1. Introduction
Traditional Chinese medicines (TCMs) are playing an increasingly crucial role for the treatment of various chronic disorders, because they are multi-component and multi-target agents, thus leading to a holistically therapeutic action for multi-factorial diseases [
1,
2,
3,
4,
5]. The efficacy of TCMs has been well-defined by more and more evidences from clinical practices; however, it is still challenging to clarify the effective material basis of a given TCM. Because exposureto a component in the circulation system is the prerequisite for efficacy in most cases, it is thereby viable to characterize the effective constituents via profiling the compounds and assessing their exposure patterns in plasma. Moreover, different pharmacokinetic profiles can usually observed between normal and pathological subjects, attributing to their different physiological status; hence, pharmacokinetic comparisons should provide direct clues for the characterization of therapeutic components. In the other words, multi-component pharmacokinetic evaluation [
6,
7,
8,
9], especially comparison between normal and diseased groups, is a feasible approach to find the ingredients being responsible for TCM efficacy.
Qishen Keli (QSKL) is derived from two well-known TCM formulas, namely
Simiaoyongan and
Zhenwu decoctions, and consists of six famous herbal medicines: Astragali Radix (Chinese name: Huangqi), Salvia Miltiorrhiza Radix et Rhizoma (Chinese name: Danshen), Lonicerae Japonicae Flos (Chinese name: Jinyinhua), Scrophulariae Radix (Chinese name: Xuanshen), Aconiti Laterlis Radix Preparata (Chinese name: Fuzi), and Glycyrrhizae Radix et Rhizoma (Chinese name: Gancao). It has been widely used in the clinic by a number of notable physicians for the treatment of coronary heart disease patients with cardiac insufficiency [
10]. In our previous studies, rats with myocardial ischemia simulated by left anterior descending coronary artery ligation havebeen proved to be a reliable heart failure model, and a set of investigations have also been carried out concerning the underlying pharmacological mechanisms for the therapeutic benefits of QSKL in pathological rats [
11,
12,
13,
14,
15]. The primary signal pathways involved in the therapeutic actions have been revealed as well [
11,
12,
13,
14,
15], however, the components responsible for the therapeutic outcomes remain unclear. Therefore, an attempt to reveal the effective components, the current study through a multi-component pharmacokinetic study of QSKL in both sham and model rats was carried out.
Ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) integrates the superior separation potential of UHPLC along with the excellent specific and sensitive detection of MS/MS; hence, it is unsurprising that UHPLC-MS/MS can serveas a workhorse for the simultaneous determination of dozens of analytes in complicated matrices, e.g., plasma [
16], feces [
17,
18], urine [
19], etc. In general, a large array of components can be generated in vivo following oral administration of a given TCM. Because of the challenges regarding chromatographic separation as well as mass spectrometric detection, only several primary components were selected as analytes in numerous previous studies [
20,
21,
22] although they aren’t able to holistically reflect the pharmacokinetic properties of the TCMs. Fortunately, some new bonding and end capping technologies, such as a tri-functional C
18 alkyl phase bonding at a ligand density and polar end capping, are recently drawing worldwide interest attributing to the significant improvements in regard of polarity range, pH span, lifespan, and peak shape have been achieved, in comparison of those conventional RP-C
18 columns [
23,
24,
25,
26], suggesting a promising tool for separating tens of components. On the other side, some of those xenobiotics may prefer positive ionization polarity, while others are better analyzed with negative polarity. In practice more than 500 milliseconds are usually required for the transformation between different ionization polarities, although some innovative techniques have been applied to the ion sources of mass spectrometers. A typical peak from UHPLC usually exhibits a width amongst 10–20 s. Generally speaking, more than 12 data-points are mandatory for reproducible quantification, and it is thereby challenging to acquire enough data-points for each peak when polarity-switching always occurs within a single analytical run. As a work-around, analysts usually measure a single sample applying positive and negative polarities in two separate runs, which represents a significant barrier for high-throughput assay [
