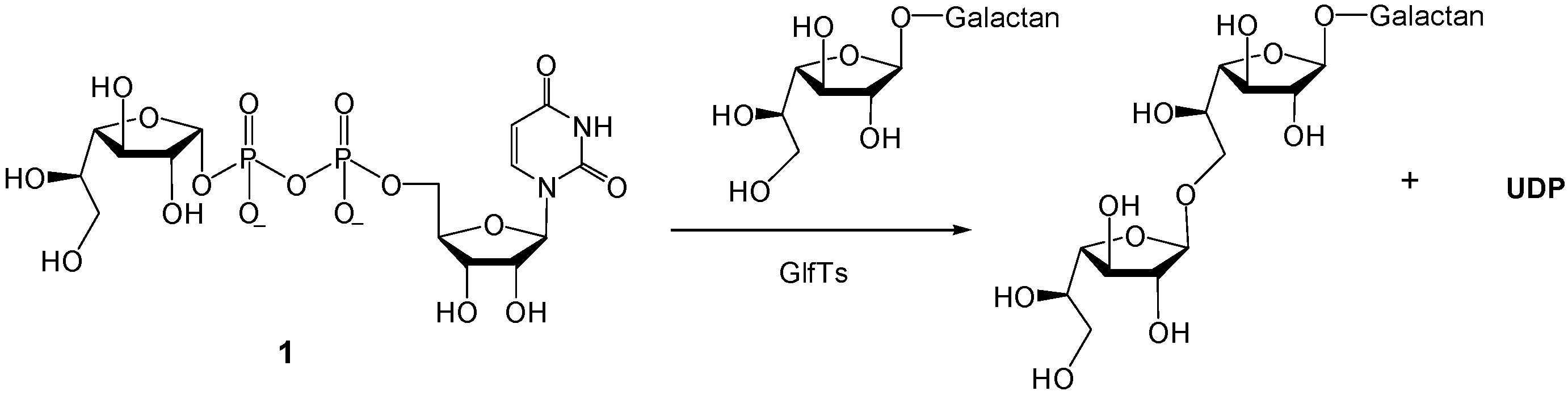
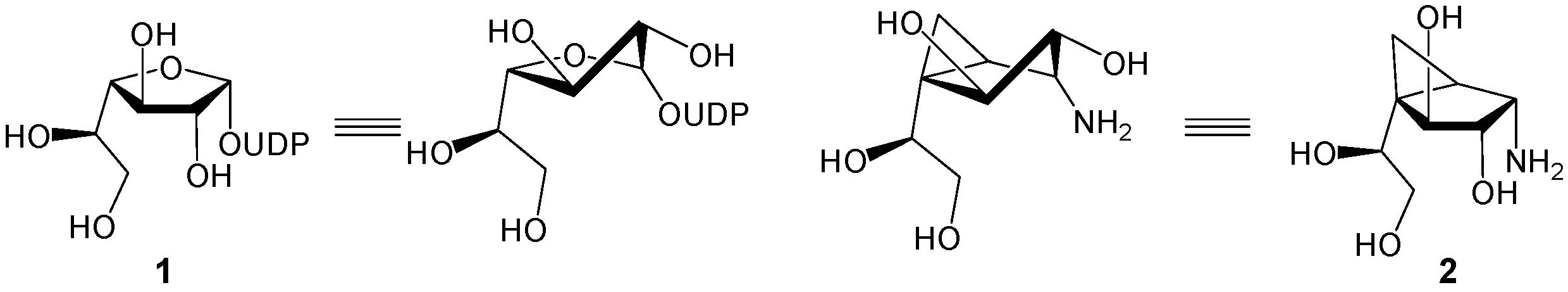
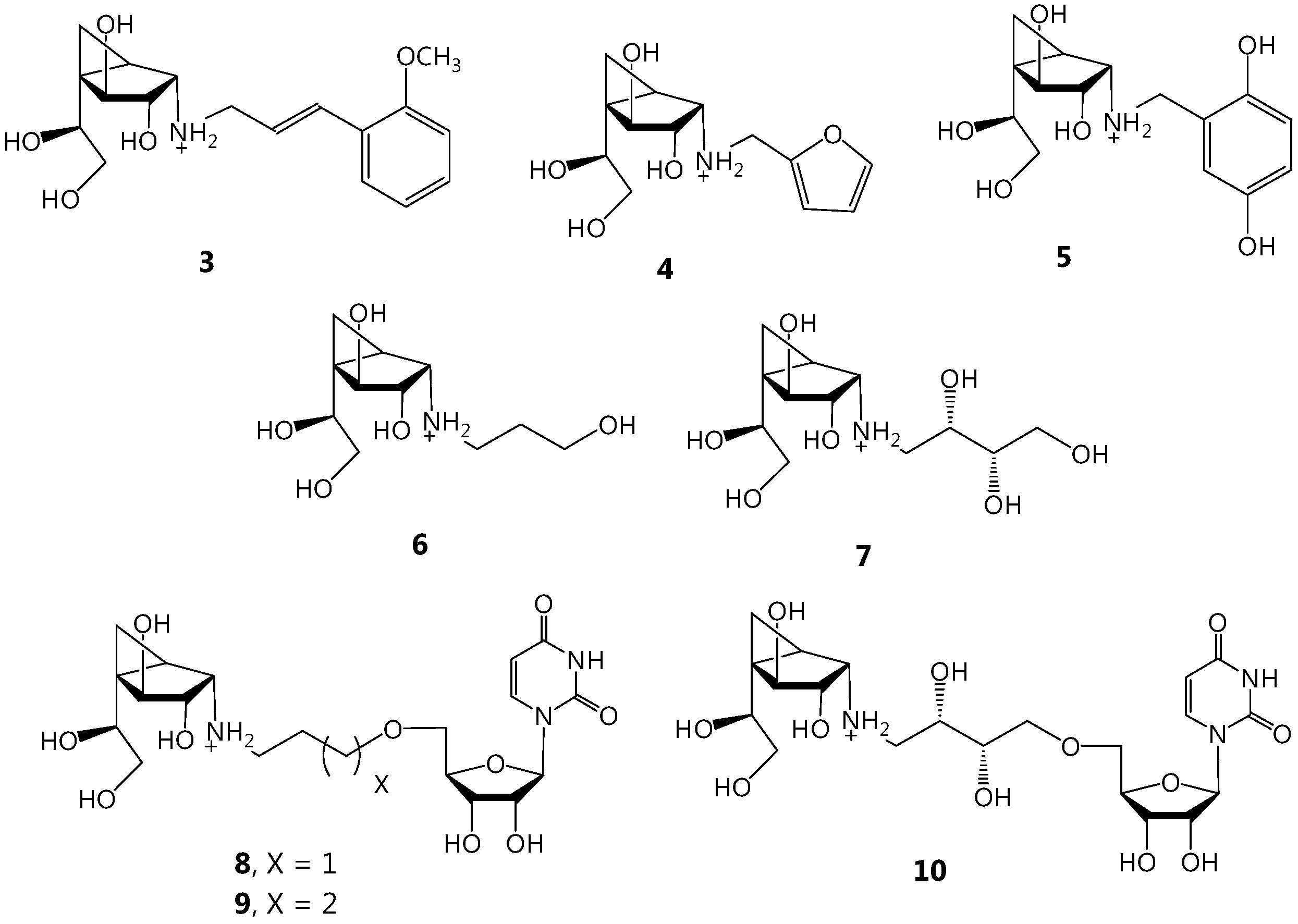
3.1. Synthesis of Target Molecules
All reagents were purchased from commercial sources and were used without further purification. Solvents used in reactions were pre-dried by PURESOLV-400 System from Innovative Technology Inc. (Amesbury, MA, USA). All reactions were monitored by TLC on silica gel G-25 UV254 (0.25 mm, Macherey–Nagel). Spots were detected under UV light and/or by charring with acidified ethanolic anisaldehyde. Solvents were evaporated under reduced pressure and below 50 °C (water bath). Column chromatography was performed on silica gel 60 (40–60 μm). The ratio between silica gel and crude product ranged from 100:1 to 20:1 (
w/
w). Iatrobeads refers to a beaded silica gel 6RS-8060, which was manufactured by Iatron laboratories (Tokyo, Japan).
1H-NMR spectra were recorded on VARIAN INOVA-NMR spectrometers (Varian, Inc., Salt Lake City, UT, USA) at 400, 500, or 600 MHz and chemical shifts are referenced to CDCl
3 (7.26, CDCl
3) or CD
3OD (4.78, CD
3OD).
13C-NMR APT spectra were recorded at 100 or 125 MHz, and chemical shifts are referenced to CDCl
3 (77.23, CDCl
3) or CD
3OD (48.9, CD
3OD).
1H-NMR data are reported as though they are first order, and the peak assignments are made by 2D-NMR spectroscopy (
1H–
1H COSY and HMQC). The numbering system used for NMR assignment is shown in
Figure 4. HRMS-ESI spectra were recorded on samples suspended in THF or CH
3OH and added NaCl. Optical rotations were measured on Perkin-Elmer 241 Polarimeter (Perkin-Elmer, Waltham, MA, USA) with sodium D line (589 nm) and are in units of deg·mL (dm·g)
−1.
