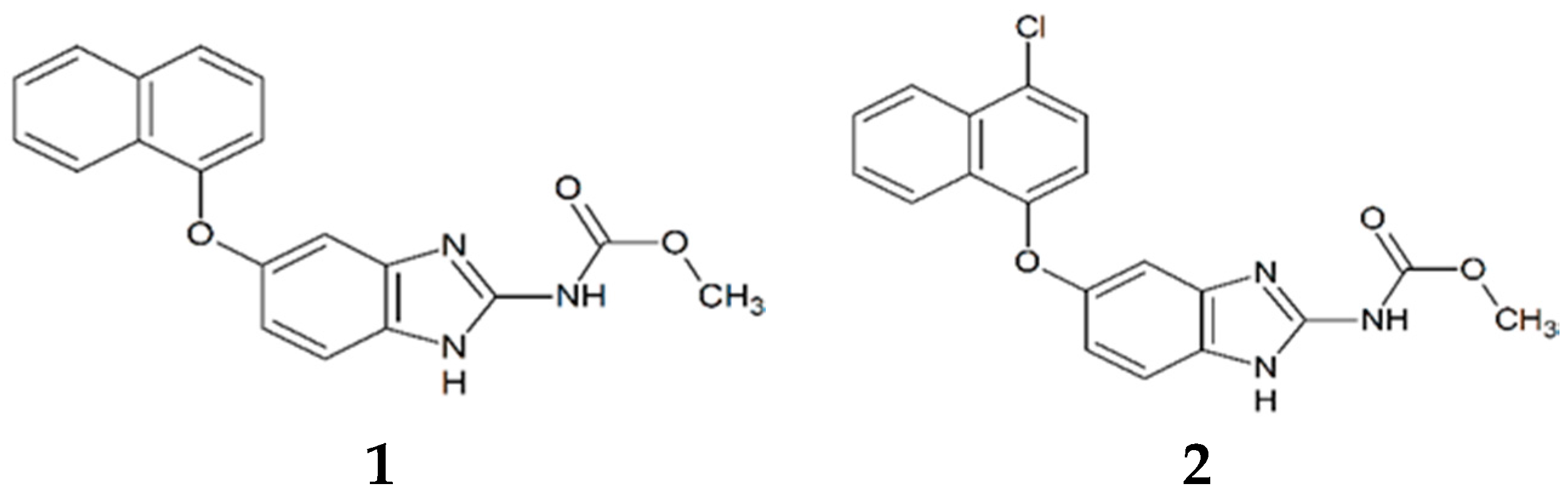
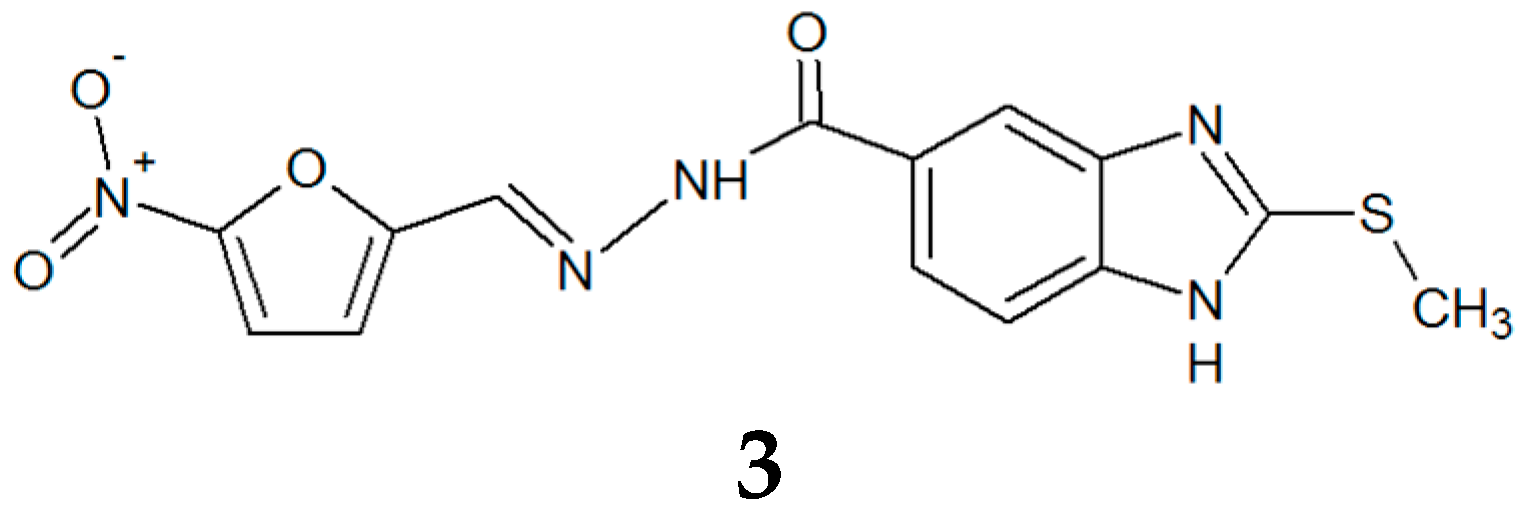
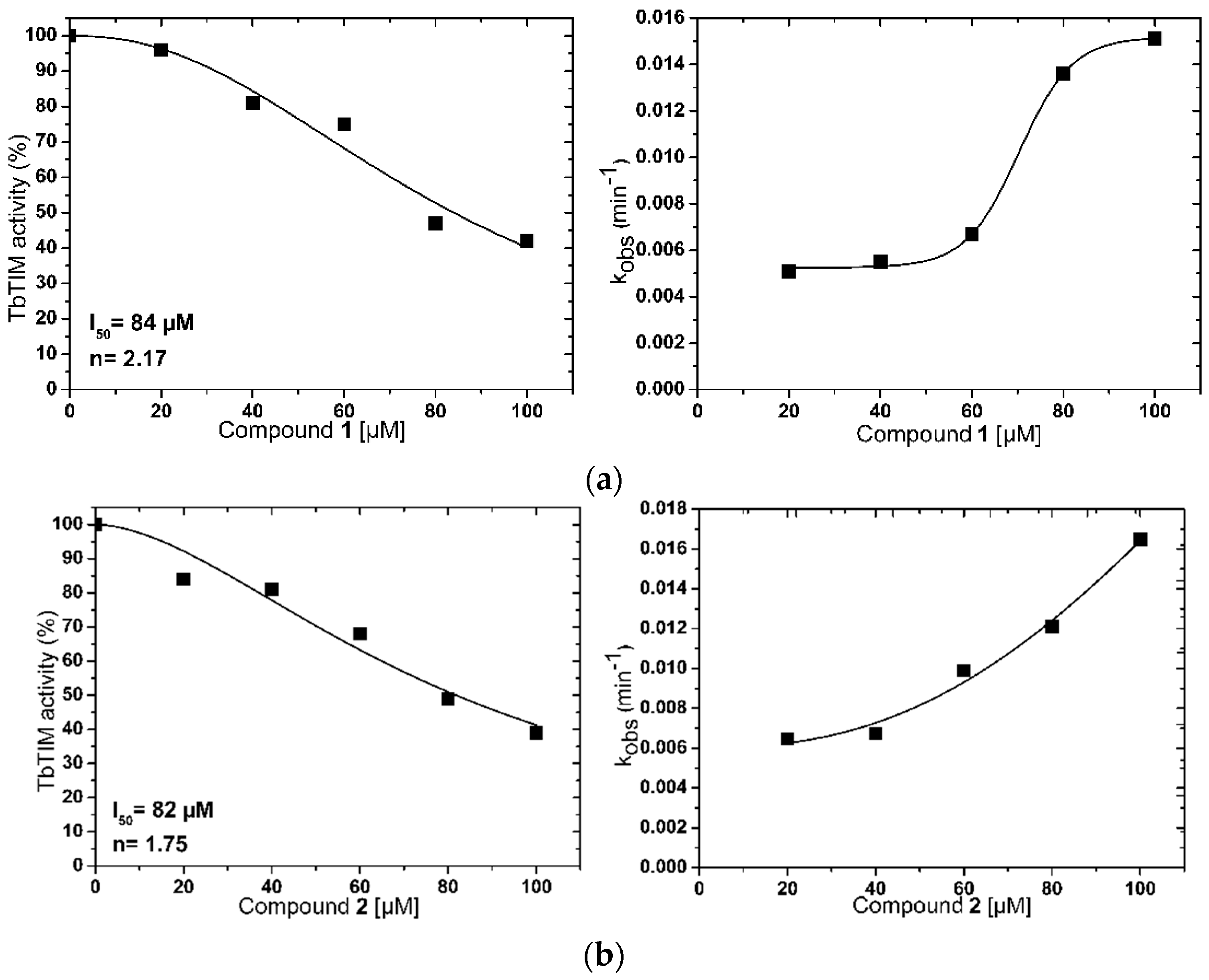
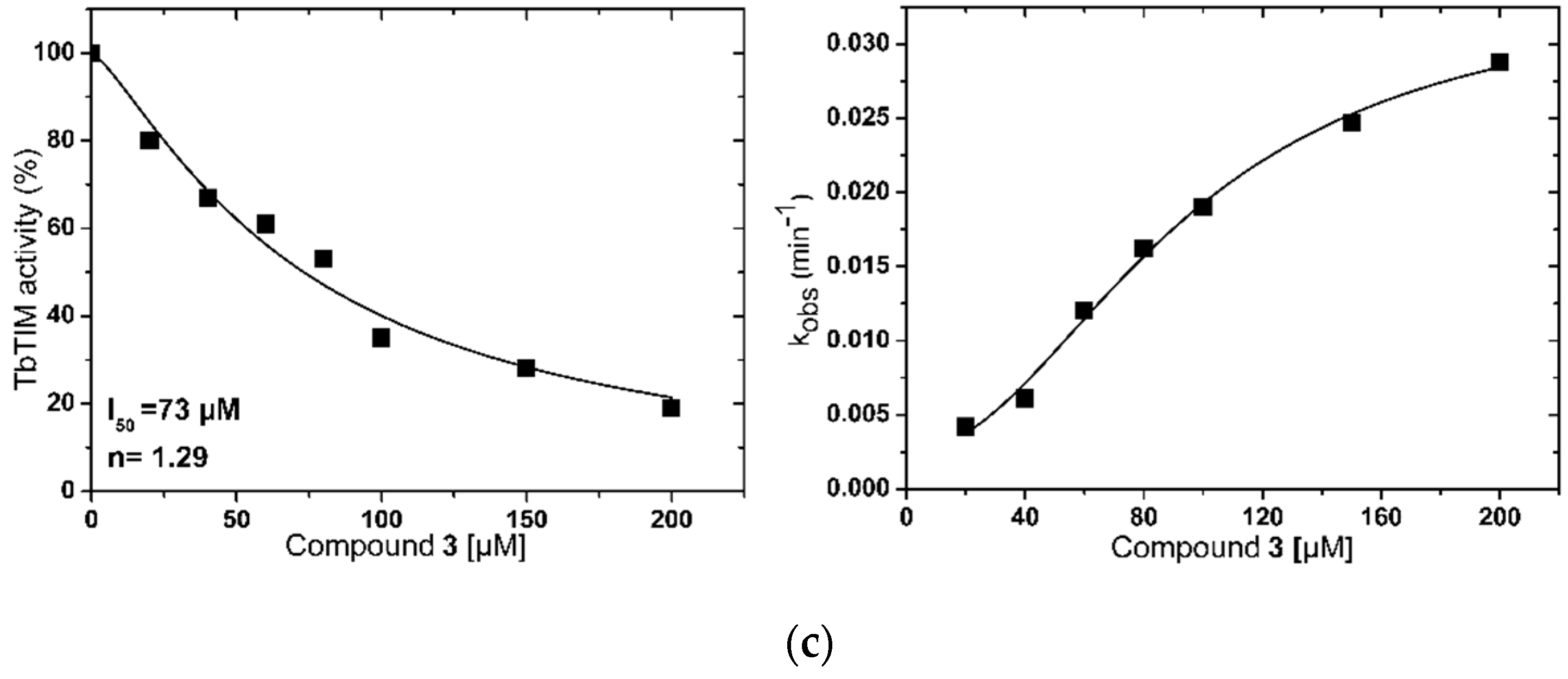
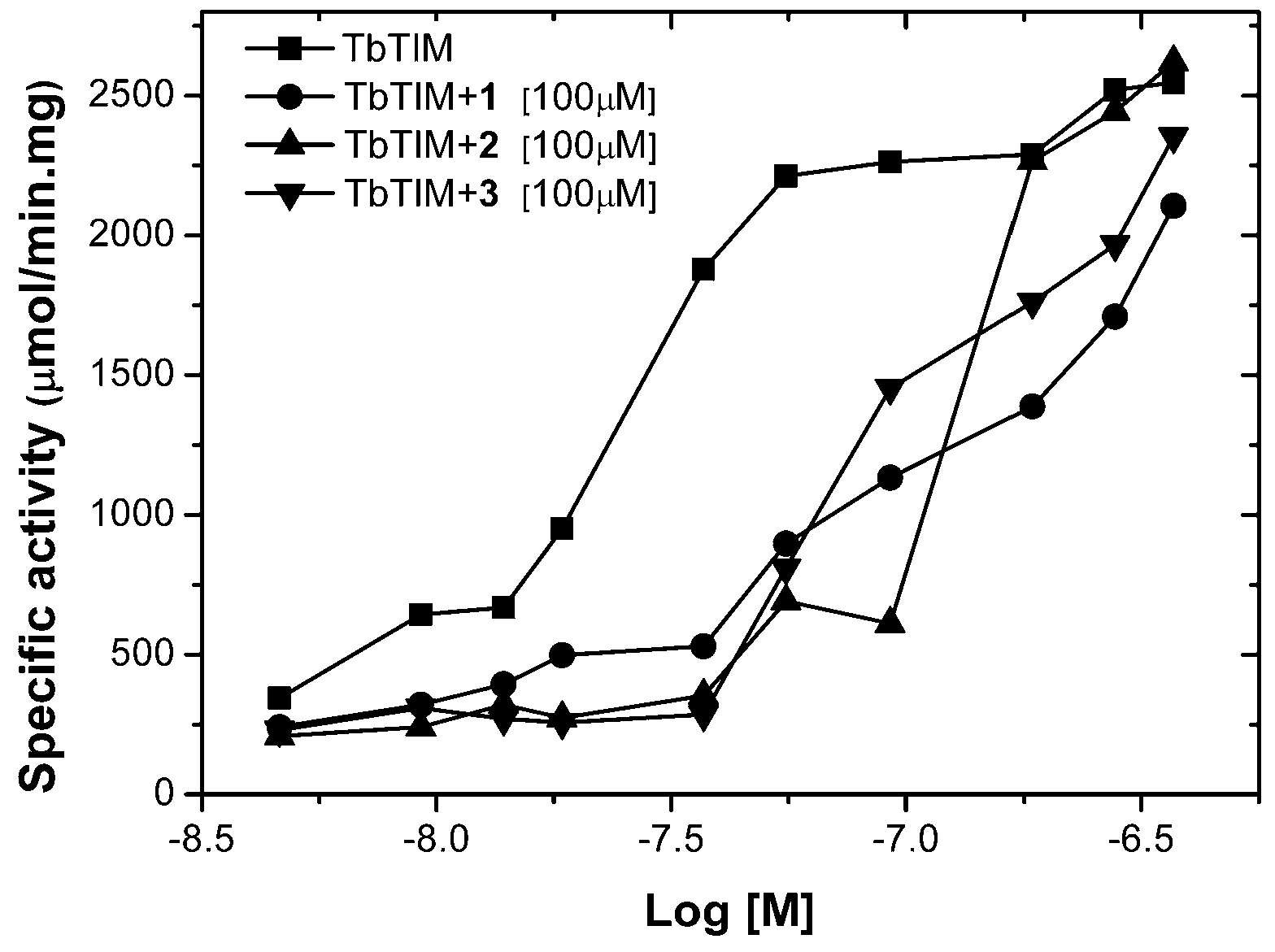
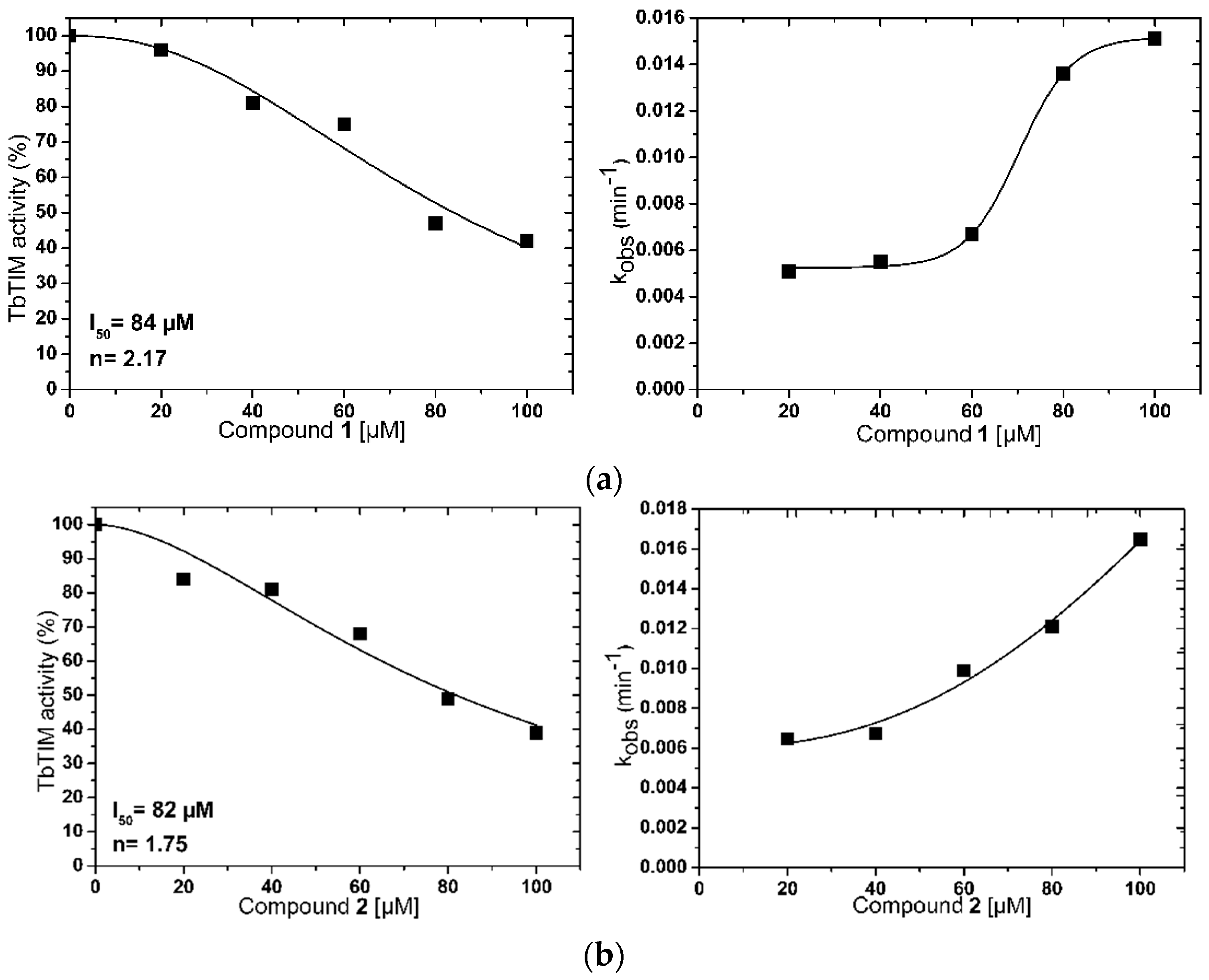
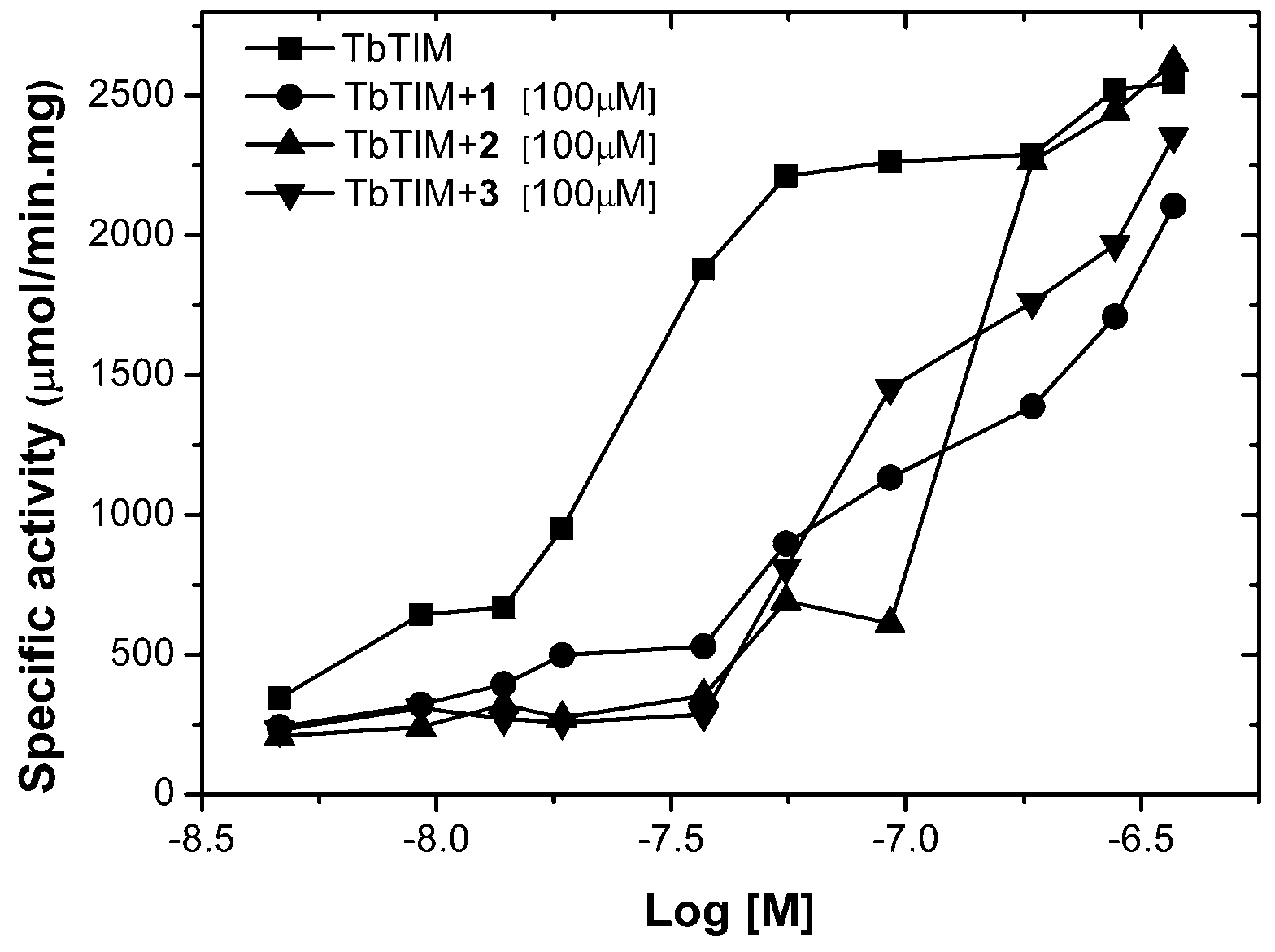
2.1. TbTIM Inactivation
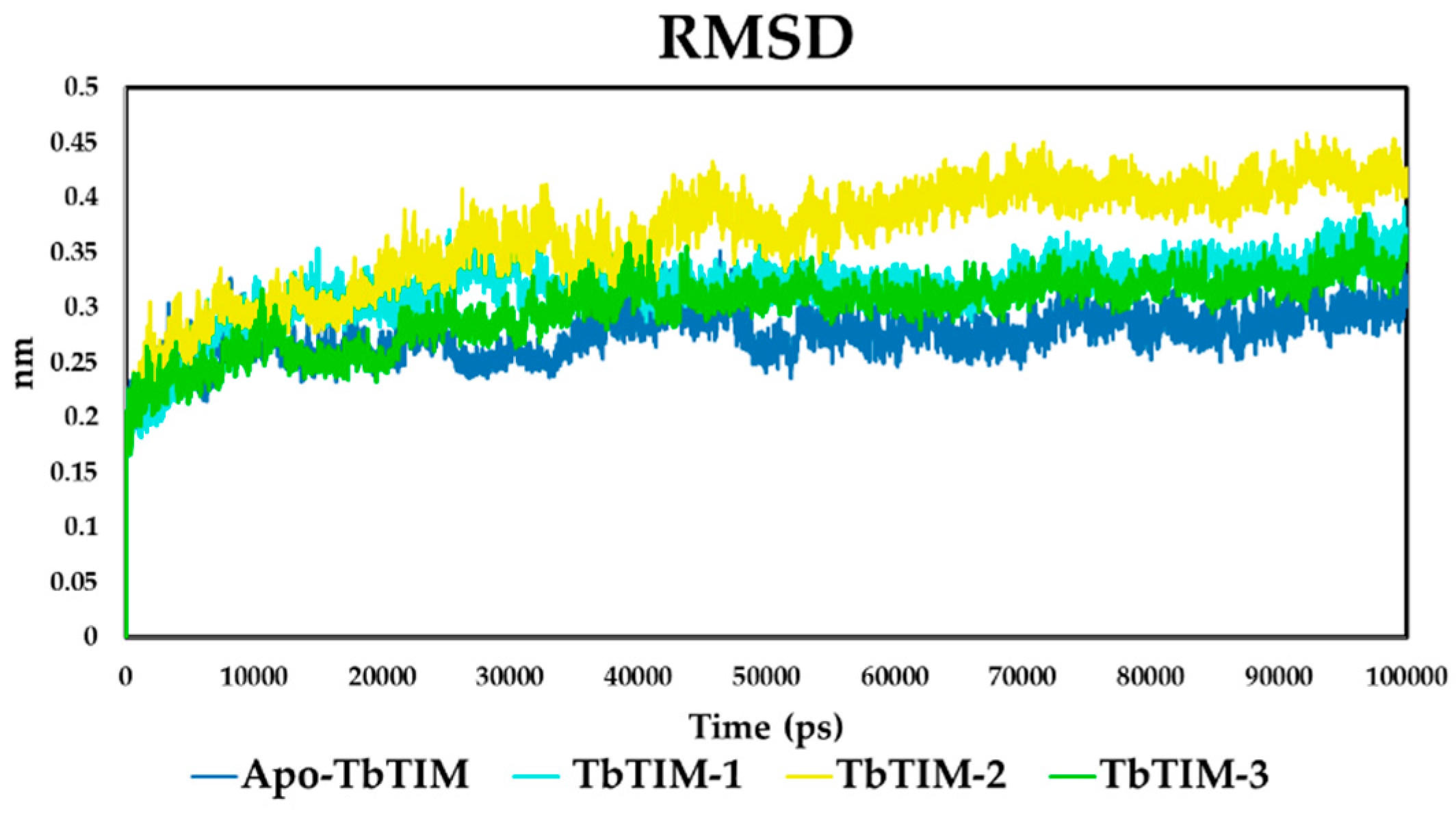
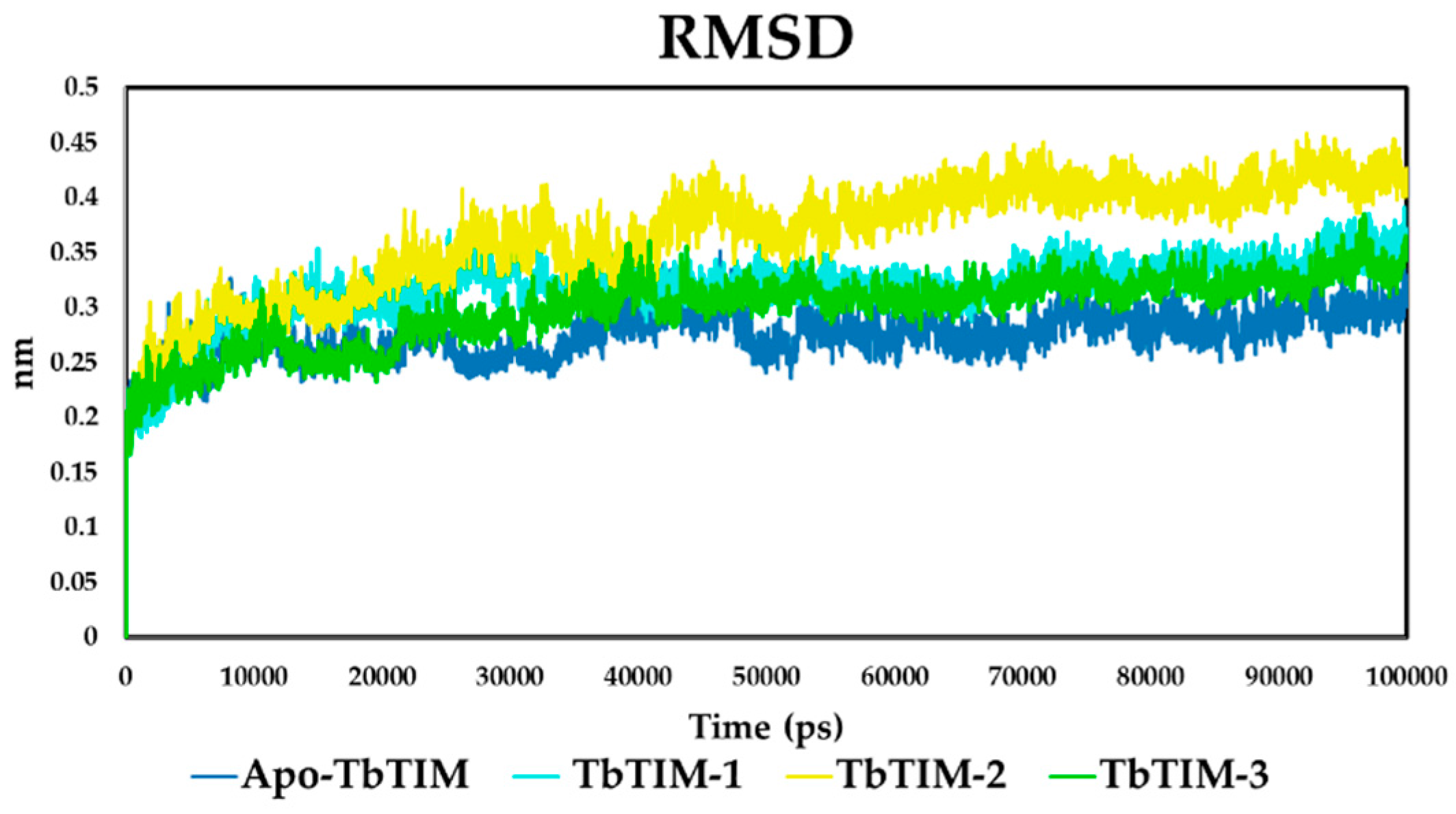
2.2. Molecular Dynamics
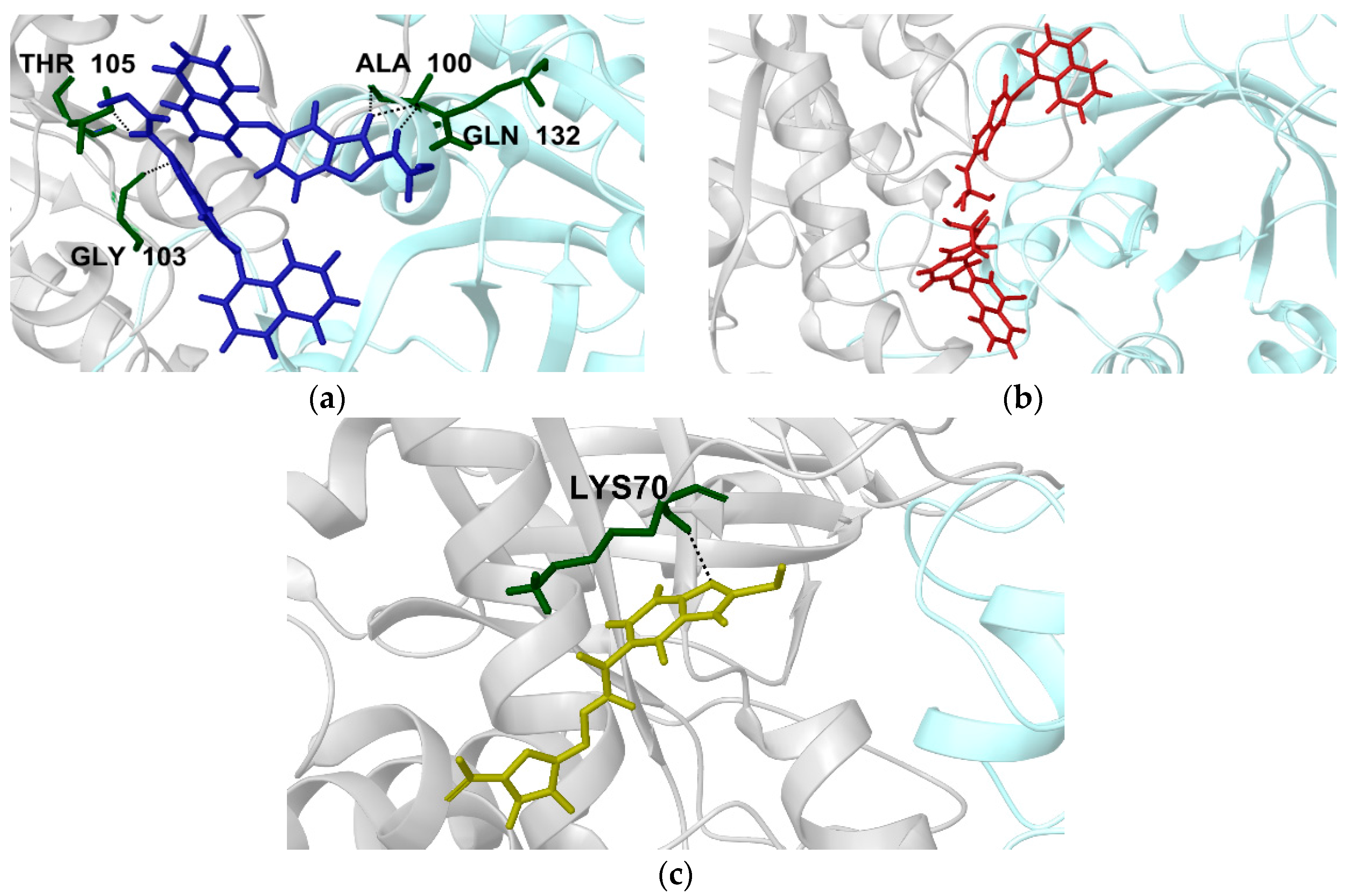
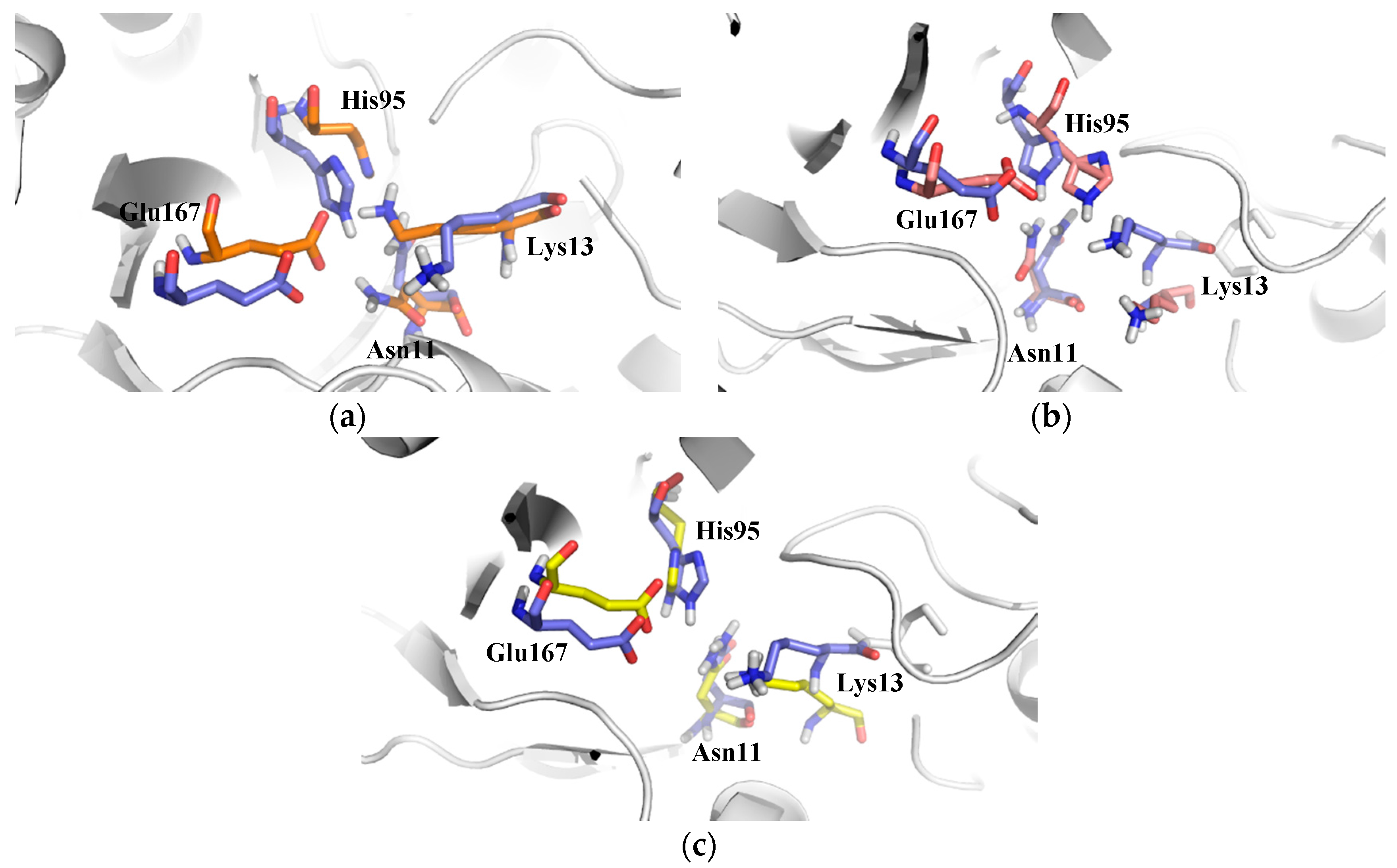
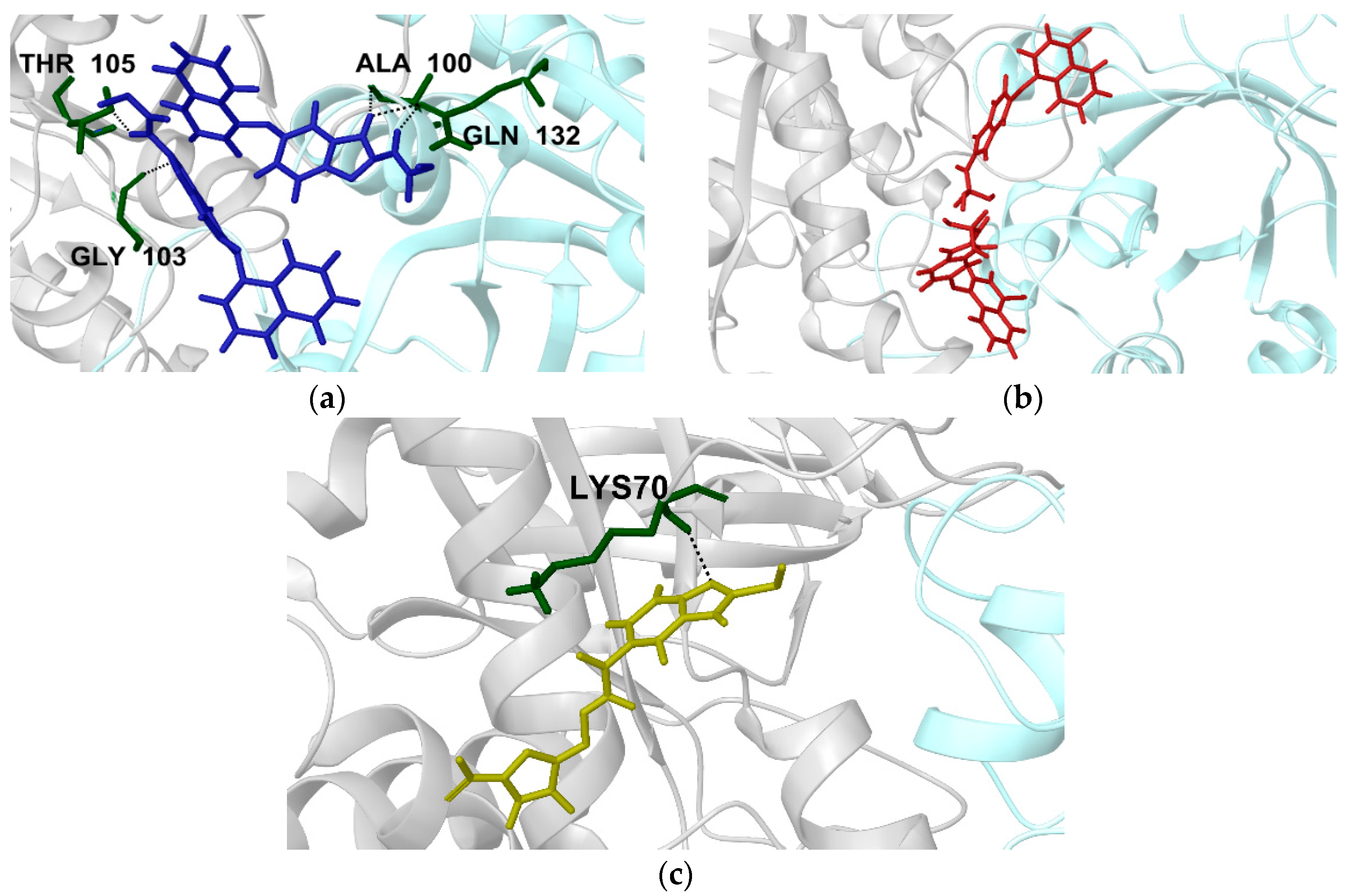
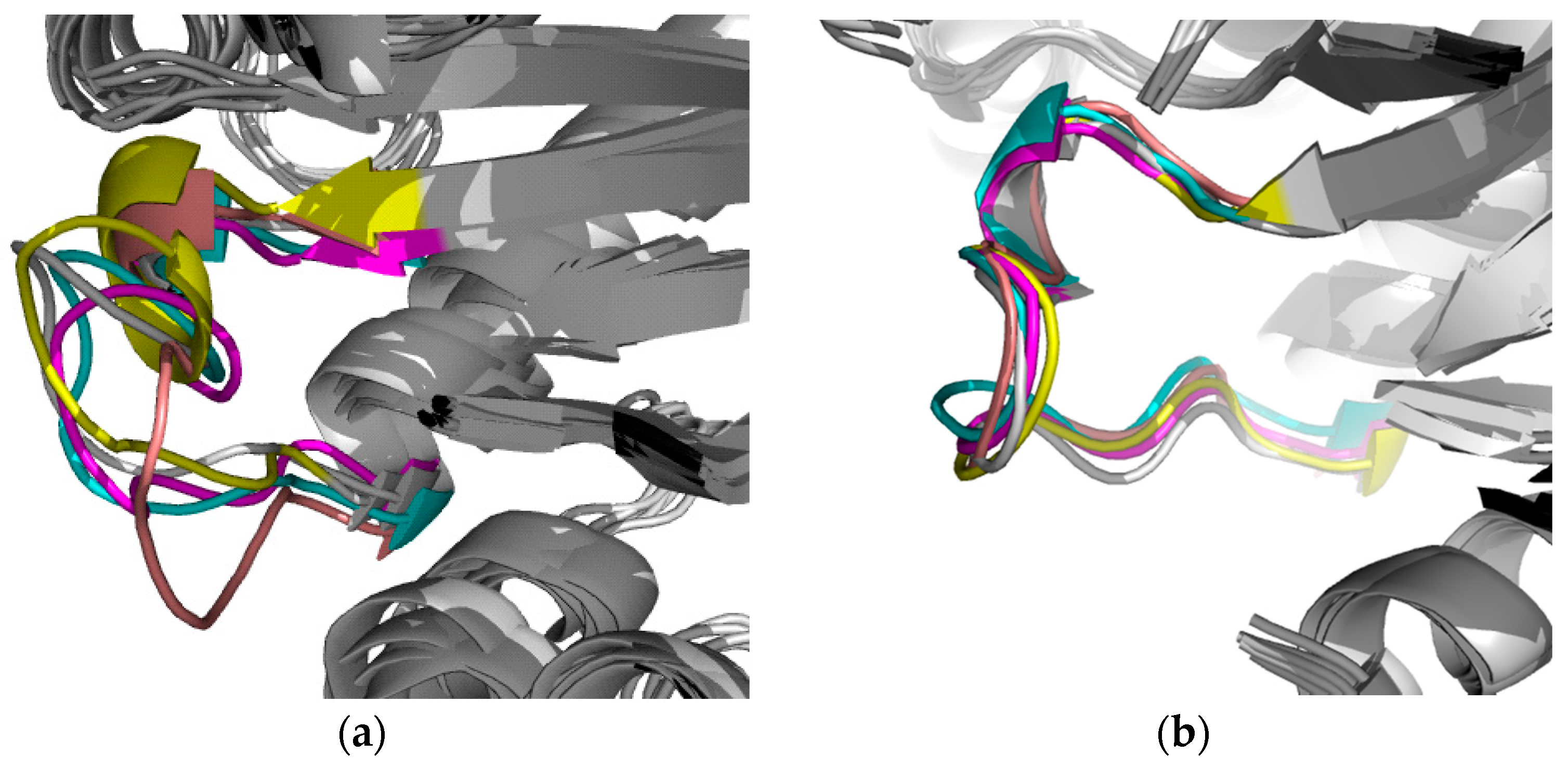
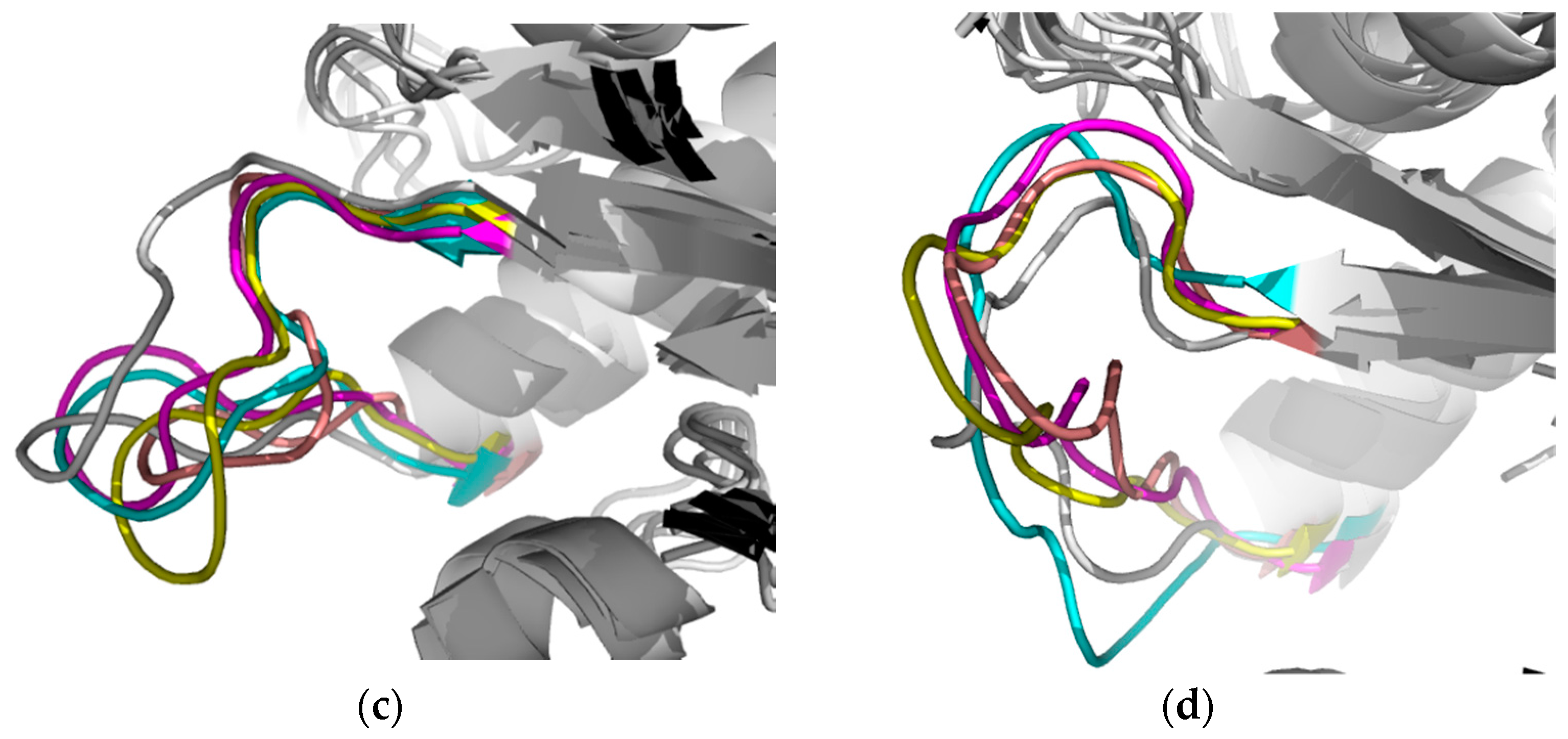
2.2.1. Structural Analyses of the TbTIM-Ligand Complex
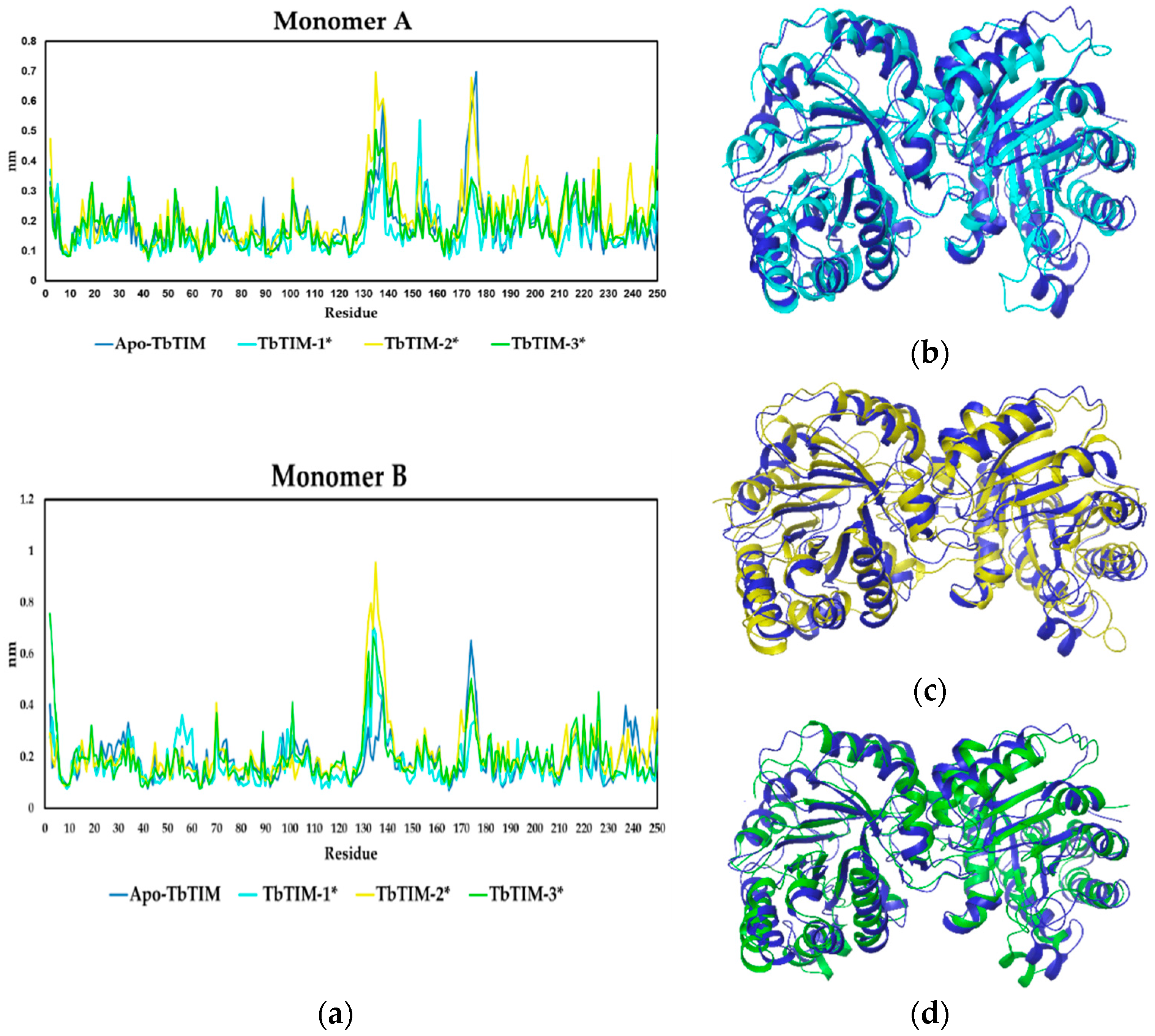
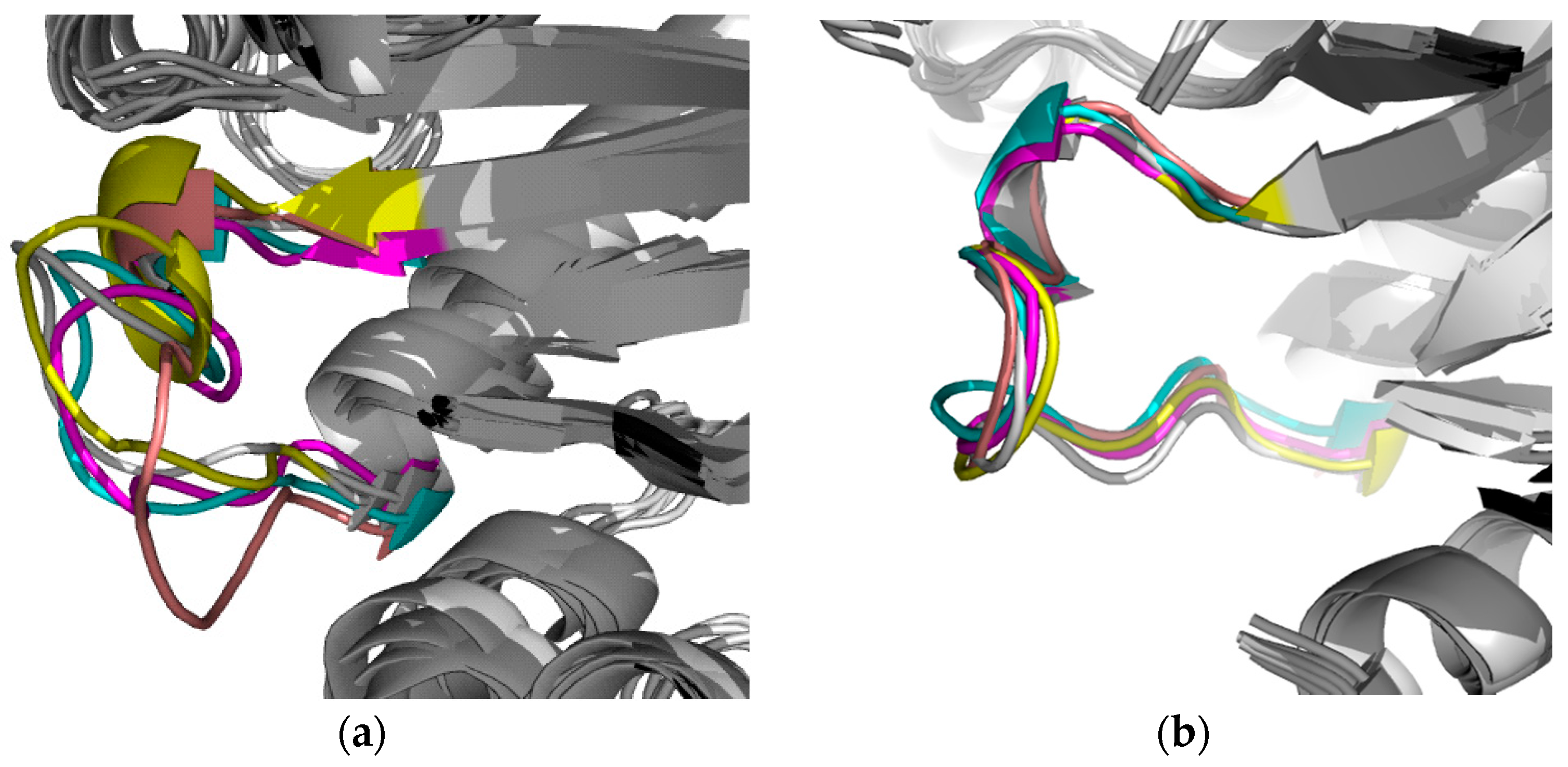
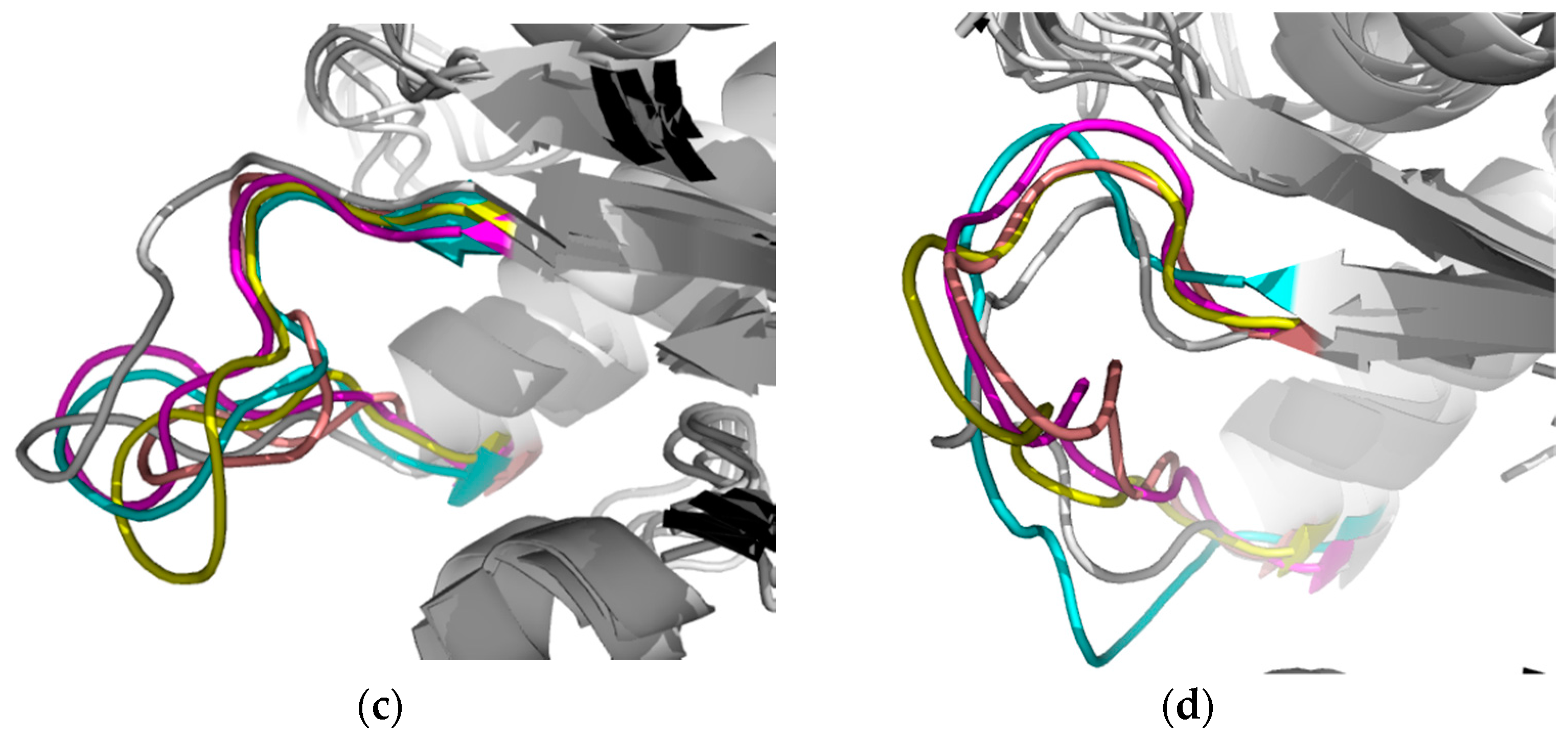
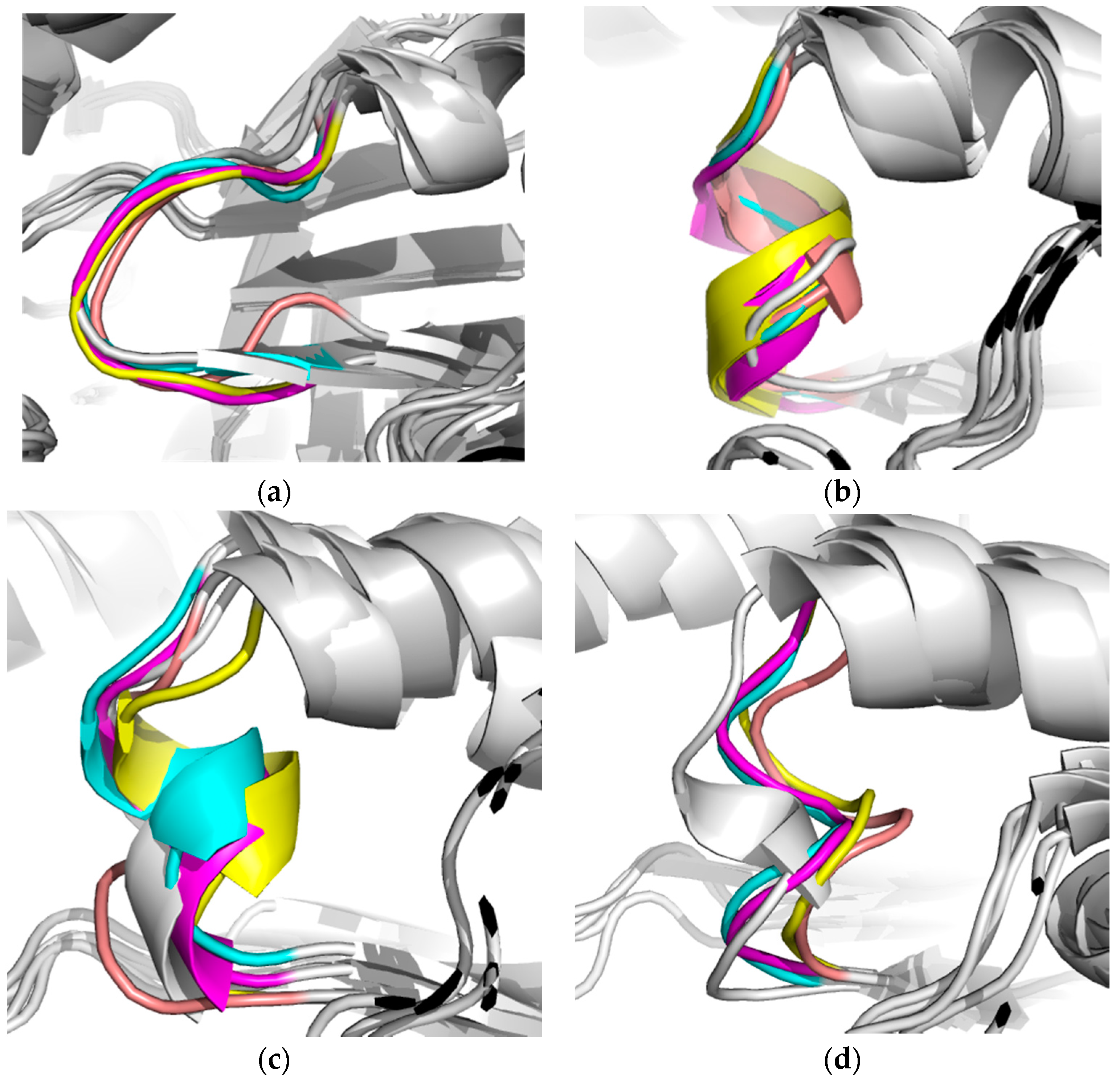
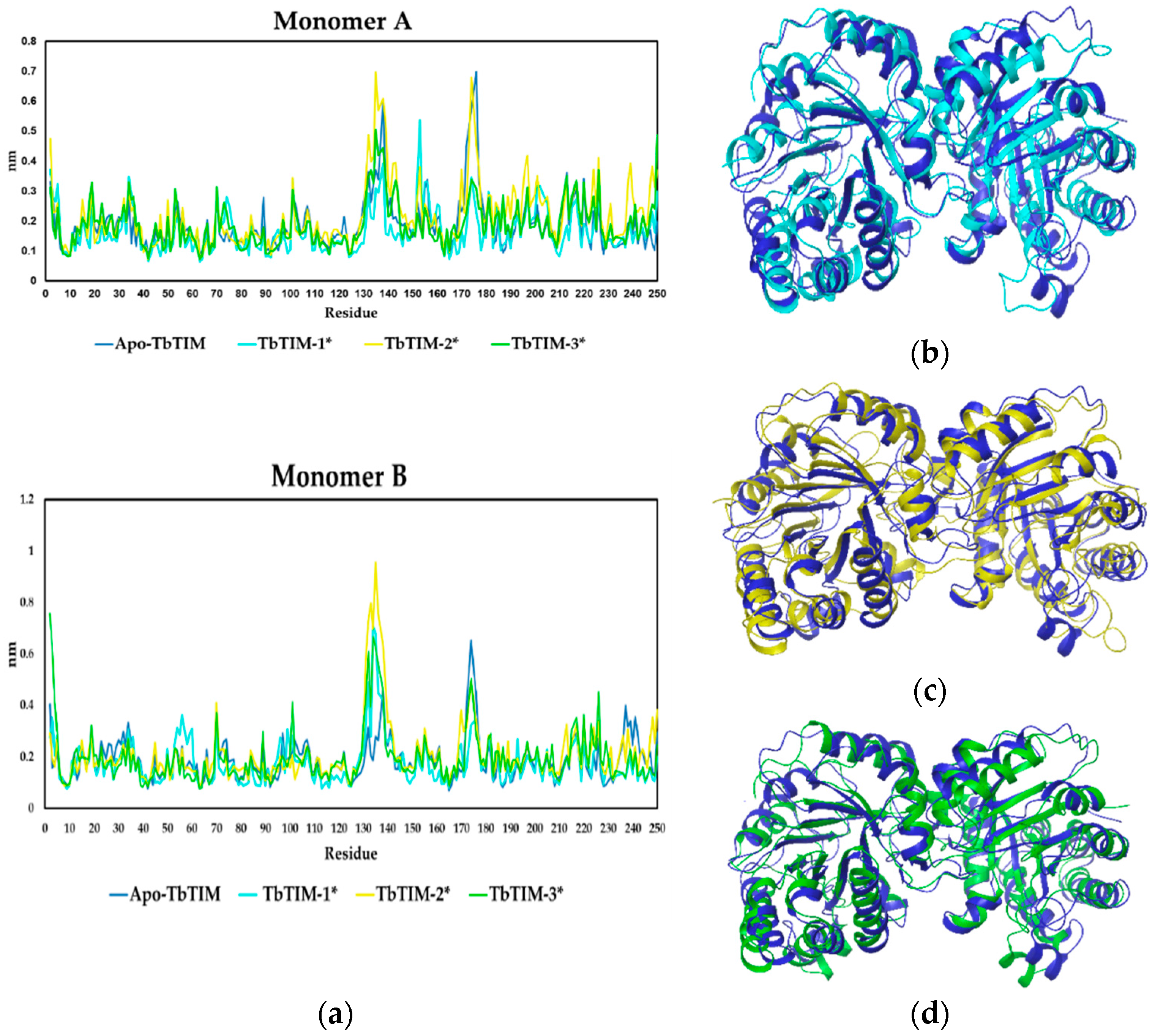
2.2.2. TbTIM Loop 6 and Loop 8 Dynamics
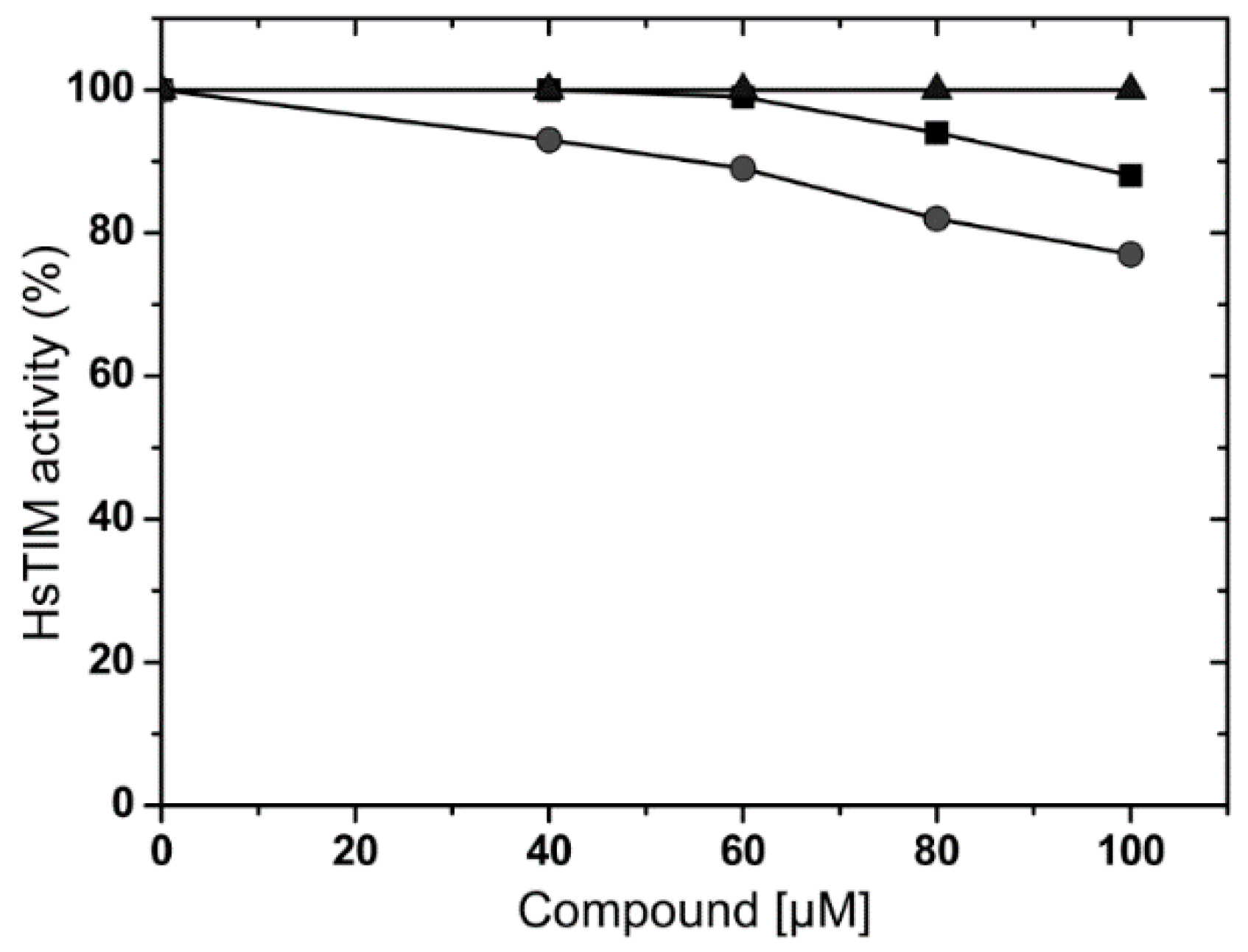
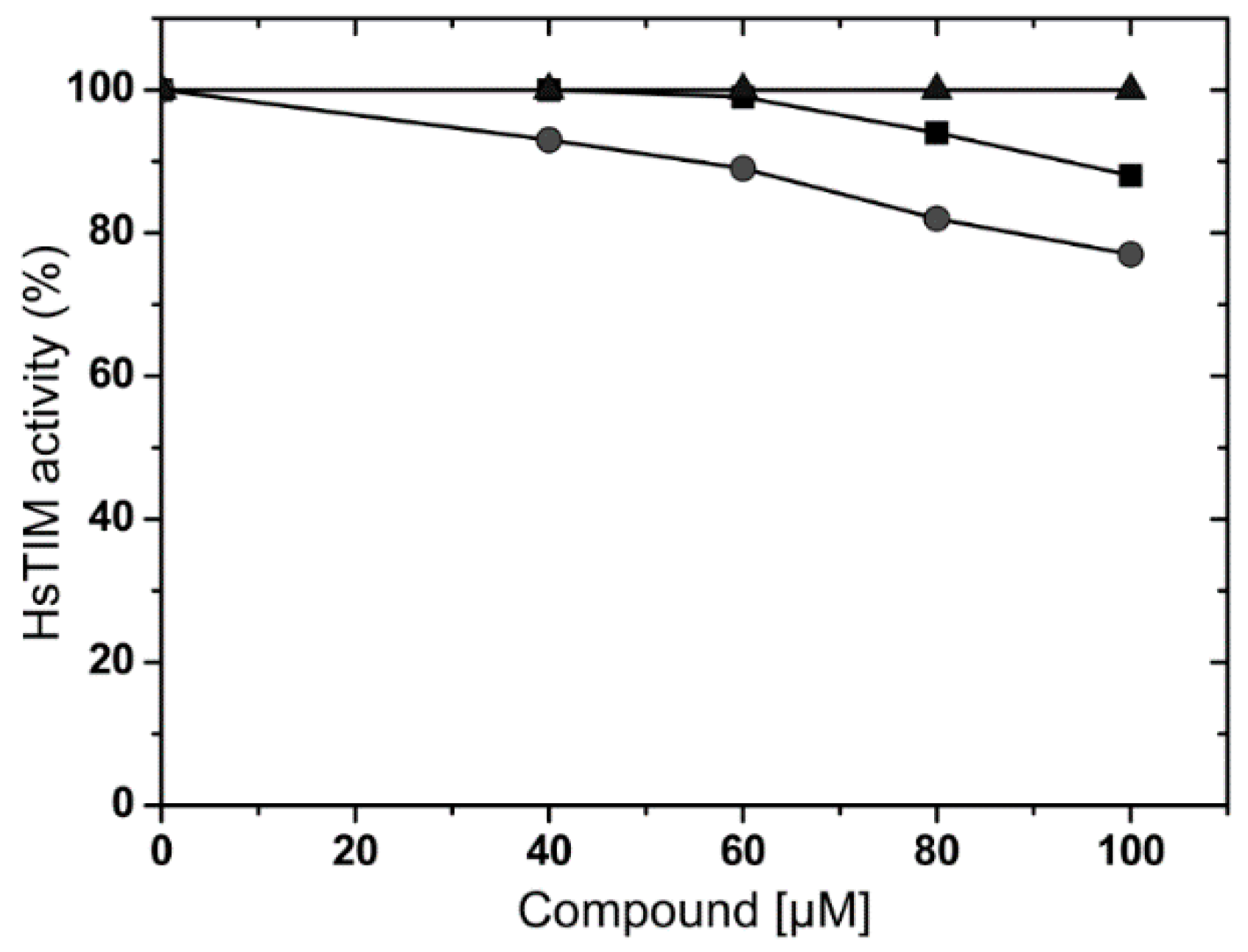
2.3. Effects of Compounds on Human Triosphosphate Isomerase (HsTIM)
2.4. In Silico Analysis of ADME-Tox Properties
3. Materials and Methods
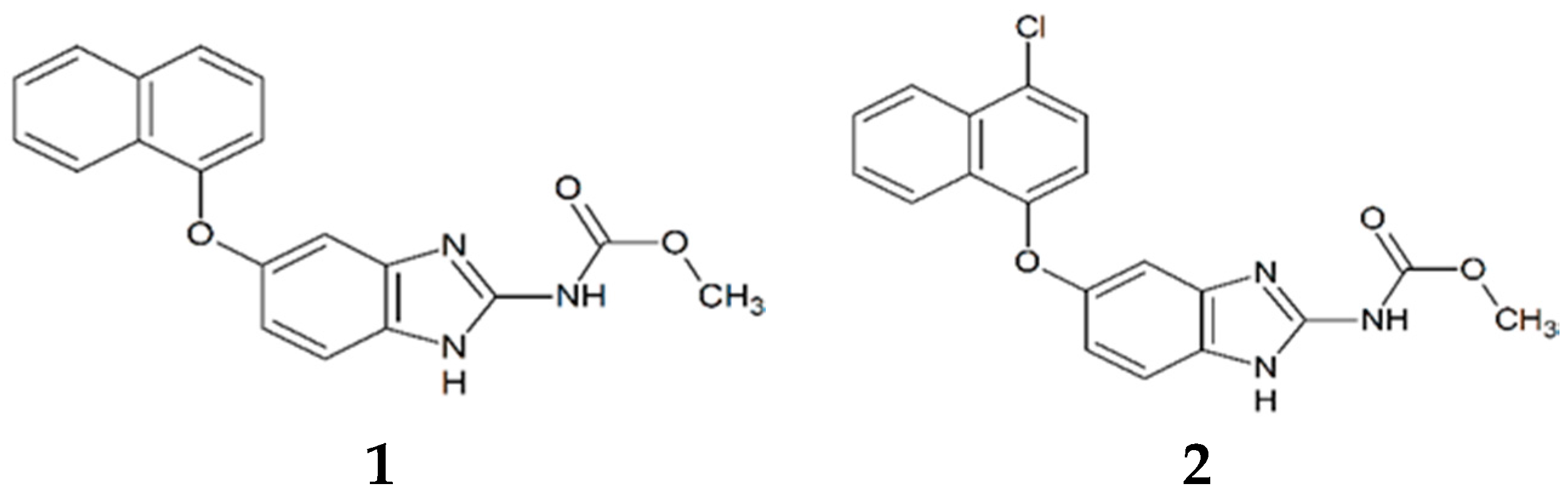
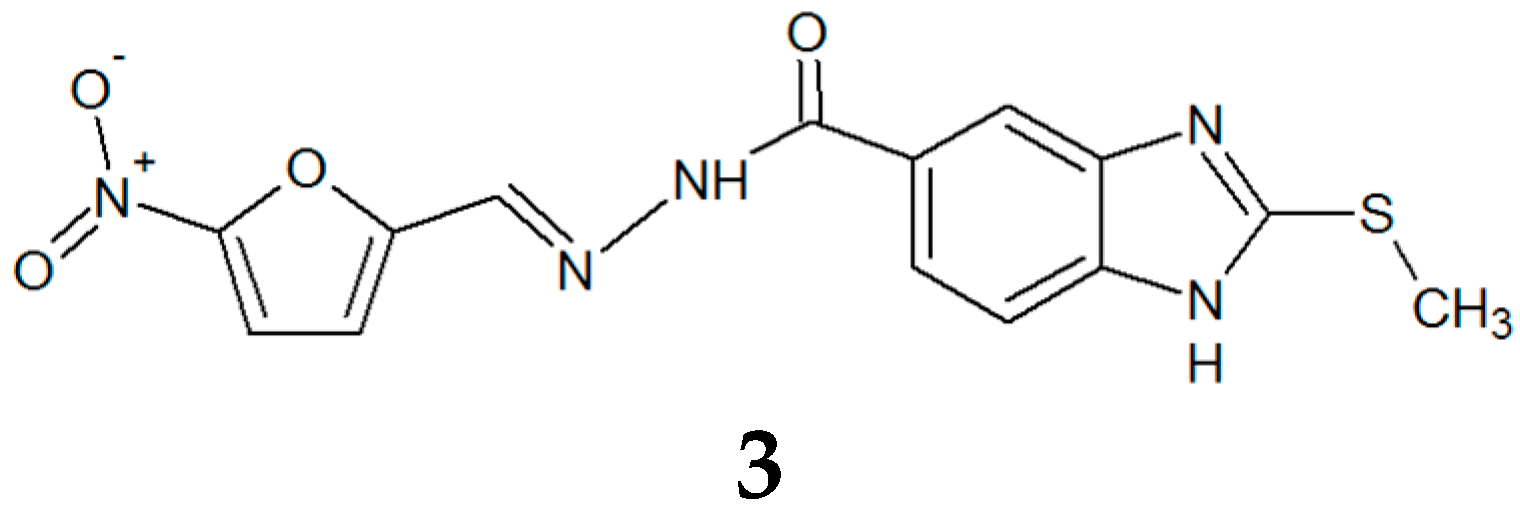
3.1. Synthesis and Molecular Characterization
3.2. Expression and Purification of TIMs
3.3. Enzymatic Activity Assays
3.4. Inactivation Assays
3.5. Molecular Docking
3.6. Molecular Dynamics
3.7. Binding Energy
3.8. Toxicological and Physicochemical Properties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- W.H.O. Trypanosomiasis, Human African (Sleeping Sickness), Epidemiological Situation. Available online: http://www.who.int/trypanosomiasis_african/country/en/ (accessed on 17 September 2017).
- Fairlamb, A.H. Chemotherapy of human African trypanosomiasis: Current and future prospects. Trends Parasitol. 2003, 19, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Likeufack, A.L.; Brun, R.; Fomena, A.; Truc, P. Comparison of the in vitro drug sensitivity of Trypanosoma brucei gambiense strains from West and Central Africa isolated in the periods 1960–1995 and 1999–2004. Acta Trop. 2006, 100, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Maina, N.; Maina, K.J.; Mäser, P.; Brun, R. Genotypic and phenotypic characterization of Trypanosoma brucei gambiense isolates from Ibba, South Sudan, an area of high melarsoprol treatment failure rate. Acta Trop. 2007, 104, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Delespaux, V.; de Koning, H.P. Drugs and drug resistance in African trypanosomiasis. Drug Resist. Updat. 