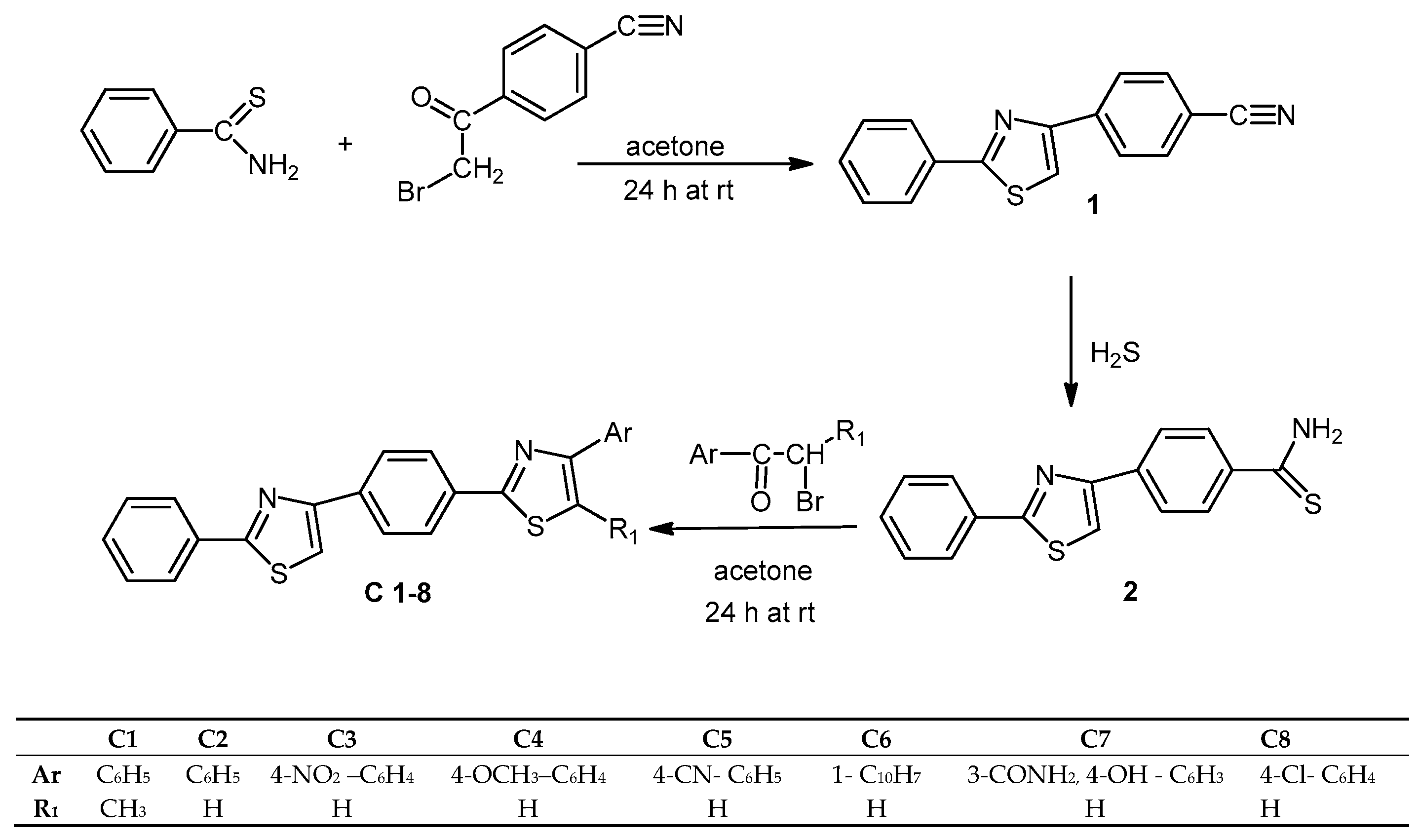
3.2. Chemistry
Synthesis of 4-(2-phenylthiazol-4-yl)benzonitrile (1). A mixture of thiobenzamide (10 mmol) and the 4-(2-bromoacetyl)benzonitrile (10 mmol) was dissolved in anhydrous acetone (50 mL) and stirred at room temperature for 24 h. The resulting solid was filtered and washed with a solution of NaHCO3 5% until free of acid. The solid was then recrystallized from methanol to yield the pure compound.