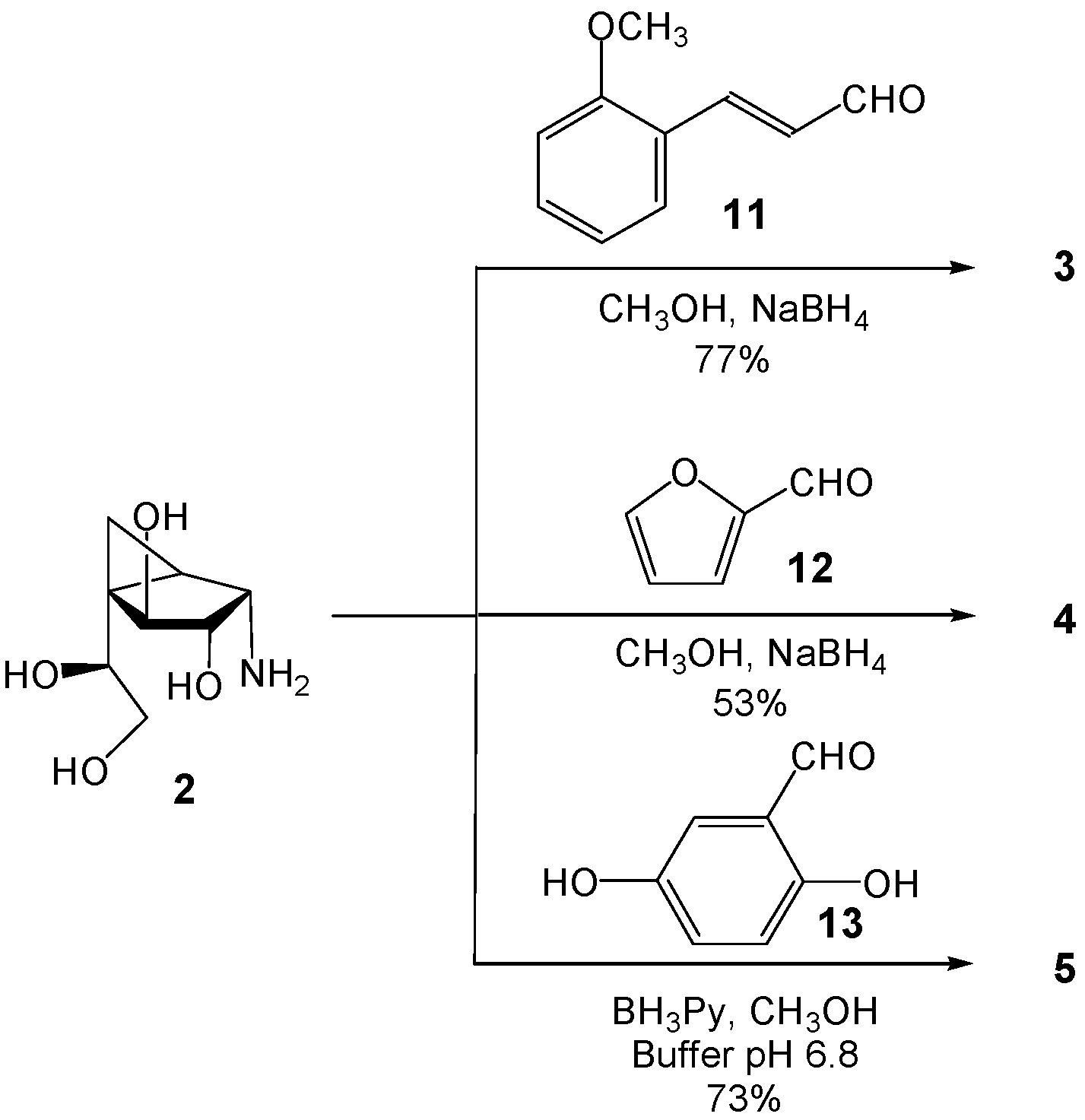
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-3,4-dihydroxy-N-((E)-3-(2-methoxyphenyl)allyl)bicyclo[3.1.0]hexan-2-ammonium acetate (3). A solution of 2 (8.8 mg, 0.046 mmol) and (E)-3-(2-methoxyphenyl) acrylaldehyde (11, 7.5 mg, 0.046 mmol) in freshly distilled CH3OH (2 mL) was stirred at rt for 1 h, before being cooled to −30 °C. NaBH4 (5.3 mg, 0.14 mmol) was added, and the solution was stirred for 5 min before being warmed to rt followed by stirring for an additional 10 min. The solution was then acidified with HOAc to pH 5 and concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:10) to give 3 (14 mg, 77%) as a white foam. Rf 0.36 (CH3OH–NH4OH 40:1); +31.8 (c 1.41, CH3OH); 1H-NMR (500 MHz, CD3OD, δH) 7.47 (dd, 1H, J = 1.3, 7.6 Hz, Ar), 7.27 (ddd, 1H, J = 1.3, 7.4, 8.6 Hz, Ar), 7.09 (d, 1H, J = 16.0 Hz, =CHPh), 6.98 (d, 1H, J = 8.4 Hz, Ar), 6.92 (dd, 1H, J = 7.4, 7.6 Hz, Ar), 6.33 (ddd, 1H, J = 7.0, 7.2, 16.0 Hz, CH2CH=), 4.29 (d, 1H, J = 6.8 Hz, H-4),3.92 (dd, 1H, J = 5.9, 6.3 Hz, H-7), 3.88–3.77 (m, 6H, NHCH2, OCH3, H-3), 3.67 (dd, 1H, J = 5.9, 11.1 Hz, H-8), 3.51 (dd, 1H, J = 6.3, 11.2 Hz, H-8), 3.49 (d, 1H, J = 6.3 Hz, H-2), 1.76 (dd, 1H, J = 3.8, 8.8 Hz, H-1), 0.91 (dd, 1H, J = 6.1, 8.8 Hz, H-6), 0.78 (dd, 1H, J = 3.8, 6.1 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 158.5 (Ar), 133.7 (CH=), 130.8 (Ar), 128.4 (Ar), 125.9 (Ar), 121.7 (Ar), 121.5 (CH=), 112.2 (Ar), 79.7 (C-4), 76.7 (C-3), 72.2 (C-7), 66.0 (C-8), 59.9 (C-2), 56.0 (CH3), 49.1 (NHCH2), 35.7 (C-5), 20.5 (C-1), 8.9 (C-6);HRMS (ESI) m/z Calcd for (M − CH3COO−) C18H26NO5: 336.1805. Found: 336.1806.
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-N-(furan-2-ylmethyl)-3,4-dihydroxy-bicyclo[3.1.0]hexan-2-ammonium acetate (4). A solution of 2 (6.7 mg, 0.035 mmol) and furan-2-carbaldehyde (12, 3.4 mg, 0.035 mmol) in freshly distilled CH3OH (2 mL) was stirred at rt for 1 h, before being cooled to −30 °C. NaBH4 (5.0 mg, 0.13 mmol) was added, and the solution was stirred for 5 min before being warmed to rt followed by stirring for an additional 10 min. The solution was then acidified with HOAc to pH 5 and concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:10) to give 4 (6.2 mg, 53%) as a white foam. Rf 0.27 (pure CH3OH); +55.0 (c 0.35, CH3OH); 1H-NMR (400 MHz, CD3OD, δH) 7.43 (d, 1H, J = 1.9 Hz, furan CH), 6.34 (dd, 1H, J = 1.9, 3.2 Hz, furan CH), 6.26 (d, 1H, J = 3.2 Hz, furan CH), 4.19 (d, 1H, J = 6.9 Hz, H-4), 3.87 (d, 1H, J = 14.4 Hz, NCH2), 3.79 (dd, 1H, J = 6.1, 6.3 Hz, H-7), 3.76 (d, 1H, J = 11.4 Hz, NCH2), 3.65 (dd, 1H, J = 6.1, 11.1 Hz, H-8), 3.59 (dd, 1H, J = 6.2, 6.9 Hz, H-3), 3.49 (dd, 1H, J = 6.3, 11.1 Hz, H-8), 2.99 (d, 1H, J = 6.2 Hz, H-2), 1.50 (dd, 1H, J = 5.1, 7.8 Hz, H-1), 0.68–0.66 (m, 2H, 2 × H-6); 13C-NMR (100 MHz, CD3OD, δC) 154.5 (C=CH), 143.3 (furan CH), 111.2 (furan CH), 108.4 (furan CH), 80.1 (C-4), 77.7 (C-3), 73.3 (C-7), 66.1 (C-8), 59.2 (C-2), 45.1 (NCH2), 34.4 (C-5), 23.9 (C-1), 8.6 (C-6); HRMS (ESI) m/z Calcd for (M − CH3COO−) C13H20NO5: 270.1336. Found: 270.1337.
(1R,2S,3S,4S,5S)-N-(2,5-Dihydroxybenzyl)-5-(1,2-dihydroxyethyl)-3,4-dihy-droxybicyclo[3.1.0]hexan-2-ammonium acetate (5). A solution of 2 (11 mg, 0.053 mmol) and 2,5-dihydroxybenzaldehyde (13, 7.3 mg, 0.053 mmol) in freshly distilled CH3OH (2 mL) was stirred at rt for 1 h. To this mixture was added phosphate buffer (0.1 M, pH 6.8, 0.2 mL) and the solution was cooled to 0 °C before BH3·pyridine (20 mL, 0.16 mmol) was added. The reaction mixture was then warmed to rt and stirred overnight before being acidified with HOAc to pH 5 and concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:10) to give compound 5 (14.4 mg, 73%) as a white foam. Rf 0.46 (CH3OH–NH4OH 20:1); +31.5 (c 0.32, CH3OH); 1H-NMR (500 MHz, CD3OD, δH) 6.77 (d, 1H, J = 2.9 Hz, Ar), 6.71 (d, 1H, J = 8.6 Hz, Ar), 6.66 (dd, 1H, J = 2.9, 8.6 Hz, Ar), 4.29 (d, 1H, J = 7.0 Hz, H-4),4.22 (d, 1H, J = 13.2 Hz, CH2N), 4.09 (d, 1H, J = 13.2 Hz, CH2N), 3.97 (dd, 1H, J = 5.7, 6.5 Hz, H-7), 3.76 (dd, 1H, J = 6.6, 7.0 Hz, H-3), 3.66 (dd, 1H, J = 5.7, 11.1 Hz, H-8), 3.66 (dd, 1H, J = 6.5, 11.1 Hz, H-8), 3.29 (d, 1H, J = 6.6 Hz, H-2),1.79 (dd, 1H, J = 4.0, 8.6 Hz, H-1), 0.84 (dd, 1H, J = 5.9, 8.6 Hz, H-6), 0.73 (dd, 1H, J = 4.0, 5.9 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 151.5 (Ar), 150.4 (Ar), 121.3 (Ar), 118.4 (Ar), 117.8 (Ar), 117.3 (Ar), 79.6 (C-4), 76.5 (C-3), 72.1 (C-7), 66.0 (C-8), 59.5 (C-2), 48.1 (CH2N), 35.5 (C-5), 20.8 (C-1), 8.6 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C15H22NO6: 312.1442. Found: 312.1442.
