
In Vitro Inhibition of Human UDP-Glucuronosyl-Transferase (UGT) Isoforms by Astaxanthin, β-Cryptoxanthin, Canthaxanthin, Lutein, and Zeaxanthin: Prediction of in Vivo Dietary Supplement-Drug Interactions



Abstract
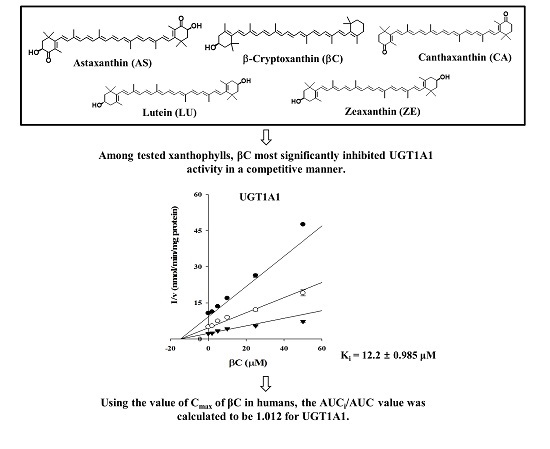
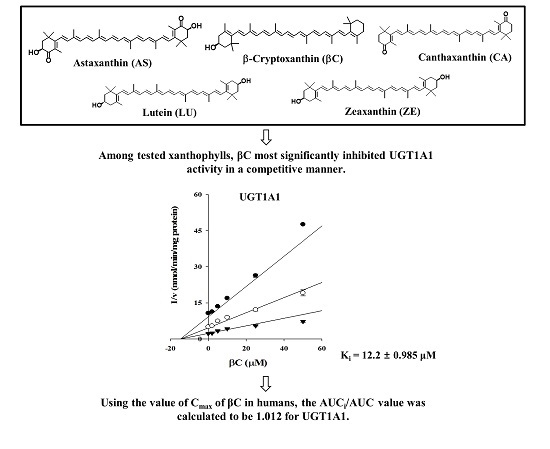
:
1. Introduction
2. Results
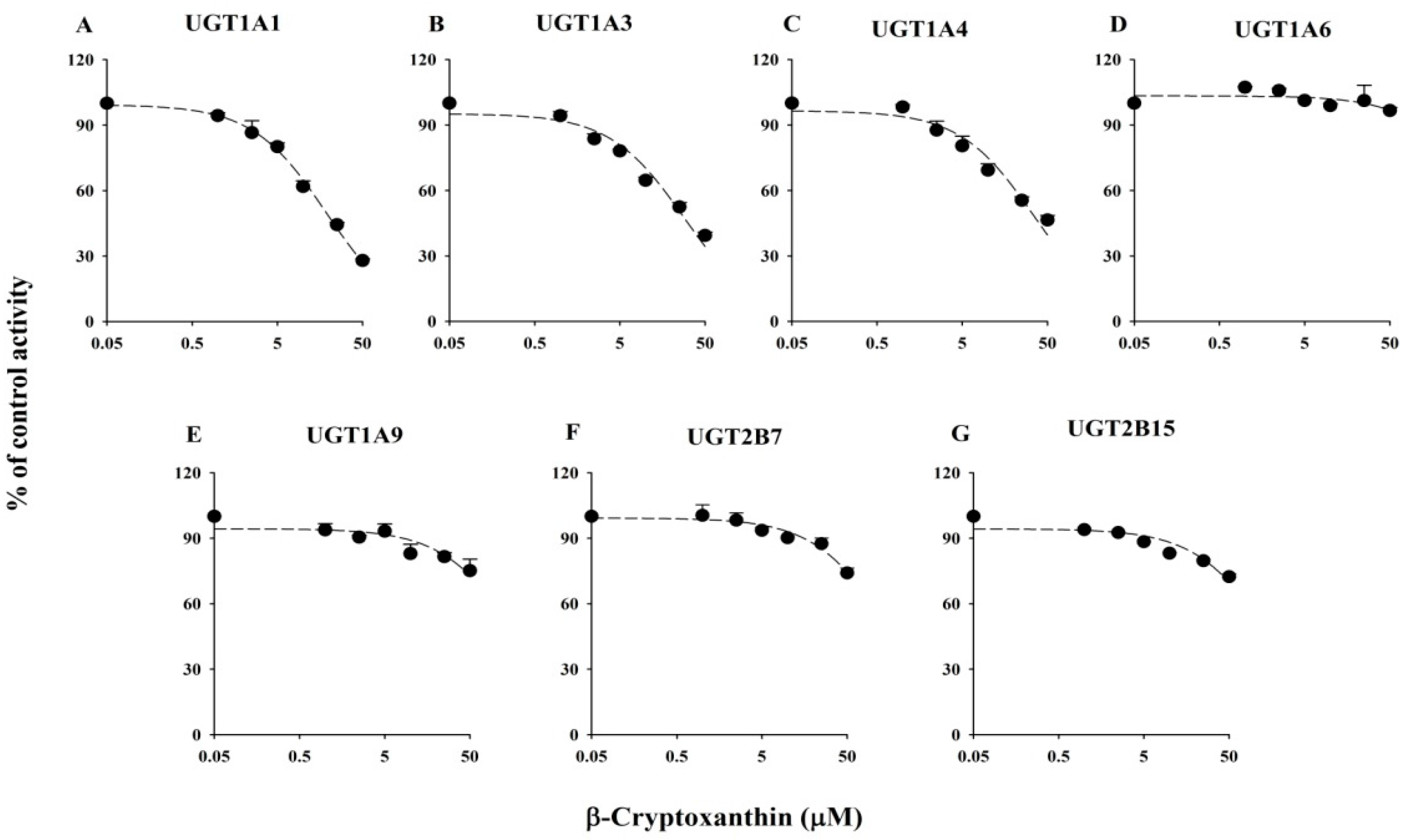
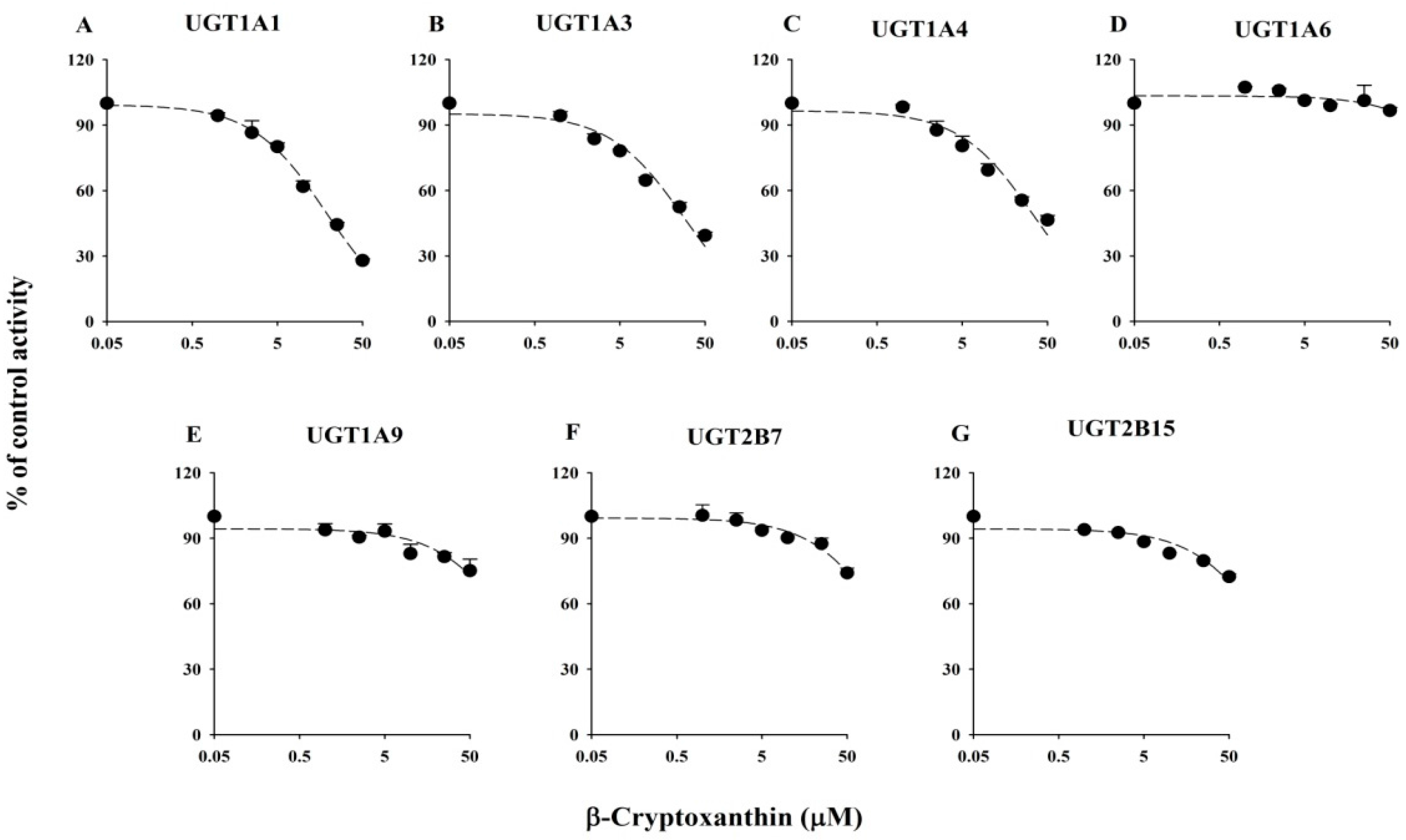
2.1. Inhibitory Effects of Five Xanthophylls on the Major UGT Isoforms in Human Liver Microsomes
2.2. Inhibitory Effects of Five Xanthophylls of UGT2B15 Activity
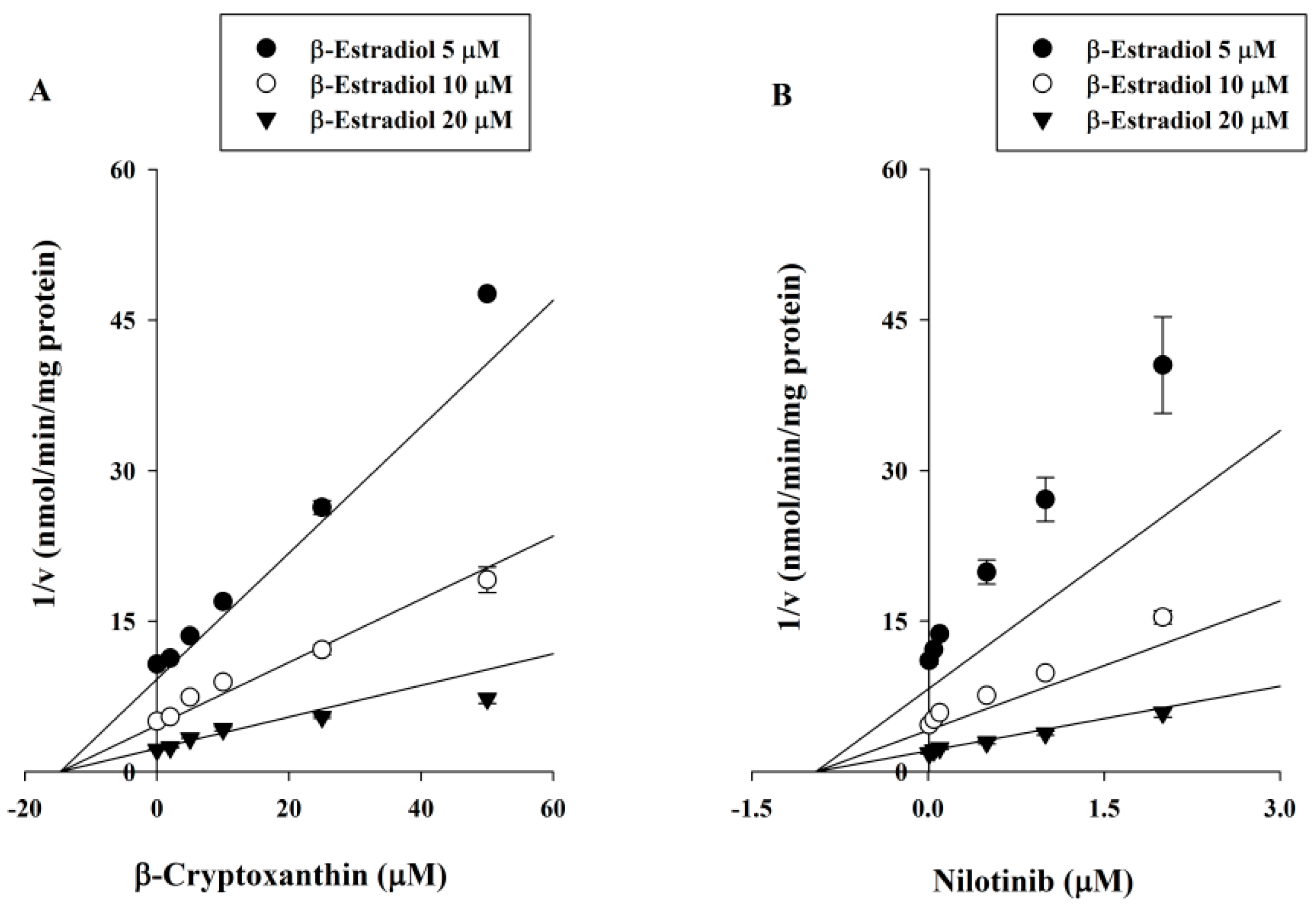
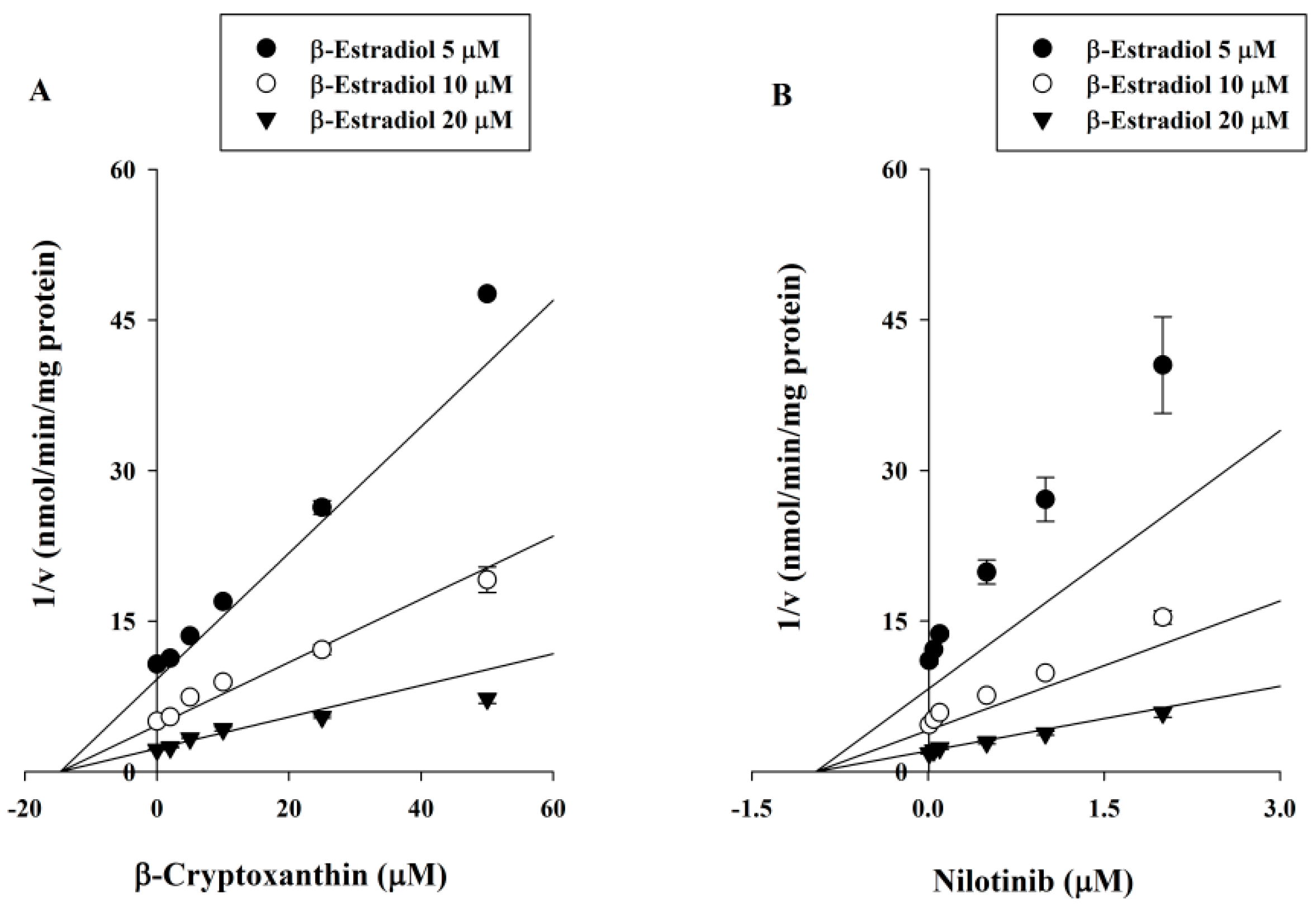
2.3. Ki Determination of βC on the UGT1A1 Activity
3. Discussion
4. Experimental Section
4.1. Materials and Reagents
4.2. Inhibitory Effects of Five Xanthophylls on the Major UGT Isoforms in Human Liver Microsomes
4.3. Inhibitory Effects of Five Xanthophylls of UGT2B15 Activity
4.4. Ki Determination of βC on the UGT1A1 Activity
4.5. LC-MS/MS Analysis
4.6. Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| UGT | UDP-glucuronosyltransferase |
| AS | astaxanthin |
| βC | β-cryptoxanthin |
| CA | canthaxanthin |
| LU | lutein |
| ZE | zeaxanthin |
| IC50 | the 50% inhibitory concentration |
| Ki | inhibitory constant |
| LC-MS/MS | liquid chromatography/tandem mass spectrometry |
| Cmax | the maximum plasma concentration |
References
