
The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
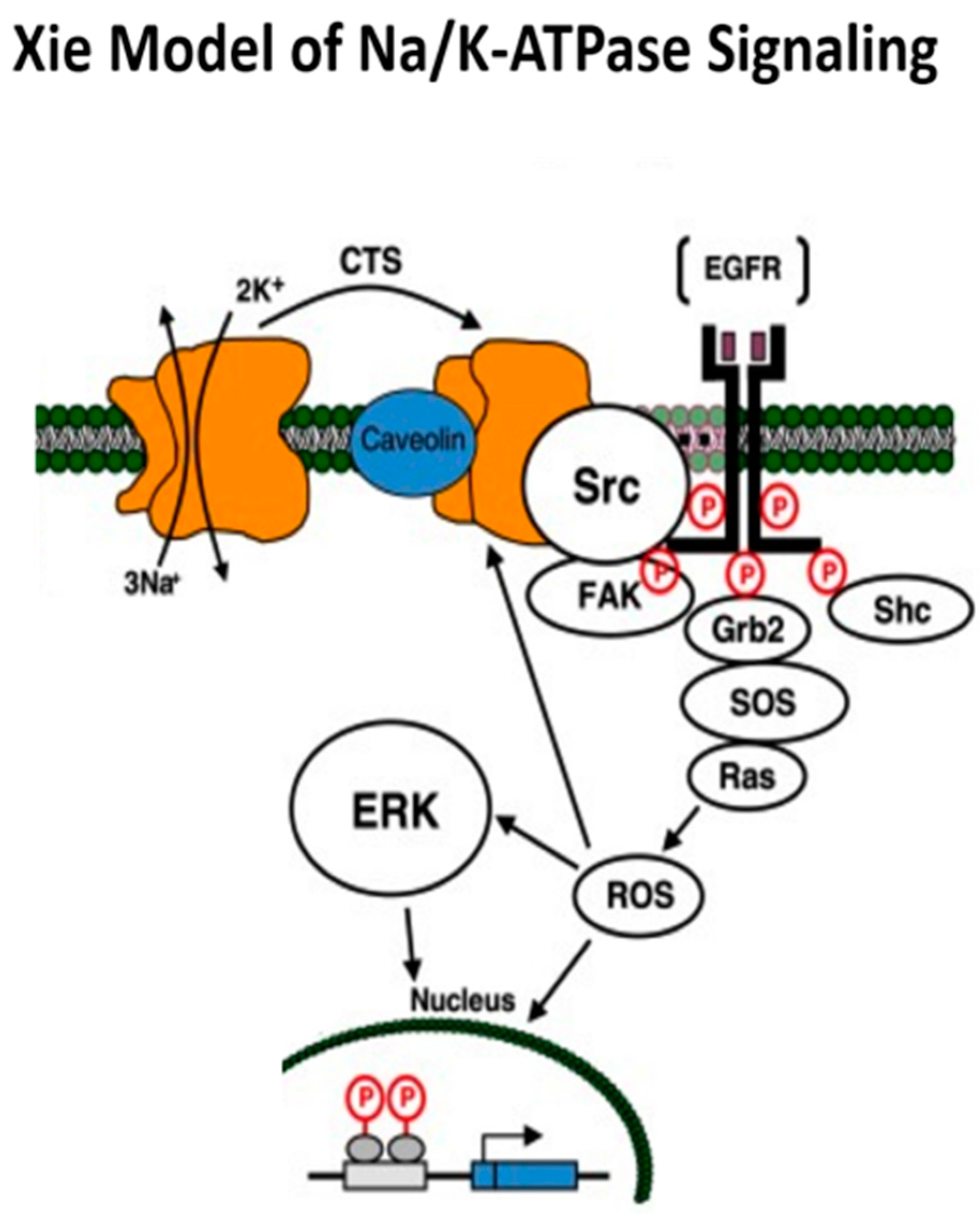
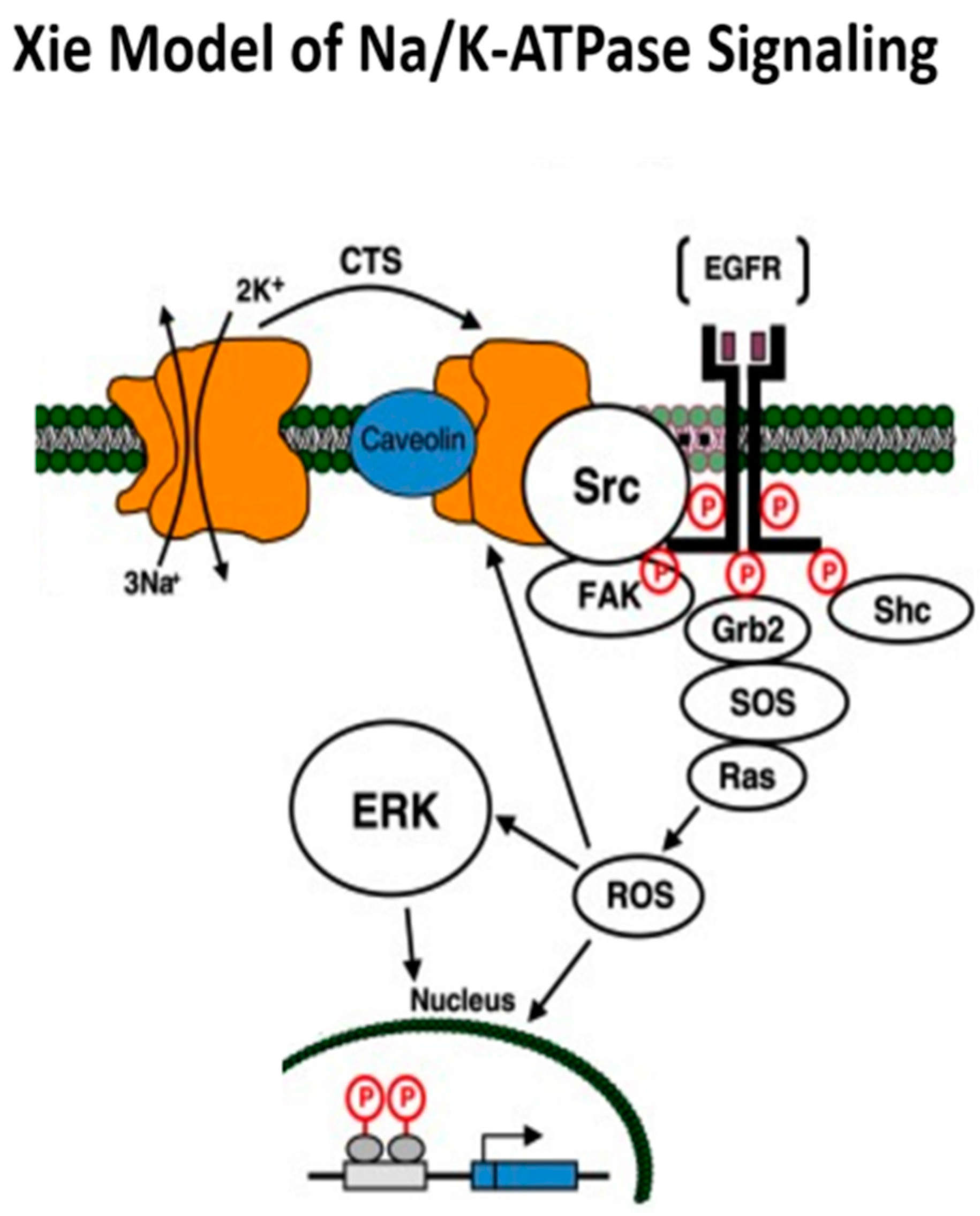
2. Na/K-ATPase: Structure, Function, and the Xie Model of Signaling
Xie Model of Na/K-ATPase Signaling
3. Oxidative Stress and Regulation of the Na/K-ATPase
4. The Development of pNaKtide
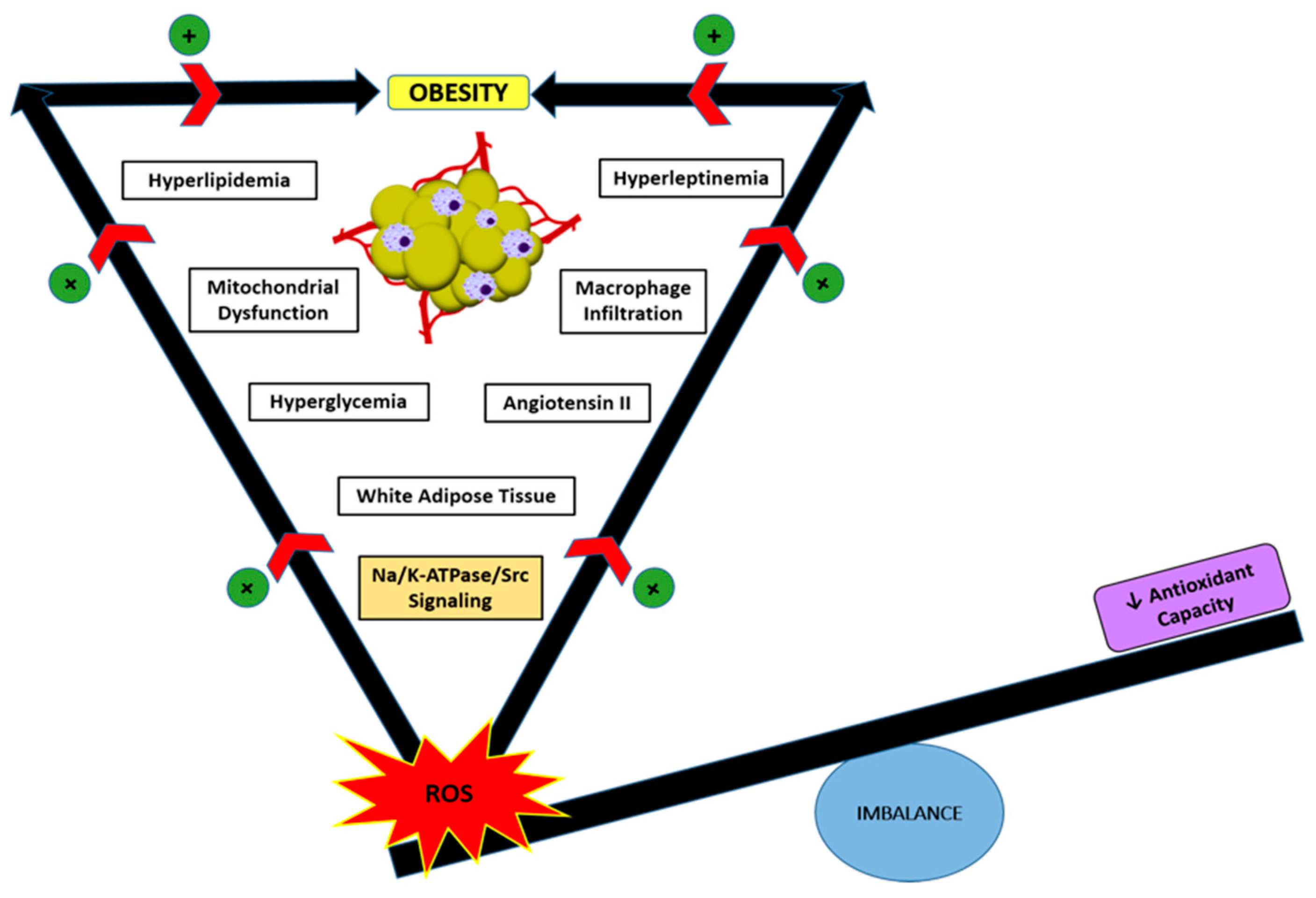
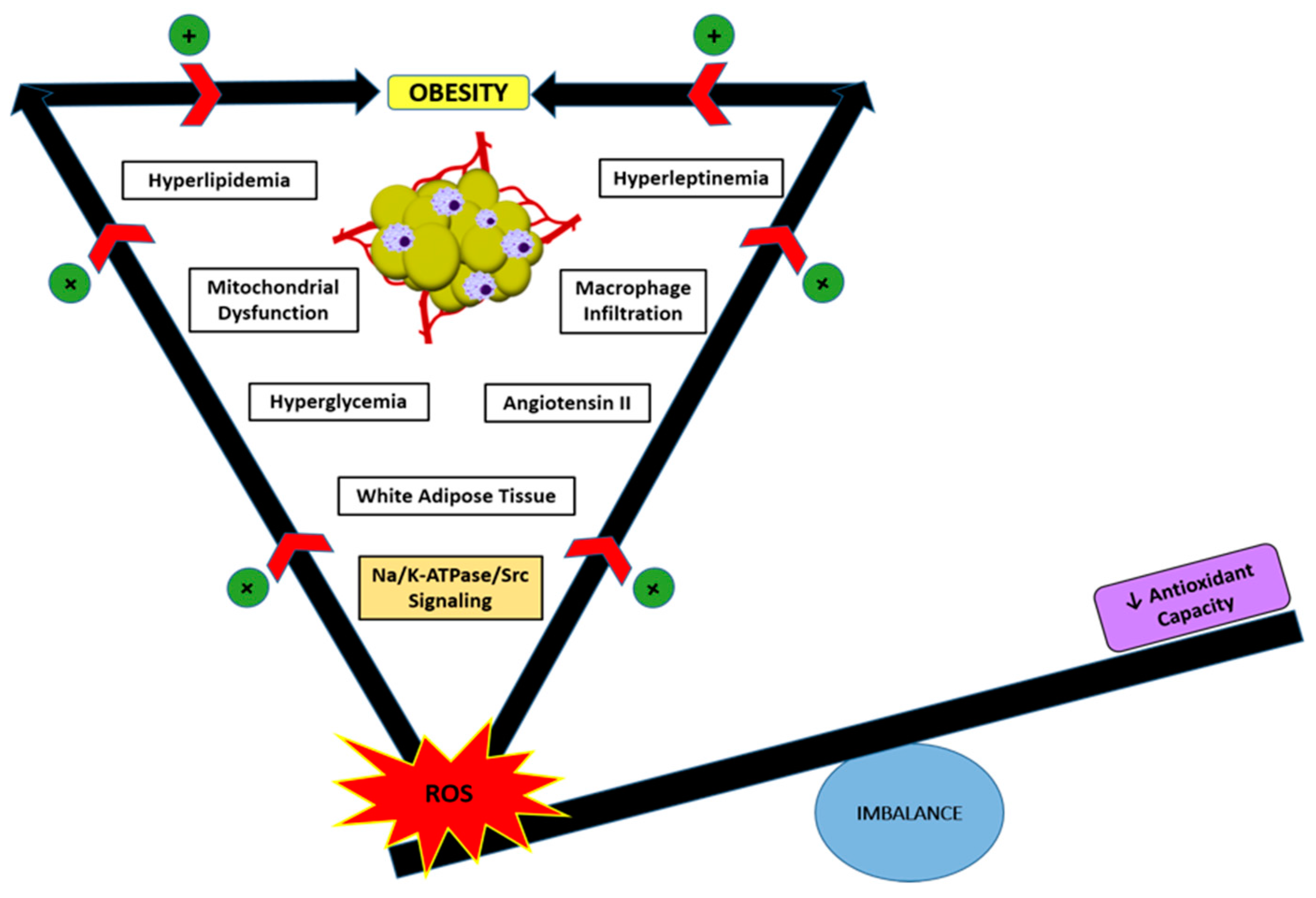
5. Role of Oxidative Stress and Na/K-ATPase/ROS Signaling in Obesity
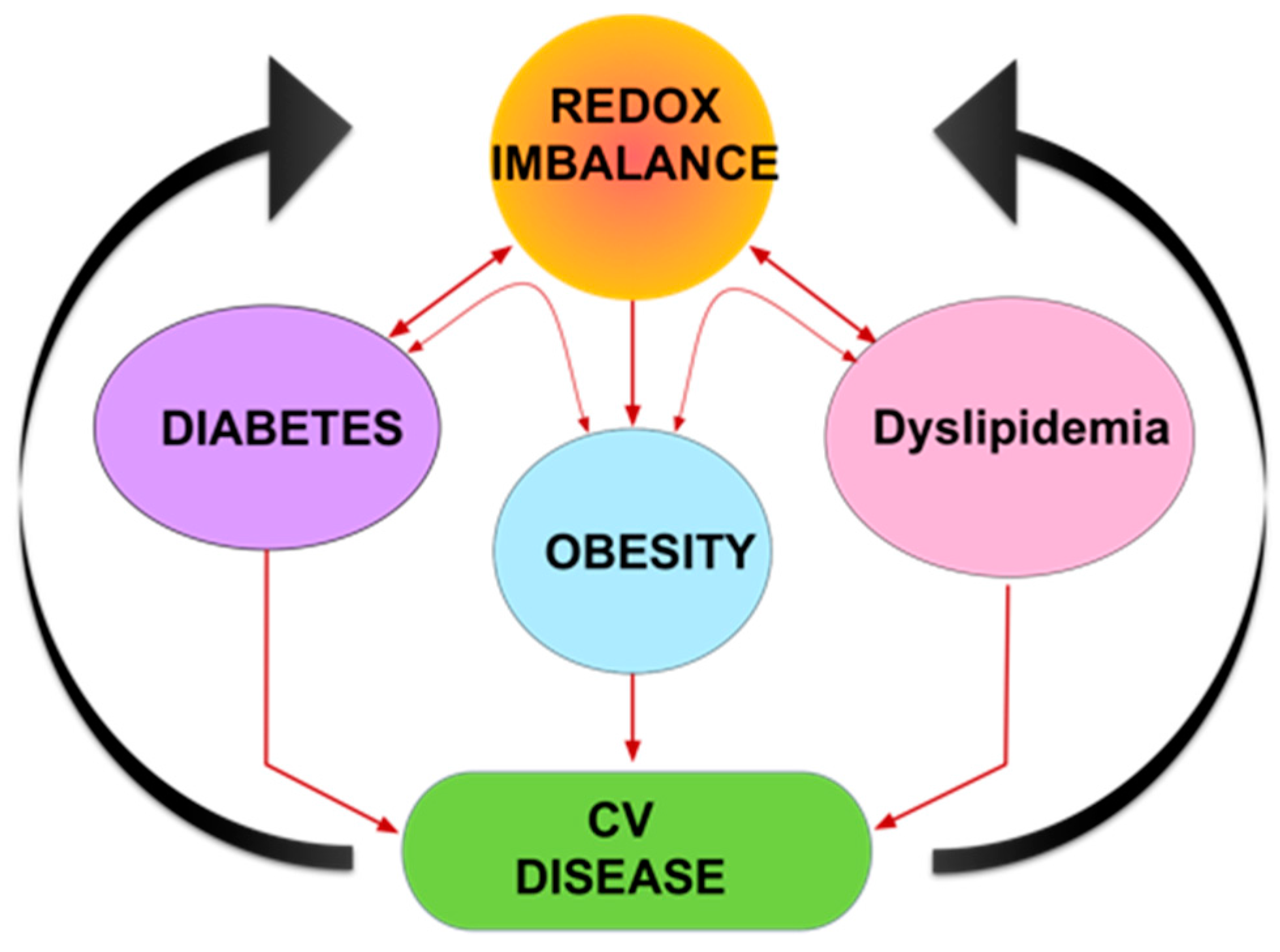
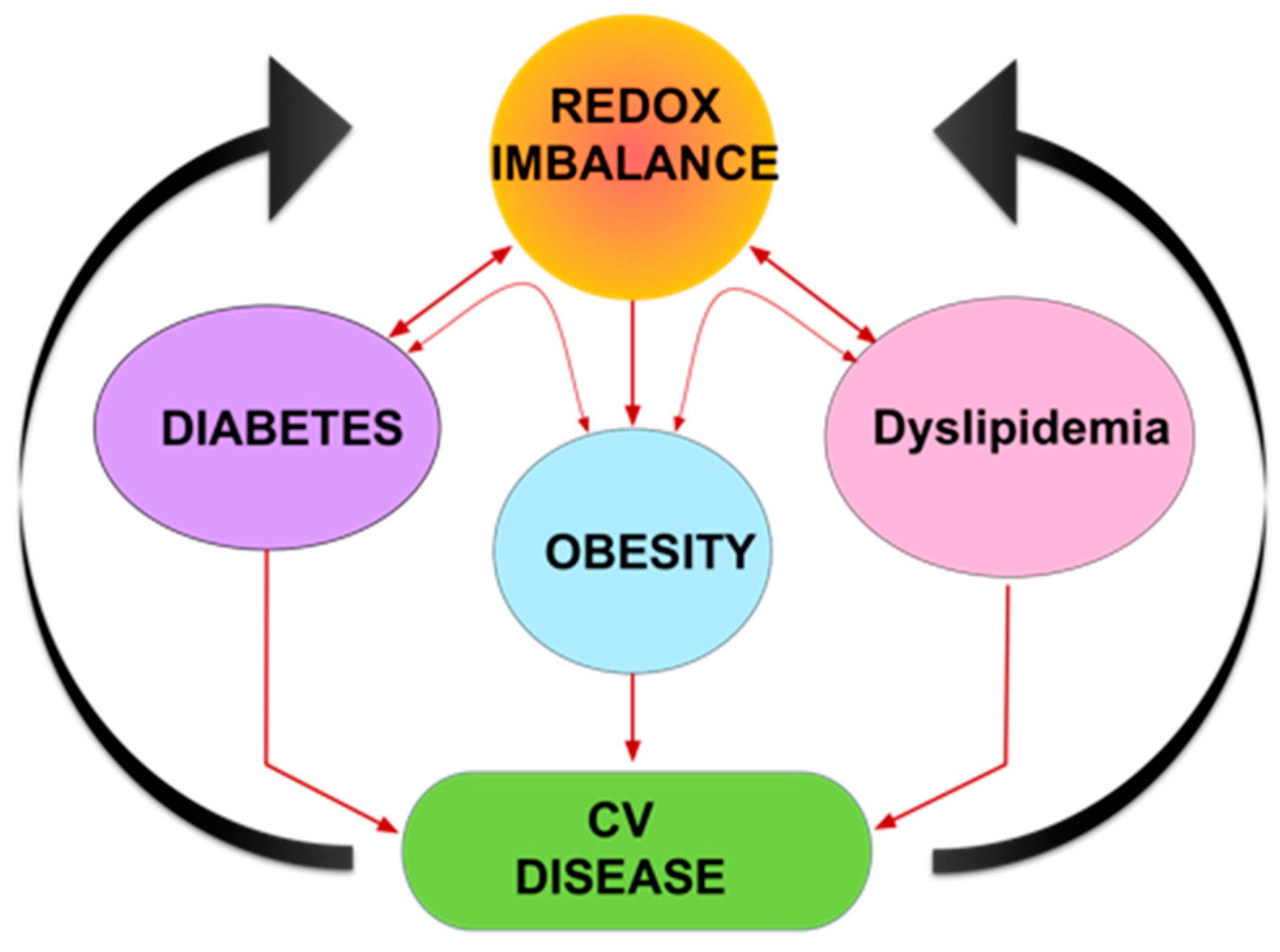
6. Role of Oxidative Stress and Na/K-ATPase/ROS signaling in Cardiovascular Disease (CVD)
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yan, Y.; Shapiro, J.I. The physiological and clinical importance of sodium potassium ATPase in cardiovascular diseases. Curr. Opin. Pharmacol. 2016, 27, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Haas, M.; Liang, M.; Cai, T.; Tian, J.; Li, S.; Xie, Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J. Biol. Chem. 2004, 279, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Kotova, O.; Al-Khalili, L.; Talia, S.; Hooke, C.; Fedorova, O.V.; Bagrov, A.Y.; Chibalin, A.V. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src- and ERK1/2-dependent mechanism. J. Biol. Chem. 2006, 281, 20085–20094. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.J. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mohammadi, K.; Aynafshar, B.; Wang, H.; Li, D.; Liu, J.; Ivanov, A.V.; Xie, Z.; Askari, A. Role of caveolae in signal-transducing function of cardiac Na+/K+-ATPase. Am. J. Physiol. Cell Physiol. 2003, 284, C1550–C1560. [Google Scholar] [CrossRef] [PubMed]