(1R,2S,3S,4S,5S)-N-(3-(Benzyloxy)propyl)-5-(1,2-dihydroxyethyl)-3,4-dihy-droxybicyclo[3.1.0]hexan-2-ammonium acetate (15). A solution of 2 (9.0 mg, 0.047 mmol) and 14 (7.8 mg, 0.047 mmol) in fresh CH3OH (2 mL) was stirred at rt for 1 h. To this mixture was added phosphate buffer (0.1 M, pH 6.8, 0.2 mL) and the solution was cooled to 0 °C before BH3·pyridine (20 mL, 0.16 mmol) was added. The reaction mixture was then warmed to rt and stirred overnight before being acidified with HOAc to pH 5 and concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH from 10:1 to 1:10) to give 15 (13.4 mg, 69%) as a white foam. Rf 0.48 (CH3OH–NH4OH 20:1); +15.4 (c 0.47, CH3OH); 1H-NMR (500 MHz, CD3OD, δH) 7.35–7.24 (m, 5H, Ar), 4.54 (d, 1H, J = 11.9 Hz, CH2Ph), 4.53 (d, 1H, J = 11.9 Hz, CH2Ph), 4.26 (d, 1H, J = 6.8 Hz, H-4), 3.94 (dd, 1H, J = 6.4, 6.2 Hz, H-7), 3.83 (dd, 1H, J = 6.4, 6.8 Hz, H-3), 3.67–3.60 (m, 3H, H-8, CH2OBn), 3.50 (dd, 1H, J = 6.4, 11.1 Hz, H-8), 3.46 (d, 1H, J = 6.4 Hz, H-2), 3.33–3.29 (m, 1H, 1 × NHCH2), 3.10 (ddd, 1H, J = 7.2, 7.2, 12.3 Hz, NHCH2), 2.11–1.97 (m, 4H, CH2CH2), 1.73 (dd, 1H, J = 4.0, 8.8 Hz, H-1), 0.88 (dd, 1H, J = 5.7, 8.8 Hz, H-6), 0.75 (dd, 1H, J = 4.0, 5.7 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 139.3 (Ar), 129.5 (2 C, Ar × 2), 129.1 (2 C, Ar × 2), 128.8 (Ar), 79.6 (C-4), 76.5 (C-3), 74.3 (CH2Ph), 71.9 (C-7), 69.5 (CH2OBn), 66.0 (C-8), 60.9 (C-2), 46.5 (NHCH2), 35.9 (C-5), 27.6 (2 C, CH2CH2), 20.2 (C-1), 8.6 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C18H28NO5: 338.1962. Found: 338.1961.
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-3,4-dihydroxy-N-(3-hydroxypropyl)-bicyclo[3.1.0]hexan-2-ammonium acetate (6). To a solution of compound 15 (13.4 mg, 0.034 mmol) in THF (4 mL) and H2O (0.5 mL) was added 10% Pd–C (4 mg), and the mixture was stirred under an H2 atmosphere for 12 h. The mixture was then filtered through Celite and concentrated. The resulting crude residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→100% CH3OH) to yield product 6 (10.3 mg, 100%) as a colorless oil. Rf 0.18 (CH3OH–NH4OH 20:1); +23.4 (c 0.28, CH3OH); 1H-NMR (400 MHz, CD3OD, δH) 4.28 (d, 1 H, J = 6.8 Hz, H-4), 3.93 (dd, 1H, J = 5.9, 6.3 Hz, H-7), 3.81 (dd, 1H, J = 6.3, 6.8 Hz, H-3),3.77–3.70 (m, 2H, CH2CH2OH), 3.66 (dd, 1H, J = 5.9, 11.1 Hz, H-8), 3.50 (dd, 1H, J = 6.3, 11.1 Hz, H-8), 3.44 (d, 1H, J = 6.3 Hz, H-2), 3.33–3.29 (m, 1H, 1 × NHCH2), 3.07 (ddd, 1H, J = 7.1, 7.1, 12.3 Hz, NHCH2), 1.99–1.86 (m, 2H, CH2), 1.73 (dd, 1H, J = 4.0, 8.8 Hz, H-1), 0.89 (dd, 1H, J = 5.7, 8.8 Hz, H-6), 0.76 (dd, 1H, J = 4.0, 5.7 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 79.6 (C-4), 76.5 (C-3), 72.1 (C-7), 65.9 (C-8), 61.2 (CH2OH), 60.8 (C-2), 46.4 (NHCH2), 35.7 (C-5), 29.9 (2 C, CH2CH2), 20.5 (C-1), 8.7 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C11H22NO5: 248.1492. Found: 248.1493.
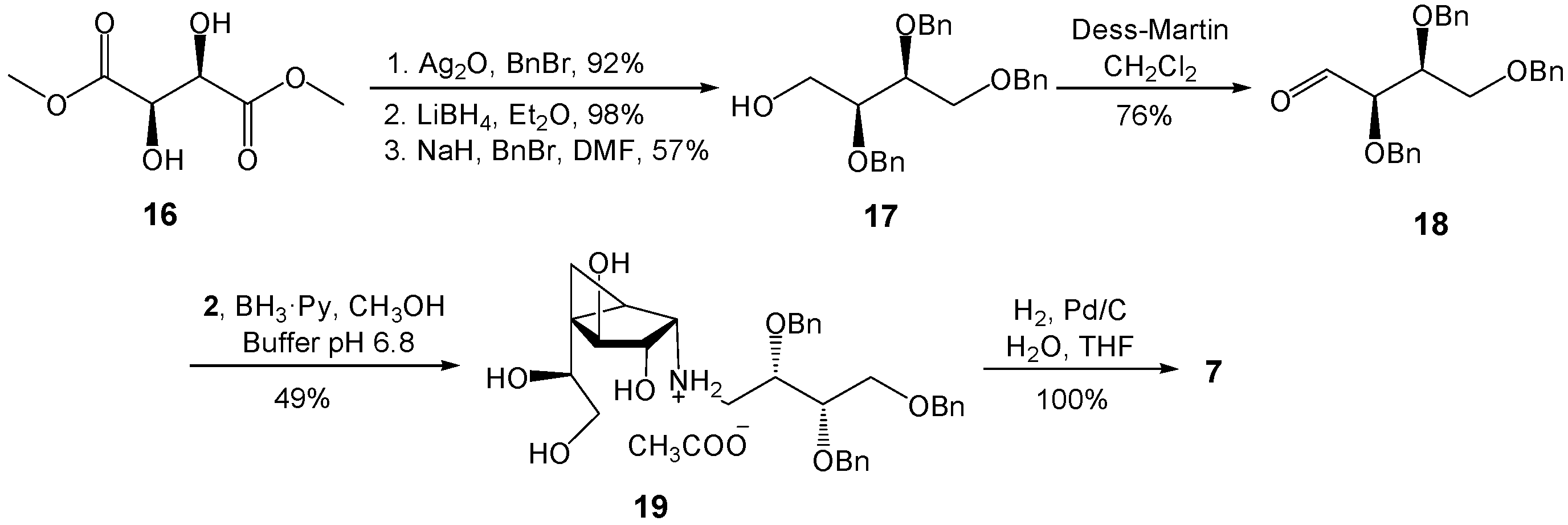
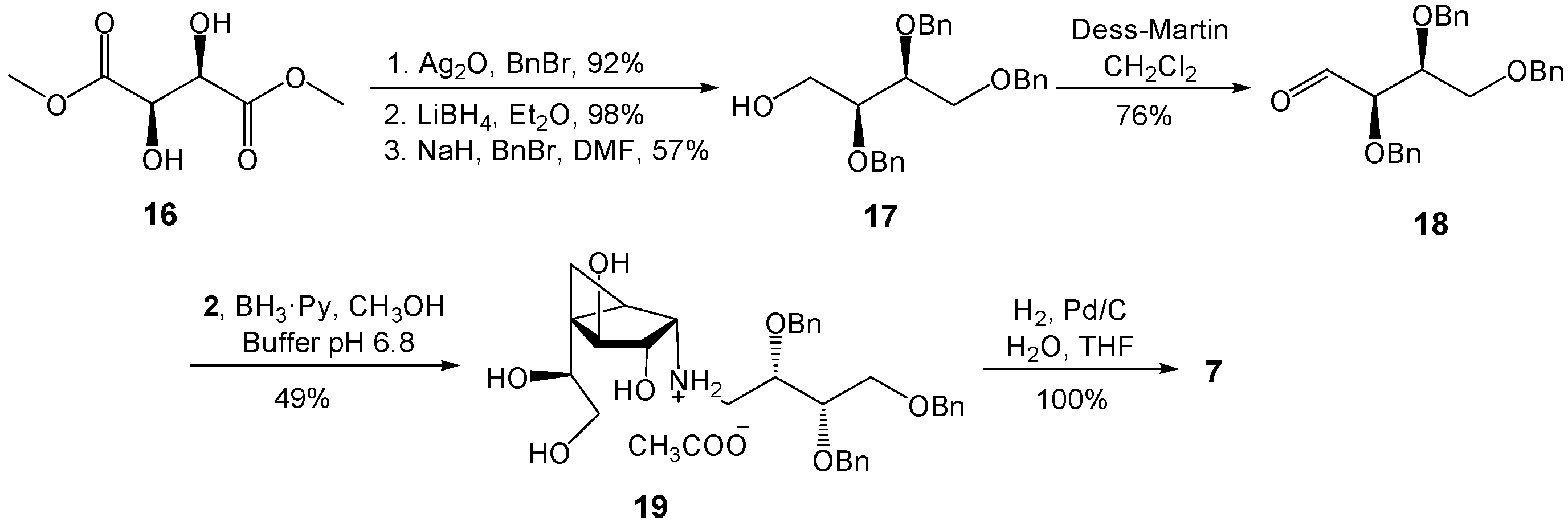
(2S,3S)-2,3,4-Tris(benzyloxy)butan-1-ol (17). To a solution of (2S,3S)-2,3-bis(benzyloxy)butane-1,4-diol (16, 0.23 g, 0.76 mmol) and benzyl bromide (0.13 g, 0.76 mmol) in DMF (4 mL) at 0 °C was added NaH (30 mg, 0.76 mmol, 60% in mineral oil). After stirring for 1 h, the reagents were quenched by the addition of H2O. The solution was extracted with Et2O twice and the organic layer was washed with brine, dried (MgSO4), and concentrated. The residue was purified by chromatography (EtOAc–Hexane 1:4) to give 17 (0.17 g, 57%) as a colorless oil. Rf 0.30 (EtOAc–Hexane 1:2); +12.3 (c 1.31, CH2Cl2); 1H-NMR (500 MHz, CDCl3, δH) 7.38–7.28 (m, 15 H, Ar), 4.74 (d, 1H, J = 11.8 Hz, CH2Ph), 4.70–4.62 (m, 3H, 3 × CH2Ph), 4.54 (s, 2H, 2 × CH2Ph), 3.84–3.63 (m, 6 H, 2 × CHOBn, 2 × CH2OBn, 2 × CH2OH), 2.23 (dd, 1H, J = 5.4, 7.0 Hz, OH); 13C-NMR (125 MHz, CDCl3, δC) 138.3 (2 C, Ar × 2), 137.9 (Ar), 128.5 (2 C, Ar × 2), 128.4 (2 C, Ar × 2), 128.3 (2 C, Ar × 2), 128.0 (2 C, Ar × 2), 127.9 (2 C, Ar × 2), 127.8 (2 C, Ar × 2), 127.7 (3 C, Ar × 3), 79.2 (CHOBn), 78.5 (CHOBn), 73.5 (CH2Ph), 72.9 (CH2Ph), 72.8 (CH2Ph), 69.5 (CH2OBn), 61.5 (CH2OH). HRMS (ESI) m/z Calcd for (M + Na+) C25H28O4: 415.1880. Found: 415.1876.
