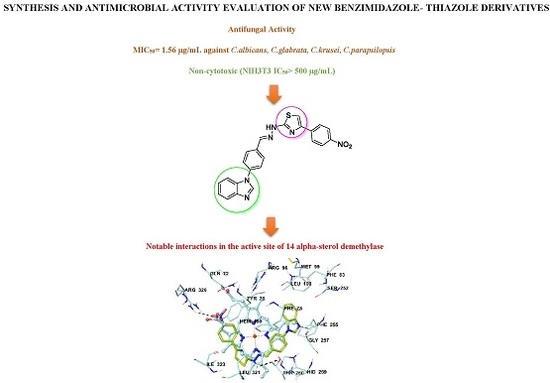
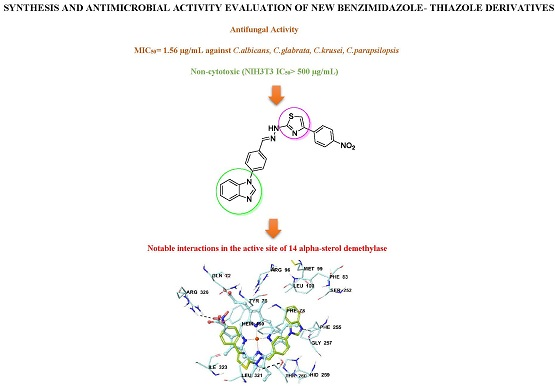
Synthesis and Anticandidal Activity Evaluation of New Benzimidazole-Thiazole Derivatives

 , and
, and
Abstract
:
1. Introduction
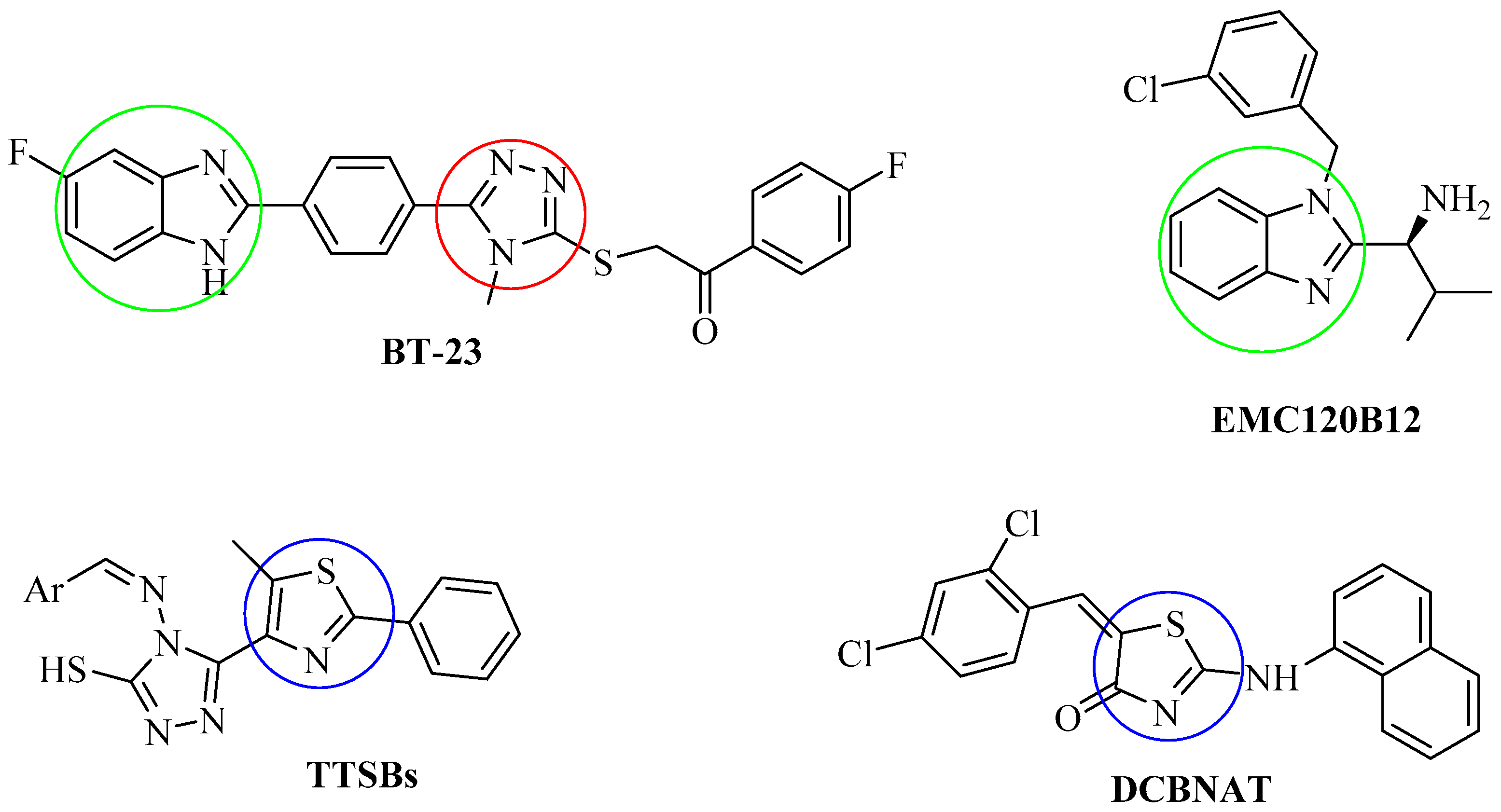
2. Results and Discussion
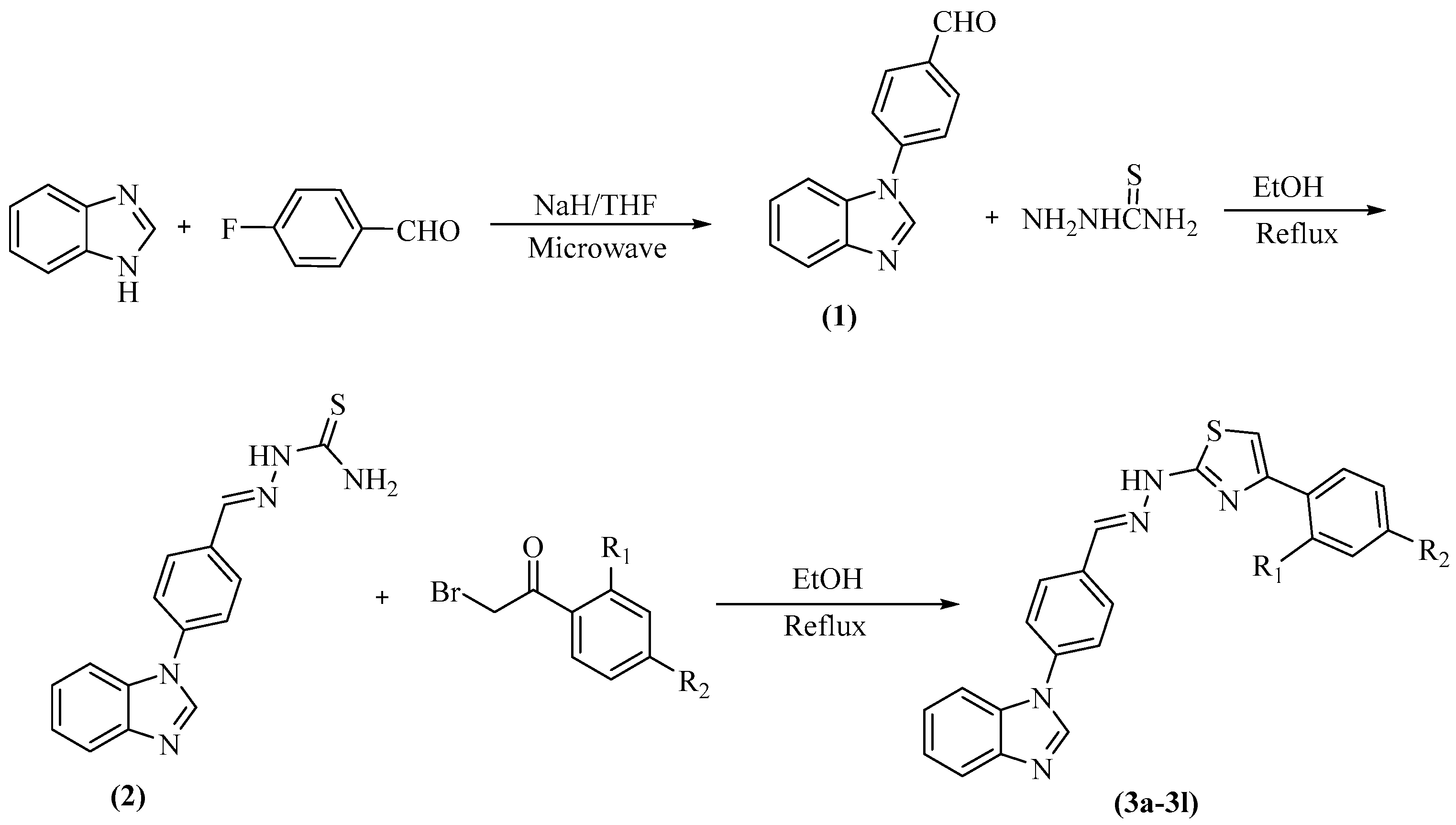
2.1. Chemistry
2.2. Antifungal Activity Assay
2.3. Quantification of Ergosterol Level
2.4. Cytotoxicity Test
2.5. Prediction of ADME Parameters
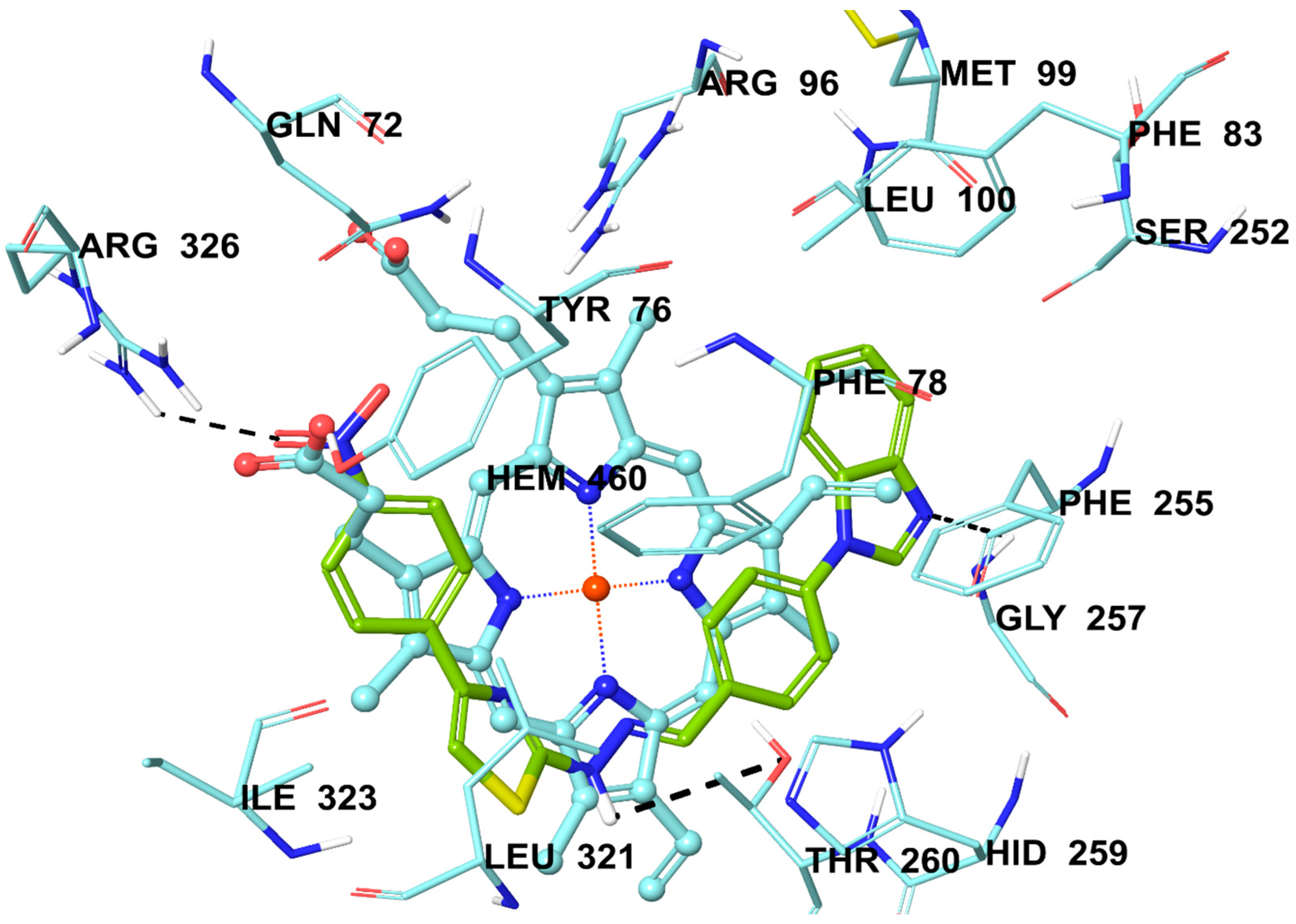
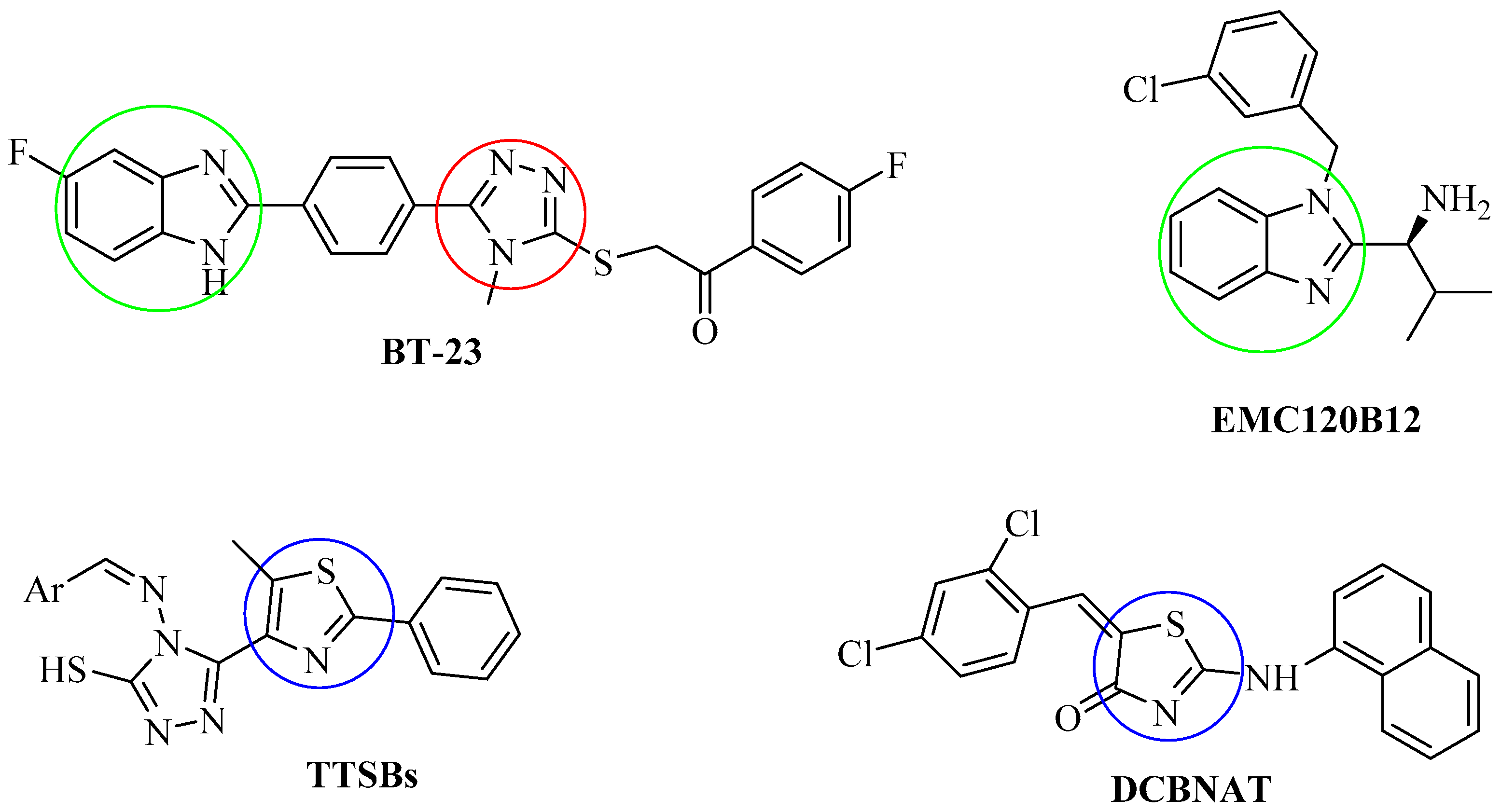
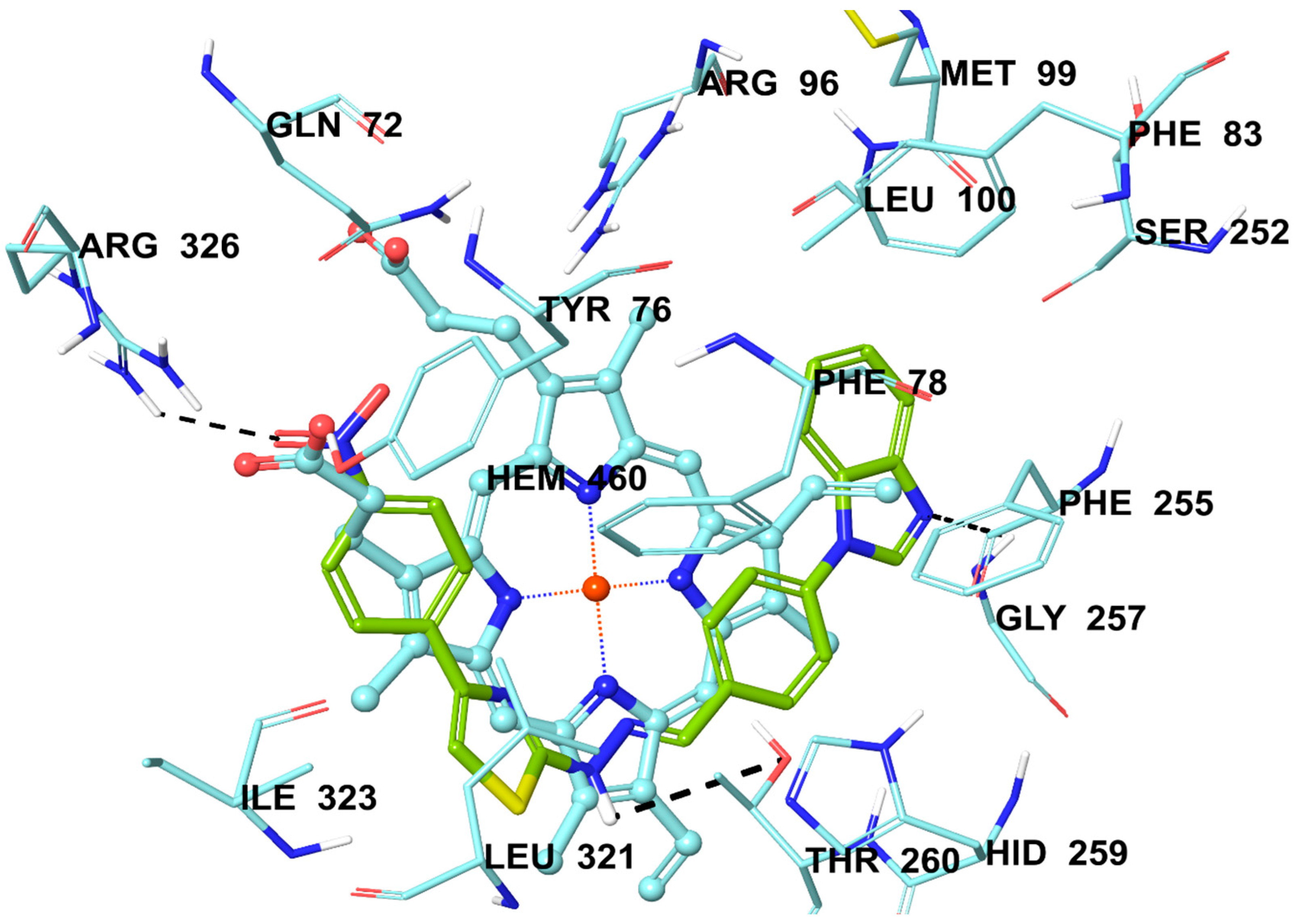
2.6. Molecular Docking Studies
3. Materials and Methods
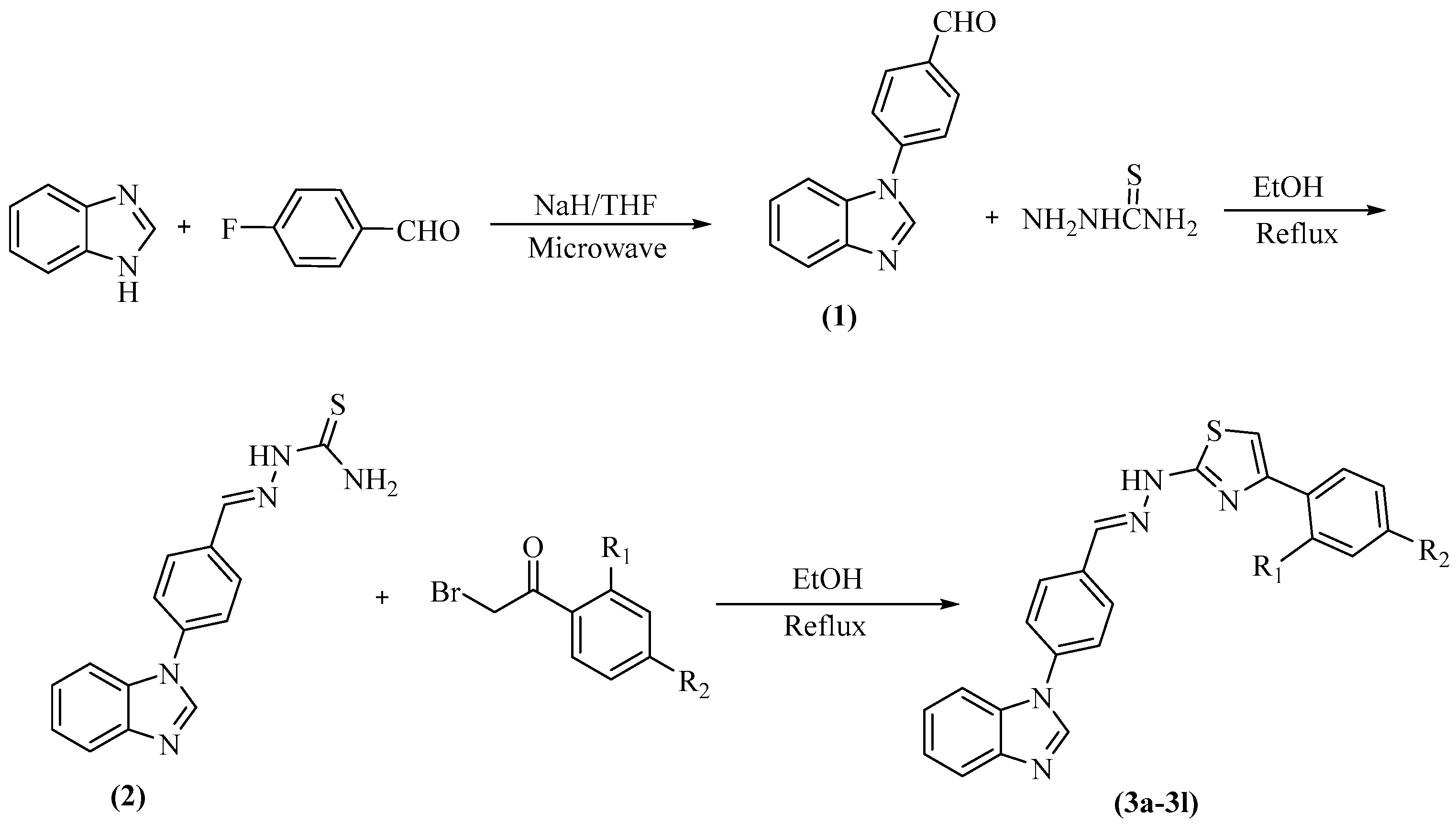
3.1. Chemistry
3.1.1. Synthesis of 4-(1H-Benzimidazol-1-yl)benzaldehyde (1)
3.1.2. Synthesis of 2-(4-(1H-Benzimidazol-1-yl)benzylidene)hydrazine-1-carbothioamide (2)
3.1.3. General Procedure for the Synthesis of the Target Compounds 3a–3l
3.2. Antifungal Activity Assays
3.3. Quantification of Ergosterol Level
3.4. Cytotoxicity Test
3.5. Prediction of ADME Parameters
3.6. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keller, P.; Müller, C.; Engelhardt, I.; Hiller, E.; Lemuth, K.; Eickhoff, H.; Wiesmüller, K.H.; Burger-Kentischer, A.; Bracher, F.; Rupp, S. An antifungal benzimidazole derivative inhibits ergosterol biosynthesis and reveals novel sterols. Antimicrob. Agents Chemother. 2015, 59, 6296–6307. [Google Scholar] [CrossRef] [PubMed]
- Nafsika, H.; Georgopapadakou, T.J.S. The fungal cell wall as a drug target. Trends Microbiol. 1995, 3, 98–104. [Google Scholar]