27,
28]. Fortunately, a more efficient approach, that is fragmenting an entire measurement into several periods is permitted by modern mass spectrometers. Therefore, a single polarity is implemented in one period and polarity conversion is only scheduled between adjacent periods [
29].
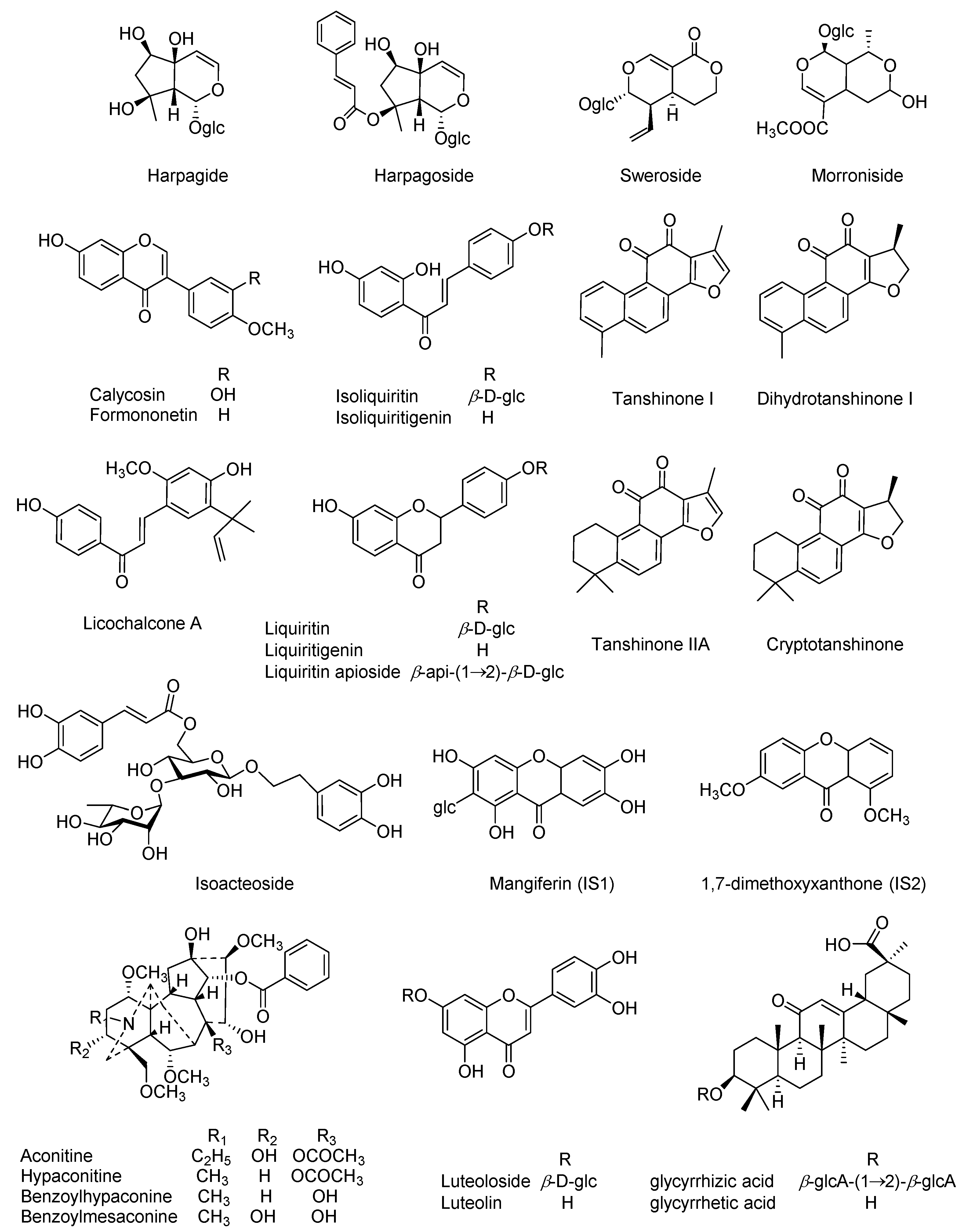
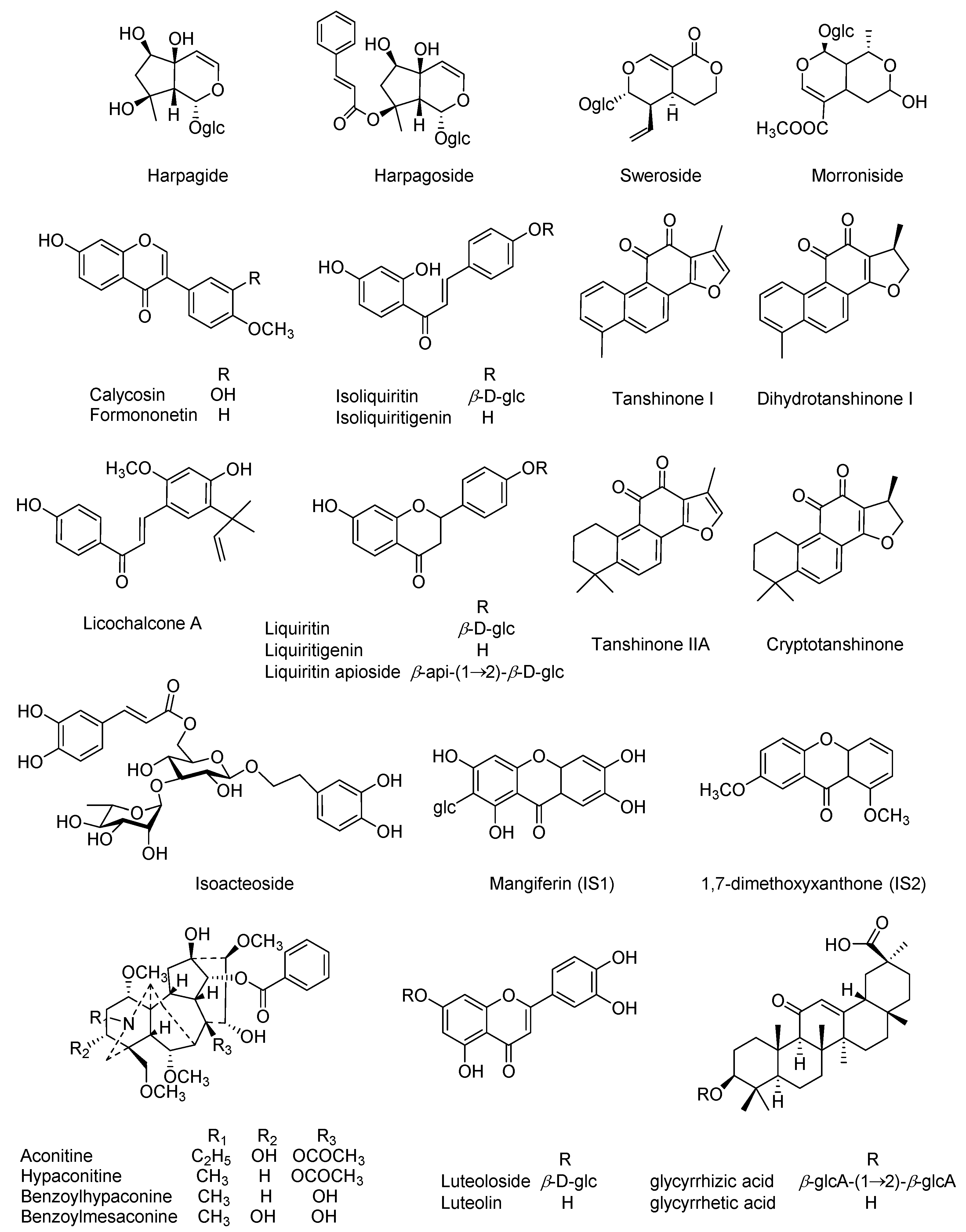
Our preliminary experiments have characterized the prototypes as well as the metabolites in rat plasma following oral administration of QSKL, and twenty-five compounds (
Figure 1) were observed as the primary QSKL-derived components. Herein, simultaneous determination of those compounds in rat plasma was attempted using UHPLC-polarity switching MS/MS (UHPLC-
psMS/MS), and the developed method was applied to characterize their kinetic patterns in both sham rats and myocardial ischemia rats. We envisioned that the obtained findings could offer a practical approach for multi-component pharmacokinetic evaluations of TCMs, while providing solid information for the clarification of the effective material basis of QSKL.
2. Results and Discussion
2.1.Method Development
It is very difficult to achieve satisfactory chromatographic profiles for TCM-treated plasma, because the complicated chemical pool contains both the components coming from the TCM and the endogenous substances found in plasma. Hence, several columns that were claimed to be robust were screened here to accomplish an acceptable separation for those twenty-five analytes, particularly between those compounds with different ionization polarities. A Waters ACQUITY UPLC HSS T3 column (100 mm × 2.1 mm, 1.8 μm, Waters, Wexford, Ireland) was ultimately employed because it offered advantages of both peak capacity and peak shape, in comparison with Kinetex-C18 core-shell column (100 mm × 2.1 mm, 2.6 μm, Phenomenex, Torrance, CA, USA), ACE UltraCore 2.5 SuperC18 column (150 mm × 3.0 mm, 2.5 μm, Advance Chromatography Technologies Ltd., Aberdeen, UK), and Waters ACQUITY UPLC BEH column (100 mm × 2.1 mm, 1.7 μm), as well as the Capcell core ADME column (150 mm × 2.1 mm, 2.7 μm, Shisheido, Tokyo, Japan), because the T3 column utilizes attractive technologies in terms of bonding and end-capping, and it was thereby able to afford satisfactory chromatographic behaviors for all compounds of interest.
Regarding manual mass parameter tuning, each analyte stock solution was diluted to an appropriate concentration (50–100 ng/mL) with 50% aqueous methanol, and directly infused into the ESI interface at a flow rate of 7 μL/min using a syringe pump. The investigated analytes along with the internal standards were firstly measured to yield both MS
1 and MS
2 spectra to ascertain their precursor-to-product ion transitions for quantitative analysis. The positive and negative ionization modes were compared, and the results proved that the positive mode could provide higher responses for some analytes, such as benzoylmesaconine, benzoylhypaconine, aconitine, hypaconitine, dihydrotanshinone I, cryptotanshinone, tanshinone I and tanshinone IIA, and IS2, whereas the other analytes including harpagide, morroniside, sweroside, liquiritin apioside, liquiritin, luteoloside, isoacteoside, isoliquiritin, harpagoside, liquiritigenin, luteolin, calycosin, isoliquiritigenin, glycyrrhizic acid, formononetin, licochalcone A, and glycyrrhetic acid, and IS1 were found to be more suitable for negative polarity. After careful manual optimization, two precursor-to-product ion transitions, including quantifier and qualifier ones, were set for each analyte, and the more sensitive one (
Table 1) served as the quantifier one. Ion source gas flows and ion source temperature adopted the typical values corresponding to the UHPLC effluent of 0.4 mL/min.