(2R,3S)-2,3,4-Tris(benzyloxy)butanal (18). To a solution of 17 (52 mg, 0.13 mmol) in CH2Cl2 (2 mL) at 0 °C was added a solution of Dess–Martin periodinane (56 mg, 0.13 mmol) in CH2Cl2 (2 mL). The mixture was stirred at 0 °C for 2 h and then poured into a cold aqueous saturated NaHCO3 solution. The organic layer was washed with brine, dried (MgSO4), concentrated, and the residue was purified by chromatography (EtOAc–Hexane 1:6) to give 18 (39 mg, 76%) as a colorless oil. Rf 0.43 (EtOAc–Hexane 1:3); +3.3 (c 0.65, CH2Cl2); 1H-NMR (400 MHz, CDCl3, δH) 9.71 (s, 1H, CHO), 7.37–7.26 (m, 15H, Ar), 4.76 (d, 1H, J = 12.0 Hz, CH2Ph), 4.64 (d, 1H, J = 11.9 Hz, CH2Ph), 4.57 (d, 1H, J = 12.0 Hz, CH2Ph), 4.56 (d, 1H, J = 11.9 Hz, CH2Ph), 4.48 (d, 1H, J = 12.0 Hz, CH2Ph), 4.46 (d, 1H, J = 12.0 Hz, CH2Ph), 4.00–3.96 (m, 2H, 2 × CHOBn), 3.71–3.64 (m, 2H, 2 × CH2OBn); 13C-NMR (100 MHz, CDCl3, δC) 202.38 (CHO), 137.8 (Ar), 137.7 (Ar), 137.2 (Ar), 128.5 (2C, Ar × 2), 128.4 (4C, Ar × 4), 128.2 (2C, Ar × 2), 128.1 (Ar), 128.0 (2C, Ar × 2), 127.8 (Ar), 127.7 (3C, Ar × 3), 82.8 (CHOBn), 77.9 (CHOBn), 73.4 (2C, 2 × CH2Ph), 72.9 (CH2Ph), 68.1 (CH2OBn). HRMS (ESI) m/z Calcd for (M + Na+) C25H26O4: 413.1679. Found: 413.1674.
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-3,4-dihydroxy-N-((2S,3S)-2,3,4-tris-(benzyloxy)butyl)bicycle[3.1.0]hexan-2-ammonium acetate (19). A solution of 2 (11.6 mg, 0.05 mmol) and 18 (21 mg, 0.05 mmol) in freshly distilled CH3OH (2 mL) was stirred at rt for 1 h. To this mixture was added phosphate buffer (0.1 M, pH 6.8, 0.2 mL) and the solution was cooled to 0 °C, before and BH3·pyridine (20 mL, 0.16 mmol) was added. The reaction mixture was then warmed to rt and stirred overnight before being acidified with HOAc to pH 5 and then concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1 to 1:1) to give 19 (14.2 mg, 49%) as a white foam. Rf 0.18 (CH3OH–CH2Cl2 1:1); +16.5 (c 0.39, CH3OH); 1H-NMR (500 MHz, CD3OD, δH) 7.35–7.24 (m, 15H, Ar), 4.68 (d, 1H, J = 11.7 Hz, CH2Ph), 4.64 (d, 1H, J = 11.3 Hz, CH2Ph),4.62 (d, 1H, J = 11.7 Hz, CH2Ph),4.57 (d, 1H, J = 11.7 Hz, CH2Ph), 4.51–4.50 (m, 2H, 2 × CH2Ph), 4.19 (d, 1H, J = 7.0 Hz, H-4), 3.86–3.81 (m, 2H, 2 × CHOBn), 3.78 (dd, 1H, J = 6.2, 6.2 Hz, H-7), 3.71 (dd, 1H, J = 3.1, 10.5 Hz, CH2OBn), 3.66–3.59 (m, 3H, H-3, 1 × CH2OBn, 1 × H-8), 3.49 (dd, 1H, J = 6.2, 11.1 Hz, H-8), 3.07 (dd, 1H, J = 2.6, 12.1 Hz, NHCH2), 2.99 (d, 1H, J = 6.3 Hz, H-2), 2.64 (dd, 1H, J = 8.3, 12.1 Hz, NHCH2), 1.50 (dd, 1H, J = 4.4, 8.6 Hz, H-1), 0.72 (dd, 1H, J = 5.8, 8.6 Hz, H-6), 0.68 (dd, 1H, J = 4.4, 5.8 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 139.7 (Ar), 139.5 (2C, Ar × 2), 129.5 (2C, Ar × 2), 129.4 (4C, Ar × 4), 129.3 (2C, Ar × 2), 129.2 (2C, Ar × 2), 129.0 (2C, Ar × 2), 128.9 (Ar), 128.8 (Ar), 128.7 (Ar), 80.0 (C-4), 79.5 (CHOBn), 79.2 (C-3), 77.5 (CHOBn), 74.4 (CH2Ph), 74.3 (CH2Ph), 73.8 (CH2Ph), 73.1 (C-7), 70.2 (CH2OBn), 66.1 (C-8), 61.2 (C-2), 49.3 (CH2NH), 34.8 (C-5), 23.4 (C-1), 8.8 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C33H42NO7: 564.2956. Found: 564.2951.
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-3,4-dihydroxy-N-((2S,3S)-2,3,4-tri-hydroxybutyl)bicyclo[3.1.0]hexan-2-ammonium acetate (7). To a solution of 19 (20 mg, 0.032 mmol) in THF (4 mL) and H2O (0.5 mL) was added 10% Pd–C (4 mg) and the reaction mixture was stirred under a H2 atmosphere for 12 h. The mixture was then filtered through Celite and concentrated to give pure 7 (11 mg, 100%) as a colorless oil. Rf 0.28 (CH3OH–NH4OH 20:1); +18.9 (c 0.24, CH3OH); 1H-NMR (500 MHz, CD3OD, δH) 4.26 (d, 1H, J = 6.8 Hz, H-4), 3.97 (ddd, 1H, J = 3.3, 3.3, 8.2 Hz, NHCH2CHOH), 3.89 (dd, 1H, J = 6.2, 6.2 Hz, H-7), 3.83 (dd, 1H, J = 6.4, 6.8 Hz, H-3), 3.67–3.58 (m, 4H, CHOHCH2OH, 1 × H-8), 3.52 (dd, 1H, J = 6.3, 11.1 Hz, H-8), 3.17 (d, 1H, J = 6.4 Hz, H-2), 3.32 (dd, 1H, J = 3.3, 12.4 Hz, NHCH2), 3.10 (dd, 1H, J = 8.2, 12.4 Hz, NHCH2), 1.71 (dd, 1H, J = 4.0, 8.9 Hz, H-1), 0.90 (dd, 1H, J = 5.7, 8.9 Hz, H-6), 0.78 (dd, 1H, J = 4.0, 5.7 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 79.7 (C-4), 76.2 (C-3), 74.4 (CHOH), 72.2 (CHOH), 68.8 (CHOH), 65.8 (CH2OH), 63.6 (CH2OH), 61.4 (C-2), 50.6 (CH2NH), 35.7 (C-5), 20.9 (C-1), 8.9 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C12H24NO7: 294.1547. Found: 294.1543.