2007, 10, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.L.; Burchmore, R.J.; Clucas, C.; Hertz-Fowler, C.; Brooks, K.; Tait, A.; MacLeod, A.; Turner, C.M.R.; De Koning, H.P.; Wong, P.E. Multiple genetic mechanisms lead to loss of functional TbAT1 expression in drug-resistant trypanosomes. Eukaryot. Cell 2010, 9, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.E.; Ludin, P.; Wenzler, T.; Kaiser, M.; Brun, R.; Pyana, P.P.; Büscher, P.; De Koning, H.P.; Horn, D.; Mäser, P. Aquaporin 2 mutations in Trypanosoma brucei gambiense field isolates correlate with decreased susceptibility to pentamidine and melarsoprol. PLoS Negl. Trop. Dis. 2013, 7, e2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Control and Surveillance of Human African Trypanosomiasis: Report of a WHO Expert Committee. 1998. Available online: http://apps.who.int/iris/bitstream/10665/42087/1/WHO_TRS_881.pdf (accessed on 17 September 2017).
- Albert, M.-A.; Haanstra, J.R.; Hannaert, V.; Van Roy, J.; Opperdoes, F.R.; Bakker, B.M.; Michels, P.A. Experimental and in silico analyses of glycolytic flux control in bloodstream form Trypanosoma brucei. J. Biol. Chem. 2005, 280, 28306–28315. [Google Scholar] [CrossRef] [PubMed]
- Helfert, S.; Estévez, A.M.; Bakker, B.; Michels, P.; Clayton, C. Roles of triosephosphate isomerase and aerobic metabolism in Trypanosoma brucei. Biochem. J. 2001, 357, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Illana, V.; Pérez-Montfort, R.; López-Calahorra, F.; Costas, M.; Rodríguez-Romero, A.; Tuena de Gómez-Puyou, M.; Gómez Puyou, A. Structural differences in triosephosphate isomerase from different species and discovery of a multitrypanosomatid inhibitor. Biochemistry 2006, 45, 2556–2560. [Google Scholar] [CrossRef] [PubMed]
- Galland, N.; de Walque, S.; Voncken, F.G.; Verlinde, C.L.; Michels, P.A. An internal sequence targets Trypanosoma brucei triosephosphate isomerase to glycosomes. Mol. Biochem. Parasitol. 2010, 171, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.E.; Michels, P. Metabolic compartmentation in African trypanosomes. Parasitol. Today 1996, 12, 465–471. [Google Scholar] [CrossRef]
- Schnackerz, K.D.; Gracy, R.W. Probing the catalytic sites of triosephosphate isomerase by 31P-NMR with reversibly and irreversibly binding substrate analogues. FEBS J. 1991, 199, 231–238. [Google Scholar] [CrossRef]
- Kursula, I.; Partanen, S.; Lambeir, A.M.; Antonov, D.M.; Augustyns, K.; Wierenga, R.K. Structural determinants for ligand binding and catalysis of triosephosphate isomerase. FEBS J. 2001, 268, 5189–5196. [Google Scholar] [CrossRef]