Moreover, mobile phase modifiers were screened among ammonium acetate, formic acid, acetic acid, and ammonium formate by comparing the overall response of allanalytes. Formic acid was finally introduced as the modifiers for both organic (solvent B) and aqueous (solvent A) solvents to strengthen the ionization of most analytes. Either contentof formic acid was optimized as 0.02% (
v/
v). The optimized chromatography and mass spectrometry parameters are discussed in
Section 4.2 and listed in
Table 1.
2.2. Method Validation
2.2.1. Selectivity and Specificity
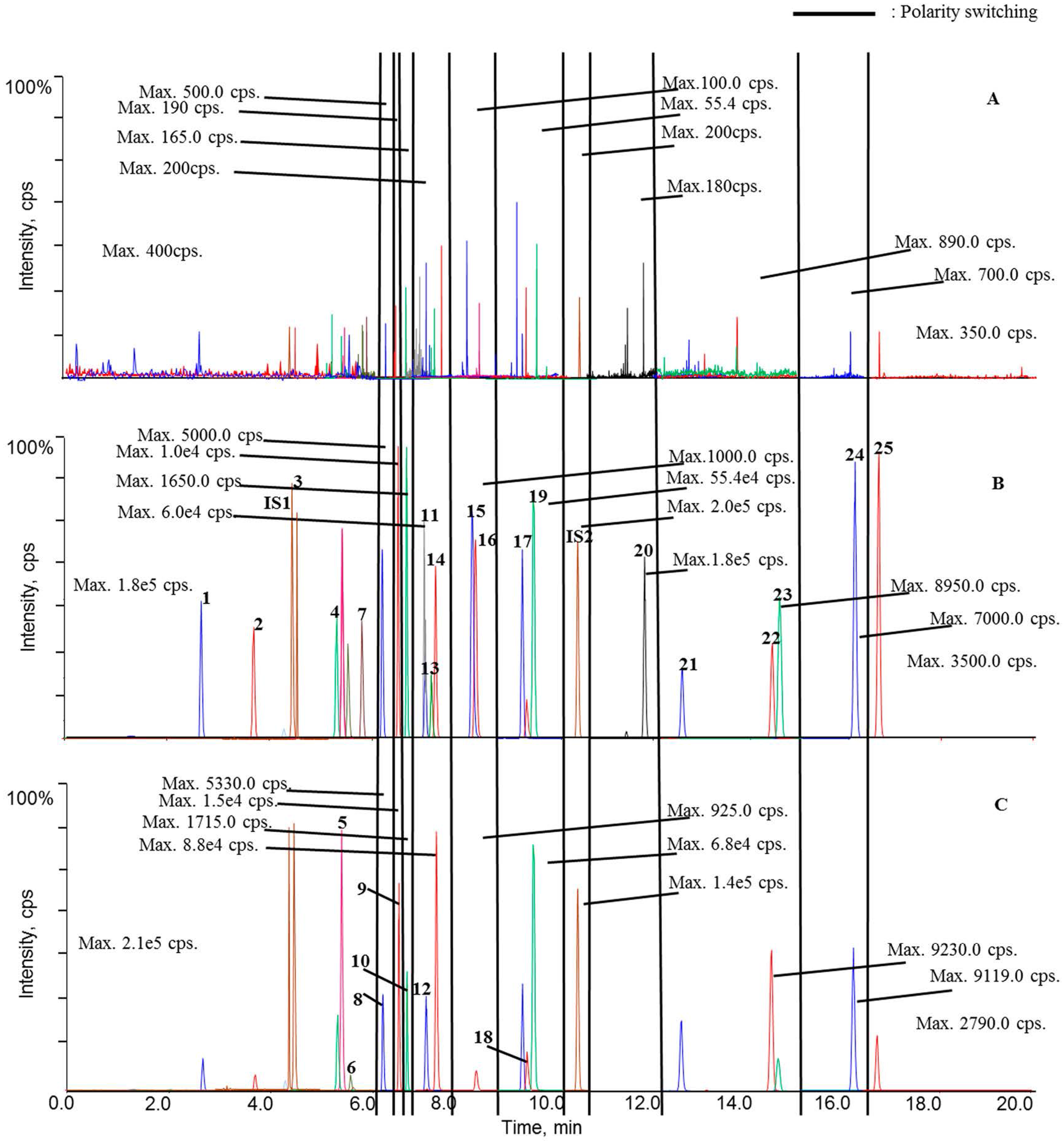
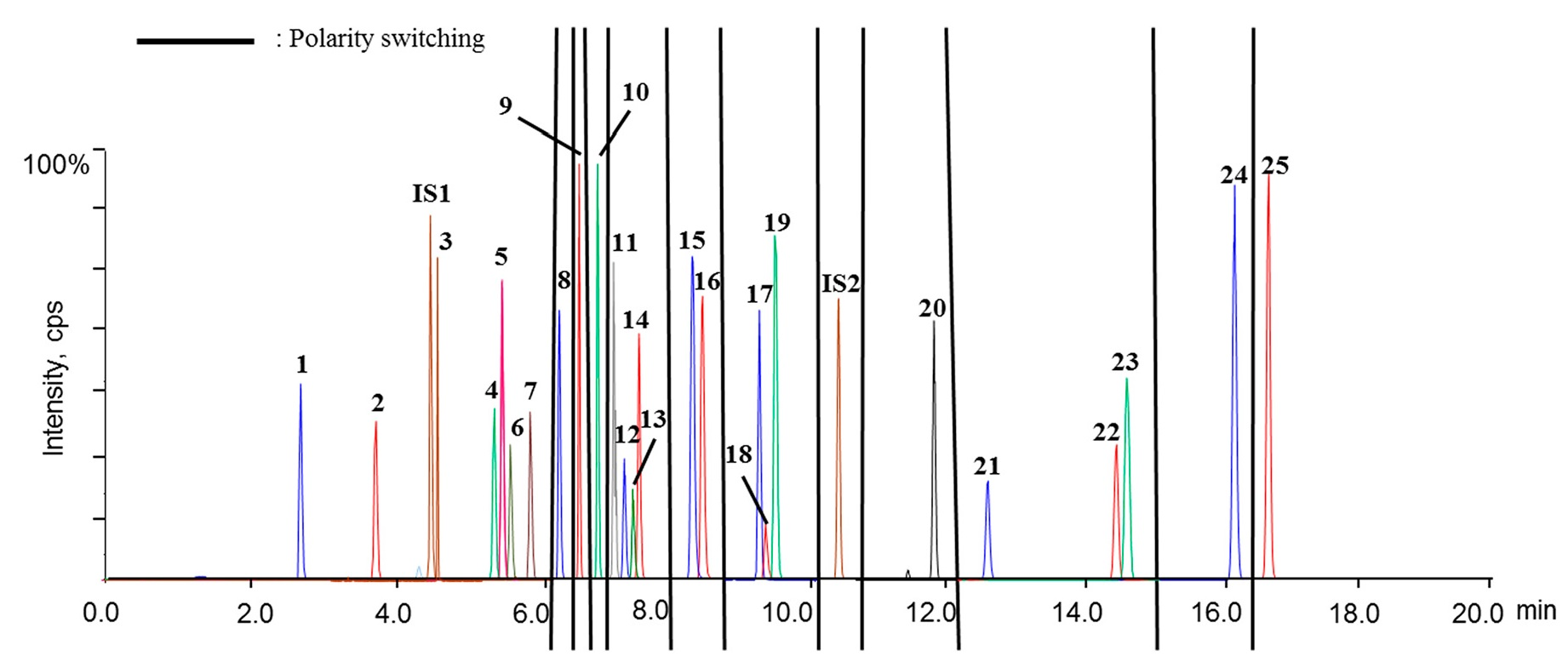
Retention times of the twenty-five analytes, IS1 and IS2 are summarized in
Table 1, while
Figure 2 displays the representative chromatograms of calibration samplesas well asplasma samples obtained from sham rat at 0 h (blank plasma) and 1 h after oral administration. Overall, satisfactory chromatographic performance was achieved for these analytes using the current UHPLC-
psMS/MS program (
Figure 2B).