4-(2-Phenylthiazol-4-yl)benzonitrile (1). White powder. m.p. 137 °C. IR (KBr) νmax cm−1: 3103 (C-H thiazole str), 3061 (C-H str arom), 2224 (str CN), 1606, 1575, 1506, 1438 (Ar ring str), 976 (C-H bend arom), 852 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.4 (s, 1H, H-thiazole), 8.10 (m, 2H, Ph2), 8 (m, 2H, Ph2), 7.9 (m, 2H, Ph1), 7.55 (m, 3H, Ph1). 13C-NMR (DMSO-d6) δ (ppm): 118.6 CN, 158.62 (thiazole C2), 148.76 (thiazole C4), 129.12 (thiazole C5), 128.2–131.3 (12 C, 2 phenyl rings). Anal. calcd. (%) for C16H10N2S (262.06): C, 73.26; H, 3.84; N, 10.68; S, 12.22. Found: C, 73.21; H, 3.89; N, 10.65; S, 12.25. MS (EI, 70 eV): m/z 262 (M+).
Synthesis of the 4-(2-phenylthiazol-4-yl)benzothioamide (2). A solution of 4-(2-phenylthiazol-4-yl)benzonitrile (8 mmol) in ethanol (10 mL) and triethylamine (1 mL) was maintained at room temperature while hydrogen sulfide gas was bubbled into the solution for 8 h. The reaction mixture was then poured in cold water and the solid formed was filtered out, washed with water and recrystallized from ethanol.
4-(2-Phenylthiazol-4-yl)benzothioamide (2). Light-yellow powder. m.p. 179 °C. IR (KBr) νmax cm−1: 3408, 3384 (N-H str), 3104 (C-H thiazole str), 3086 (C-H str arom), 1625 (N-H bend), 1603, 1570, 1506, 1459 (Ar ring str), 1133 (C=S str), 975 (C-H bend arom), 850 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.55 (s, 2H, thioamide), 8.45 (s, 1H, H-thiazole), 8 (m, 2H, Ph2), 7.9 (m, 2H, Ph1), 7.55 (m, 3H, Ph1), 7.45 (m, 2H, Ph2). 13C-NMR (DMSO-d6) δ (ppm): 188.2 C=S, 158.92 (thiazole C2), 148.96 (thiazole C4), 128.38 (thiazole C5), 128.2–143.3 (12 C, 2 phenyl rings). Anal. calcd. (%) for C16H12N2S2 (296.04): C, 64.83; H, 4.08; N, 9.45; S, 21.64. Found: C, 64.88.21; H, 4.03; N, 9.41; S, 21.69. MS (EI, 70 eV): m/z 296 (M+).
General Procedure for the Synthesis of the 4-Aryl-2-(4-(2-phenylthiazol-4-yl)phenyl)thiazoles (C1–8). A mixture of 4-(2-phenylthiazol-4-yl)benzothioamide (1 mmol) and the corresponding α-bromoketone (1 mmol) was dissolved in anhydrous acetone (5 mL) and stirred at room temperature for 24 h. The resulting solid was filtered and washed with a solution of NaHCO3 5% until free of acid. The compounds were then recrystallized from methanol to yield the pure compounds.
5-Methyl-4-phenyl-2-(4-(2-phenylthiazol-4-yl)phenyl)thiazole (C1). Off-white powder. m.p. 235 °C. IR (KBr) νmax cm−1: 3106 (C-H thiazole str), 3080 (C-H str arom), 2920 (C-H str CH3), 1602, 1572, 1503, 1439 (Ar ring str), 978 (C-H bend arom), 851 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.33 (s, 1H, H-thiazole), 8.23 (m, 2H, Ph2), 8.18 (m, 2H, Ph2), 8.05–7.5 (m, 10H, Ph1,3), 2.75 (s, 3H, CH3). 13C-NMR (DMSO-d6) δ (ppm): 166.32 (thiazole-1 C2), 158.5 (thiazole-2 C2), 152.96 (thiazole-1 C4), 152.1 (thiazole-2 C4), 129.8 (thiazole-1 C5), 120.81 (thiazole-2 C5), 17.45 (CH3 thiazole), 126–130.9 (18C, 3 phenyl rings). Anal. calcd. (%) for C25H18N2S2 (410.09): C, 73.14; H, 4.42; N, 6.82; S, 15.62. Found: C, 73.01; H, 4.45; N, 6.89; S, 15.65. MS (EI, 70 eV): m/z 410 (M+).