- Wierenga, R.; Noble, M.; Vriend, G.; Nauche, S.; Hol, W. Refined 1.83 Å structure of trypanosomal triosephosphate isomerase crystallized in the presence of 2.4 M-ammonium sulphate: A comparison with the structure of the trypanosomal triosephosphate isomerase-glycerol-3-phosphate complex. J. Mol. Biol. 1991, 220, 995–1015. [Google Scholar] [CrossRef]
- Téllez-Valencia, A.; Ávila-Ríos, S.; Pérez-Montfort, R.; Rodríguez-Romero, A.; de Gómez-Puyou, M.T.; López-Calahorra, F.; Gómez-Puyou, A. Highly specific inactivation of triosephosphate isomerase from Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2002, 295, 958–963. [Google Scholar] [CrossRef]
- Zomosa-Signoret, V.; Hernández-Alcántara, G.; Reyes-Vivas, H.; Martínez-Martínez, E.; Garza-Ramos, G.; Pérez-Montfort, R.; Tuena de Gómez-Puyou, M.; Gómez-Puyou, A. Control of the reactivation kinetics of homodimeric triosephosphate isomerase from unfolded monomers. Biochemistry 2003, 42, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Gayosso-De-Lucio, J.; Torres-Valencia, M.; Rojo-Domínguez, A.; Nájera-Peña, H.; Aguirre-López, B.; Salas-Pacheco, J.; Avitia-Domínguez, C.; Téllez-Valencia, A. Selective inactivation of triosephosphate isomerase from Trypanosoma cruzi by brevifolin carboxylate derivatives isolated from Geranium bellum Rose. Bioorg. Med. Chem. Lett. 2009, 19, 5936–5939. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, D.A.; Osowski, R.; Schudok, M.; Wierenga, R.K.; Müller, K.; Kessler, H.; Opperdoes, F.R. Inhibition of triosephosphate isomerase from Trypanosoma brucei with cyclic hexapeptides. FEBS J. 1992, 207, 441–447. [Google Scholar] [CrossRef]
- Alvarez, G.; Martínez, J.; Aguirre-Lopez, B.; Cabrera, N.; Pérez-Díaz, L.; de Gómez-Puyou, M.T.; Gómez-Puyou, A.; Pérez-Montfort, R.; Garat, B.; Merlino, A. New chemotypes as Trypanosoma cruzi triosephosphate isomerase inhibitors: A deeper insight into the mechanism of inhibition. J. Enzym. Inhib. Med. Chem. 2014, 29, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Chung, K.-H.; You, H.-J.; Choi, I.H.; Chae, M.J.; Han, J.-Y.; Jung, O.-J.; Kang, S.-J.; Ryu, C.-K. Synthesis and biological evaluation of benzimidazole-4,7-diones that inhibit vascular smooth muscle cell proliferation. Bioorg. Med. Chem. Lett. 2004, 14, 3563–3566. [Google Scholar] [CrossRef] [PubMed]