- Gable, M.E.; Abdallah, S.L.; Najjar, S.M.; Liu, L.; Askari, A. Digitalis-induced cell signaling by the sodium pump: On the relation of Src to Na(+)/K(+)-ATPase. Biochem. Biophys. Res. Commun. 2014, 446, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.Q.; Li, Z.C.; Tian, J.; Xie, J.X.; Liu, L.J.; Xie, Z.J. Identification of a potential receptor that couples ion transport to protein kinase activity. J. Biol. Chem. 2011, 286, 6225–6232. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ivanov, A.V.; Gable, M.E.; Jolivel, F.; Morrill, G.A.; Askari, A. Comparative properties of caveolar and noncaveolar preparations of kidney Na+/K+-ATPase. Biochemistry 2011, 50, 8664–8673. [Google Scholar] [CrossRef] [PubMed]
- Weigand, K.M.; Swarts, H.G.; Fedosova, N.U.; Russel, F.G.; Koenderink, J.B. Na,K-ATPase activity modulates Src activation: A role for ATP/ADP ratio. Biochim. Biophys. Acta 2012, 1818, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Yosef, E.; Katz, A.; Peleg, Y.; Mehlman, T.; Karlish, S.J. Do Src Kinase and Caveolin Interact Directly with Na,K-ATPase? J. Biol. Chem. 2016, 291, 11736–11750. [Google Scholar] [CrossRef] [PubMed]
- Clifford, R.J.; Kaplan, J.H. Human breast tumor cells are more resistant to cardiac glycoside toxicity than non-tumorigenic breast cells. PLoS ONE 2013, 8, e84306. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009, 457, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kennedy, D.J.; Yan, Y.; Shapiro, J.I. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. Int. J. Nephrol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Akera, T. O2 free radicals: Cause of ischemia-reperfusion injury to cardiac Na+-K+-ATPase. Am. J. Physiol. 1987, 252, H252–H257. [Google Scholar] [PubMed]