- Kotake-Nara, E.; Nagao, A. Absorption and metabolism of xanthophylls. Mar. Drugs 2011, 9, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Jaswir, I.; Noviendri, D.; Hasrini, R.F.; Octavianti, F. Carotenoids: Sources, medicinal properties and their application in food and nutraceutical industry. J. Med. Plants Res. 2011, 5, 7119–7131. [Google Scholar]
- Miller, N.J.; Sampson, J.; Candeias, L.P.; Bramley, P.M.; Rice-Evans, C.A. Antioxidant activities of carotenes and xanthophylls. FEBS Lett. 1996, 384, 240–242. [Google Scholar] [CrossRef]
- Tanaka, T.; Shnimizu, M.; Moriwaki, H. Cancer chemoprevention by carotenoids. Molecules 2012, 17, 3202–3042. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Ciapara, I.; Felix-Valenzuela, L.; Goycoolea, F.M. Astaxanthin: A review of its chemistry and applications. Crit. Rev. Food Sci. Nutr. 2006, 46, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Yoshida, H.; Kondo, K. Potential anti-atherosclerotic properties of astaxanthin. Mar. Drugs 2016, 14, E35. [Google Scholar] [CrossRef] [PubMed]
- Breithaupt, D.E.; Weller, P.; Wolters, M.; Hahn, A. Plasma response to a single dose of dietary beta-cryptoxanthin esters from papaya (Carica papaya L.) or non-esterified beta-cryptoxanthin in adult human subjects: A comparative study. Br. J. Nutr. 2003, 90, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Burri, B.J.; La Frano, M.R.; Zhu, C. Absorption, metabolism, and functions of beta-cryptoxanthin. Nutr. Rev. 2016, 74, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, C.J.; Eto, I.; Juliana, M.M.; Whitaker, L.M. Effect of canthaxanthin on chemically induced mammary carcinogenesis. Oncology 1991, 48, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Palozza, P.; Maggiano, N.; Calviello, G.; Lanza, P.; Piccioni, E.; Ranelletti, F.O.; Bartoli, G.M. Canthaxanthin induces apoptosis in human cancer cell lines. Carcinogenesis 1998, 19, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yan, S.F.; Huang, Y.M.; Lu, X.R.; Qian, F.; Pang, H.L.; Xu, X.R.; Zou, Z.Y.; Dong, P.C.; Xiao, X.; et al. Effect of lutein and zeaxanthin on macular pigment and visual function in patients with early age-related macular degeneration. Ophthalmology 2012, 119, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Scripsema, N.K.; Hu, D.N.; Rosen, R.B. Lutein, zeaxanthin, and meso-zeaxanthin in the clinical management of eye disease. J. Ophthalmol. 2015, 2015, 865179. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Keunen, J.E.; Bird, A.C.; van Kuijk, F.J. Fruits and vegetables that are sources for lutein and zeaxanthin: The macular pigment in human eyes. Br. J. Ophthalmol. 1998, 82, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sweet, B.V. Lutein and zeaxanthin for macular degeneration. Am. J. Health Syst. Pharm. 2008, 65, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Lesko, L.J. Drug-drug, drug-dietary supplement, and drug-citrus fruit and other food interactions: What have we learned? J. Clin. Pharmacol. 2004, 44, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.E.; Frye, R.F. Effects of herbal supplements on drug glucuronidation. Review of clinical, animal, and in vitro studies. Planta Med. 2011, 77, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Nisly, N.L.; Zimmerman, M.B.; Gryzlak, B.M.; Wallace, R.B. Use