
Efficient MW-Assisted Synthesis, Spectroscopic Characterization, X-ray and Antioxidant Properties of Indazole Derivatives

 ,
,  ,
,  , and
, and
Abstract
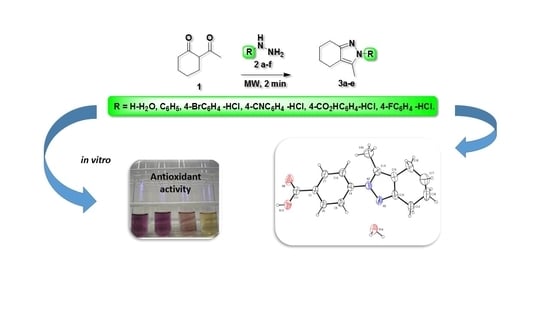
:
1. Introduction
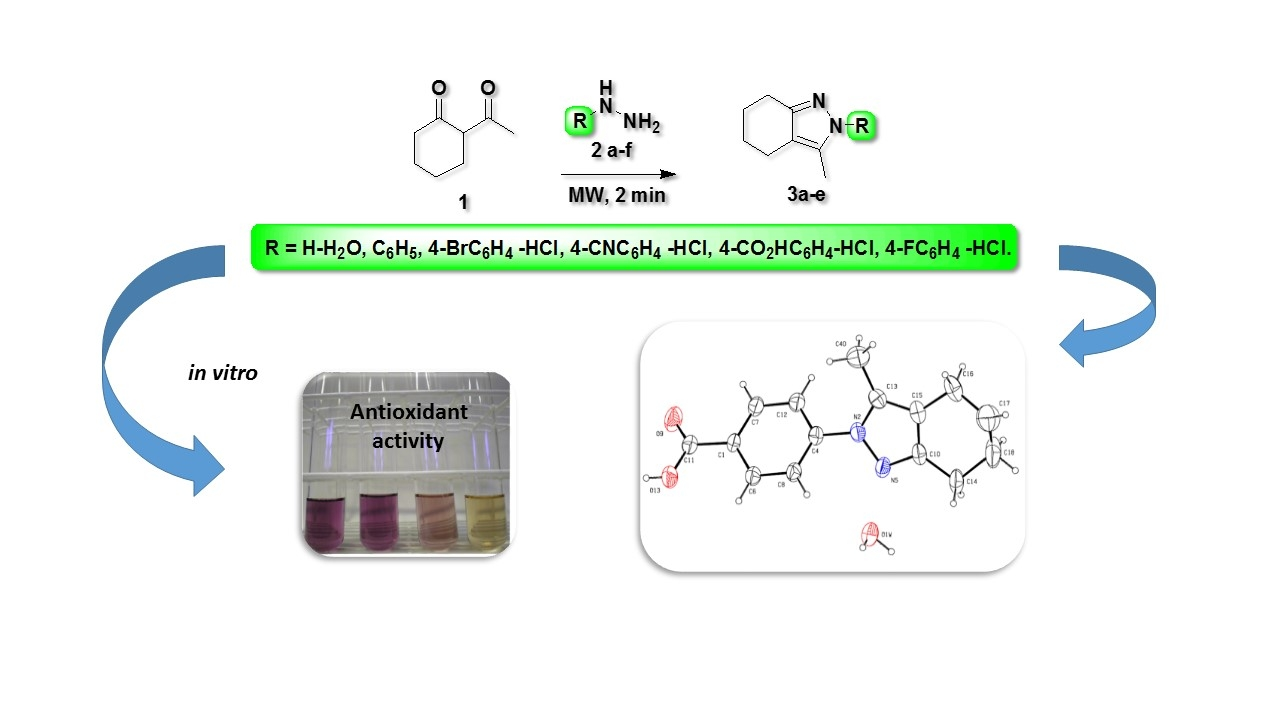
2. Results and Discussions
3. Materials and Methods
3.1. General Information
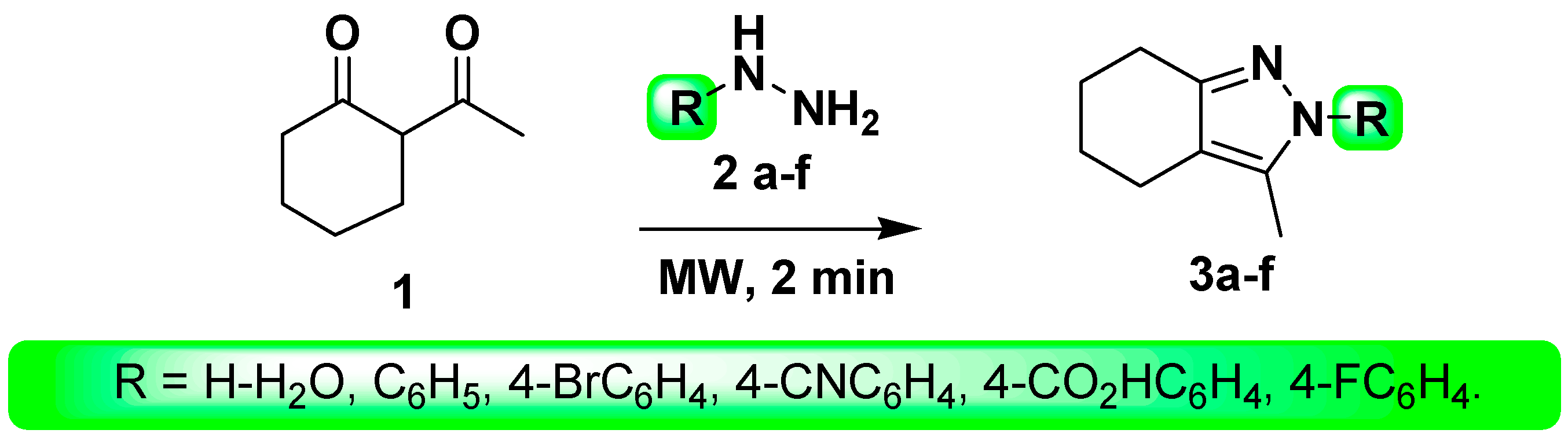
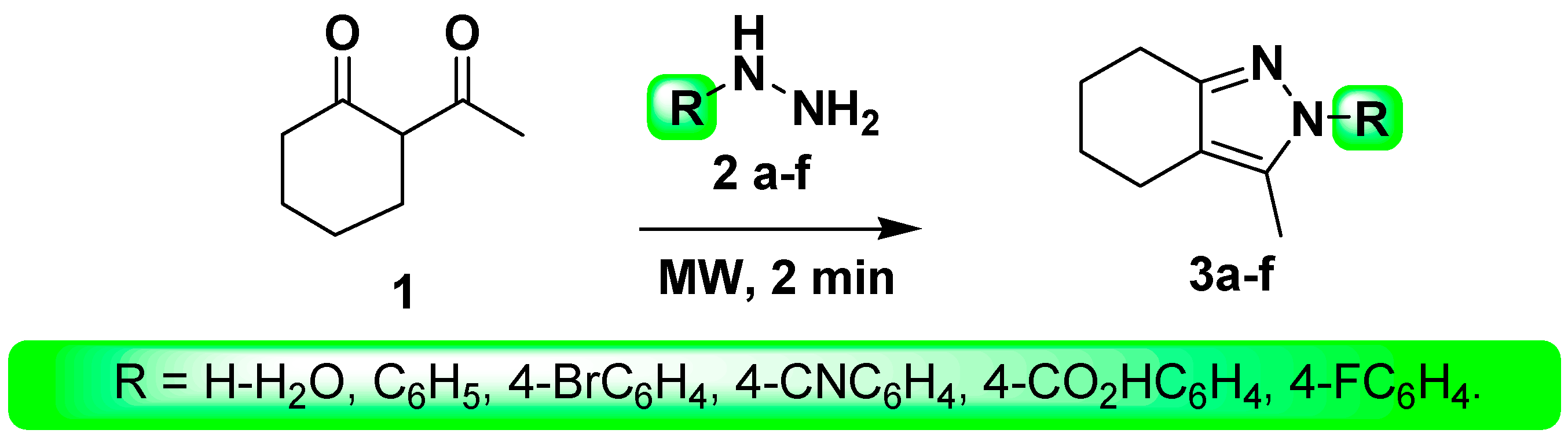
3.2. Synthesis
- Method A (Reflux)
- (a)
- A mixture of 2-acetylcyclohexanone (1, 1 mmol), and hydrazines 2 (1.0 mmol) in DMF (10 mL) was heated under reflux for 6–8 h. The precipitated solid product was filtered, washed with ethanol (EtOH), dried and finally recrystallized from DMF.
- (b)
- 2-Acetylcyclohexanone (1, 1 mmol) and hydrazines 2 (1.0 mmol) in AcOH (10 mL) were stirred at 80 °C for the given times. After completion of the reaction, the mixture was cooled to room temperature and the precipitate was filtered and purified by recrystallization from EtOH.