The signals were assigned to analytes and internal standards by comparing retention times, response ratios between the quantifier and qualifier ion transitions, and MS2 spectra with those of authentic compounds. Although minor responses (lower than 100 cps for each) were observed for some endogenous substances around the signals of certain analytes, those weak signals would not significantly affect the reliable quantitation of those analytes.
2.2.2. Linearity, LOQ and LOD
Regressive linear equations, correlation coefficients, linear ranges, LOQs, and LODs of the twenty-five analytes are summarized in
Table 2. All calibration curves showed good linearity within individually concentration ranges with correlation coefficients (
r) ranged from 0.9978 to 0.9995. LOQs and LODs of analytes varied amongst the ranges 7.11 pg/mL–6.60 ng/mL and 0.900 pg/mL–1.89 ng/mL, respectively. Above all, the linear properties along with sensitive features could fulfill the requirements of the multi-component pharmacokinetic studies.
2.2.3. Precision and Accuracy
Inter- and intra-day precisions of the method for the simultaneous determination of twenty-five analytes were studied using QC samples, and the RSD% values, which ranged from 1.9% to 15.5%, are presented in
Table 3. REs of all concentration levels in the linearity range of all analytes were between 85.3–115.0%, indicating that the accuracy could also meet the requirements of the pertinent guidelines. Those findings proved the newly developed method to be precise and accurate.
2.2.4. Matrix Effect and Recovery
The overall method recovery was governed by two terms such as matrix effect and recovery, and the overall method recovery played a pivotal role for the assessment of the method validity. Matrix effect and recovery assays were utilized to evaluate the interferences for the targeted components from inherent substances in the matrices and the influence of extraction process, respectively. Following the preparation of those three sets (sets A–C) of samples, comparisons were performed among their quantitative results. The overall method recoveries were calculated among 58.4–91.8% (RSD% among 2.6–16.3%), while the recovery (