- Xie, Z.J.; Wang, Y.H.; Askari, A.; Huang, W.H.; Klaunig, J.E.; Askari, A. Studies on the specificity of the effects of oxygen metabolites on cardiac sodium pump. J. Mol. Cell Cardiol. 1990, 22, 911–920. [Google Scholar] [CrossRef]
- Huang, W.H.; Wang, Y.; Askari, A. (Na+ + K+)-ATPase: Inactivation and degradation induced by oxygen radicals. Int. J. Biochem. 1992, 24, 621–626. [Google Scholar] [PubMed]
- Huang, W.H.; Wang, Y.; Askari, A.; Zolotarjova, N.; Ganjeizadeh, M. Different sensitivities of the Na+/K(+)-ATPase isoforms to oxidants. Biochim. Biophys. Acta 1994, 1190, 108–114. [Google Scholar] [CrossRef]
- Xie, Z.; Jack-Hays, M.; Wang, Y.; Periyasamy, S.M.; Blanco, G.; Huang, W.H.; Askari, A. Different oxidant sensitivities of the alpha 1 and alpha 2 isoforms of Na+/K(+)-ATPase expressed in baculovirus-infected insect cells. Biochem. Biophys. Res. Commun. 1995, 207, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zolotarjova, N.; Ho, C.; Mellgren, R.L.; Askari, A.; Huang, W.H. Different sensitivities of native and oxidized forms of Na+/K(+)-ATPase to intracellular proteinases. Biochim. Biophys. Acta 1994, 1192, 125–131. [Google Scholar] [CrossRef]
- Thevenod, F.; Friedmann, J.M. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K(+)-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 1999, 13, 1751–1761. [Google Scholar] [PubMed]
- Figtree, G.A.; Liu, C.C.; Bibert, S.; Hamilton, E.J.; Garcia, A.; White, C.N.; Chia, K.K.; Cornelius, F.; Geering, K.; Rasmussen, H.H. Reversible oxidative modification: A key mechanism of Na+-K+ pump regulation. Circ. Res. 2009, 105, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Liu, C.C.; Figtree, G.A.; Garcia, A.; Hamilton, E.J.; Marassi, F.M.; Sweadner, K.J.; Cornelius, F.; Geering, K.; Rasmussen, H.H. FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its beta1 subunit. J. Biol. Chem. 2011, 286, 18562–18572. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.J.; Abraham, N.G.; et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.Y.; Rothblum, L.I.; Moorman, J.R.; Tucker, A.L.; Song, J.; Ahlers, B.A.; Carl, L.L.; Wang, J.; Zhang, X.Q. Regulation of cardiac Na+/Ca2+ exchanger by phospholemman. Ann. N. Y. Acad. Sci. 2007, 1099, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Petrushanko, I.Y.; Hernansanz-Agustin, P.; Martinez-Ruiz, A. “Oxygen Sensing” by Na,K-ATPase: These Miraculous Thiols. Front. Physiol. 2016, 7, 314. [Google Scholar] [CrossRef] [PubMed]
- Siems, W.G.; Hapner, S.J.; van Kuijk, F.J. 4-hydroxynonenal inhibits Na(+)-K(+)-ATPase. Free Radic. Biol. Med. 1996, 20, 215–223. [Google Scholar] [CrossRef]
- Morel, P.; Tallineau, C.; Pontcharraud, R.; Piriou, A.; Huguet, F. Effects of 4-hydroxynonenal, a lipid peroxidation product, on dopamine transport and Na+/K+ ATPase in rat striatal synaptosomes. Neurochem. Int. 1998, 33, 531–540. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagashima, R.; Yoneyama, M.; Shiba, T.; Ogita, K. Disruption of ion-trafficking system in the cochlear spiral ligament prior to permanent hearing loss induced by exposure to intense noise: Possible involvement of 4-hydroxy-2-nonenal as a mediator of oxidative stress. PLoS ONE 2014, 9, e102133. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, J. L.; Shapiro, I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [PubMed]
- Kraker, A.J.; Hartl, B.G.; Amar, A.M.; Barvian, M.R.; Showalter, H.D.; Moore, C.W. Biochemical and cellular effects of c-Src kinase-selective pyrido[2, 3-d]pyrimidine tyrosine kinase inhibitors. Biochem. Pharmacol. 2000, 60, 885–898. [Google Scholar] [CrossRef]