- Method B (MW)
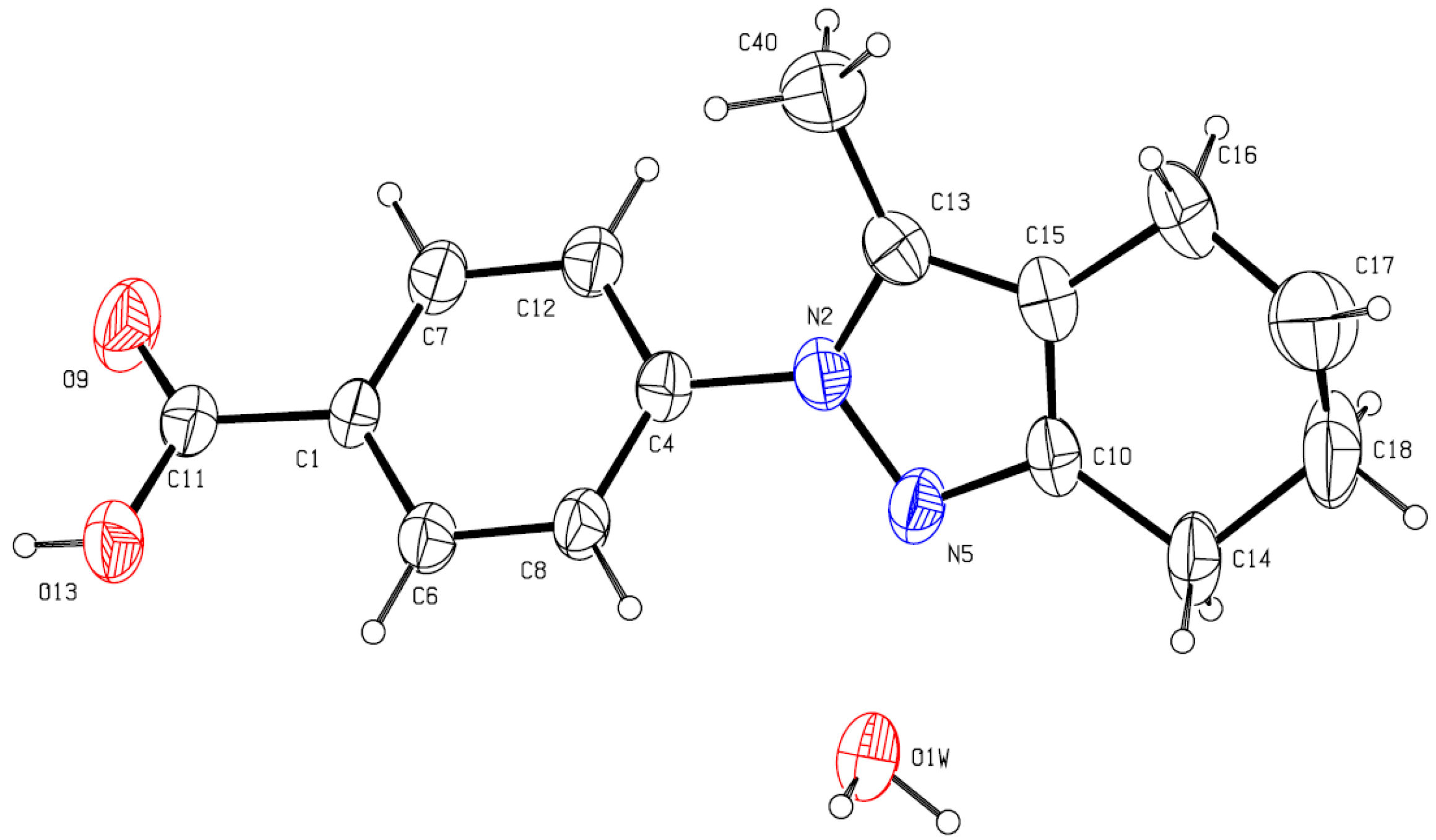
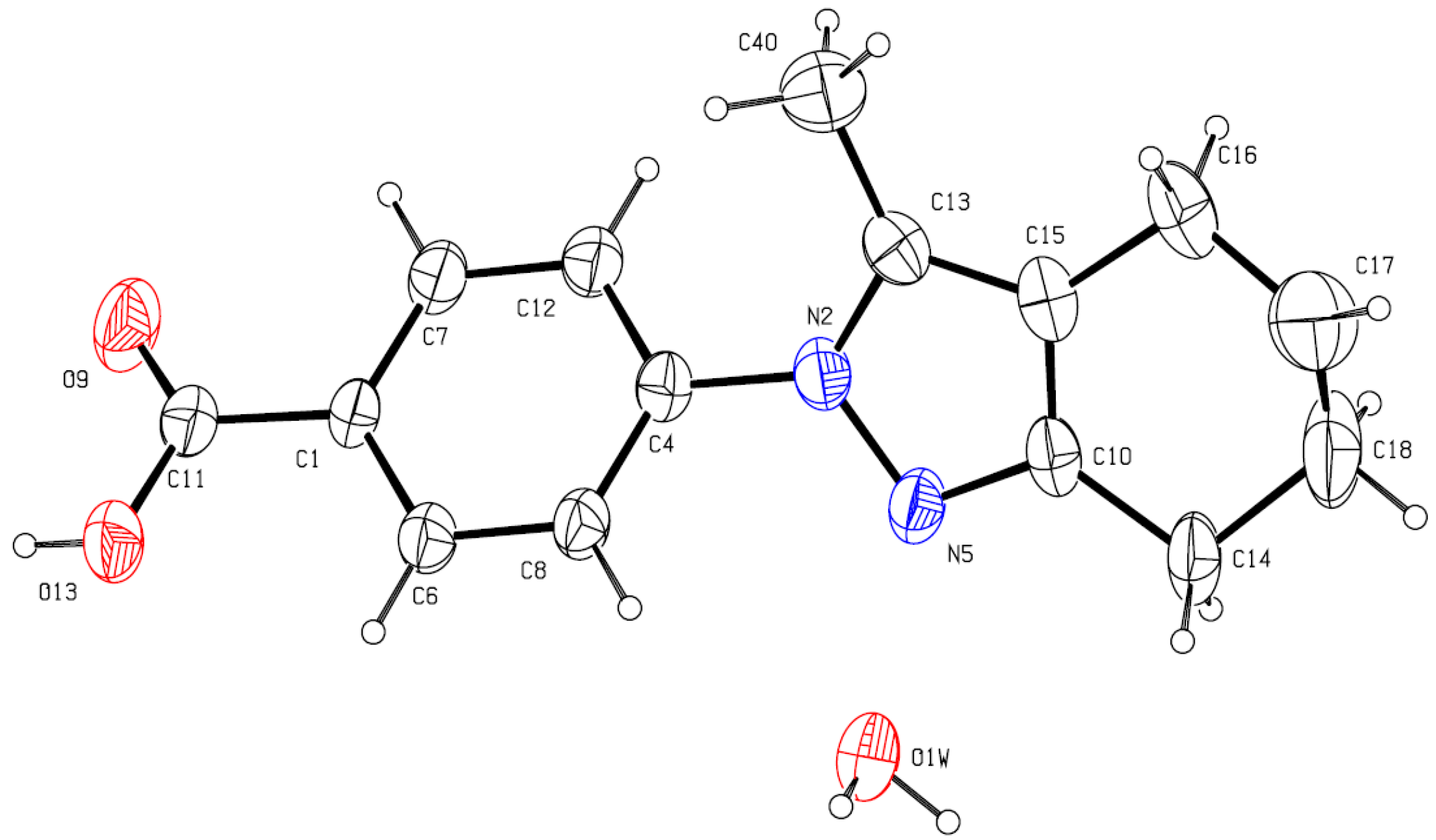
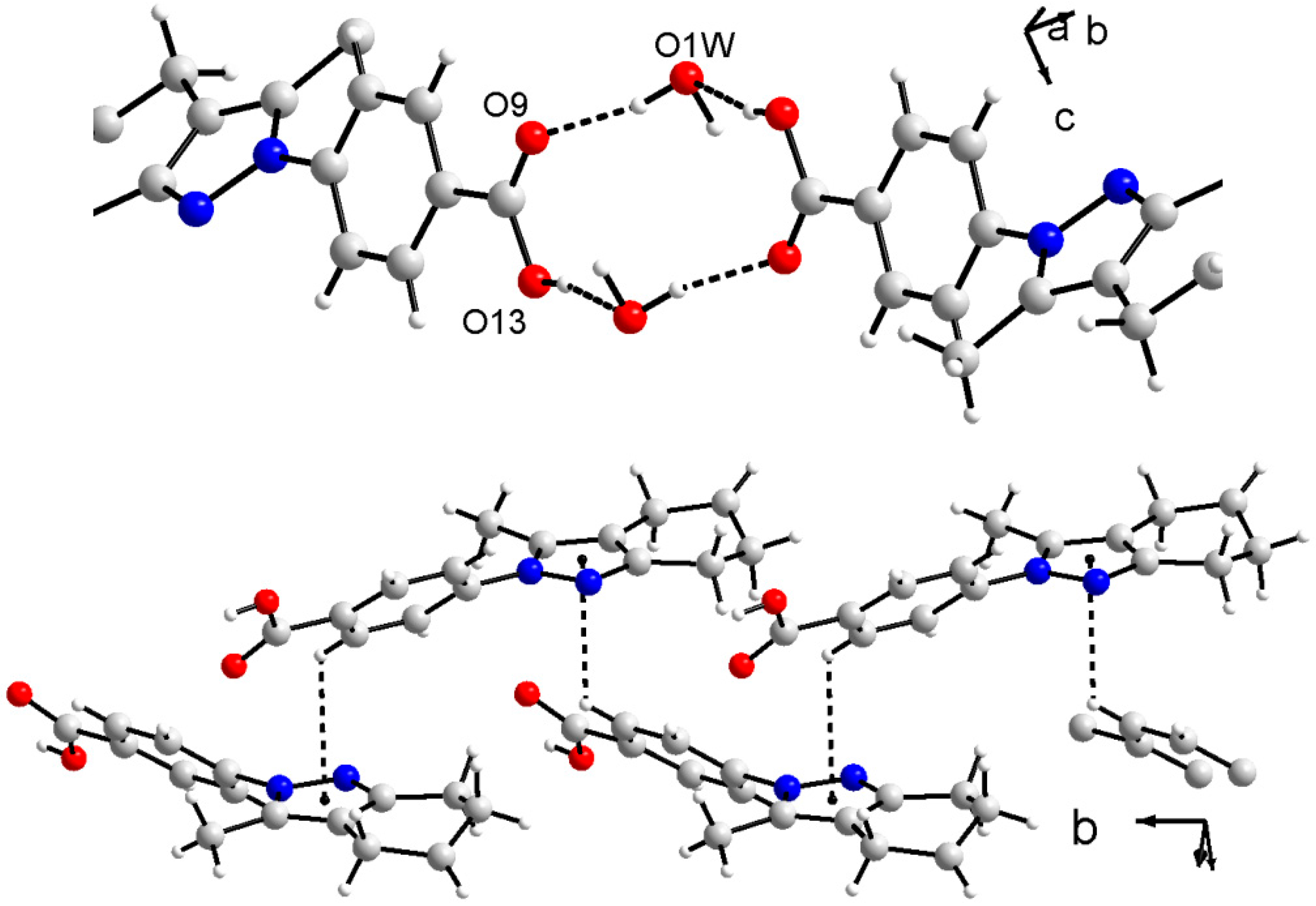
3.3. X-ray Crystallography
3.4. Biological Activity
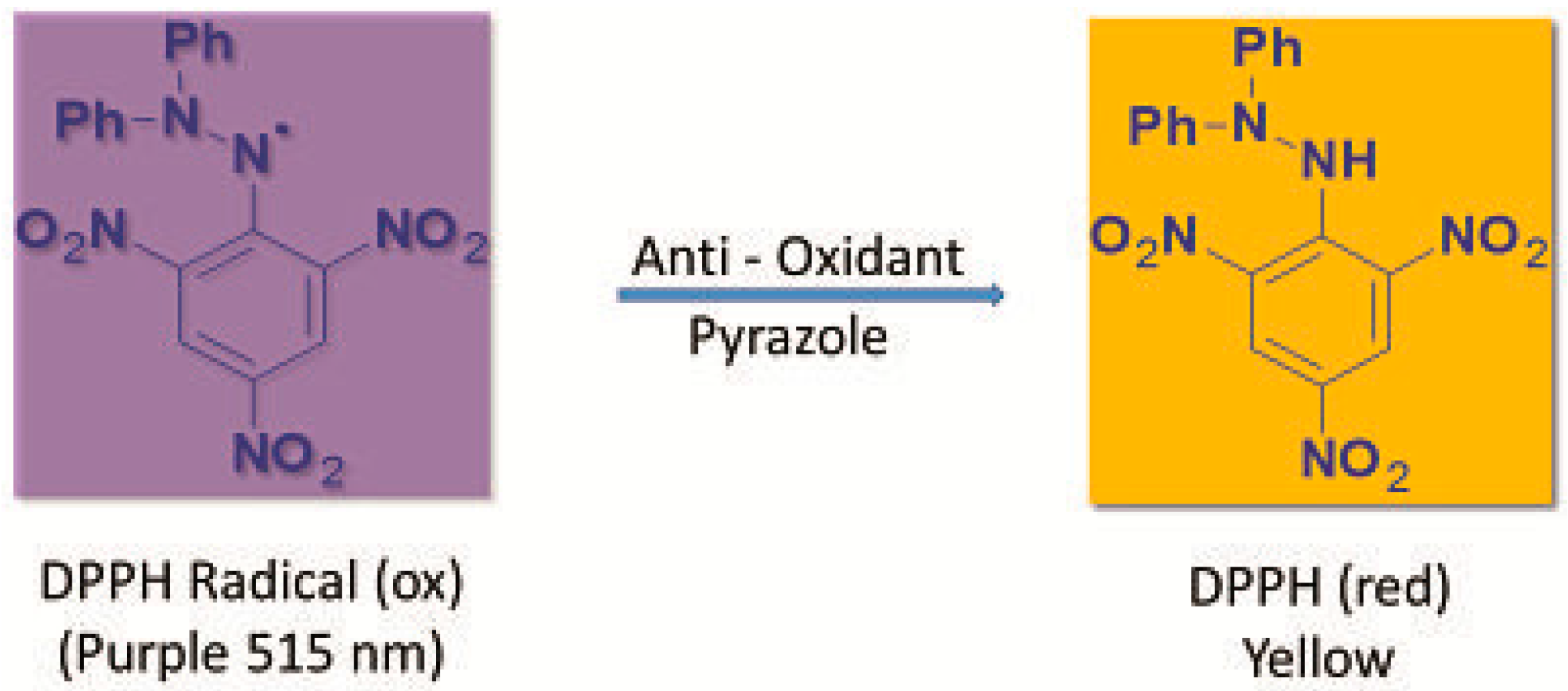
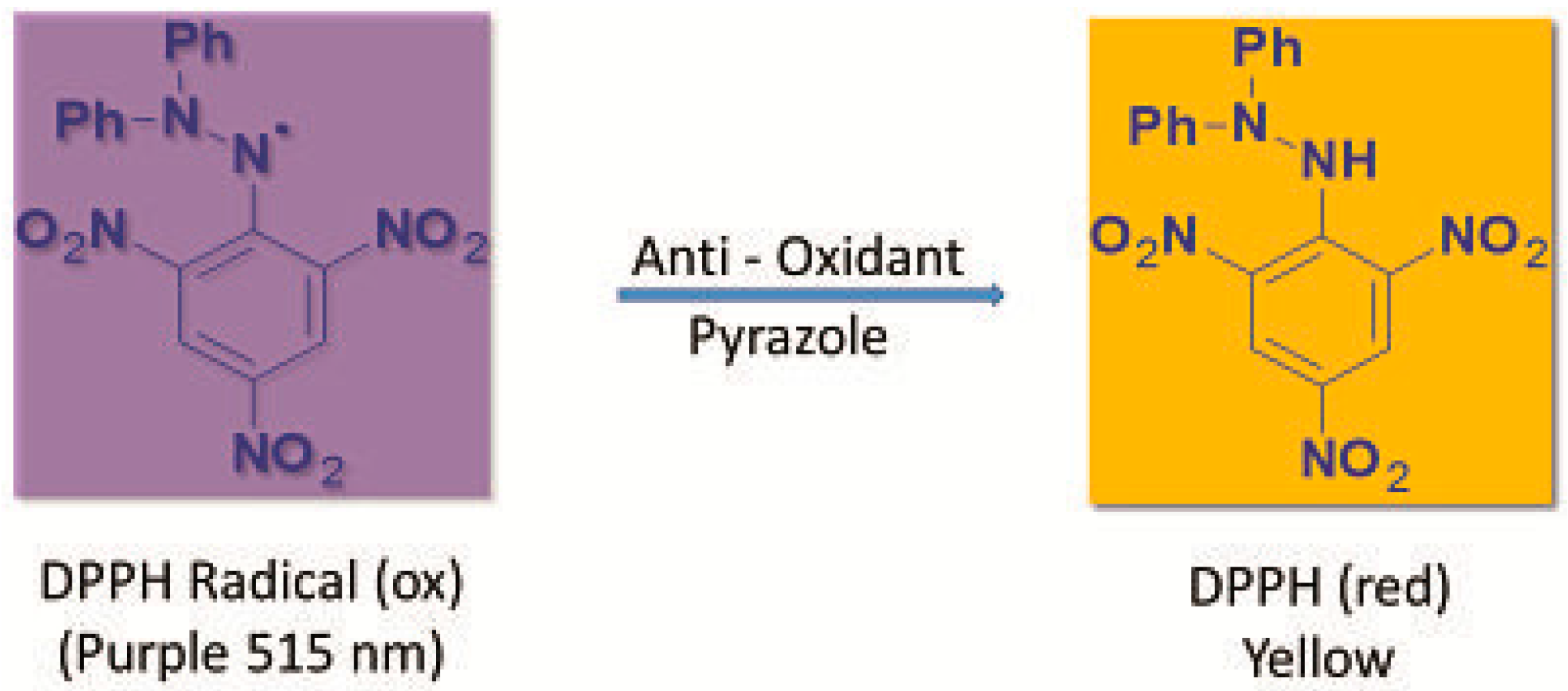
3.4.1. Measurement of DPPH Radical Scavenging Activity
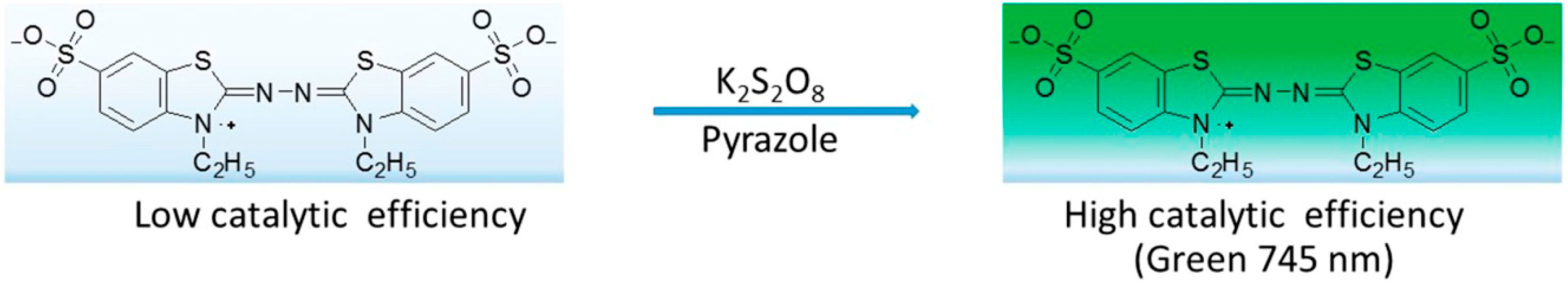
3.4.2. Measurement of ABTS Radical Scavenging Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Surati, M.A.; Jauhari, S.; Desai, K.R. A brief review: Microwave assisted organic reaction. Arch. Appl. Sci. Res. 2012, 4, 645–661. [Google Scholar]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis—A review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Wathey, B.; Tierney, J.; Lidström, P.; Westman, J. The impact of microwave-assisted organic chemistry on drug discovery. Drug Discov. Today 2002, 6, 373–380. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Dallinger, D. Controlled microwave heating in modern organic synthesis: Highlights from the 2004–2008 literature. Mol. Divers. 2009, 13, 71–193. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Bougrin, K.; Loupy, A.; Soufiaoui, M. Microwave-assisted solvent-free heterocyclic synthesis. J. Photochem. Photobiol. C 2005, 6, 139–167. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Singh, S. Microwave-assisted heterocyclic synthesis. ARKIVOC 2003, 13, 68–86. [Google Scholar]