- Kuş, C.; Ayhan-Kılcıgil, G.; Özbey, S.; Kaynak, F.B.; Kaya, M.; Çoban, T.; Can-Eke, B. Synthesis and antioxidant properties of novel N-methyl-1,3,4-thiadiazol-2-amine and 4-methyl-2H-1,2,4-triazole-3(4H)-thione derivatives of benzimidazole class. Bioorg. Med. Chem. 2008, 16, 4294–4303. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Narasimhan, B.; Kumar, P.; Judge, V.; Narang, R.; De Clercq, E.; Balzarini, J. Synthesis, antimicrobial and antiviral activity of substituted benzimidazoles. J. Enzym. Inhib. Med. Chem. 2009, 24, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Hosamani, K.M.; Seetharamareddy, H.R.; Keri, R.S.; Hanamanthagouda, M.S.; Moloney, M.G. Microwave assisted, one-pot synthesis of 5-nitro-2-aryl substituted-1H-benzimidazole libraries: Screening in vitro for antimicrobial activity. J. Enzym. Inhib. Med. Chem. 2009, 24, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Abonia, R.; Cortés, E.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Synthesis of novel 1,2,5-trisubstituted benzimidazoles as potential antitumor agents. Eur. J. Med. Chem. 2011, 46, 4062–4070. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F.; Turner, E.M.; Gudmundsson, K.S.; Jenkinson, S.; Spaltenstein, A.; Thomson, M.; Wheelan, P. Novel N-substituted benzimidazole CXCR4 antagonists as potential anti-HIV agents. Bioorg. Med. Chem. 2010, 20, 2125–2128. [Google Scholar] [CrossRef] [PubMed]
- Flores-Carrillo, P.; Velázquez-López, J.M.; Aguayo-Ortiz, R.; Hernández-Campos, A.; Trejo-Soto, P.J.; Yépez-Mulia, L.; Castillo, R. Synthesis, antiprotozoal activity, and chemoinformatic analysis of 2-(methylthio)-1H-benzimidazole-5-carboxamide derivatives: Identification of new selective giardicidal and trichomonicidal compounds. Eur. J. Med. Chem. 2017, 137, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.J.; Pyun, Y.M.; Kim, J.Y.; Pagire, H.S.; Kim, K.Y.; Kim, K.R.; Dal Rhee, S.; Jung, W.H.; Song, J.S.; Bae, M.A. Synthesis and biological evaluation of aminobenzimidazole derivatives with a phenylcyclohexyl acetic acid group as anti-obesity and anti-diabetic agents. Bioorg. Med. Chem. 2013, 23, 4713–4718. [Google Scholar] [CrossRef] [PubMed]
- Soria-Arteche, O.; Hernández-Campos, A.; Yépez-Mulia, L.; Trejo-Soto, P.J.; Hernández-Luis, F.; Gres-Molina, J.; Maldonado, L.A.; Castillo, R. Synthesis and antiprotozoal activity of nitazoxanide–N-methylbenzimidazole hybrids. Bioorg. Med. Chem. 2013, 23, 6838–6841. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ramos, M.; Ibarra-Velarde, F.; Hernández-Campos, A.; Vera-Montenegro, Y.; Jung-Cook, H.; Cantó-Alarcón, G.J.; del Rivero, L.M.; Castillo, R. A highly water soluble benzimidazole derivative useful for the treatment of fasciolosis. Bioorg. Med. Chem. 2014, 24, 5814–5817. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Pérez-Villanueva, J.; Hernández-Campos, A.; Castillo, R.; Meurice, N.; Medina-Franco, J.L. Chemoinformatic characterization of activity and selectivity switches of antiprotozoal compounds. Future Med. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Matadamas-Martínez, F.; Castillo, R.; Hernández-Campos, A.; Méndez-Cuesta, C.; de Souza, W.; Gadelha, A.P.; Nogueda-Torres, B.; Hernández, J.M.; Yépez-Mulia, L. Proteomic and ultrastructural analysis of the effect of a new nitazoxanide-N-methyl-1H-benzimidazole hybrid against Giardia intestinalis. Res. Vet. Sci. 2016, 105, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ramos, M.; Ibarra-Velarde, F.; Jung-Cook, H.; Hernández-Campos, A.; Vera-Montenegro, Y.; Castillo, R. Novel triclabendazole prodrug: A highly water soluble alternative for the treatment of fasciolosis. Bioorg. Med. Chem. 2017, 27, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Romo-Mancillas, A.; Téllez-Valencia, A.; Yépez-Mulia, L.; Hernández-Luis, F.; Hernández-Campos, A.; Castillo, R. The design and inhibitory profile of new benzimidazole derivatives against triosephosphate isomerase from Trypanosoma cruzi: A problem of residue motility. J. Mol. Graph. Model. 2011, 30, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Chiguer, D.L.; Hernández-Luis, F.; Nogueda-Torres, B.; Castillo, R.; Reynoso-Ducoing, O.; Hernández-Campos, A.; Ambrosio, J.R. JVG9, a benzimidazole derivative, alters the surface and cytoskeleton of Trypanosoma cruzi bloodstream trypomastigotes. Mem. Inst. Oswaldo Cruz 2014, 109, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-López, J.M.; Hernández-Campos, A.; Yépez-Mulia, L.; Téllez-Valencia, A.; Flores-Carrillo, P.; Nieto-Meneses, R.; Castillo, R. Synthesis and trypanocidal activity of novel benzimidazole derivatives. Eur. J. Med. Chem. Lett. 2016, 26, 4377–4381. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, D.W. Inactivation of Escherichia coli glycerol kinase by 5,5′-dithiobis (2-nitrobenzoic acid) and N-ethylmaleimide: Evidence for nucleotide regulatory binding sites. Biochemistry 1986, 25, 4711–4718. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.; Aguirre-López, B.; Cabrera, N.; Marins, E.B.; Tinoco, L.; Batthyány, C.I.; de Gómez-Puyou, M.T.; Puyou, A.G.; Pérez-Montfort, R.; Cerecetto, H. 1,2,4-thiadiazol-5(4H)-ones: A new class of selective inhibitors of Trypanosoma cruzi triosephosphate isomerase. Study of the mechanism of inhibition. J. Enzym. Inhib. Med. Chem. 2013, 28, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Minini, L.; Álvarez, G.; González, M.; Cerecetto, H.; Merlino, A. Molecular docking and molecular dynamics simulation studies of Trypanosoma cruzi triosephosphate isomerase inhibitors. Insights into the inhibition mechanism and selectivity. J. Mol. Graph. Model. 2015, 58, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kurkcuoglu, Z.; Findik, D.; Akten, E.D.; Doruker, P. How an inhibitor bound to subunit interface alters triosephosphate isomerase dynamics. Biophys. J. 2015, 109, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Valencia, A.; Olivares-Illana, V.; Hernández-Santoyo, A.; Pérez-Montfort, R.; Costas, M.; Rodríguez-Romero, A.; López-Calahorra, F.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Inactivation of triosephosphate isomerase from Trypanosoma cruzi by an agent that perturbs its dimer interface. J. Mol. Biol. 2004, 341, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Exploring the possible binding sites at the interface of triosephosphate isomerase dimer as a potential target for anti-tripanosomal drug design. Bioorg. Med. Chem. 2004, 14, 3151–3154. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Structural considerations for the rational design of selective anti-trypanosomal agents: The role of the aromatic clusters at the interface of triosephosphate isomerase dimer. Biochem. Biophys. Res. Commun. 2005, 328, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Zabori, S.; Rudolph, R.; Jaenicke, R. Folding and association of triose phosphate isomerase from rabbit muscle. Z. Naturforsch. C 1980, 35, 999–1004. [Google Scholar] [PubMed]
- Pompliano, D.L.; Peyman, A.; Knowles, J.R. Stabilization of a reaction intermediate as a catalytic device: Definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry 1990, 29, 3186–3194. [Google Scholar] [CrossRef] [PubMed]
- Borchert, T.V.; Kishan, K.R.; Zeelen, J.P.; Schliebs, W.; Thanki, N.; Abagyan, R.; Jaenicke, R.; Wierenga, R.K. Three new crystal structures of point mutation variants of mono TIM: Conformational flexibility of loop-1, loop-4 and loop-8. Structure 1995, 3, 669–679. [Google Scholar] [CrossRef]