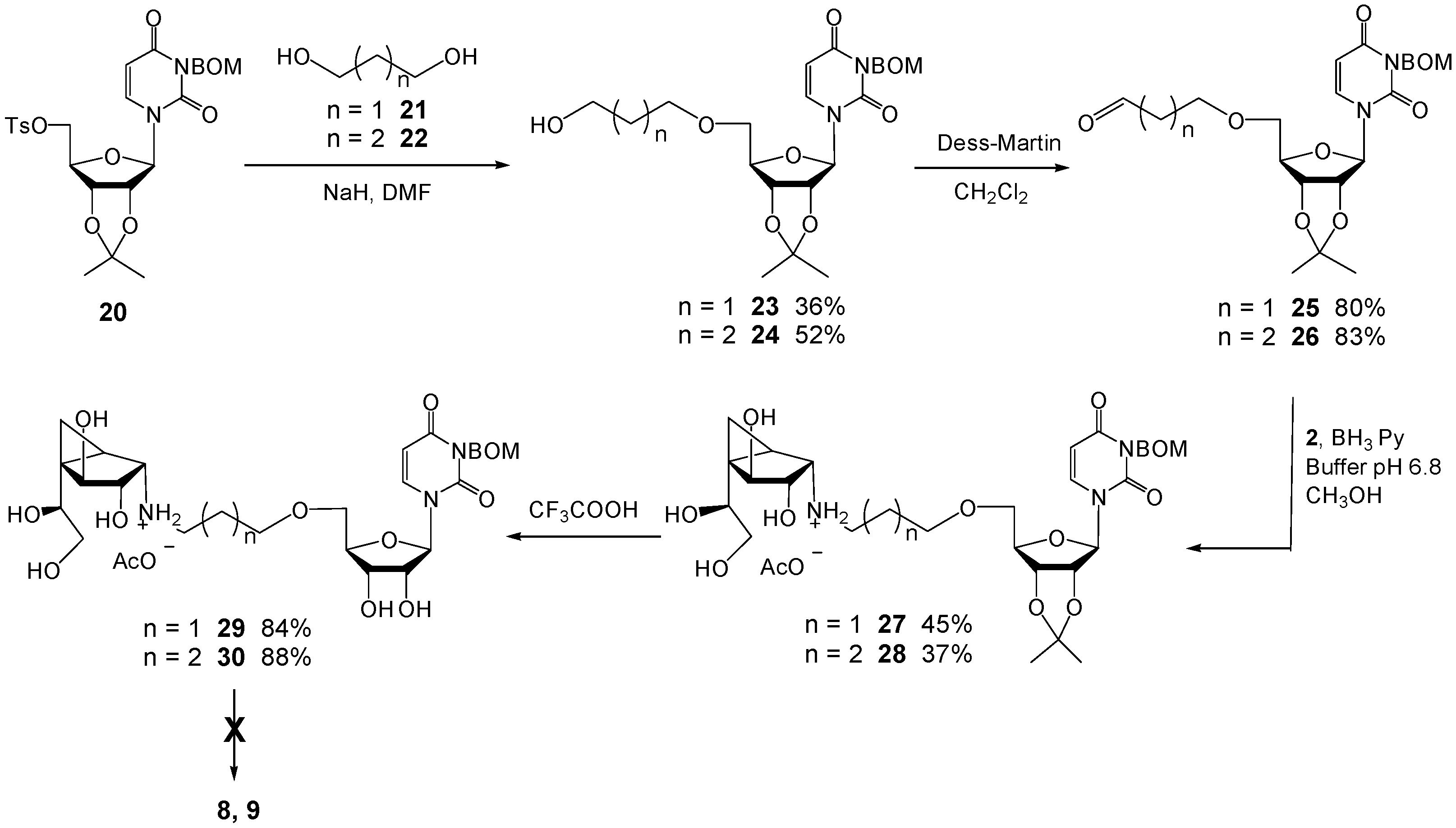
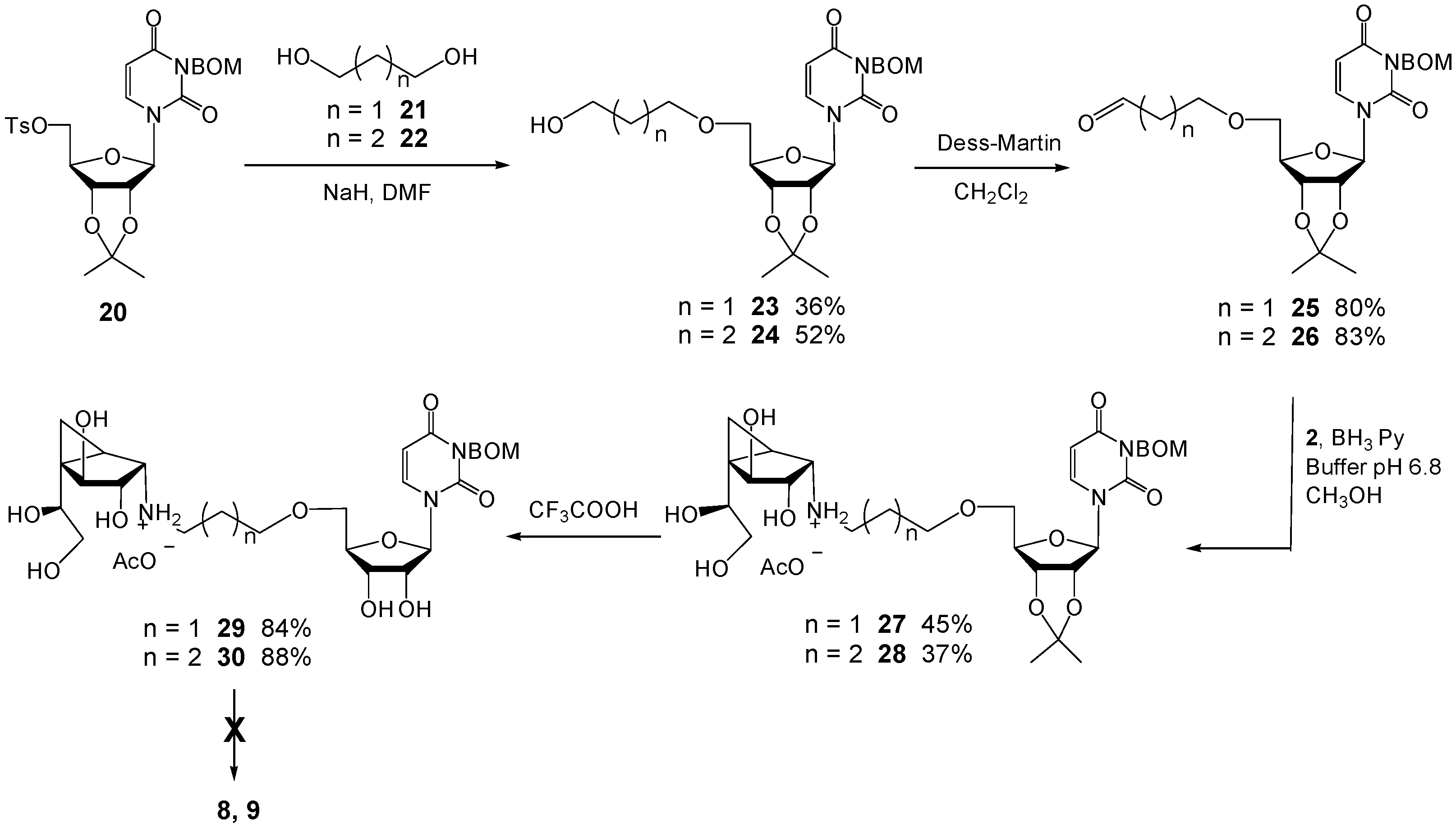
2′,3′-O-Isopropylidene-5′-O-tosyl-3-(benzyloxymethyl)uridine (20). To a solution of 2′,3′-O-isopropylidene-3-(benzyloxylmethyl)uridine (4.0 g, 10 mmol) in pyridine (25 mL) was added p-toluenesulfonyl chloride (2.26 g, 12 mmol) at rt. The reaction was stirred overnight and then the excess reagents were quenched by the addition of CH3OH (3 mL). The solution was then concentrated and the residue was purified by chromatography (EtOAc–Hexane 1:2) to give 20 (3.94 g, 71%) as a foam. Rf 0.54 (EtOAc–Hexane 1:1); +23.6 (c 1.33, CH2Cl2); 1H-NMR (500 MHz, CDCl3, δH) 7.77–7.75 (m, 2H, Ar), 7.37–7.25 (m, 7H, Ar), 7.16 (d, 1H, J = 8.1 Hz, CH=), 5.71 (d, 1H, J = 8.1 Hz, CH=), 5.60 (d, 1H, J = 2.0 Hz, H-1), 5.46 (d, 1H, J = 9.8 Hz, NCH2OBn), 5.35 (d, 1H, J = 9.8 Hz, NCH2OBn), 4.88 (dd, 1H, J = 2.0, 6.4 Hz, H-2), 4.79 (dd, 1H, J = 3.7, 6.4 Hz, H-3), 4.68 (s, 2H, CH2Ph), 4.35 (ddd, 1H, J = 3.7, 4.6, 4.6 Hz, H-4), 4.27 (d, 2H, J = 4.6 Hz, 2 × H-5), 2.42 (s, 3H, CH3), 1.55 (s, 3H, CH3), 1.34 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3, δC) 162.3 (C=O), 150.7 (C=O), 145.2 (Ar), 140.6 (CH=), 137.8 (Ar), 132.6 (Ar), 129.9 (2C, Ar × 2), 128.3 (2C, Ar × 2), 127.9 (2C, Ar × 2), 127.7 (Ar), 127.6 (2C, Ar × 2), 114.5 (C(CH3)2), 102.3 (CH=), 95.6 (C-1), 85.1 (C-4), 84.4 (C-2), 80.8 (C-3), 72.4 (CH2Ph), 70.3 (NCH2OBn), 69.2 (C-5), 27.0 (CH3), 25.2 (CH3), 21.6 (CH3). HRMS (ESI) m/z Calcd for (M + Na+) C27H30N2O9S: 581.1562. Found: 581.1558.
2′,3′-O-Isopropylidene-5′-O-(3-hydroxylpropyl)-3-(benzyloxymethyl)uridine (23). To a solution of 20 (0.50 g, 0.9 mmol) and 1,3-propanediol (21) (0.69 g, 9.0 mmol) in DMF (4 mL) at 0 °C was added NaH (72 mg, 1.8 mmol, 60% in mineral oil). The solution was stirred at rt for 20 h and then H2O (10 mL) was added and the mixture was extracted with Et2O. The organic layer was washed with brine, dried (MgSO4), and concentrated. The residue was purified by chromatography (EtOAc–Hexane 2:1) to give 23 (0.15 g, 36%) as a colorless oil. Rf 0.14 (EtOAc–Hexane 2.5:1); −3.3 (c 0.96, CH2Cl2); 1H-NMR (400 MHz, CDCl3, δH) 7.44 (d, 1H, J = 8.2 Hz, CH=), 7.38–7.23 (m, 5H, Ar), 5.77 (d, 1H, J = 1.9 Hz, H-1), 5.73 (d, 1H, J = 8.2 Hz, CH=), 5.50 (d, 1H, J = 9.7 Hz, NCH2OBn), 5.47 (d, 1H, J = 9.7 Hz, NCH2OBn), 4.81–4.77 (m, 2H, H-2, H-3), 4.70 (s, 2H, CH2Ph), 4.37 (ddd, 1H, J = 3.0, 3.0, 4.5 Hz, H-4),3.73–3.69 (m, 3H, H-5, CH2OH), 3.67–3.58 (m, 3H, H-5, CH2O), 1.83–1.77 (m, 2H, CH2), 1.58 (s, 3H, CH3), 1.37 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3, δC) 162.6 (C=O), 150.8 (C=O), 139.9 (CH=), 137.9 (Ar), 128.3 (2C, Ar × 2), 127.6 (3C, Ar × 3), 114.1 (C(CH3)2), 101.6 (CH=), 94.4 (C-1), 85.9 (C-2), 85.2 (C-4), 80.9 (C-3), 72.3 (CH2Ph), 70.9 (NCH2OBn), 70.3 (C-5), 69.6 (CH2OCH2), 60.6 (CH2OH), 32.2 (CH2), 27.2 (CH3), 25.4 (CH3). HRMS (ESI) m/z Calcd for (M + Na+) C23H30N2O8: 485.1894. Found: 485.1894.