of herbs among adults based on evidence-based indications: Findings from the National Health Interview Survey. Mayo Clin. Proc. 2007, 82, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.W.; Kelly, J.P.; Rosenberg, L.; Anderson, T.E.; Mitchell, A.A. Recent patterns of medication use in the ambulatory adult population of the United States: The Slone survey. JAMA 2002, 287, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Klotz, U. Drug Interactions with herbal medicines. Clin. Pharmacokinet. 2012, 51, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Gradelet, S.; Astorg, P.; Leclerc, J.; Chevalier, J.; Vernevaut, M.F.; Siess, M.H. Effects of canthaxanthin, astaxanthin, lycopene and lutein on liver xenobiotic-metabolizing enzymes in the rat. Xenobiotica 1996, 26, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Kistler, A.; Liechti, H.; Pichard, L.; Wolz, E.; Oesterhelt, G.; Hayes, A.; Maurel, P. Metabolism and CYP-inducer properties of astaxanthin in man and primary human hepatocytes. Arch. Toxicol. 2002, 75, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.F.; Bae, S.H.; Kwon, M.J.; Park, J.B.; Choi, H.D.; Shin, W.G.; Bae, S.K. Inhibitory effects of astaxanthin, beta-cryptoxanthin, canthaxanthin, lutein, and zeaxanthin on cytochrome P450 enzyme activities. Food Chem. Toxicol. 2013, 59, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Ther. 2005, 106, 97–132. [Google Scholar] [CrossRef] [PubMed]