- Kathiravan, M.K.; Salake, A.B.; Chothe, A.S.; Dudhe, P.B.; Watode, R.P.; Mukta, M.S.; Gadhwe, S. The biology and chemistry of antifungal agents: A review. Bioorg. Med. Chem. 2012, 20, 5678–5698. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.V.; Butassi, E.; Monteiro, M.C.; Svetaz, L.A.; Vicente, F.; Susana, A.; Zacchino, S.A. Novel antifungal agents: A patent review (2011-present). Expert Opin. Ther. Pat. 2014, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pianalto, K.M.; Alspaugh, J.A. New horizons in antifungal therapy. J. Fungi 2016, 2, 26. [Google Scholar] [CrossRef]
- Perfect, J.R. Is there an emerging need for new antifungals? Expert Opin. Emerg. Drugs 2016, 21, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.S.; Robbins, N.; Cowen, L.E. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev. 2011, 75, 213–267. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Wright, G.D.; Cowen, L.E. Antifungal drugs: The current armamentarium and development of new agents. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.D. Changing patterns and trends in systemic fungal infections. J. Antimicrob. Chemother. 2007, 56, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Merkamm, M.; Manning, N.J.; Pompon, D.; Kelly, S.L.; Kelly, D.E. Differential azole antifungal efficacies contrasted using a Saccharomyces cerevisiae strain humanized for sterol 14α-demethylase at the homologous locus. Antimicrob. Agents Chemother. 2008, 52, 3597–3603. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Martel, C.M.; Parker, J.E.; Melo, N.; Lamb, D.C.; Nes, D.; Kelly, D.E.; Kelly, S.L. Azole binding properties of Candida albicans sterol 14α-demethylase (CaCYP51). Antimicrob. Agents Chemother. 2011, 54, 4235–4245. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.L.; Lamb, D.C.; Jackson, C.J.; Warrilow, A.G.S.; Kelly, D.E. The biodiversity of microbial cytochromes P450. Adv. Microb. Physiol. 2013, 47, 131–186. [Google Scholar]