Table 4) and the matrix effect (
Table 5) ranged from 61.3% to 90.4% (RSD% among 1.3–13.9%) and from 91.6% to 107.0% (RSD% among 1.0–10.1%), respectively. Because 25% of the plasma sample was deserted to completely remove those precipitates during sample preparation, the recovery as well as the overall method was approximately 75%. Given the parallel sample preparation between calibration samples and real samples, the relative low recovery could not affect the acquisition of reliable results. Above all, the overall method recovery could not obstruct reliable quantitation.
2.2.5. Stability
Stability of the twenty-five analytes under conditions of room temperature for 24 h, three freeze-thaw cycles, and −80 °C for 30 days were assayed. The results are summarized in
Table 6, and it showed the deviations (RE) between measured values and nominal values were among 81.6–117.0%, the RSD% values ranged from 0.5% to 15.9%. Therefore, all analytes kept intact in all stability assays.
2.3. Application in Pharmacokinetic Study
As aforementioned, our preliminarystudy disclosed that twenty-five ingredients in QSKL could be detectable in the circulation system following oral administration; hence, these components were employed as markers to reflect the pharmacokinetic pattern of the entire QSKL formula. The validated method was applied for pharmacokinetic investigations of QSKL in the sham and model rats. Among thetwenty-five analytes, trace distributions were observed for four compounds, such as isoacteoside, luteolin, aconitine, and licochalcone A, and these four compoundsofinterestwere unquantifiable in most QSKL-treated samples even that the dosage was set at 8.0 g/kg (