- Driowya, M.; Saber, A.; Marzag, H.; Demange, L.; Benhida, R.; Bougrin, K. Review microwave-assisted synthesis of bioactive six-membered heterocycles and their fused analogues. Molecules 2016, 21, 492–547. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Silva, A.; Pinto, D.; Cavaleiro, J. Efficient microwave-assisted synthesis of tetrahydroindazoles and their oxidation to indazoles. Synlett 2006, 9, 1369–1373. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S.; Pinto, D.C.G.A.; Elguero, J.; Cavaleiro, J.A.S. Synthesis of new 1H-indazoles through Diels–Alder transformations of 4-styrylpyrazoles under microwave irradiation conditions. Eur. J. Org. Chem. 2009, 26, 4468–4479. [Google Scholar] [CrossRef]
- Scala, A.; Piperno, A.; Risitano, F.; Cirmi, S.; Navarra, M.; Grassi, G. Efficient synthesis of highly substituted tetrahydroindazolone derivatives. Mol. Divers. 2015, 19, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Burch, J.D.; Lau, K.; Barker, J.J.; Brookfield, F.; Chen, Y.; Chen, Y.; Eigenbrot, C.; Ellebrandt, C.; Ismaili, M.H.A.; Johnson, A.; et al. Property- and structure-guided discovery of a tetrahydroindazole series of interleukin-2 inducible T-Cell kinase inhibitors. J. Med. Chem. 2014, 57, 5714–5727. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.H.; Veal, J.M.; Fadden, R.P.; Rice, J.W.; Eaves, J.; Strachan, J.-P.; Barabasz, A.F.; Foley, B.E.; Barta, T.E.; Ma, W.; et al. Discovery of novel 2-Aminobenzamide inhibitors of heat shock protein 90 as potent, selective and orally active antitumor agents. J. Med. Chem. 2009, 52, 4288–4305. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.E.; Harries, M.; Aldegheri, L.; Austin, N.E.; Ballantine, S.; Ballini, E.; Bradley, D.M.; Bax, B.D.; Clarke, B.P.; Harris, A.J.; et al. Integration of lead optimization with crystallography for a membrane-bound ion