- Ritter, J.K. Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem. Biol. Interact. 2000, 129, 171–193. [Google Scholar] [CrossRef]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef] [PubMed]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Mackenzie, P.I.; Knights, K.M. The prediction of drug-glucuronidation parameters in humans: UDP-glucuronosyltransferase enzyme-selective substrate and inhibitor probes for reaction phenotyping and in vitro-in vivo extrapolation of drug clearance and drug-drug interaction potential. Drug Metab. Rev. 2010, 42, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Mackenzie, P.I.; Elliot, D.J.; Miners, J.O. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2006, 34, 449–456. [Google Scholar] [PubMed]
- Ouzzine, M.; Barré, L.; Netter, P.; Magdalou, J.; Fournel-Gigleux, S. The human UDP-glucuronosyltransferases: Structural aspects and drug glucuronidation. Drug Metab. Rev. 2003, 35, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, J.B.; Kim, D.; Bae, S.H.; Chin, Y.W.; Oh, E.; Bae, S.K. In vitro selective inhibition of human UDP-glucuronosyltransferase (UGT) 1A4 by finasteride, and prediction of in vivo drug-drug interactions. Toxicol. Lett. 2015, 232, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H. Isoform-selective probe substrates for in vitro studies of human UDP-glucuronosyltransferases. Meth. Enzymol. 2005, 400, 104–116. [Google Scholar] [PubMed]
- Uchaipichat, V.; Mackenzie, P.I.; Guo, X.H.; Gardner-Stephen, D.; Galetin, A.; Houston, J.B.; Miners, J.O. Human UDP-glucuronosyltransferases: Isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab. Dispos. 2004, 32, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [Google Scholar] [PubMed]