- Lamb, D.C.; Kelly, D.E.; Waterman, M.R.; Stromstedt, M.; Rozman, D.; Kelly, S.L. Characteristics of the heterologously expressed human lanosterol 14α-demethylase (other names: P45014DM, CYP51, P45051) and inhibition of the purified human and Candida albicans CYP51 with azole antifungal agents. Yeast 1996, 15, 755–763. [Google Scholar] [CrossRef]
- Andrew, G.; Warrilow, J.E.; Parker, D.E.; Kelly, S.L.; Kelly, D.E. Azole affinity of sterol 14α-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2013, 57, 1352–1360. [Google Scholar]
- Pasqualotto, A.C.; Denning, D.W. New and emerging treatments for fungal infections. J. Antimicrob. Chemother. 2008, 61, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Asai, K.; Tsuchimori, N.; Okonogi, K.; Perfect, J.R.; Gotoh, O.; Yoshida, Y. Formation of azole-resistant Candida albicans by mutation of sterol 14α-demethylase P450. Antimicrob. Agents Chemother. 1999, 43, 1163–1169. [Google Scholar] [PubMed]
- Sanglard, D.; Odds, F.C. Resistance of Candida species to antifungal agents: Molecular mechanisms and clinical consequences. Lancet Infect. Dis. 2002, 2, 73–85. [Google Scholar] [CrossRef]
- Goetz, A.K.; Dix, D.J. Mode of action for reproductive and hepatic toxicity inferred from a genomic study of triazole antifungals. Toxicol. Sci. 2009, 110, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Kinast, S.; Burger-Kentischer, A.; Finkelmeier, D.; Kleymann, G.; Abu