Figure 2C). Meanwhile, significant distributions were detected for the other twenty analytes, including harpagide, morroniside, sweroside, liquiritin apioside, liquiritin, luteoloside, benzoylmesaconine, isoliquiritin, benzoylhypaconine, liquiritigenin, calycosin, hypaconitine, isoliquiritigenin, glycyrrhizic acid, formononetin, dihydrotanshinone I, cryptotanshinone, tanshinone I, glycyrrhetic acid, and tanshinone IIA, in rat plasma after a single oral dose of QSKL because they were quantifiable in most samples (
Figure 2C).
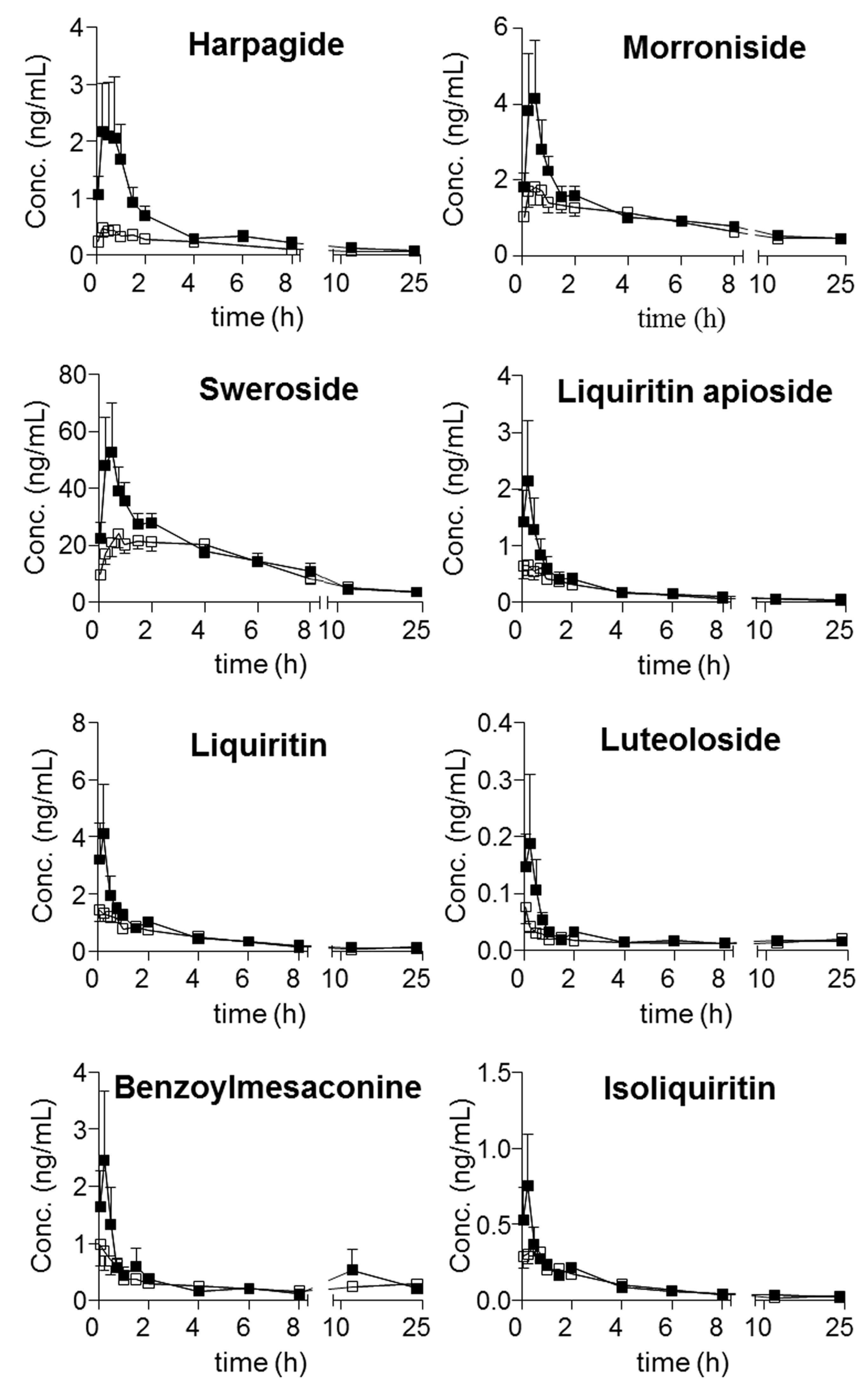
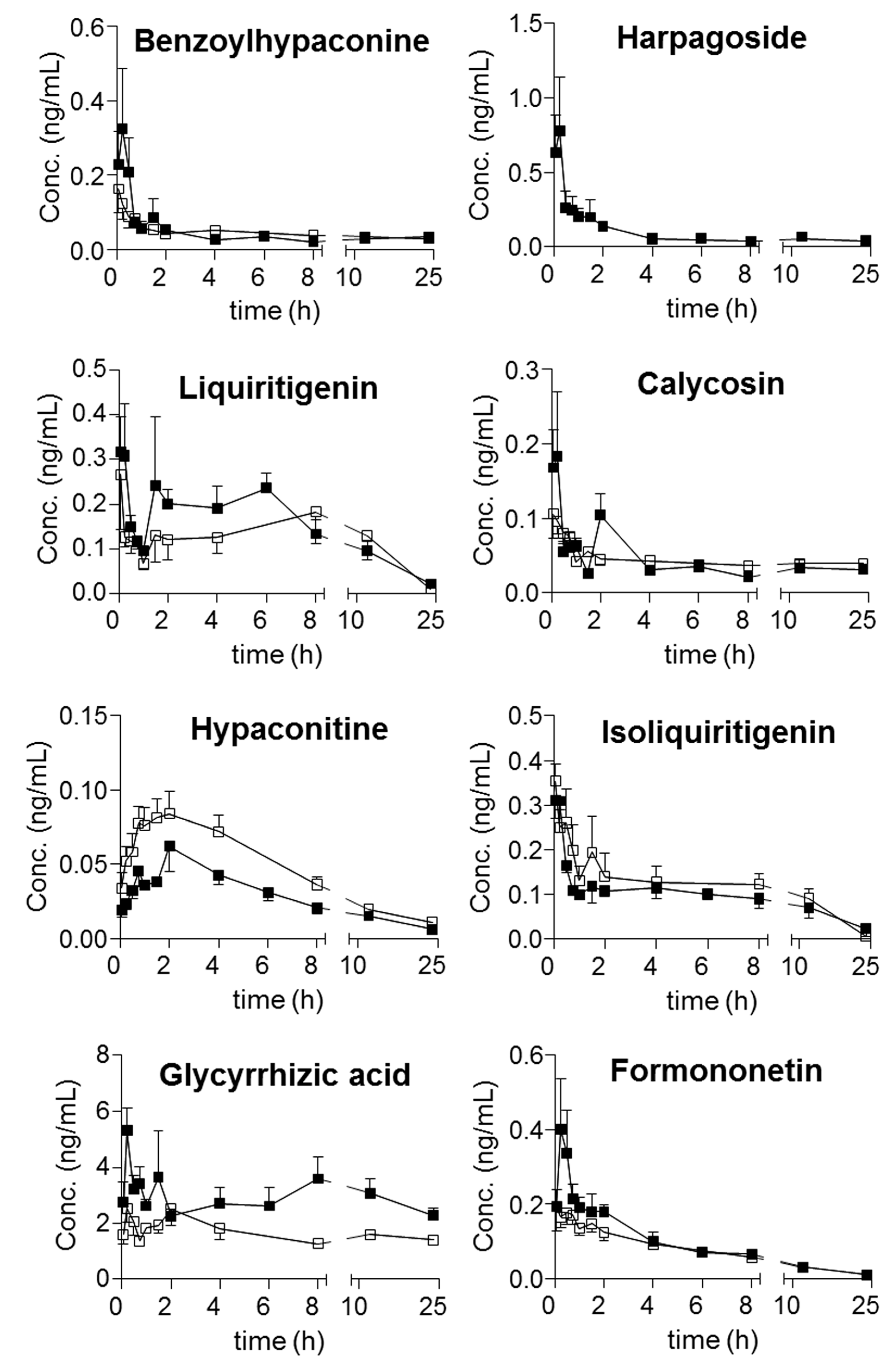
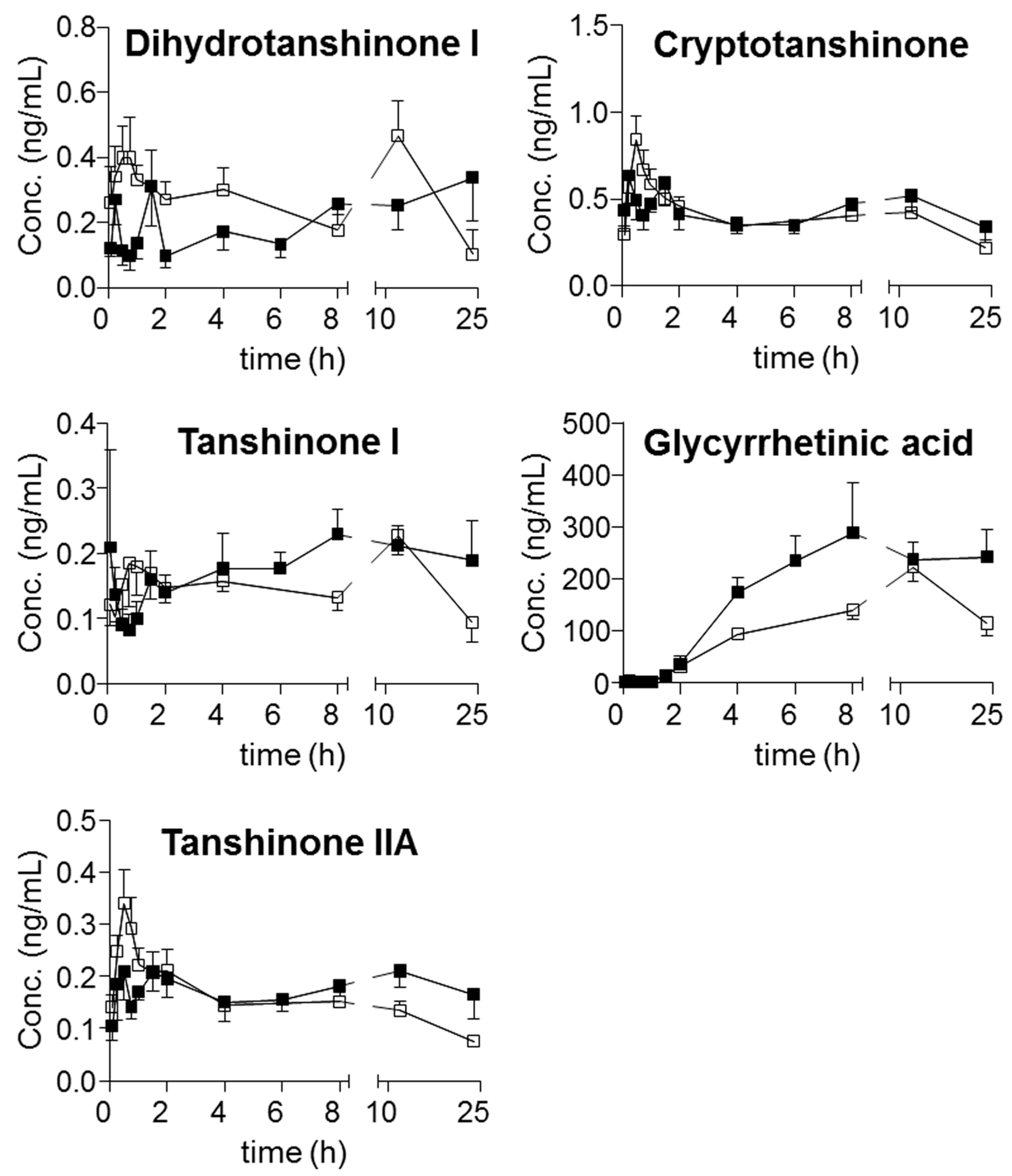
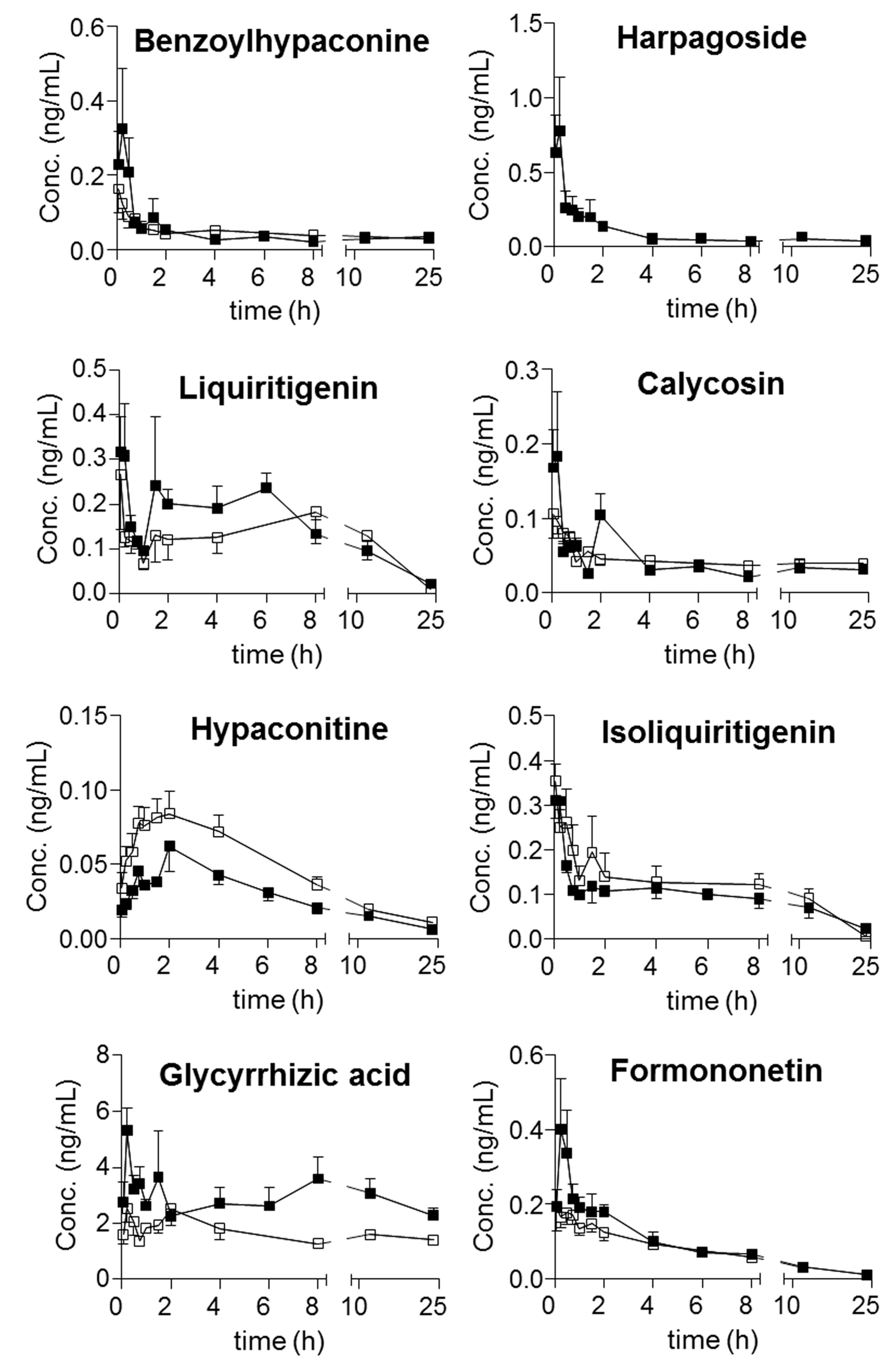
Moreover, harpagoside was always quantifiable in the plasma of the model group, whereas most of the concentrations in the samples from the sham group were lower than its LOQ. Following the achievement of simultaneous determination of the twenty-one compounds in plasma, their trajectories in all QSKL-administrated rats were obtained. Mean plasma concentration-time profiles of the twenty-one analytes are illustrated in
Figure 3, and the main pharmacokinetic parameters that were calculated using non-compartmental model are summarized in
Table 7. Overall, most compounds reached their maximum concentrations within 2 h, whereas slow absorption was observed for the four diterpenequinonederivatives (T
max, 8 h). e.g., dihydrotanshinone I, cryptotanshinone, tanshinone I, and tanshinone IIA, as well as glycyrrhetic acid (T
max greater than 12 h). Except glycyrrhizic acid and glycyrrhetic acid, relatively rapid elimination (
t1/2 less than 24 h) occurred for the other components after oral dosing QSKL. Multiple-peak profiles occurred for liquiritigenin, isoliquiritigenin, glycyrrhetinic acid, and glycyrrhizic acid.
3. Discussion
The pharmacokinetic profiles of QSKL were compared between the sham and model groups. C
max of harpagoside in model rats (1.02 ± 0.692 ng/mL) was quite greater than that of sham rats (
Table 7), and significant differences were observed for AUC
0→t of glycyrrhizic acid (sham: 37.9 ± 7.11 vs. model: 72.8 ± 27.8, ng·h/mL), glycyrrhetic acid (sham: 3205 ± 298 vs. model: 5038 ± 1857, ng·h/mL), and tanshinone IIA (sham: 3.18 ± 0.664 vs. model: 4.28 ± 0.678, ng·h/mL) (
Table 7).