2’,3’-O-Isopropylidene-5’-O-(3-oxopropyl)-3-(benzyloxymethyl)uridine (25). A solution of 23 (32 mg, 0.07 mmol) in CH2Cl2 (2 mL) was added to a solution of Dess–Martin periodinane (35 mg, 0.08 mmol) in CH2Cl2 (4 mL) at 0 °C. The reaction was stirred for 3 h at 0 °C and poured into an ice-cold saturated aqueous NaHCO3 solution. The organic layer was washed with H2O, brine, dried (MgSO4), and concentrated. The residue was purified by chromatography (EtOAc–Hexane 1:1) to give 25 (25 mg, 80%) as a colorless oil. Rf 0.26 (EtOAc–Hexane 2:1); −12.6 (c 0.68, CH2Cl2); 1H-NMR (400 MHz, CDCl3, δH) 9.75 (dd, 1H, J = 1.6, 1.6 Hz, CHO), 7.38 (d, 1H, J = 8.2 Hz, CH=), 7.36–7.23 (m, 5H, Ar), 5.81 (d, 1H, J = 2.3 Hz, H-1), 5.75 (d, 1H, J = 8.2 Hz, CH=), 5.50 (d, 1H, J = 9.7 Hz, NCH2OBn), 5.46 (d, 1H, J = 9.7 Hz, NCH2OBn), 4.75 (dd, 2H, J = 3.2, 6.2 Hz, H-3), 4.74 (dd, 2H, J = 2.3, 6.2 Hz, H-2), 4.70 (s, 2H, CH2Ph), 4.35 (ddd, 1H, J = 2.8, 3.2, 4.2 Hz, H-4), 3.82–3.79 (m, 2H, CH2OCH2), 3.74 (dd, 1H, J = 2.8, 10.6 Hz, H-5), 3.62 (dd, 1H, J = 4.2, 10.6 Hz, H-5), 2.69–2.65 (m, 2H, CH2), 1.58 (s, 3H, CH3), 1.36 (s, 3H, CH3); 13C-NMR (100 MHz, CDCl3, δC) 199.9 (CHO), 162.6 (C=O), 150.9 (C=O), 139.6 (CH=), 137.9 (Ar), 128.3 (2C, Ar × 2), 127.6 (3C, Ar × 3), 114.1 (C(CH3)2), 101.7 (CH=), 93.9 (C-1), 85.7 (C-4), 85.2 (C-2), 80.7 (C-3), 72.3 (CH2Ph), 71.0 (CH2OBn), 70.3 (C-5), 64.8 (CH2OCH2), 43.7 (CH2), 27.2 (CH3), 25.3 (CH3). HRMS (ESI) m/z Calcd for (M + Na+) C23H28N2O8: 483.1738. Found: 483.1838.
(1R,2S,3S,4S,5S)-N-(3-(2′,3′-O-Isopropylidene-3-(benzyloxymethyl)uridin)-propyl)-5-(1,2-dihydroxyethyl)-3,4-dihydroxybicyclo[3.1.0]hexan-2-ammonium acetate (27). To a mixture of 2 (8 mg, 0.043 mmol) and 25 (20 mg, 0.043 mmol) in freshly distilled CH3OH (2 mL) at 0 °C was added BH3·pyridine (20 mL, 0.16 mmol) and phosphate buffer (0.1 M, pH 6.8, 0.4 mL). The mixture was stirred overnight at rt and then acidified with HOAc to pH 5 before being concentrated. The resulting residue was purified by chromatography on C18 silica gel (H2O–CH3OH 10:1→1:10) to give 27 (13.3 mg, 45%) as a white foam. Rf 0.11 (pure CH3OH; C18 silica gel TLC); +15.4 (c 1.32, CH3OH); 1H-NMR (300 MHz, CD3OD, δH) 7.71 (d, 1H, J = 8.1 Hz, CH=), 7.37–7.21 (m, 5H, Ar), 5.80 (d, 1H, J = 1.4 Hz, H-1′), 5.73 (d, 1H, J = 8.1 Hz, CH=),5.45 (s, 2H, NCH2OBn), 4.81 (m, 2H, H-2′, H-3′), 4.66 (s, 2H, CH2Ph), 4.38–4.37 (m, 1H, H-4′), 4.17 (d, 1H, J = 6.9 Hz, H-4), 3.75–3.47 (m, 8H, H-3, H-7, H-8, H-8, H-5’, H-5’, CH2OC-5′), 2.91 (d, 1H, J = 6.1 Hz, H-2), 2.80 (ddd, 1H, J = 6.8, 7.0, 11.6 Hz, NHCH2), 2.56 (ddd, 1H, J = 7.0, 7.1, 11.6 Hz, NHCH2), 1.79–1.71 (m, 2H, CH2), 1.54 (s, 3H, CH3), 1.49 (dd, 1H, J = 6.0, 6.0 Hz, H-1), 1.36 (s, 3H, CH3), 0.68–0.67 (m, 2H, 2 × H-6); 13C-NMR (125 MHz, CD3OD, δC) 164.9 (C=O), 152.3 (C=O), 142.3 (CH=), 139.5 (Ar), 129.3 (2C, Ar × 2), 128.7 (Ar), 128.6 (2C, Ar × 2), 114.8 (C(CH3)2), 101.7 (CH=), 95.6 (C-1′), 87.6 (C-4′), 86.6 (C-2′), 82.6 (C-3′), 80.0 (C-4), 77.7 (C-3), 73.3 (C-7), 73.2 (CH2Ph), 72.0 (C-5′ or CH2OCH2), 71.6 (NCH2OBn), 70.8 (C-5’ or CH2OCH2), 66.1 (C-8), 60.7 (C-2), 46.6 (NHCH2), 34.5 (C-5), 30.6 (CH2), 27.5 (CH3), 25.5 (CH3), 24.3 (C-1), 8.9 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C31H44N3O11: 634.2970. Found: 634.2969.
(1R,2S,3S,4S,5S)-N-(3-(3-(Benzyloxymethyl)uridin)propyl)-5-(1,2-dihydroxy-ethyl)-3,4-dihydroxybicyclo[3.1.0]hexan-2-ammonium 2,2,2-trifluoroacetate (29). Compound 27 (14 mg, 0.02 mmol) was dissolved in TFA (2 mL) at 0 °C. The solution was then stirred at rt for 12 h and concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:3) to give the 29 (12 mg, 84%) as a white foam. Rf 0.24 (CH3OH–NH4OH 40:1); +14.9 (c 0.15, CH3OH); 1H-NMR (400 MHz, CD3OD, δH) 7.76 (d, 1H, J = 8.2 Hz, CH=), 7.33–7.24 (m, 5H, Ar), 5.78 (d, 1H, J = 2.8 Hz, H-1′), 5.76 (d, 1H, J = 8.2 Hz, CH=), 5.45 (s, 2H, NCH2OBn), 4.66 (s, 2H, CH2Ph), 4.25 (d, 1H, J = 6.7 Hz, H-4), 4.15 (dd, 1 H, J = 2.8, 4.8 Hz, H-2′), 4.10–4.06 (m, 2H, H-3′, H-4′), 3.89–3.80 (m, 3H, H-7, H-5′, CH2OC-5′), 3.72–3.62 (m, 3H, H-3, H-5′, CH2OC-5′), 3.64 (dd, 1H, J = 5.9, 11.1 Hz, H-8), 3.54 (d, 1H, J = 6.9 Hz, H-2), 3.51 (dd, 1H, J = 6.2, 11.1 Hz, H-8), 3.27–3.12 (m, 2H, NHCH2), 2.05–1.96 (m, 2H, CH2), 1. 69 (dd, 1H, J = 4.0, 9.0 Hz, H-1), 0.93 (dd, 1H, J = 5.8, 9.0, Hz, H-6), 0.79 (dd, 1H, J = 4.0, 5.8 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 164.8 (C=O), 152.4 (C=O), 141.6 (CH=), 139.4 (Ar), 129.4 (2C, Ar × 2), 128.8 (Ar), 128.7 (2C, Ar × 2), 102.1 (CH=), 93.0 (C-1′), 84.0 (C-4′), 79.6 (C-4), 76.2 (C-3), 75.3 (C-3’), 73.2 (CH2Ph), 71.9 (C-7), 71.6 (NCH2OBn), 71.6 (C-5′ or CH2OCH2), 71.1 (CH), 70.4 (C-5’ or CH2OCH2), 65.8 (C-8), 61.0 (C-2), 45.8 (NHCH2), 35.9 (C-5), 27.3 (CH2), 19.8 (C-1), 9.0 (C-6). HRMS (ESI) m/z Calcd for (M − CF3COO−) C28H40N3O11: 594.2657. Found: 594.2651.