- Wen, Z.; Tallman, M.N.; Ali, S.Y.; Smith, P.C. UDP-glucuronosyltransferase 1A1 is the principal enzyme responsible for etoposide glucuronidation in human liver and intestinal microsomes: Structural characterization of phenolic and alcoholic glucuronides of etoposide and estimation of enzyme kinetics. Drug Metab. Dispos. 2007, 35, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Winner, L.K.; Mackenzie, P.I.; Elliot, D.J.; Williams, J.A.; Miners, J.O. Quantitative prediction of in vivo inhibitory interactions involving glucuronidated drugs from in vitro data: The effect of fluconazole on zidovudine glucuronidation. Br. J. Clin. Pharmacol. 2006, 61, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Gaganis, P.; Elliot, D.J.; Mackenzie, P.I.; Knights, K.M.; Miners, J.O. Binding of inhibitory fatty acids is responsible for the enhancement of UDP glucuronosyltransferase 2B7 activity by albumin: Implications for in vitro-in vivo extrapolation. J. Pharmacol. Exp. Ther. 2007, 321, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Park, J.B.; Yoon, K.D.; Bae, S.K. Evaluation of the in vitro/in vivo potential of five berries (bilberry, blueberry, cranberry, elderberry, and raspberry ketones) commonly used as herbal supplements to inhibit uridine diphospho-glucuronosyltransferase. Food Chem. Toxicol. 2014, 72, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Zhu, L.; Yang, L.; Ge, G.; Cao, Y.; Liu, Y.; Fang, Z.; Zhang, Y. Selectivity for inhibition of nilotinib on the catalytic activity of human UDP-glucuronosyltransferases. Xenobiotica 2014, 44, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, A.S.; Caron, P.; Harvey, M.; Zimmerman, P.A.; Mehlotra, R.K.; Guillelmette, C. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine. Drug Metab. Dispos. 2009, 37, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Trottier, J.; Verreault, M.; Grepper, S.; Monté, D.; Bélanger, J.; Kaeding, J.; Caron, P.; Inaba, T.T.; Barbier, O. Human UDP-glucuronosyltransferase (UGT)1A3 enzyme conjugates chenodeoxycholic acid in the liver. Hepatology 2006, 44, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cao, Y.F.; Fang, Z.Z.; Zhang, Y.Y.; Hu, C.M.; Sun, X.Y.; Huang, T.; Zeng, J.; Fan, X.R.; Mo, H. Strong inhibition of deoxyschizandrin and schisantherin A toward UDP-glucuronosyltransferase (UGT) 1A3 indicating UGT inhibition-based herb-drug interaction. Fitoterapia 2012, 83, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Bowalgaha, K.; Elliot, D.J.; Baranczewski, P.; Knights, K.M. Characterization of niflumic acid as a selective inhibitor of human liver microsomal UDP-glucuronosyltransferase 1A9: Application to the reaction phenotyping of acetaminophen glucuronidation. Drug Metab. Dispos. 2011, 39, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Duan, S.X.; Guillemette, C.; Journault, K.; Krishnaswamy, S.; von Moltke, L.L.; Greenblatt, D.J. Stereoselective conjugation of oxazepam by human UDP-glucuronosyltransferases (UGTS): S-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug Metab. Dispos. 2002, 30, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.