Multiple peak phenomena have been widely reported for a number oforal drugs, and several mechanisms have been proposed for the phenomenon, such as enterohepatic recycling, and the presence of absorption windows along the gastrointestinal tract. Moreover, the complicated chemical correlations among the ingredients in TCMs can also contribute to this phenomenon, for instance the generation of aglycones viahydrolysis of the glycosides. In current study, multiple peak profiles were observed for liquiritigenin along with isoliquiritigenin, attributing to the hydrolysis of their glycoside. On the other side, the unique pharmacokinetic curves of glycyrrhetinic acid and glycyrrhizic acid might be attributed to the mutual transformation between these compounds which was governed by enterohepatic recycling [
30,
31,
32,
33], hydrolysis of glycyrrhizic acid, and glucuronidation of glycyrrhetinic acid.
Disorders usually result in changes of enzyme expressions and various organs, for instance the modification of gut flora, the damage of intestinal mucosa; hence, the absorption, distribution, metabolism, and excretion (ADME) profiles of those absorbable components might change, due to the changes of the interactions between the components and relevant proteins. In other words, the differences of pharmacokinetic profiles might provide meaningful clues for searching effective components. Moreover, significant exposureshould be afforded by a given component in plasma if it exhibits activity in vivo. Therefore, the pharmacokinetic differences of components, especially those primary ones, between normal and disease animals could provide meaningful information for seeking the effective components. Although the greatest content was observed for sweroside in QSKL extract, more abundant occurrencewas detected for glycyrrhetic acid (C
max: 230 ± 57.1 ng/mL for sham group and 376 ± 196 ng/mL for model group) than sweroside (C
max: 28.9 ± 14.3 ng/mL for sham group and 54.2 ± 38.9 ng/mL for model group). In addition to sweroside along with glycyrrhetic acid, the exposure of glycyrrhizic acid (AUC
0→t: 37.9 ± 7.11 ng·h/mL in sham group and 72.8 ± 27.8, ng·h/mL in model group) was also quite abundant. Moreover, different from sweroside, significant differences occurred between the kinetic profiles, in particular AUC
0→t values of glycyrrhizic acid as well as glycyrrhetic acid between the sham and model groups. In addition, significant differences also occurred for C
max of harpagoside as well as AUC
0→t of tanshinone IIA; however, relative low contents were observed for these two compounds. As a consequence, glycyrrhizic acid together with glycyrrhetic acid might play primary roles in response to myocardial ischemia induced by LAD coronary artery ligation in rats. Actually, the efficacy of these two compounds has been well-defined on cardiac performance [
34,
35], and the modification of the interactions between these two compounds and their targets should play key roles for those significant differences.
4. Materials and Methods
4.1. Materials and Reagents
Authentic compounds, including aconitine, hypaconitine, benzoylmesaconine, benzoyl-hypaconine, dihydrotanshinone I, harpagide, isoacteoside, harpagoside, morroniside, sweroside, glycyrrhizic acid, liquiritigenin, isoliquiritigenin, liquiritin, isoliquiritin, liquiritin apioside, licochalcone A, formononetin, calycosin, luteolin, and luteoloside, were purchased from Shanghai Standard Biotech Co. Ltd. (Shanghai, China), while tanshinone IIA, cryptotanshinone, and tanshinone I, as well as glycyrrhetic acid, were obtained from the National Institutes for Food and Drug Control (Beijing, China). All chemical structures (
Figure 1), 25 in total, were further verified by
1H-,
13C-NMR, and MS analyses, and the purity of each authentic compound was determined to be greater than 98% by normalization of the peak areas detected by UHPLC-IT-TOF-MS (Shimadzu, Kyoto, Japan). Two additional compounds such as mangiferin (IS1) [
36] and 1,7-dimethoxyxanthone (IS2) [
37] which were previously purified from
Polygala tenuifolia Willd in our laboratory served as internal standards for positive and negative polarities, respectively.
Formic acid, dimethylsulfoxide (DMSO), and acetonitrile (ACN) were of LC-MS grade and purchased from Merck (Darmstadt, Germany). Ultrapure water was prepared in-house with a Milli-Q system (Millipore, Bedford, MA, USA). The other chemicals were of analytical grade and obtained commercially from Beijing Chemical Works (Beijing, China). Freeze-dried QSKL extract powders were prepared in our group following the previous protocol [
15] and the contents of those twenty-five compounds including harpagide (2.7%), morroniside (5.3%), sweroside (17.6%), liquiritin apioside (5.3%), liquiritin (9.2%), luteoloside (0.7%), isoacteoside (0.09%), benzoylmesaconine (4.8%), isoliquiritin (1.7%), benzoylhypaconine (0.7%), harpagoside (2.1%), liquiritigenin (0.9%), luteolin (0.02%), calycosin (0.7%), aconitine (0.07%), hypaconitine (0.2%), isoliquiritigenin (0.4%), glycyrrhizic acid (14.2%), formononetin (0.8%), licochalcone A (0.009%), dihydrotanshinone I (0.2%), cryptotanshinone (0.2%), tanshinone I (0.2%), glycyrrhetic acid (9.7%), and tanshinone IIA (0.4%) were quantified using UHPLC-MS/MS.
4.2. Method Development
An LC-20AD LC system (Shimadzu, Kyoto, Japan) consisting of two LC-20ADXR solvent delivery units, a SIL-20ACXR auto-sampler, a CTO-20AC column oven, a DGU-20-A3R degasser, and a CBM-20A controller, was directly connected with an ABSciex 5500 Q-trap mass spectrometer (Foster City, CA, USA) via an electrospray ionization (ESI) interface. Chromatographic separations were conducted on theACQUITY UPLC HSS T3 column. The mobile phase was composed of 0.02% aqueous formic acid (v/v, A) and ACN containing 0.02% formic acid (v/v, B), the gradient elution was programmed as follows: 0–10 min, 5–50% B; 10–15 min, 50–70% B; 15–17 min, 70–95% B; 17–20 min, 95% B; and flow rate, 0.4 mL/min. Re-equilibration of the entire system was achieved by delivering 5% B for 5 min after each program. Column oven was maintained at 35 °C, and the injection volume was set at 10 μL. The auto-sampler chamber was maintained at 4°C.
Regarding the mass spectrometer domain, mass spectrometric detection was operated in multiple reaction monitoring (MRM) mode with programmed polarity switching (
Figure 2). Nitrogen acted as the nebulizer (GS1), heater (GS2), and curtain (CUR), as well as collision gas. MS parameters were set as follows: GS1, 55 psi; GS2, 55 psi; CUR, 35 psi; ion spray needle voltage, 5500 V/−4500 V (5500 V and −4500 V for positive and negative polarities, respectively); heater gas temperature, 550 °C; entrance potential (EP), 10 V/−10 V; collision cell exit potential (CXP), 13 V/−16 V. The MRM ion transitions, declustering potential (DP) levels, and collision energy (CE) values, as well as the polarity-switching schedule, are summarized in
Table 1. Moreover, MRM mode also acted as survey experiment to trigger two separate enhanced product ion (EPI) scans via information dependent acquisition (IDA)algorithm with a threshold criterion as 200 cps. Key parameters for EPI experiments, such as CE and collision energy spread (CES), were set as ±30 eV and 20 eV, respectively.
4.3. Method Validation
The developed method was validated in terms of selectivity, linearity, precision, and accuracy following the FDA Guidance [
38].
4.3.1. Linearity and Sensitivity
Based on our preliminary experiments, stock solutions of all authentic compounds were separately prepared with DMSO to appropriate concentrations. Mixed standard stock solution was obtained by pooling all stock solutions of authentic compounds. The solutions were stored at −20 °C until usage.