2′,3′-O-Isopropylidene-5′-O-(4-hydroxylbutyl)-3-(benzyloxymethyl)uridine (24). To a solution of 20 (0.89 g, 1.6 mmol) and 1,4-butanediol (22) (1.62 g, 19.4 mmol) in DMF (4 mL) at 0 °C was added NaH (128 mg, 3.2 mmol, 60% in mineral oil). The reaction was stirred at rt for 20 h and then H2O (10 mL) was added. The mixture was extracted with Et2O and the organic layer was washed with brine, dried (MgSO4) and concentrated. The resulting residue was purified by chromatography (EtOAc–Hexane 2:1) to give 24 (0.40 g, 52%) as a colorless oil. Rf 0.16 (EtOAc–Hexane 2.5:1); −3.7 (c 2.43, CH2Cl2); 1H-NMR (500 MHz, CDCl3, δH) 7.53 (d, 1H, J = 8.1 Hz, CH=), 7.36–7.23 (m, 5H, Ar), 5.83 (s, 1H, H-1), 5.71 (d, 1H, J = 8.1 Hz, CH=), 5.59 (d, 1H, J = 9.8 Hz, NCH2OBn), 5.45 (d, 1H, J = 9.8 Hz, NCH2OBn), 4.78–4.74 (m, 2H, H-2, H-3), 4.70 (s, 2H, CH2Ph), 4.38 (ddd, 1H, J = 2.6, 2.6, 3.9 Hz, H-4), 3.69 (dd, 1H, J = 2.6, 10.7 Hz, H-5), 3.61–3.57 (m, 3H, H-5, CH2OH), 3.59–3.46 (m, 2H, CH2), 1.64–1.55 (m, 4H, CH2CH2), 1.57 (s, 3H, CH3), 1.36 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3, δC) 162.7 (C=O), 150.9 (C=O), 139.7 (CH=), 137.9 (Ar), 128.3 (2C, Ar × 2), 127.6 (3C, Ar × 3), 114.0 (C(CH3)2), 101.4 (CH=), 94.0 (C-1), 85.9 (C-4), 85.4 (C-2), 80.9 (C-3), 72.3 (CH2Ph), 71.6 (CH2OCH2), 70.7 (C-5), 70.3 (NCH2OBn), 62.4 (CH2OH), 29.5 (CH2), 27.2 (CH3), 26.2 (CH2), 25.4 (CH3). HRMS (ESI) m/z Calcd for (M + Na+) C24H32N2O8: 499.2051. Found: 499.2046.
2′,3′-O-Isopropylidene-5′-O-(4-oxobutyl)-3-(benzyloxymethyl)uridine (26). A solution of 24 (73 mg, 0.15 mmol) in CH2Cl2 (4 mL) was added to a solution of Dess-Martin periodinane (78 mg, 0.18 mmol) in CH2Cl2 (8 mL) at 0 °C. The reaction was stirred for 3 h at 0 °C and poured into an ice cold saturated aqueous NaHCO3 solution. The organic layer was washed with H2O, brine, dried (MgSO4) and concentrated. The residue was purified by chromatography (EtOAc–Hexane 1:1) to give 26 (60 mg, 83%) as a colorless oil. Rf 0.28 (EtOAc–Hexane 2:1); +17.1 (c 0.70, CH2Cl2); 1H-NMR (400 MHz, CDCl3, δH) 9.72 (dd, 1H, J = 1.5, 1.5 Hz, CHO), 7.44 (d, 1H, J = 8.2 Hz, CH=), 7.43–7.24 (m, 5H, Ar), 5.80 (d, 1H, J = 2.0 Hz, H-1), 5.73 (d, 1H, J = 8.2 Hz, CH=), 5.50 (d, 1 H, J = 9.7 Hz, NCH2OBn), 5.48 (d, 1 H, J = 9.7 Hz, NCH2OBn), 4.78–4.74 (m, 2H, H-3, H-2), 4.71 (s, 2H, CH2Ph), 4.35 (ddd, 1H, J = 3.0, 4.4, 4.4 Hz, H-4), 3.67 (dd, 1H, J = 4.4, 10.7 Hz, H-5), 3.59 (dd, 1H, J = 4.4, 10.7 Hz, H-5), 3.53–3.45 (m, 2H, CH2), 2.50–2.46 (m, 2H, CH2), 1.92–1.87 (m, 2H, CH2), 1.59 (s, 3H, CH3), 1.37 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3, δC) 201.4 (CHO), 162.6 (C=O), 150.9 (C=O), 139.7 (CH=), 137.9 (Ar), 128.3 (2C, Ar × 2), 127.6 (3C, Ar × 3), 114.2 (C(CH3)2), 101.6 (CH=), 94.2 (C-1), 85.9 (C-4), 85.2 (C-2), 80.7 (C-3),72.3 (CH2Ph), 70.8 (CH2OBn), 70.6 (C-5), 70.3 (CH2OCH2), 40.6 (CH2), 27.2 (CH3), 25.3 (CH3), 22.2 (CH2). HRMS (ESI) m/z Calcd for (M + Na+) C24H30N2O8: 497.1894. Found: 497.1894.
(1R,2S,3S,4S,5S)-N-(4-(2’,3’-O-Isopropylidene-3-(benzyloxymethyl)uridin)-butyl)-5-(1,2-dihydroxyethyl)-3,4-dihydroxybicyclo[3.1.0]hexan-2-ammonium acetate (28). To a mixture of 2 (7.2 mg, 0.038 mmol) and 26 (18 mg, 0.038 mmol) in freshly distilled CH3OH (2 mL) at 0 °C was added BH3·pyridine (20 mL, 0.16 mmol) and phosphate buffer (0.1 M, pH 6.8, 0.4 mL). The mixture was stirred overnight at rt and then acidified with HOAc to pH 5 before being concentrated. The residue was purified by chromatography on C18 silica gel (H2O–CH3OH 10:1→1:10) to give the product 28 (10 mg, 37%) as a white foam. Rf 0.11 (Pure CH3OH; C18 silica gel TLC); +10.9 (c 1.00, CH3OH); 1H-NMR (500 MHz, CD3OD, δH) 7.74 (d, 1H, J = 8.1 Hz, CH=), 7.31–7.22 (m, 5H, Ar), 5.80 (d, 1H, J = 1.4 Hz, H-1’), 5.72 (d, 1H, J = 8.1 Hz, CH=), 5.46 (s, 2H, NCH2OBn), 4.81 (m, 2H, H-2′, H-3′), 4.66 (s, 2H, CH2Ph), 4.39–4.37 (m, 1H, H-4′), 4.17 (d, 1H, J = 6.8 Hz, H-4), 3.74 (dd, 1H, J = 6.3, 6.4 Hz, H-7), 3.69 (dd, 1H, J = 3.1, 10.7 Hz, 1 × CH2OCH2), 3.65 (dd, 1H, J = 6.4, 11.2 Hz, H-8), 3.61–3.57 (m, 2H, H-3, 1 × CH2OCH2), 3.50–3.47 (m, 3H, 2 × H-5’, 1 × H-8), 2.91 (d, 1H, J = 6.2 Hz, H-2), 2.73 (ddd, 1H, J = 6.2, 7.7, 11.6 Hz, NHCH2), 2.56 (ddd, 1H, J = 6.4, 7.6, 11.6 Hz, NHCH2), 1.61–1.49 (m, 5H, H-1, CH2CH2), 1.54 (s, 3H, CH3), 1.35 (s, 3H, CH3), 0.68–0.67 (m, 2H, 2 × H-6); 13C-NMR (125 MHz, CD3OD, δC) 164.9 (C=O), 152.3 (C=O), 142.4 (CH=), 139.5 (Ar), 129.4 (2C, Ar × 2), 128.7 (Ar), 128.6 (2C, Ar × 2), 114.8 (C(CH3)2), 101.7 (CH=), 95.7 (C-1′), 87.6 (C-4′), 86.6 (C-2’), 82.7 (C-3′), 79.9 (C-4), 77.5 (C-3), 73.3 (CH2Ph), 73.2 (C-7), 72.2 (C-5’ or CH2OCH2), 71.9 (NCH2OBn), 71.6 (C-5’ or CH2OCH2), 66.0 (C-8), 60.6 (C-2), 48.7 (NHCH2), 34.7 (C-5), 28.3 (CH2), 27.5 (CH3), 26.9 (CH2), 25.5 (CH3), 23.5 (C-1), 8.9 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C32H46N3O11: 648.3127. Found: 648.3135.
(1R,2S,3S,4S,5S)-N-(4-(3-(Benzyloxymethyl)uridin)butyl)-5-(1,2-dihydroxyet-hyl)-3,4-dihydroxybicyclo[3.1.0]hexan-2-ammonium 2,2,2-trifluoroacetate (30). Compound 28 (13 mg, 0.019 mmol) was dissolved in TFA (2 mL) at 0 °C, stirred at rt for 12 h and then concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:3) to give 30 (12 mg, 88%) as a white foam. Rf 0.19 (CH3OH–NH4OH 40:1); +16.0 (c 0.15, CH3OH); 1H-NMR (400 MHz, CD3OD, δH) 7.90 (d, 1H, J = 8.2 Hz, CH=), 7.34–7.22 (m, 5H, Ar), 5.83 (d, 1H, J = 2.9 Hz, H-1′), 5.74 (d, 1H, J = 8.2 Hz, CH=), 5.45 (s, 2H, NCH2OBn), 4.66 (s, 2H, CH2Ph), 4.27 (d, 1H, J = 6.9 Hz, H-4), 4.13–4.09 (m, 3H, H-2′, H-3′, H-4′), 3.90–3.80 (m, 3H, H-7, H-3, H-5′), 3.67–3.39 (m, 6H, H-5′, 2 × H-8, H-2, CH2OC-5′), 3.20–3.07 (m, 2H, NHCH2), 1.87–1.69 (m, 5H, CH2CH2, H-1), 0.94 (dd, 1H, J = 5.8, 8.7 Hz, H-6), 0.79 (dd, 1H, J = 4.0, 5.8 Hz, H-6); 13C-NMR (100 MHz, CD3OD, δC) 164.9 (C=O), 152.5 (C=O), 141.3 (CH=), 139.4 (Ar), 129.4 (2C, Ar × 2), 128.8 (Ar), 128.7 (2C, Ar × 2), 101.9 (CH=), 92.3 (C-1′), 84.4 (C-4′), 79.6 (C-4), 76.3 (C-3), 75.8 (C-3’), 73.2 (CH2 Ph), 71.9 (C-7), 71.7 (CH2OC-5′), 71.6 (C-5’), 71.1 (C-2′), 70.9 (NCH2OBn), 65.8 (C-8), 60.8 (C-2), 47.0 (NHCH2), 35.9 (C-5), 27.8 (CH2), 24.3 (CH2), 19.9 (C-1), 9.1 (C-6). HRMS (ESI) m/z Calcd for (M–CF3COO−) C29H42N3O11: 608.2814. Found: 608.2809.