channel target: discovery of a new class of AMPA receptor positive allosteric modulators. J. Med. Chem. 2011, 54, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Song, Y.; Huang, Q.; Yuan, H.; Wan, B.; Wang, Y.; He, R.; Beconi, M.G.; Franzblau, S.G.; Kozikowski, A.P. Identification, synthesis, and pharmacological evaluation of tetrahydroindazole based ligands as novel antituberculosis agents. J. Med. Chem. 2010, 53, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Chaudhary, R.P. Synthesis, structure and antimicrobial evaluation of new 3,3a,4,5-tetrahydro-2H-benzo[g]indazol-2-yl-thiazol-4(5H)-ones. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2015, 135, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, R.M.; López, C.; Pérez-Medina, C.; Pinilla, E.; Torres, M.R.; Elguero, J. Synthesis and structuralstudy of tetrahydroindazolones. Tetrahedron 2006, 62, 11704–11713. [Google Scholar] [CrossRef]
- Wiley, R.H.; Behr, L.C.; Fusco, R.; Jarboe, C.H. Indazoles and condensed types. In Chemistry of Heterocyclic Compounds; John Wiley&Sons, Inc.: New York, NY, USA, 1967; pp. 289–382. [Google Scholar]
- Ainsworth, C. Indazole. Org. Synth. 1959, 39, 27. [Google Scholar]
- Li, J.J. Paal–Knorr pyrrole synthesis. In Name Reactions—A Collection of Detailed Reaction Mechanisms, 1st ed.; Cham, Springer: Berlin, Germany, 2014; pp. 454–455. [Google Scholar]
- Fadeeva, Y.A.; Safonova, P.J. Dissociation Constant of Acetic Acid in (N,N-Dimethylformamide + Water) Mixtures at the Temperature 298.15 K. J. Solut. Chem. 2011, 40, 980–988. [Google Scholar] [CrossRef]