- Thakur, S.S.; Deepalakshmi, P.; Gayathri, P.; Banerjee, M.; Murthy, M.; Balaram, P. Detection of the protein dimers, multiple monomeric states and hydrated forms of Plasmodium falciparum triosephosphate isomerase in the gas phase. Protein Eng. Des. Sel. 2009, 22, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Alahuhta, M.; Salin, M.; Casteleijn, M.G.; Kemmer, C.; El-Sayed, I.; Augustyns, K.; Neubauer, P.; Wierenga, R.K. Structure-based protein engineering efforts with a monomeric TIM variant: The importance of a single point mutation for generating an active site with suitable binding properties. Protein Eng. Des. Sel. 2008, 21, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Berlow, R.B.; Loria, J.P. The role of loop-loop interactions in coordinating motions and enzymatic function in triosephosphate isomerase. Biochemistry 2009, 48, 4548. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, R.; Kapetaniou, E.; Venkatesan, R. Triosephosphate isomerase: A highly evolved biocatalyst. Cell Mol. Life Sci. 2010, 67, 3961–3982. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Go, M.K.; O’Donoghue, A.C.; Amyes, T.L.; Pegan, S.D.; Wang, Y.; Loria, J.P.; Mesecar, A.D.; Richard, J.P. Enzyme architecture: The effect of replacement and deletion mutations of loop 6 on catalysis by triosephosphate isomerase. Biochemistry 2014, 53, 3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, T.V.; Pratt, K.; Zeelen, J.P.; Callens, M.; Noble, M.E.; Opperdoes, F.R.; Michels, P.A.; Wierenga, R.K. Overexpression of trypanosomal triosephosphate isomerase in Escherichia coli and characterisation of a dimer-interface mutant. FEBS J. 1993, 211, 703–710. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Casteleijn, M.G.; Alahuhta, M.; Groebel, K.; El-Sayed, I.; Augustyns, K.; Lambeir, A.-M.; Neubauer, P.; Wierenga, R.K. Functional role of the conserved active site proline of triosephosphate isomerase. Biochemistry 2006, 45, 15483–15494. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Schuèttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.; van Gunsteren, W. The GROMOS software package for biomolecular simulations. METECC 1995, 95, 397–434. [Google Scholar]
- Kony, D.B.; Hünenberger, P.H.; Van Gunsteren, W.F. Molecular dynamics simulations of the native and partially folded states of ubiquitin: Influence of methanol cosolvent, pH, and temperature on the protein structure and dynamics. Protein Sci. 2007, 16, 1101–1118. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Punkvang, A.; Saparpakorn, P.; Hannongbua, S.; Wolschann, P.; Beyer, A.; Pungpo, P. Investigating the structural basis of arylamides to improve potency against M. tuberculosis strain through molecular dynamics simulations. Eur. J. Med. Chem. 2010, 45, 5585–5593. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Méndez-Lucio, O.; Medina-Franco, J.L.; Castillo, R.; Yépez-Mulia, L.; Hernández-Luis, F.; Hernández-Campos, A. Towards the identification of the binding site of benzimidazoles to β-tubulin of Trichinella spiralis: Insights from computational and experimental data. J. Mol. Graph. Model. 2013, 41, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Faf-Drugs4 Server. Available online: http://fafdrugs3.mti.univ-paris-diderot.fr/ (accessed on 20 July 2017).
- PROTOX—Prediction of Rodent Oral TOXicity. Available online: http://tox.charite.de/tox/ (accessed on 20 July 2017).
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Energy (kcal/mol) | Hydrogen Bond | |||||
|---|---|---|---|---|---|---|---|
| (VLJ)bound | (VLJ)free | (VCL)bound | (VCL)free | ∆Gbind | Range | Average | |
| TbTIM-1 | −29.45 | −6.53 | −4.45 | −1.28 | −5.71 | 0–7 | 2 |
| TbTIM-2 | −38.90 | −2.67 | −0.92 | −0.33 | −6.82 | 0–6 | 0 |
| TbTIM-3 | −27.52 | −8.49 | −44.83 | −43.59 | −4.05 | 1–7 | 1 |
| Compound | cLogP 1 | tPSA 1 | HB-D 1 | HB-A 1 | VEBER Rules 1 | EGAN Rules 1 | Predicted LD50 2 (mg/kg) |
|---|---|---|---|---|---|---|---|
| 1 | 4.1256 | 76.24 | 2 | 6 | Good | Good | 911 |
| 2 | 5.3882 | 53.08 | 1 | 6 | Good | Good | 990 |
| 3 | 1.9686 | 154.4 | 2 | 9 | Good | Good | 1000 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Raygoza, A.; Cano-González, L.; Velázquez-Martínez, I.; Trejo-Soto, P.J.; Castillo, R.; Hernández-Campos, A.; Hernández-Luis, F.; Oria-Hernández, J.; Castillo-Villanueva, A.; Avitia-Domínguez, C.; et al. Species-Specific Inactivation of Triosephosphate Isomerase from Trypanosoma brucei: Kinetic and Molecular Dynamics Studies. Molecules 2017, 22, 2055. https://doi.org/10.3390/molecules22122055
Vázquez-Raygoza A, Cano-González L, Velázquez-Martínez I, Trejo-Soto PJ, Castillo R, Hernández-Campos A, Hernández-Luis F, Oria-Hernández J, Castillo-Villanueva A, Avitia-Domínguez C, et al. Species-Specific Inactivation of Triosephosphate Isomerase from Trypanosoma brucei: Kinetic and Molecular Dynamics Studies. Molecules. 2017; 22(12):2055. https://doi.org/10.3390/molecules22122055
Chicago/Turabian StyleVázquez-Raygoza, Alejandra, Lucia Cano-González, Israel Velázquez-Martínez, Pedro Josué Trejo-Soto, Rafael Castillo, Alicia Hernández-Campos, Francisco Hernández-Luis, Jesús Oria-Hernández, Adriana Castillo-Villanueva, Claudia Avitia-Domínguez, and et al. 2017. "Species-Specific Inactivation of Triosephosphate Isomerase from Trypanosoma brucei: Kinetic and Molecular Dynamics Studies" Molecules 22, no. 12: 2055. https://doi.org/10.3390/molecules22122055