- Abe, J.; Takahashi, M.; Ishida, M.; Lee, J.D.; Berk, B.C. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef] [PubMed]
- Burkard, C.; Verheije, M.H.; Haagmans, B.L.; van Kuppeveld, F.J.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. ATP1A1-mediated Src signaling inhibits coronavirus entry into host cells. J. Virol. 2015, 89, 4434–4448. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am. J. Pathol. 2009, 175, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Li, M.; Vanella, L.; Kim, D.H.; Rezzani, R.; Rodella, L.; Sodhi, K.; Canestraro, M.; Martasek, P.; Peterson, S.J.; et al. Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice. Hypertension 2010, 56, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kim, D.H.; Tsenovoy, P.L.; Peterson, S.J.; Rezzani, R.; Rodella, L.F.; Aronow, W.S.; Ikehara, S.; Abraham, N.G. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008, 57, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. 2014, 40, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.B.; Diaz-Lagares, A.; Carreira, M.C.; Amil, M.; Casanueva, F.F. Oxidative stress associated to dysfunctional adipose tissue: A potential link between obesity, type 2 diabetes mellitus and breast cancer. Free Radic. Res. 2013, 47, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, H.; Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Morioka, K.; Maruyama, N.; Kitagawa, N.; Tanaka, T.; Hori, Y.; et al. Oxidative stress is associated with adiposity and insulin resistance in men. J. Clin. Endocrinol. Metab. 2003, 88, 4673–4676. [Google Scholar] [CrossRef] [PubMed]