The mixed stock solution was sequentially diluted with DMSO to yield a series of solutions with desired concentration levels, and a 2 μL aliquot of each one was added into 98 μL of pooled rat plasma that was obtained from six drug-free male Spargue-Dawley rats to generate a set of calibration samples. After thoroughly mixing, protein precipitation was accomplished for each calibration sample by adding three volumes of ACN containing both internal standards (200 ng/mL for either). The precipitates were excluded through 12,000 rpm centrifugation for 10 min at 4 °C. A 300 μL aliquot of the supernatant was transferred into another Eppendorf tube and then concentrated to dryness with nitrogen blow at 40 °C. The residues were reconstituted with 100 μL of 50% aqueous ACN (v/v) and centrifugation at 12,000 rpm for 10 min was performed to remove the precipitates. Finally, a 10 μL aliquot of the supernatant was injected into UHPLC-psMS/MS system. Each calibration sample was assayed in triplicate.
To gain acceptable deviations for all concentration levels, the standard curves were fitted by a 1/x weighed least squares linear regression method through plotting the peak area ratios of each analyte to respective IS versus the theoretical plasma concentrations over the calibration concentration range. The acceptance criterion for each calibration curve was a correlation coefficient (r) of 0.99 or better and a back-calculated standard concentration within a 15% deviation from the nominal value except at the lower limit of quantification (LOQ). The term of LOQ of each analyte was defined as the lowest concentration in the calibration curve with inter-day imprecision less than 20% as well as inter-day inaccuracy less than ±20%, whereas the limit of detection (LOD) of each analyte corresponded to the lowest detective concentration with signal-to-noise ratio (S/N) of about 3. Moreover, quality control (QC) samples with three concentration levels (high, medium, and low) were prepared based on the linear ranges of all analytes.
4.3.2. Selectivity and Specificity
Selectivity and specificity assays were carried out to check the potential chromatographic interferences from endogenous substances around the signals of the analytes as well as the internal standards. Chromatographic peaks from plasma samples were compared with the authentic standards in terms of the retention times and MS2 spectra obtained by EPI scans.
4.3.3. Accuracy and Precision
Accuracy and intra- and inter-day precisions were assessed by analyzing three consecutive batches of calibration samples and six replicates of each QC level (low, medium or high concentration level), respectively. The accuracy was expressed as ratio earning (RE, %) which was calculated according to (observed concentration/nominal concentration) × 100% and acceptable at the case of RE within 100 ± 15%, while the relative standard deviations (RSDs, %) within ±15% was acceptable for precision assays.
4.3.4. Matrix Effect and Recovery Assays
Process efficiency (overall method recovery), recovery (extraction yield), and matrix effect were assessed by combined experiments following the descriptions archived in the literature [
39]. In brief, independent experiments using pooled drug-free rat plasma along with three diluted mixed standard solutions, corresponding to those QC samples, were performed on three different days with threefold analysis repetitions at each day. The process efficiency (overall method recovery) was determined by comparing the analyte signals (peak areas) obtained from plasma samples spiked prior to extraction (set C) with signals from spiked sample preparation solution (set A). The recovery was determined by comparing analyte responses obtained from set C with signals obtained from plasma samples spiked after the extraction process (set B). Moreover, the matrix effect was evaluated by comparing the analyte peak areas obtained from set A with the ones from set B. The process efficiency (%) is calculated as C/A×100, while the recovery (%) as C/B×100 and the matrix effect (%) as B/A×100, respectively.
4.3.5. Stability Assays
Stability was assayed according to maintaining those QC samples in different conditions. The short-term stability was assessed within the exposure of the QC samples at room temperature for 24 h and measurements were carried out every three hours. The freeze-thaw stability was conducted by evaluating the effects from three freeze-thaw cycles from −80 °C to room temperature (22 °C). The long-term stability was assessed by maintaining QC samples at −80 °C for 30 days.
4.4. Pharmacokinetic Study
Twelve male Spargue-Dawley rats (200 ± 20 g) were supplied by Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). The study protocol was approved by the Committee on the Ethics of Animal Experiments of Beijing University of Chinese Medicine (IACUC approval number 2010-D-013). The rats were acclimated under a humidity (50%) controlled room at 22–25 °C with a 12 h light-dark cycle, and standard diet and water were provided ad libitum for one week.
Afterwards, all rats were randomly divided into two groups, namely sham and model groups. The myocardial ischemia model was simulated by left anterior descending (LAD) coronary artery ligation [
12], whilst the rats belonging to the sham group received the similar surgery, however no actual ligation in LAD. All animals were fasted overnight with free access to water prior to oral dosing. Aliquots (250 μL) of blood were sampled at 0 (pre-dose), 0.08, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 12, and 24 h from venous around eye socket into heparinized tubes after a single oral dosing of 8.0 g/kg of QSKL and the collected blood samples were immediately subjected for 4000 rpm centrifugation for 10 min at 4 °C to harvest the plasma samples. Each 100 μL aliquot of plasma sample was transferred into another Eppendorf tube and stored at −80 °C till use.
When analysis, 300 μL of internal standards (200 ng/mL for either internal standard) fortified ACN, corresponding to three volumes of plasma, was spiked into each thawed plasma sample and mixed thoroughly. Then, the resulting samples were processed as those calibration samples.
Individual animal plasma concentrations versus time dataset were subjected to a non-compartmental pharmacokinetic analysis using WinNonlin software (ver. 6.4, Pharsight Corp., Sunnyvale, CA, USA). The pharmacokinetic parameters of each analyte including the maximum plasma concentration (Cmax), the time at which Cmax was achieved (Tmax), the terminal elimination half-life (t1/2), and the area under the plasma concentration versus time curve (AUC0→t along with AUC0→∞) were calculated. Moreover, Lz corresponding to the ratio of the dosage and AUC0→∞, as well as mean residence time (MRT) that was defined as the ratio of the area under the first moment curve from time 0 to infinity (AUMC0→∞/AUC0→∞) was also calculated. All results are expressed as mean ± SD. Comparisons regarding the pharmacokinetic parameters were carried out between sham and model groups using Student’s t-test, and p values less than 0.05 were considered to be significant.
5. Conclusions
In the present study, a reliable and sensitive method using UHPLC-psMS/MS was developed and validated for the simultaneous determination of as many as twenty-five compounds in rat plasma. Scheduled polarity switching was programed for the ion source of the mass spectrometer to meet the demands of simultaneous monitoring both positive ionization-and negative ionization-favored compounds, while a robust ACQUITY UPLC HSS T3 column afforded satisfactory chromatographic performance in terms of measurement time, peak shape and resolution. Formic acid was found to be suitable for improving the peak shapes, and thereby used as the additive in the mobile phase. A variety of method validation assays demonstrated the method to be specific, sensitive, and accurate. Except those four unquantifiable components (isoacteoside, luteolin, aconitine, and licochalcone A) as well as harpagoside, the pharmacokinetic profiles of the other twenty analytes in both sham and model rats were acquired by applying the validated method for QSKL-treated plasma samples. Significant different pharmacokinetic patternswere observed for glycyrrhizic acid and glycyrrhetic acid between the sham and model rats, whereas mild differences occurred for the other analytes. Hence, the contributions of these twenty-five components, in particular glycyrrhizic acid and glycyrrhetic acid, for the therapeutic outcomes of QSKL can be plausibly claimed. Overall, the information obtained from this study might provide some meaningful clues and evidences for the further studies regarding the clarification of the effective material basis of QSKL and is alsobeneficial for its clinical applications.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}