2-Phenyl-4-(4-(4-phenylthiazol-2-yl)phenyl)thiazole (C2). Off-white powder. m.p. 205 °C. IR (KBr) νmax cm−1: 3108 (C-H thiazole str), 3069 (C-H str arom), 1599, 1576, 1502, 1458 (Ar ring str), 980 (C-H bend arom), 851 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.53 (s, 1H, H-thiazole), 8.35 (s, 1H, H-thiazole), 8.25 (m, 2H, Ph2), 8.15 (m, 2H, Ph2), 8.02–7.4 (m, 10H, Ph1,3). 13C-NMR (DMSO-d6) δ (ppm): 166.1 (thiazole-1 C2), 159 (thiazole-2 C2), 152.66 (thiazole-1 C4), 152.3 (thiazole-2 C4), 129.2 (thiazole-1 C5), 121.11 (thiazole-2 C5), 126.4–130 (18C, 3 phenyl rings). Anal. calcd. (%) for C24H16N2S2 (396.53): C, 72.70; H, 4.07; N, 7.06; S, 16.17. Found: C, 72.55; H, 4.12; N, 7.13; S, 16.2. MS (EI, 70 eV): m/z 396 (M+).
4-(4-Nitrophenyl)-2-(4-(2-phenylthiazol-4-yl)phenyl)thiazole (C3). Yellow powder. m.p. 238 °C. IR (KBr) νmax cm−1: 3110 (C-H thiazole str), 3081 (C-H str arom), 1597, 1577 (Ar ring str), 1519 (N-O NO2 str), 1448 (Ar ring str), 1346 (N-O NO2 str), 980 (C-H bend arom), 842 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.6 (s, 1H, H-thiazole), 8.4 (s, 1H, H-thiazole), 8.33 (dd, 2H, Ph-NO2), 8.25 (dd, 2H, Ph-NO2), 8.2 (m, 2H, Ph2), 8.10 (m, 2H, Ph2), 8 (m, 2H, Ph1), 7.55 (m, 3H, Ph1). 13C-NMR (DMSO-d6) δ (ppm): 165.82 (thiazole-1 C2), 158.7 (thiazole-2 C2), 153.26 (thiazole-1 C4), 151.8 (thiazole-2 C4), 127.8 (thiazole-1 C5), 119.81 (thiazole-2 C5), 126–147.2 (18C, 3 phenyl rings). Anal. calcd. (%) for C24H15N3O2S2 (441.06): C, 65.29; H, 3.42; N, 9.52; S, 14.52. Found: C, 65.33; H, 3.40; N, 9.54; S, 14.49. MS (EI, 70 eV): m/z 441(M+).
4-(4-Methoxyphenyl)-2-(4-(2-phenylthiazol-4-yl)phenyl)thiazole (C4). Yellowish—brown powder. m.p. 223 °C. IR (KBr) νmax cm−1: 3109 (C-H thiazole str), 3057 (C-H str arom), 1608, 1578, 1503, 1460 (Ar ring str), 1251 (C-O-C assym str), 1030 (C-O-C sym str), 981 (C-H bend arom), 838 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.65 (s, 1H, H-thiazole), 8.38 (s, 1H, H-thiazole), 8.3 (m, 2H, Ph2), 8.20 (m, 2H, Ph2), 8 (m, 2H, Ph-OCH3) 8.06 (m, 2H, Ph1), 7.60 (m, 3H, Ph1), 7.05 (dd, 2H, Ph-OCH3), 3.85 (s, 3H, OCH3). 13C-NMR (DMSO-d6) δ (ppm): 166.92 (thiazole-1 C2), 157.9 (thiazole-2 C2), 152.66 (thiazole-1 C4), 152.18 (thiazole-2 C4), 128.2 (thiazole-1 C5), 120.21 (thiazole-2 C5), 56.92 (OCH3), 126–159.04 (18C, 3 phenyl rings). Anal. calcd. (%) for C25H18N2OS2 (426.09): C, 70.39; H, 4.25; N, 6.57; S, 15.03. Found: C, 70.44; H, 4.21; N, 6.59; S, 15.13. MS (EI, 70 eV): m/z 426 (M+).
4-(2-(4-(2-Phenylthiazol-4-yl)phenyl)thiazol-4-yl)benzonitrile (C5). Yellow powder. m.p. 242 °C. IR (KBr) νmax cm−1: 3110 (C-H thiazole str), 3061 (C-H str arom), 2225 (str CN), 1605, 1575, 1503, 1448 (Ar ring str), 980 (C-H bend arom), 844 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.55 (s, 1H, H-thiazole), 8.45 (dd, 2H, Ph-CN), 8.30 (s, 1H, H-thiazole), 8.25 (m, 2H, Ph2), 8.15 (m, 2H, Ph2), 7.9 (m, 2H, Ph1), 7.77 (dd, 2H, Ph-CN), 7.50 (m, 3H, Ph1). 13C-NMR (DMSO-d6) δ (ppm): 165.62 (thiazole-1 C2), 157.85 (thiazole-2 C2), 152.91 (thiazole-1 C4), 151.81 (thiazole-2 C4), 128.8 (thiazole-1 C5), 118.6 CN, 119.91 (thiazole-2 C5), 126–130.8 (18C, 3 phenyl rings). Anal. calcd. (%) for C25H15N3S2 (421.07): C, 71.23; H, 3.59; N, 9.97; S, 15.21. Found: C, 71.4; H, 3.64; N, 9.70; S, 15.26. MS (EI, 70 eV): m/z 421 (M+).