- Vincent, H.K.; Taylor, A.G. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int. J. Obes. (Lond.) 2006, 30, 400–418. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative stress and metabolic pathologies: From an adipocentric point of view. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talior, I.; Yarkoni, M.; Bashan, N.; Eldar-Finkelman, H. Increased glucose uptake promotes oxidative stress and PKC-delta activation in adipocytes of obese, insulin-resistant mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E295–E302. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Executive Summary: Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Invest. 2004, 114, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, E.; Baldassari, F.; Bononi, A.; Wieckowski, M.R.; Pinton, P. Oxidative stress in cardiovascular diseases and obesity: Role of p66Shc and protein kinase C. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Srikanthan, K.; Feyh, A.; Visweshwar, H.; Shapiro, J.I.; Sodhi, K. Systematic Review of Metabolic Syndrome Biomarkers: A Panel for Early Detection, Management, and Risk Stratification in the West Virginian Population. Int. J. Med. Sci. 2016, 13, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.V.; Padmaja, G.; Kuppusamy, P.; Kutala, V.K. Oxidative stress in cardiovascular disease. Indian J. Biochem. Biophys. 2009, 46, 421–40. [Google Scholar] [PubMed]
- Fruchart, J.C.; Nierman, M.C.; Stroes, E.S.; Kastelein, J.J.; Duriez, P. New risk factors for atherosclerosis and patient risk assessment. Circulation 2004, 109. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.; Tengattini, S.; Fabiano, A.; Bianchi, R.; Rezzani, R. Atherosclerosis and oxidative stress. Histol. Histopathol. 2008, 23, 381–390. [Google Scholar] [PubMed]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef]