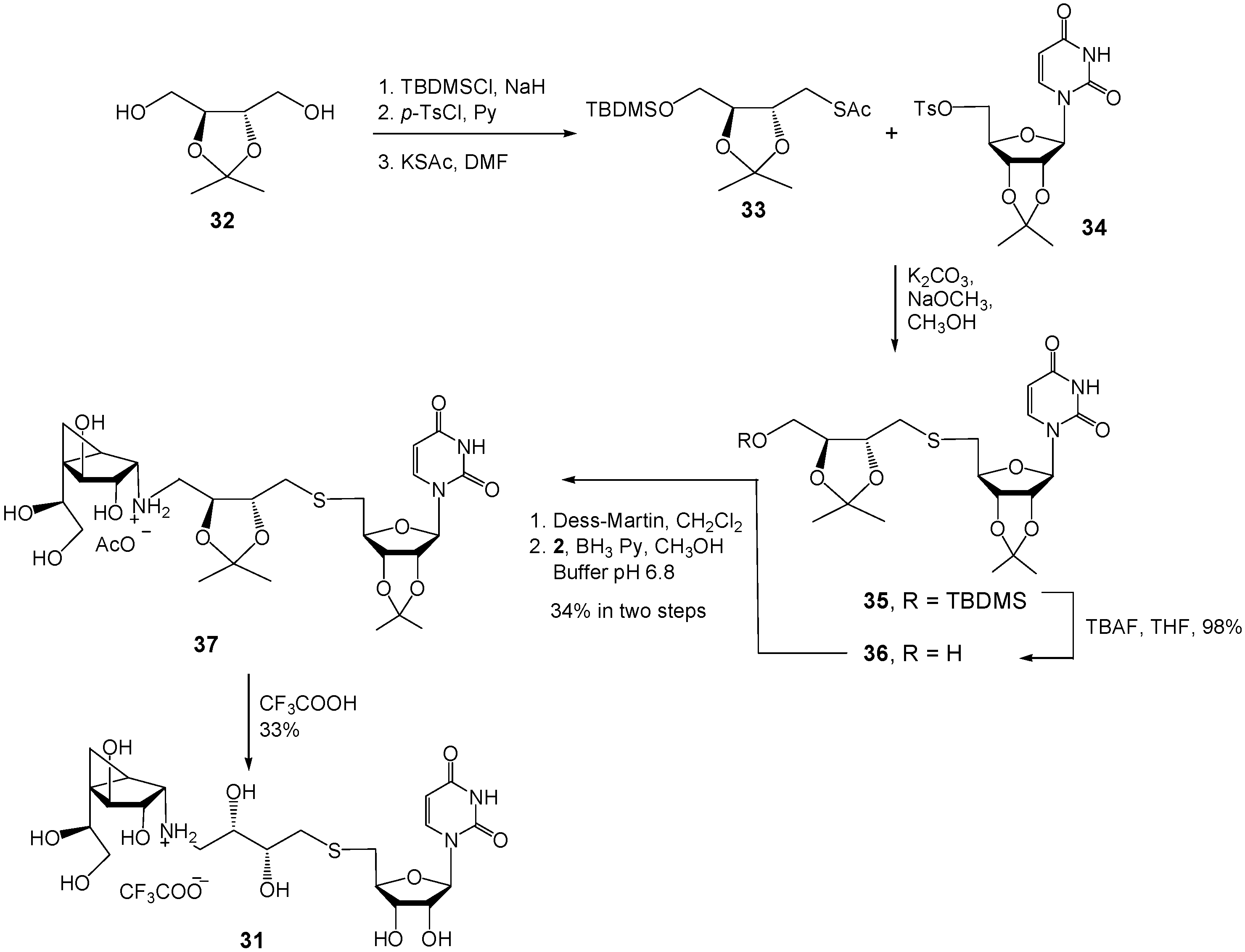
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-N-((2S,3R)-2,3-O-isopropylidene-4-(2′,3′-O-iso-propylidene-5′-thiouridin)-2,3-dihydroxylbutyl)-3,4-dihydroxybicyclo[3.1.0]hexan-2-ammonium acetate (37). A solution of 36 (30 mg, 0.067 mmol) in CH2Cl2 (4 mL) was added to a solution of Dess-Martin periodinane (29 mg, 0.067 mmol) in CH2Cl2 (8 mL) at 0 °C. The solution was stirred for 3 h and then poured into an ice cold saturated aqueous NaHCO3 solution. The organic layer was washed with H2O, brine, dried (MgSO4), and concentrated. The resulting aldehyde was dissolved in freshly distilled CH3OH (2 mL), and was added to 2 (12.7 mg, 0.067 mmol). The mixture was stirred for 1 h, cooled to 0 °C, and then BH3·pyridine (20 mL, 0.16 mmol) and phosphate buffer (0.1 M, pH 6.8, 0.4 mL) were added. After stirring overnight at rt, the solution was acidified with HOAc to pH 5 and then concentrated. The resulting residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:10) to give 37 (10 mg, 34%) as a white foam. Rf 0.25 (Pure CH3OH); +13.9 (c 1.28, CH3OH); 1H-NMR (400 MHz, CD3OD, δH) 7.65 (d, 1H, J = 8.1 Hz, CH=), 5.74 (d, 1H, J = 2.3 Hz, H-1′), 5.69 (d, 1H, J = 8.1 Hz, CH=), 5.05 (dd, 1H, J = 2.3, 6.6 Hz, H-2′), 4.79 (dd, 1H, J = 4.1, 6.6 Hz, H-3′), 4.25–4.21 (m, 2H, H-4′, H-4), 4.05–3.97 (m, 1H, OCH), 3.96–3.91 (m, 1H, OCH), 3.82 (dd, 1H, J = 6.1, 6.3 Hz, H-7), 3.70 (dd, 1H, J = 6.4, 6.7 Hz, H-3), 3.66 (dd, 1H, J = 6.1, 11.1 Hz, H-8), 3.50 (dd, 1H, J = 6.3, 11.1 Hz, H-8), 3.19–3.15 (m, 2H, H-2, 1 × NHCH2), 2.97–2.95 (m, 2H, 2 × H-5′), 2.88–2.80 (m, 3H, 1 × NHCH2, CH2S), 1.59 (dd, 1H, J = 4.3, 8.6 Hz, H-1), 1.53 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.39 (s, 3H, CH3), 1.34 (s, 3H, CH3), 0.77 (dd, 1H, J = 5.8, 8.6 Hz, H-6), 0.72 (dd, 1H, J = 4.3, 5.8 Hz, H-6); 13C-NMR (125 MHz, CD3OD, δC) 166.1 (C=O), 151.9 (C=O), 144.8 (CH=), 115.5 (C(CH3)2), 110.9 (C(CH3)2), 103.0 (CH=), 95.3 (C-1′), 88.5 (C-4′), 85.5 (C-2′), 84.7 (C-3′), 80.3 (C-4), 80.2 (OCH), 80.0 (OCH), 77.4 (C-3), 72.9 (C-7), 66.1 (C-8), 60.9 (C-2), 51.4 (NHCH2), 36.1 (SCH2), 35.7 (SCH2), 34.9 (C-5), 27.6 (CH3), 27.5 (2C, 2 × CH3), 25.5 (CH3), 23.0 (C-1), 8.9 (C-6). HRMS (ESI) m/z Calcd for (M − CH3COO−) C27H42N3O11S: 616.2534. Found: 616.2525.
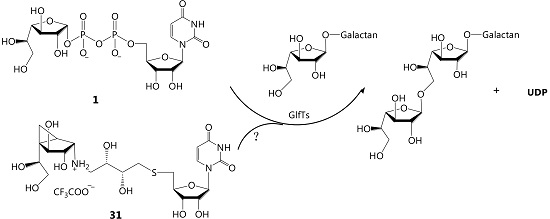
(1R,2S,3S,4S,5S)-5-(1,2-Dihydroxyethyl)-N-((2S,3R)-4-(2’,3’-O-isopropylidene-5’-thiouridin)-2,3-dihydroxybutyl)-3,4-dihydroxybicyclo[3.1.0]hexan-2-ammonium 2,2,2-trifluoroacetate (31). Compound 37 (10 mg, 0.015 mmol) was dissolved in TFA (2 mL) at 0 °C and the solution was then stirred at rt for 12 h and concentrated. The residue was purified by chromatography on Iatrobeads (CH2Cl2–CH3OH 10:1→1:3) to give the 31 (3 mg, 33%) as a white foam. Rf 0.3 (CH3OH–NH4OH 20:1); +23.2 (c 0.34, CH3OH); 1H-NMR (400 MHz, D2O, δH) 7.73 (d, 1H, J = 8.1 Hz, CH=), 5.90 (d, 1H, J = 8.1 Hz, CH=), 5.86 (d, 1H, J = 4.2 Hz, H-1′), 4.40 (dd, 1H, J = 4.2, 4.6 Hz, H-2′), 4.34 (d, 1H, J = 7.2 Hz, H-4), 4.20–4.18 (m, 2H, H-3′, H-4′), 4.03 (ddd, 1H, J = 3.1, 3.1, 9.0 Hz, H-c), 3.94–3.91 (m, 2H, H-3, H-7), 3.81–3.75 (m, 2H, H-b, H-8), 3.57 (d, 1H, J = 6.4 Hz, H-2), 3.50 (dd, 1H, J = 7.7, 11.6 Hz, H-8), 3.26 (dd, 1H, J = 3.1, 12.7 Hz, H-d), 3.13–3.02 (m, 2H, H-d, H-5′), 2.95 (dd, 1H, J = 6.7, 14.2 Hz, H-5’), 2.86 (dd, 1H, J = 5.0, 13.8 Hz, H-a), 2.78 (dd, 1H, J = 8.2, 13.8 Hz, H-a), 1.71 (dd, 1H, J = 4.1, 8.9 Hz, H-5), 0.92 (dd, 1H, J = 5.6, 8.9 Hz, H-6), 0.84 (dd, 1H, J = 4.1, 5.6 Hz, H-6); 13C-NMR (100 MHz, D2O, δC) 166.6 (C=O), 151.9 (C=O), 142.4 (CH=), 102.7 (CH=), 90.5 (C-1′),83.1(C-4′), 77.9 (C-2′), 75.1 (C-3′), 73.3, 72.2, 71.7, 71.3, 63.9, 59.5 (C-2), 49.4 (C-d), 35.3 (C-5), 34.0 (SCH2), 33.1 (SCH2), 23.5 (C-1), 8.5 (C-6); HRMS (ESI) m/z Calcd for (M − CF3COO−) C21H34N3O11S: 536.1909. Found: 536.1906.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}