4-(Naphthalen-1-yl)-2-(4-(2-phenylthiazol-4-yl)phenyl)thiazole (C6). Off-white powder. m.p. 237 °C. IR (KBr) νmax cm−1: 3109 (C-H thiazole str), 3061 (C-H str arom), 1603, 1571, 1504, 1455 (Ar ring str), 979 (C-H bend arom), 839 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 8.6 (s, 1H, H-thiazole), 8.42 (s, 1H, H-thiazole), 8.25 (m, 2H, Ar2), 8.15 (m, 2H, Ph2), 8.06–8.03 (m, 2H, naph), 7.9 (m, 2H, Ph1), 7.56–7.53 (m, 4H, naph), 7.45 (m, 3H, Ph1). 13C-NMR (DMSO-d6) δ (ppm): 166.32 (thiazole-1 C2), 158.5 (thiazole-2 C2), 152.96 (thiazole-1 C4), 152.1 (thiazole-2 C4), 128.8 (thiazole-1 C5), 120.61 (thiazole-2 C5), 126–140.6 (22 C, 2 phenyl and 1 naphtyl rings). Anal. calcd. (%) for C28H18N2S2 (446.09): C, 75.30; H, 4.06; N, 6.27; S, 14.36. Found: C, 75.26; H, 4.18; N, 6.31; S, 14.25. MS (EI, 70 eV): m/z 446 (M+).
2-Hydroxy-5-{2-[4-(2-phenyl-1,3-thiazol-4-yl)phenyl]-1,3-thiazol-4-yl}benzamide (C7) Light-grey. m.p. 225 °C. IR (KBr) νmax cm−1: 3443 (O-H str phenol), 3326, 3230 (N-H str amide), 3108 (C-H thiazole str), 3059 (C-H str arom), 1666 (C=O str amide), 1619 (N-H bend amide), 1601, 1505, 1447 (Ar ring str), 983 (C-H bend arom), 840 (C-H def arom). 1H-NMR (DMSO-d6, ppm): δ 13.2 (s, 1H, OH), 8.63 (s, 1H, H-thiazole), 8.56 (d, 1H, Ph3), 8.35 (s, 1H, H-thiazole), 8.23 (m, 2H, Ph2), 8.18 (m, 2H, Ph2), 8.13 (m, 1H, Ph3), 8.06 (m, 2H, Ph1), 8.02 (s, 2H, CONH2), 7.55 (m, 3H, Ph1), 7.02 (d, 1H, Ph3). 13C-NMR (DMSO-d6) δ (ppm): 167.66 (amide C=O), 166.32 (thiazole-1 C2), 158.5 (thiazole-2 C2), 152.96 (thiazole-1 C4), 152.1 (thiazole-2 C4), 129.8 (thiazole-1 C5), 120.41 (thiazole-2 C5), 126-159.6 (18 C, 3 phenyl rings). Anal. calcd. (%) for C25H17N3O2S2 (455.08): C, 65.91; H, 3.76; N, 9.22; S, 14.08. Found: C, 65.8; H, 3.6; N, 9.3; S, 14.4. MS (EI, 70 eV): m/z 455 (M+).
4-(4-Chlorophenyl)-2-(4-(2-phenylthiazol-4-yl)phenyl)thiazole (C8) Off-white powder. m.p. 232 °C. IR (KBr) νmax cm−1: 3111 (C-H thiazole str), 3059 (C-H str arom), 1597, 1572, 1503, 1462 (Ar ring str), 981 (C-H bend arom), 843 (C-H def arom), 826 (C-Cl str). 1H-NMR (DMSO-d6, ppm): δ 8.56 (s, 1H, H-thiazole), 8.35 (s, 1H, H-thiazole), 8.30 (m, 2H, Ph2), 8.20 (m, 2H, Ph2), 7.9 (m, 2H, Ph1), 7.60 (dd, 2H, Ph-Cl), 7.55 (dd, 2H, Ph-Cl), 7.45 (m, 3H, Ph1). 13C-NMR (DMSO-d6) δ (ppm): 165.32 (thiazole-1 C2), 159.5 (thiazole-2 C2), 151.96 (thiazole-1 C4), 151.1 (thiazole-2 C4), 128.38 (thiazole-1 C5), 120.81 (thiazole-2 C5), 126–132.89 (18 C, 3 phenyl rings). Anal. calcd. (%) for C24H15 ClN2S2 (430.04): C, 66.89; H, 3.51; N, 6.50; S, 14.88. Found: C, 66.80; H, 3.55; N, 6.52; S, 14.91. MS (EI, 70 eV): m/z 430 (M+).