Rayyan, W.; Schröppel, K.; Singh, A.; Jung, G.; Wiesmüller, K.H.; et al. High-throughput-screening-based identification and structure-activity relationship characterization defined (S)-2-(1-aminoisobutyl)-1-(3-chlorobenzyl)benzimidazole as a highly antimycotic agent nontoxic to cell lines. J. Med. Chem. 2011, 54, 6993–6997. [Google Scholar] [CrossRef] [PubMed]
- Ates-Alagoz, Z. Antimicrobial activities of 1H-benzimidazole-based molecules. Curr. Top. Med. 2016, 16, 2953–2962. [Google Scholar] [CrossRef]
- Burger-Kentischer, A.; Finkelmeier, D.; Keller, P.; Bauer, J.; Eickhoff, H.; Kleymann, G.; Abu Rayyan, W.; Singh, A.; Schröppel, K.; Lemuth, K.; et al. Screening assay based on host-pathogen interaction models identifies a set of novel antifungal benzimidazole derivatives. Antimicrob. Agents Chemother. 2011, 55, 4789–4801. [Google Scholar] [CrossRef] [PubMed]
- Karaca-Gençer, H.; Acar-Çevik, U.; Levent, S.; Sağlık, B.N.; Korkut, B.; Özkay, Y.; Ilgın, S.; Öztürk, Y. New benzimidazole-1,2,4-triazole hybrid compounds: Synthesis, anticandidal activity and cytotoxicity evaluation. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Pereira de Sa, N.; Lino, C.I.; Fonseca, N.C.; Borelli, B.M.; Ramos, J.P.; Souza-Fagundes, E.M.; Rosa, C.A.; Santos, D.A.; de Oliveira, R.B.; Johann, S. Thiazole compounds with activity against Cryptococcus gattii and Cryptococcus neoformans in vitro. Eur. J. Med. Chem. 2015, 102, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Enache, A.; Vodnar, D.C.; Nastasă, C.; Benedec, D.; Ionuț, I.; Login, C.; Marc, G.; Oniga, O.; Tiperciuc, B. New thiazolyl-triazole schiff bases: Synthesis and evaluation of the anti-candida potential. Molecules 2016, 21, 1595. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Vodnar, D.C.; Tamaian, R.; Pîrnău, A.; Vlase, L.; Ionuț, I.; Oniga, O.; Tiperciuc, B. Design, synthesis and antifungal activity evaluation of new thiazolin-4-ones as potential lanosterol 14α-demethylase inhibitors. Int. J. Mol. Sci. 2017, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Tudela, J.L.; Arendrup, M.C.; Barchiesi, F.; Bille, J.; Chryssanthou, E.; Cuenca-Estrella, M.; Dannaoui, E.; Denning, D.W.; Donnelly, J.P.; Dromer, F.; et al. EUCAST definitive document EDef 7.1: Method for the determination of broth dilution MICs of antifungal agents for fermentative yeasts: Subcommittee on Antifungal Susceptibility Testing (AFST) of the ESCMID European Committee for Antimicrobial Susceptibility Testing (EUCAST)*. Clin. Microbiol. Infect. 2008, 14, 398–405. [Google Scholar]
- Borra, R.C.; Lotufo, M.A.; Gagioti, S.M.; Barros, F.D.M.; Andrade, P.M. A simple method to measure cell viability in proliferation and cytotoxicity assays. Braz. Oral Res. 2009, 23, 255–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino, J.C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portaels, F. Resazurin microtiter assay plate: Simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.A.; Al-Thabaiti, S.A.; Malik, M.A. Synthesis, structure optimization and antifungal screening of novel tetrazole ring bearing acyl-hydrazones. Int. J. Mol. Sci. 2012, 13, 10880–10898. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Cramer, R.A. Regulation of sterol biosynthesis in the human fungal pathogen aspergillus fumigatus: Opportunities for therapeutic development. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lupetti, A.; Danesi, R.; Campa, M.; Del Tacca, M.; Kelly, S. Molecular basis of resistance to azole antifungals. Trends Mol. Med. 2002, 8, 76–81. [Google Scholar] [CrossRef]