- Vogiatzi, G.; Tousoulis, D.; Stefanadis, C. The role of oxidative stress in atherosclerosis. Hellenic J. Cardiol. 2009, 50, 402–409. [Google Scholar] [PubMed]
- Saavedra, W.F.; Paolocci, N.; St John, M.E.; Skaf, M.W.; Stewart, G.C.; Xie, J.S.; Harrison, R.W.; Zeichner, J.; Mudrick, D.; Marban, E.; et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ. Res. 2002, 90, 297–304. [Google Scholar] [CrossRef] [PubMed]
- De Biase, L.; Pignatelli, P.; Lenti, L.; Tocci, G.; Piccioni, F.; Riondino, S.; Pulcinelli, F.M.; Rubattu, S.; Volpe, M.; Violi, F. Enhanced TNF alpha and oxidative stress in patients with heart failure: Effect of TNF alpha on platelet O2-production. Thromb. Haemost. 2003, 90, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Bundgaard, H.; Muller-Ehmsen, J.; Kjeldsen, K. The Na, K-ATPase in the failing human heart. Cardiovasc. Res. 2003, 57, 913–920. [Google Scholar] [CrossRef]
- Kennedy, D.; Omran, E.; Periyasamy, S.M.; Nadoor, J.; Priyadarshi, A.; Willey, J.C.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003, 14, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Xie, M.; Periyasamy, S.M.; Xie, Z.; Han, C.; Basrur, V.; Mutgi, K.; Fedorov, V.; Malhotra, D.; et al. Ouabain decreases sarco(endo)plasmic reticulum calcium ATPase activity in rat hearts by a process involving protein oxidation. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H3003–H3011. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Shidyak, A.; Periyasamy, S.M.; Haller, S.; Taleb, M.; El-Okdi, N.; Elkareh, J.; Gupta, S.; Gohara, S.; Fedorova, O.V.; et al. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension 2009, 54, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Elkareh, J.; Shidyak, A.; Shapiro, A.P.; Smaili, S.; Mutgi, K.; Gupta, S.; Tian, J.; Morgan, E.; Khouri, S.; et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am. J. Physiol. Renal Physiol. 2008, 294, F450–F454. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Kennedy, D.J.; Shidyak, A.; Budny, G.V.; Malhotra, D.; Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am. J. Hypertens 2012, 25, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Briones, A.M. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens Res. 2011, 34, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Garvin, J.L.; Ortiz, P.A. The role of reactive oxygen species in the regulation of tubular function. Acta Physiol. Scand. 2003, 179, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L.; Touyz, R.M. Multiple actions of angiotensin II in hypertension: Benefits of AT1 receptor blockade. J. Am. Coll. Cardiol. 2003, 42, 911–913. [Google Scholar] [CrossRef]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid. Redox. Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A Critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srikanthan, K.; Shapiro, J.I.; Sodhi, K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease. Molecules 2016, 21, 1172. https://doi.org/10.3390/molecules21091172
Srikanthan K, Shapiro JI, Sodhi K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease. Molecules. 2016; 21(9):1172. https://doi.org/10.3390/molecules21091172
Chicago/Turabian StyleSrikanthan, Krithika, Joseph I. Shapiro, and Komal Sodhi. 2016. "The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease" Molecules 21, no. 9: 1172. https://doi.org/10.3390/molecules21091172