 ,
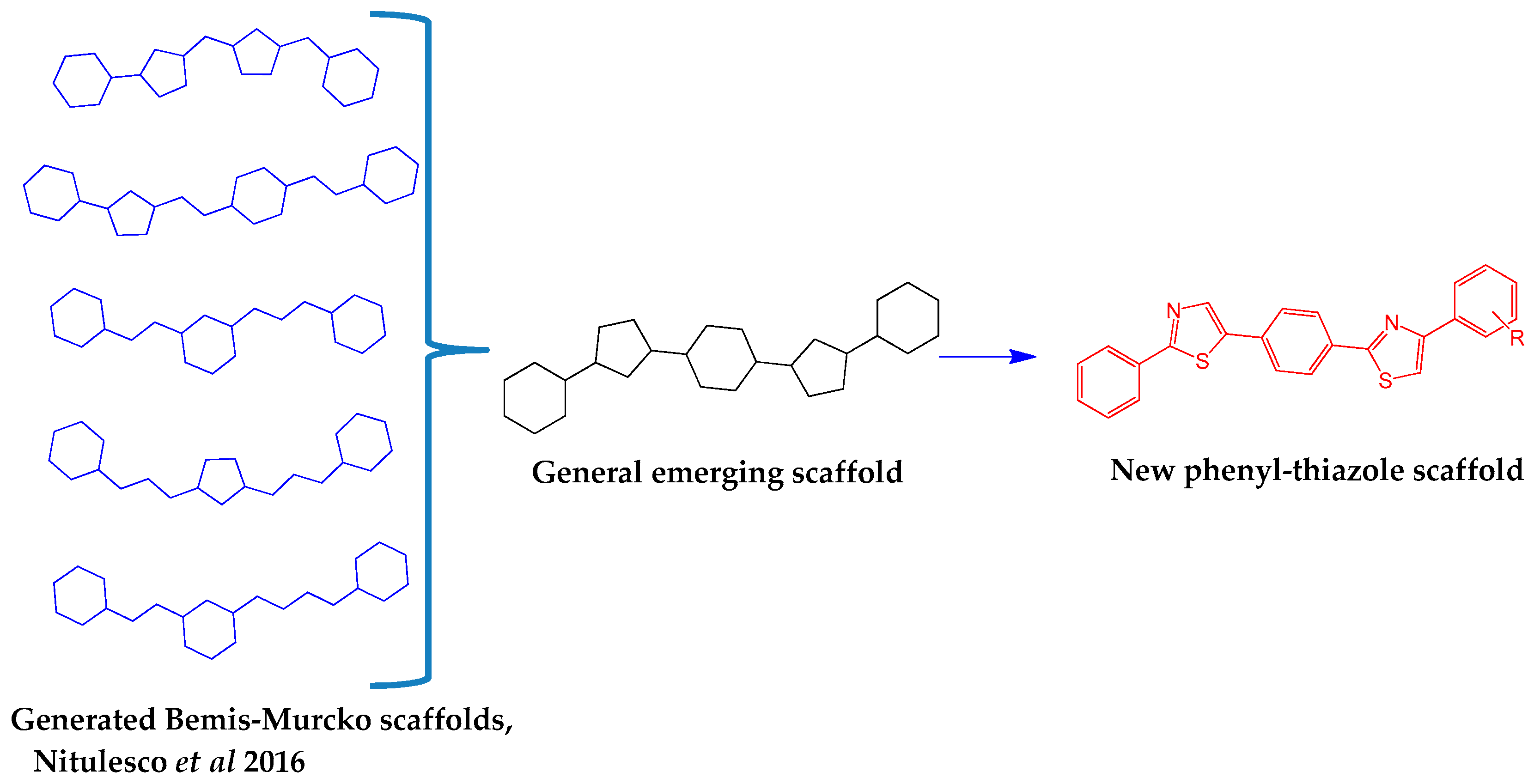
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