- International Organization for Standardization. Biological Evaluation of Medical Devices-Part 5: Tests for In Vitro Cytotoxicity ISO-10993-5, 3rd ed.; International Organization for Standardization: Geneva, Switzerland, 2009. [Google Scholar]
- De Waterbeemd, H.V.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2013, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- QikProp, version 4.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Lipinski Christopher, A.; Franco, L.; Dominy Beryl, W.; Feeney Paul, J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Larissa, M.P.; Thomas, L.P.; Michael, R. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 3068–3073. [Google Scholar]
- Saeed, B.E.; Touba, I.; Hamid, F.; Alireza, F.; Mehraban, A.K.; Mahtab, S.; Somaye, S. Imidazolylchromanones containing alkyl side chain as lanosterol 14α-demethylase inhibitors: Synthesis, antifungal activity and docking study. J. Enzym. Inhib. Med. Chem. 2014, 29, 263–271. [Google Scholar]
- Gonzalez-Chavez, R.; Martinez, R.; Torre-Bouscoulet, M.E.; Gallo, M.; Gonzalez-Chavez, M.M. De novo design of non-coordinating indolones as potential inhibitors for lanosterol 14-α-demethylase (CYP51). Chem. Pharm. Bull. 2014, 62, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Reena, G.; Subhash, A.; Sudhir, A.K. Modeling and interactions of Aspergillus fumigatus lanosterol 14-α demethylase ‘A’ with azole antifungals. Bioorg. Med. Chem. 2004, 12, 2937–2950. [Google Scholar]
