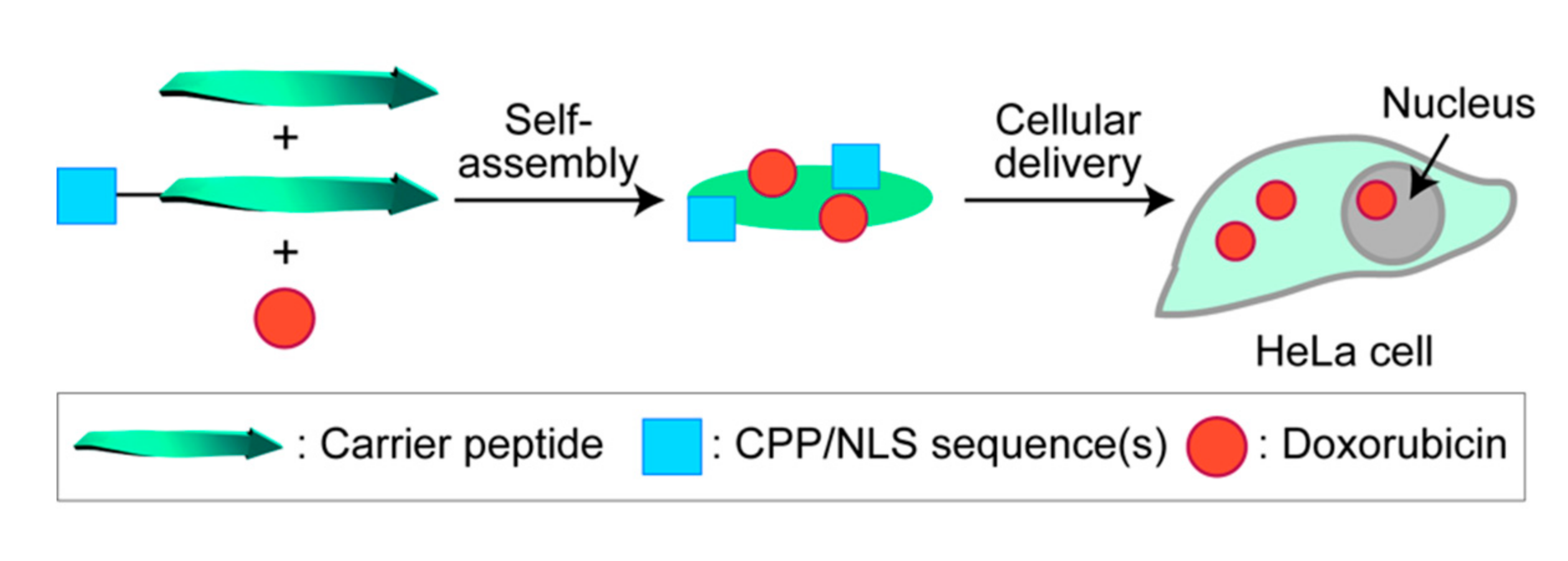
Non-Covalent Loading of Anti-Cancer Doxorubicin by Modularizable Peptide Self-Assemblies for a Nanoscale Drug Carrier




Abstract
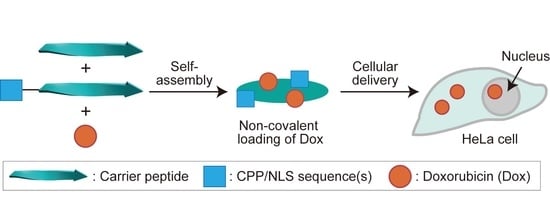
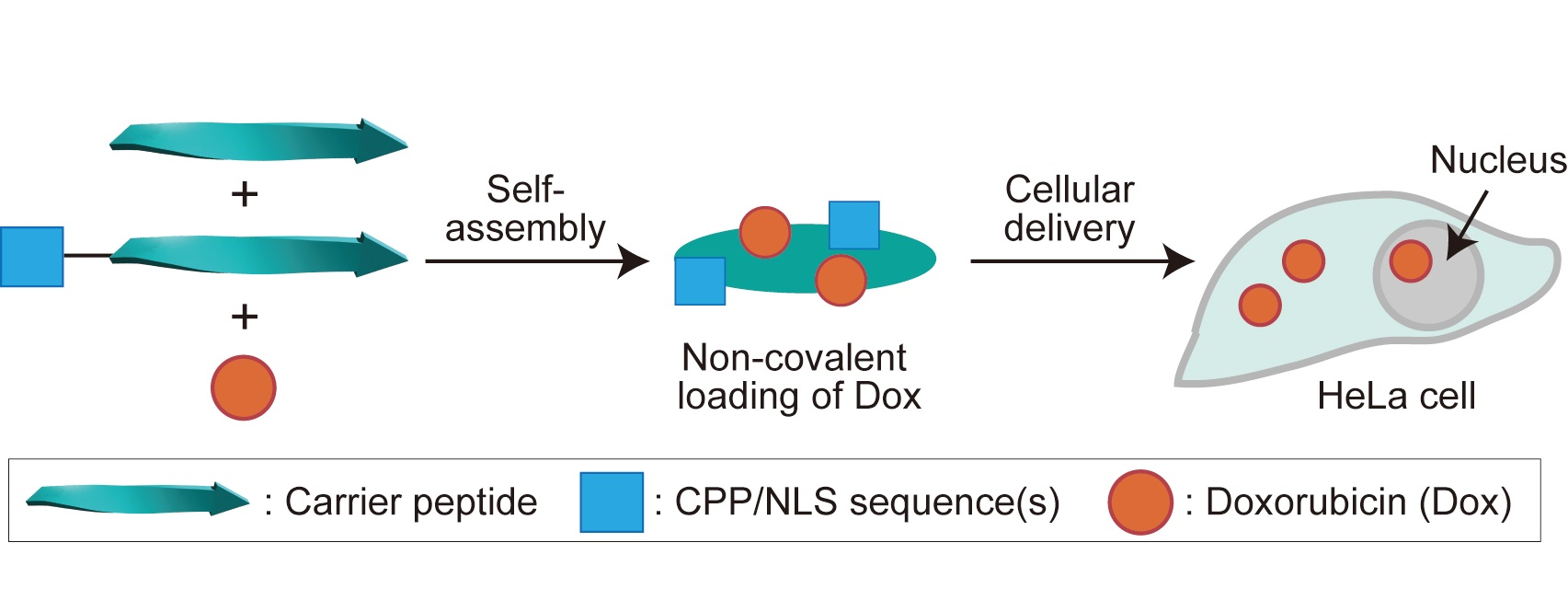
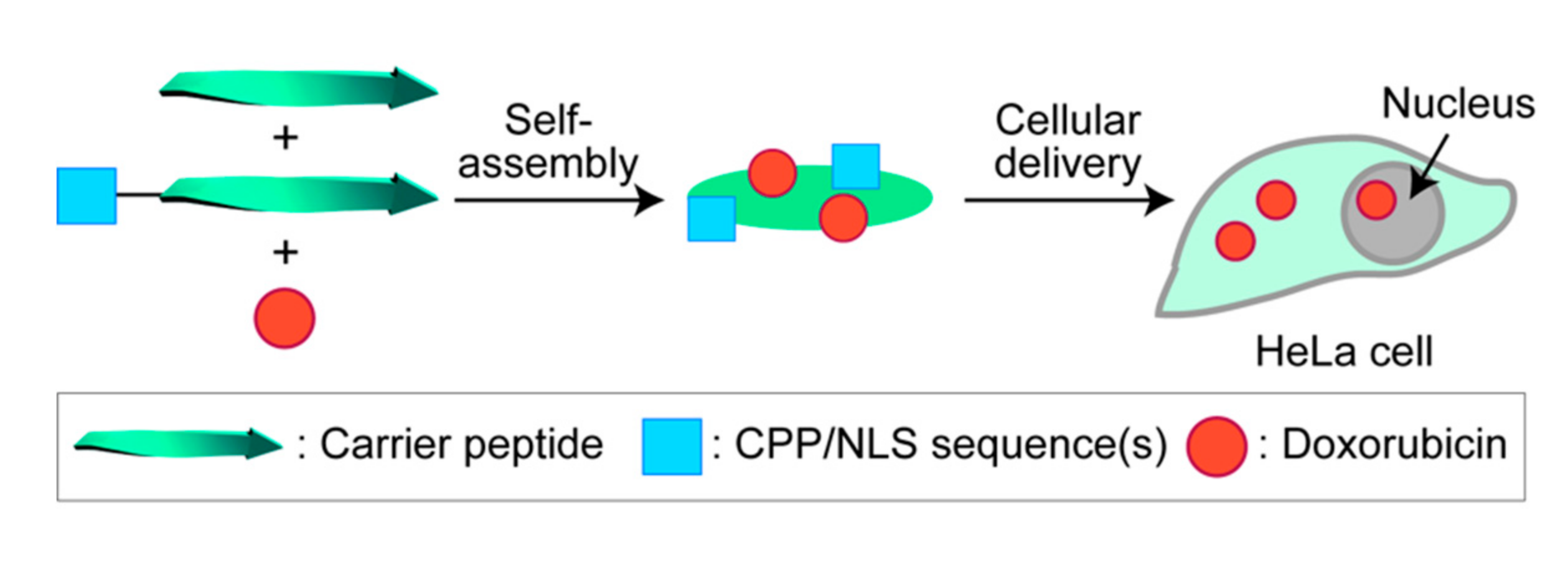
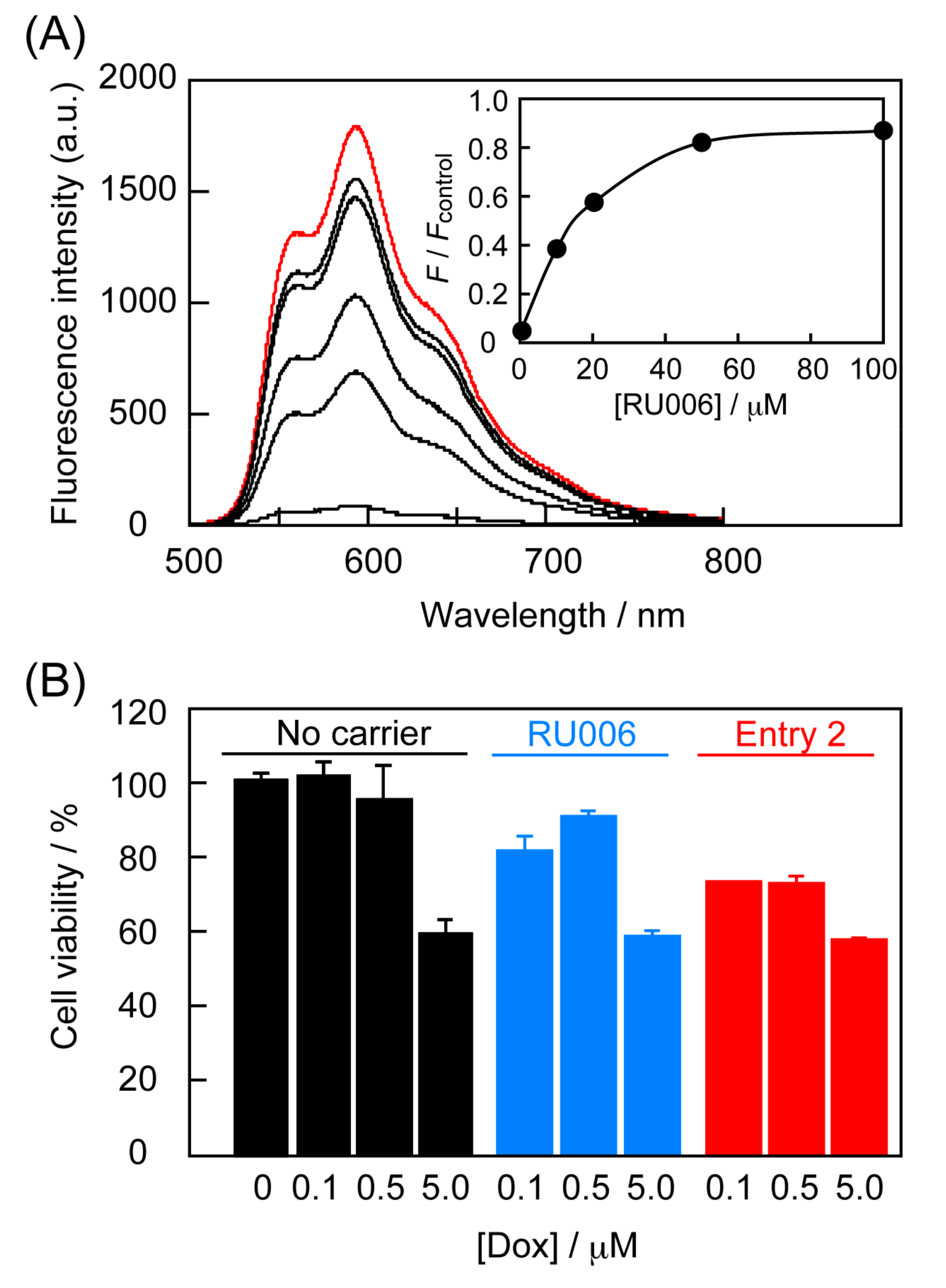
:
{kind=link}
{kind=link}
{kind=link}
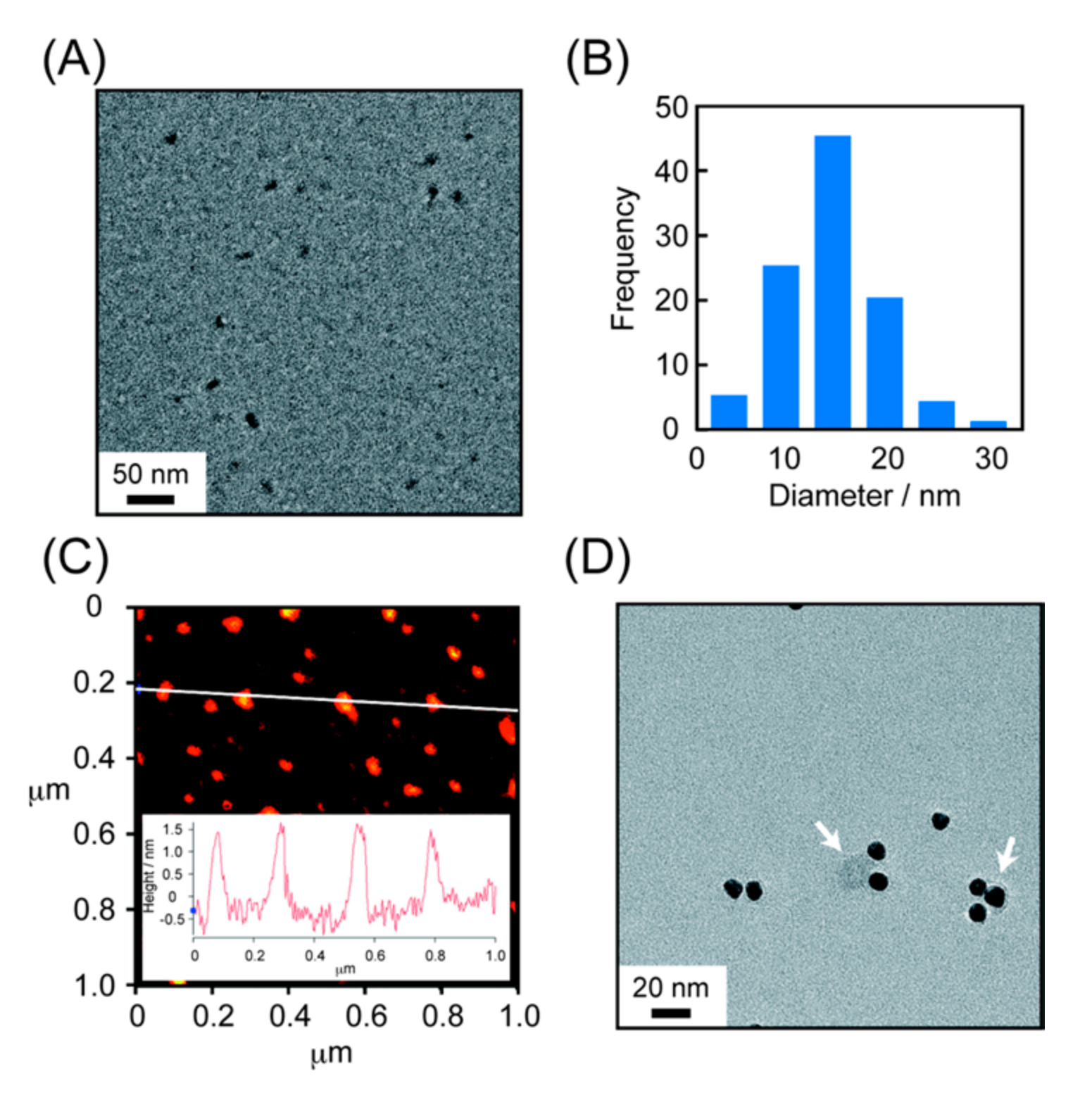{kind=link}
{kind=link}
{kind=link}
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van der Aa, M.A.; Mastrobattista, E.; Oosting, R.S.; Hennink, W.E.; Koning, G.A.; Grommelin, D.J.A. The nuclear pore complex: The gateway to successful nonviral gene delivery. Pharm. Res. 2006, 23, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Kubitscheck, U.; Grünwald, D.; Hoekstra, A.; Rohleder, D.; Kues, T.; Siebrasse, J.P.; Peters, R.J. Nuclear transport of single molecules: Dwell times at the nuclear pore complex. Cell Biol. 2005, 168, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.; Sahoo, S.K. Intracellular trafficking of nuclear localization signal