{kind=link}
{kind=link}
{kind=link}
| UGTs | IC50 Values (μM) | ||||||
|---|---|---|---|---|---|---|---|
| Known Potent Inhibitors | AS | βC | CA | LU | ZE | ||
| UGT1A1 | Nilotinib | 1.55 ± 0.118 | >50 1 | 18.8 ± 2.07 | 38.5 ± 4.65 | 45.5 ± 4.01 | >50 |
| UGT1A3 | Deoxyschizandrin | 5.17 ± 0.700 | >50 | 28.3 ± 4.40 | 41.2 ± 3.14 | >50 | >50 |
| UGT1A4 | Hecogenin | 8.07 ± 2.60 | >50 | 34.9 ± 5.98 | >50 | 28.7 ± 3.79 | >50 |
| UGT1A6 | Diclofenac | 106 ± 26.6 | >50 | >50 | >50 | >50 | >50 |
| UGT1A9 | Niflumic acid | 0.390 ± 0.0256 | >50 | >50 | >50 | >50 | >50 |
| UGT2B7 | Efavirenz | 37.8 ± 2.37 | >50 | >50 | >50 | >50 | >50 |
| UGT2B15 2 | Amitriptyline | 69.4 ± 10.6 | >50 | >50 | >50 | >50 | >50 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.F.; Min, J.S.; Kim, D.; Park, J.B.; Choi, S.-W.; Lee, E.S.; Na, K.; Bae, S.K. In Vitro Inhibition of Human UDP-Glucuronosyl-Transferase (UGT) Isoforms by Astaxanthin, β-Cryptoxanthin, Canthaxanthin, Lutein, and Zeaxanthin: Prediction of in Vivo Dietary Supplement-Drug Interactions. Molecules 2016, 21, 1052. https://doi.org/10.3390/molecules21081052
Zheng YF, Min JS, Kim D, Park JB, Choi S-W, Lee ES, Na K, Bae SK. In Vitro Inhibition of Human UDP-Glucuronosyl-Transferase (UGT) Isoforms by Astaxanthin, β-Cryptoxanthin, Canthaxanthin, Lutein, and Zeaxanthin: Prediction of in Vivo Dietary Supplement-Drug Interactions. Molecules. 2016; 21(8):1052. https://doi.org/10.3390/molecules21081052
Chicago/Turabian StyleZheng, Yu Fen, Jee Sun Min, Doyun Kim, Jung Bae Park, Sung-Wook Choi, Eun Seong Lee, Kun Na, and Soo Kyung Bae. 2016. "In Vitro Inhibition of Human UDP-Glucuronosyl-Transferase (UGT) Isoforms by Astaxanthin, β-Cryptoxanthin, Canthaxanthin, Lutein, and Zeaxanthin: Prediction of in Vivo Dietary Supplement-Drug Interactions" Molecules 21, no. 8: 1052. https://doi.org/10.3390/molecules21081052
APA StyleZheng, Y. F., Min, J. S., Kim, D., Park, J. B., Choi, S.-W., Lee, E. S., Na, K., & Bae, S. K. (2016). In Vitro Inhibition of Human UDP-Glucuronosyl-Transferase (UGT) Isoforms by Astaxanthin, β-Cryptoxanthin, Canthaxanthin, Lutein, and Zeaxanthin: Prediction of in Vivo Dietary Supplement-Drug Interactions. Molecules, 21(8), 1052. https://doi.org/10.3390/molecules21081052