- Rossello, A.; Bertini, S.; Lapucci, A.; Macchia, M.; Martinelli, A.; Rapposelli, S.; Herreros, E.; Macchia, B. Synthesis, antifungal activity, and molecular modeling studies of new inverted oxime ethers of oxiconazole. J. Med. Chem. 2002, 45, 4903–4912. [Google Scholar] [CrossRef] [PubMed]
- Breivik, O.N.; Owades, J.L. Spectrophotometric semi-microdetermination of ergosterol in yeast. Agric. Food Chem. 1957, 5, 360–363. [Google Scholar] [CrossRef]
- Karaca, G.H.; Acar, Ç.U.; Kaya, Ç.B.; Sağlık, B.N.; Levent, S.; Atlı, Ö.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, and evaluation of novel 2-phenylpropionic acid derivatives as dual COX inhibitory-antibacterial agents. J. Enzym. Inhib. Med. Chem. 2017, 32, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Can, Ö.D.; Osmaniye, D.; Demir, Ö.Ü.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Baysal, M.; Özkay, Y.; Kaplancıklı, Z.A. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur. J. Med. Chem. 2017, 131, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Demir, Ö.Ü.; Can, Ö.D.; Sağlık, B.N.; Acar, Ç.U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Gheewala, N.; Suthar, A.; Shah, A. In-vitro cytotoxicity activity of Solanum nigrum extract against Hela cell line and Vero cell line. Int. J. Pharm. Pharm. Sci. 2009, 1, 38–46. [Google Scholar]
- Maestro, version 10.6; Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger Suite, version 2016-2; Schrödinger, LLC: New York, NY, USA, 2016.
- LigPrep, version 3.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Glide, version 7.1; Schrödinger, LLC: New York, NY, USA, 2016.