conjugated nanoparticles for cancer therapy. Eur. J. Pharm. Sci. 2010, 39, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Nakielny, S.; Dreyfuss, G. Transport of proteins and RNAs in and out of the nucleus. Cell 1999, 99, 677–690. [Google Scholar] [CrossRef]
- Patel, S.S.; Belmont, B.J.; Sante, J.M.; Rexach, M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T. The molecular architecture of the nuclear pore complex. Nature 2007, 450, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Qin, S.-Y.; Ren, S.; Jiang, X.-J.; Zhou, R.-X.; Zhang, X.-Z. Peptide-based vector of VEGF plasmid for efficient gene delivery in vitro and vessel formation in vivo. Bioconjugate Chem. 2013, 24, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Y.A.; Raschle, P.S.; Wennemers, H. Effect of preorganized charge-display on the cell-penetrating properties of cationic peptides. Angew. Chem. Int. Ed. 2017, 56, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, S.; Laufer, S.D.; Restle, T. Recent developments in peptide-based nucleic acid delivery. Int. J. Mol. Sci. 2008, 9, 1276–1320. [Google Scholar] [CrossRef] [PubMed]
- Dostmann, W.R.; Taylor, M.S.; Nickl, C.K.; Brayden, J.E.; Frank, R.; Tegge, W.J. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc. Natl. Acad. Sci. USA 2000, 97, 14772–14777. [Google Scholar] [CrossRef] [PubMed]
- Säälik, P.; Elmquist, A.; Hansen, M.; Padari, K.; Saar, K.; Viht, K.; Langel, Ü Pooga, M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjugate Chem. 2004, 15, 1246–1253. [Google Scholar]
- Torchilin, V.P. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv. Drug Deliv. Rev. 2008, 60, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Medintz, L.; Pons, T.; Delehanty, J.B.; Susumu, K.; Brunel, F.M.; Dawson, P.E.; Mattoussi, H. Intracellular delivery of quantum dot-protein cargos mediated by cell penetrating peptides. Bioconj. Chem. 2008, 19, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, G.R.; Tulumello, D.V.; Kelley, S.O. Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 2013, 8, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Raad, M.D.; Teunissen, E.A.; Mastrobattista, E. Peptide vectors for gene delivery: From single peptides to multifunctional peptide nanocarriers. Nanomedicine 2014, 9, 2217–2232. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chen, Y.; Tong, Y.W. Delivery of therapeutics and molecules using self-assembled peptides. Curr. Med. Chem. 2014, 21, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.P.; Rathi, B.; Rodrigues, J.; Gorobets, N.Y. Self-assembled peptide nanoarchitectures: Applications and future aspects. Curr. Top. Med. Chem. 2015, 15, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Kamaly, N.; Memic, A.; Shaflee, H. Self-assembled nanstructures: Smart nanomaterials toward targeted drug delivery. Nano Today 2016, 11, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.; Guerin, T.; Toth, I.; Stephenson, R.J. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Adv. Drug Deliv. Rev. 2017, 110–111, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Tomizaki, K.-Y.; Wakizaka, S.; Yamaguchi, Y.; Kobayashi, A.; Imai, T. Ultrathin gold nanoribbons synthesized within the interior cavity of a self-assembled peptide nanoarchitecture. Langmuir 2014, 30, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Tomizaki, K.-Y.; Kishioka, K.; Kobayashi, H.; Kobayashi, A.; Yamada, N.; Kataoka, S.; Imai, T.; Kasuno, M. Roles of aromatic side chains and template effects of the hydrophobic cavity of a self-assembled peptide nanoarchitecture for anisotropic growth of gold nanocrystals. Bioorg. Med. Chem. 2015, 23, 7282–7291. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; White, P.D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: New York, NY, USA, 2000; pp. 41–76. [Google Scholar]
- Chen, G.; Xie, Y.; Peltier, R.; Lei, H.; Wang, P.; Chen, J.; Hu, Y.; Wang, F.; Yao, X.; Sun, H. Peptide-decorated gold nanoparticles as functional nano-capping agent of mesoporous silica container for targeting drug delivery. ACS Appl. Mater. Interfaces 2016, 8, 11204–11209. [Google Scholar] [CrossRef] [PubMed]
- Bremner, K.H.; Seymour, L.W.; Logan, A.; Read, M.L. Factors influencing the ability of nuclear localization sequence peptides to enhance nonviral gene delivery. Bioconjugate Chem. 2004, 15, 152–161. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds studied in this research are available from the authors. |


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomizaki, K.-y.; Kishioka, K.; Kataoka, S.; Miyatani, M.; Ikeda, T.; Komada, M.; Imai, T.; Usui, K. Non-Covalent Loading of Anti-Cancer Doxorubicin by Modularizable Peptide Self-Assemblies for a Nanoscale Drug Carrier. Molecules 2017, 22, 1916. https://doi.org/10.3390/molecules22111916
Tomizaki K-y, Kishioka K, Kataoka S, Miyatani M, Ikeda T, Komada M, Imai T, Usui K. Non-Covalent Loading of Anti-Cancer Doxorubicin by Modularizable Peptide Self-Assemblies for a Nanoscale Drug Carrier. Molecules. 2017; 22(11):1916. https://doi.org/10.3390/molecules22111916
Chicago/Turabian StyleTomizaki, Kin-ya, Kohei Kishioka, Shunsuke Kataoka, Makoto Miyatani, Takuya Ikeda, Mami Komada, Takahito Imai, and Kenji Usui. 2017. "Non-Covalent Loading of Anti-Cancer Doxorubicin by Modularizable Peptide Self-Assemblies for a Nanoscale Drug Carrier" Molecules 22, no. 11: 1916. https://doi.org/10.3390/molecules22111916