Sample Availability: Samples of the compounds 1, 2 and 3a–3l are available from the authors. |
| Compounds | R1 | R2 |
|---|---|---|
| 3a | –H | –H |
| 3b | –H | –CH3 |
| 3c | –H | –NO2 |
| 3d | –H | –CN |
| 3e | –H | –OCH3 |
| 3f | –H | –F |
| 3g | –H | –Cl |
| 3h | –H | –Br |
| 3i | –H | –CF3 |
| 3j | –CH3 | –CH3 |
| 3k | –F | –F |
| 3l | –Cl | –Cl |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | C. albicans | C. glabrata | C. krusei | C. parapsilopsis |
|---|---|---|---|---|
| 3a | 100 | 25 | 100 | 100 |
| 3b | 100 | 25 | 100 | 100 |
| 3c | 1.56 | 1.56 | 1.56 | 1.56 |
| 3d | 3.12 | 1.56 | 1.56 | 3.12 |
| 3e | 6.25 | 100 | 100 | 100 |
| 3f | 6.25 | 25 | 6.25 | 6.25 |
| 3g | 6.25 | 12.5 | 6.25 | 6.25 |
| 3h | 100 | 12.5 | 100 | 100 |
| 3i | 100 | 100 | 100 | 100 |
| 3j | 50 | 100 | 100 | 100 |
| 3k | 6.25 | 6.25 | 12.5 | 6.25 |
| 3l | 6.25 | 12.5 | 6.25 | 6.25 |
| Ketoconazole | 0.78 | 1.56 | 1.56 | 1.56 |
| Fluconazole | 0.78 | 1.56 | 1.56 | 0.78 |
| Compounds | Concentrations (µg/mL) | ||
|---|---|---|---|
| 0.78 | 1.56 | 3.12 | |
| 3c | 56.83 ± 2.96 | 65.81 ± 3.88 | 79.14 ± 4.29 |
| 3d | 48.25 ± 3.17 | 58.77 ± 4.03 | 66.58 ± 3.27 |
| Ketoconazole | 60.99 ± 2.94 | 73.12 ± 4.16 | 84.56 ± 3.01 |
| Fluconazole | 61.74 ± 1.70 | 70.12 ± 3.22 | 82.13 ± 4.45 |
| Compounds | IC50 (µg/mL) | R2 |
|---|---|---|
| 3c | >500 | 0.9754 |
| 3d | >500 | 0.9839 |
| Compounds | MW | RB | MV | DHB | AHB | PSA | log P | VRT | VRF |
|---|---|---|---|---|---|---|---|---|---|
| 3a | 395.48 | 4 | 1263.60 | 1 | 5.5 | 51.98 | 5.33 | 1 | 1 |
| 3b | 409.51 | 4 | 1322.54 | 1 | 5.5 | 51.98 | 5.63 | 1 | 1 |
| 3c | 440.48 | 5 | 1346.00 | 1 | 6.5 | 100.64 | 4.62 | 0 | 1 |
| 3d | 420.49 | 5 | 1330.30 | 1 | 7 | 77.77 | 4.56 | 0 | 1 |
| 3e | 425.51 | 5 | 1331.38 | 1 | 6.25 | 60.46 | 5.37 | 1 | 1 |
| 3f | 413.47 | 4 | 1279.71 | 1 | 5.5 | 51.98 | 5.56 | 1 | 1 |
| 3g | 429.93 | 4 | 1307.72 | 1 | 5.5 | 51.98 | 5.82 | 1 | 1 |
| 3h | 474.38 | 4 | 1316.63 | 1 | 5.5 | 51.98 | 5.90 | 1 | 1 |
| 3i | 463.48 | 4 | 1360.43 | 1 | 5.5 | 51.98 | 6.31 | 1 | 1 |
| 3j | 423.53 | 4 | 1346.88 | 1 | 5.5 | 47.09 | 5.80 | 1 | 1 |
| 3k | 431.46 | 4 | 1289.37 | 1 | 5.5 | 50.54 | 5.72 | 1 | 1 |
| 3l | 464.37 | 4 | 1316.53 | 1 | 5.5 | 48.57 | 6.00 | 1 | 1 |
| Ketoconazole | 530.45 | 5 | 1511.39 | 0 | 6.75 | 55.75 | 4.91 | 1 | 0 |
| Fluconazole | 306.27 | 6 | 883.50 | 1 | 6.75 | 72.55 | 3.55 | 0 | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplancıklı, Z.A.; Levent, S.; Osmaniye, D.; Sağlık, B.N.; Çevik, U.A.; Çavuşoğlu, B.K.; Özkay, Y.; Ilgın, S. Synthesis and Anticandidal Activity Evaluation of New Benzimidazole-Thiazole Derivatives. Molecules 2017, 22, 2051. https://doi.org/10.3390/molecules22122051
Kaplancıklı ZA, Levent S, Osmaniye D, Sağlık BN, Çevik UA, Çavuşoğlu BK, Özkay Y, Ilgın S. Synthesis and Anticandidal Activity Evaluation of New Benzimidazole-Thiazole Derivatives. Molecules. 2017; 22(12):2051. https://doi.org/10.3390/molecules22122051
Chicago/Turabian StyleKaplancıklı, Zafer Asım, Serkan Levent, Derya Osmaniye, Begüm Nurpelin Sağlık, Ulviye Acar Çevik, Betül Kaya Çavuşoğlu, Yusuf Özkay, and Sinem Ilgın. 2017. "Synthesis and Anticandidal Activity Evaluation of New Benzimidazole-Thiazole Derivatives" Molecules 22, no. 12: 2051. https://doi.org/10.3390/molecules22122051