- Shah, J.J.; Mohanraj, K. Comparison of conventional and microwave-assisted synthesis of benzotriazole derivatives. Indian J. Pharm. Sci. 2014, 76, 46–53. [Google Scholar] [PubMed]
- Mistry, B.D.; Desai, K.R.; Patel, J.A.; Patel, N.I. Conventional and microwave-assisted synthesis of pyrazole derivatives and screening of their antibacterial and antifungal activities. Indian J. Chem. Sect. B 2012, 51, 746–751. [Google Scholar] [CrossRef]
- Allen, F.H.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Typical interatomic distances: Organic compounds. In International Tables for Crystallography; Prince, E., Ed.; Wiley: New York, NY, USA, 2006; Volume C, Chapter 9.5; pp. 790–811. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Portilla, J.; Quiroga, J.; Cobo, J.; Glidewell, C. 2-tert-Butyl-5-methyl-7,8-dihydro-6H-cyclopenta[e]pyrazolo[1,5-a]pyrimidine: Molecular stacks built from C-H—p(pyrazole) hydrogen bonds and [pi]-[pi] stacking interactions. Acta Crystallogr. Sect. C: Cryst. Struct. Commun. C 2008, 64, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Nunes, G.B.; Paola, R.; Policarpo, P.R.; Costa, L.M.; Da Silva, T.G.; Militão, G.C.; Câmara, C.A.; Barbosa Filho, J.M.; Gutierrez, S.J.; Islam, M.T.; et al. In vitro antioxidant and cytotoxic activity of some synthetic riparin-derived compounds. Molecules 2014, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Urbani, P.; Ramunno, A.; Filosa, R.; Pinto, A.; Popolo, A.; Bianchino, E.; Piotto, P.; Saturnino, C.; De Prisco, R.; Nicolaus, B.; et al. Antioxidant activity of diphenylpropionamide derivatives: Synthesis, biological evaluation and computational analysis. Molecules 2008, 13, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Thorat, I.D.; Jagtap, D.D.; Mohapatra, D.; Joshi, D.C.; Sutar, R.F.; Kapdi, S.S. Antioxidants, their properties, uses in food products and their legal implications. IJFS 2013, 2, 81–104. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Duthie, G.; Campbell, F.; Bestwick, C.; Stephen, S.; Russell, W. Antioxidant effectiveness of vegetable powders on the lipid and protein oxidative stability of cooked turkey meat patties: Implications for health. Nutrients 2013, 5, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.M.; Torres, B.G.; Spohr, P.R.; Machado, P.; Bonacorso, H.G.; Zanatta, N.; Martins, M.A.; Emanuelli, T. Antioxidant potential of new pyrazoline derivatives to prevent oxidative damage. Basic Clin. Pharmacol. Toxicol. 2008, 104, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Karrouchi, K.; Chemlal, L.; Doudach, L.; Taoufik, J.; Cherrah, Y.; Radi, S.; Faouzi, M.E.; Ansar, M.H. Synthesis, anti-inflammatory and antioxidant activities of some new pyrazole derivatives. J. Pharm. Res. 2014, 8, 1171–1177. [Google Scholar]
- Viveka, S.; Madhu, L.N.; Nagaraja, G.K. Synthesis of new pyrazole derivatives via multicomponent reaction and evaluation of their antimicrobial and antioxidant activities. Med. Chem. Res. 2014, 23, 2916–2929. [Google Scholar]
- Durgamma, S.; Muralikrishna, A.; Padmavathi, V.; Adivireddy Padmaja, A. Synthesis and antioxidant activity of amido-linked benzoxazolyl/benzothiazolyl/benzimidazolyl-pyrroles and pyrazoles. Med. Chem. Res. 2014, 23, 2916–2929. [Google Scholar] [CrossRef]
- Mubeen, M.; Kini, S.G.; Pai, K.S.R. Design, synthesis, antioxidant and anticancer activity of novel pyrazole derivatives. Der. Pharma. Chem. 2015, 7, 215–223. [Google Scholar]
- Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-catalyzed hydroamination of alkynes and allenes with parent hydrazine. Angew. Chem. Int. Ed. 2011, 50, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Rahmatzadeh, S.S.; Karami, B.; Khodabakhshi, S.J. A modified and practical synthetic route to indazoles and pyrazoles using tungstate sulfuric acid. Chin. Chem. Soc. 2015, 62, 17–20. [Google Scholar] [CrossRef]
- Curini, M.; Rosati, O.; Campagna, V.; Montanari, F.; Cravotto, G.; Boccalini, M. Layered Zirconium sulfophenyl phosphonate as heterogeneous catalyst in the synthesis of pyrazoles and 4,5,6,7-Tetrahydro-1(2)H-indazoles. Synlett 2005, 19, 2927–2930. [Google Scholar] [CrossRef]
- Bruker. SMART, SAINTPLUS and SADABS; Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. SHELXL-97; Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J. Sci. Technol. 2004, 26, 211–219. [Google Scholar]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yong, M.; Rice-Evas, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Rad. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3a–f are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | Reflux | Microwave (MW) | M.p. °C | ||
|---|---|---|---|---|---|---|
| DMF | Acid Acetic | 300 W/150 °C | ||||
| Time Reaction (h)/Yield (%) | Time Reaction (min)/Yield (%) | |||||
| 3a | H | 6/55 | 2/28 | 2 | 90 | Oil |
| 3b | C6H5 | 5/60 | 2.5/46 | 2 | 90 | Oil |
| 3c | 4-BrC6H4 | 7/47 | 3/41 | 2 | 90 | 150–152 |
| 3d | 4-CNC6H4 | 6/28 | 2/22 | 2 | 37 | 178–180 |
| 3e | 4-CO2HC6H4 | 8/33 | 2/25 | 2 | 40 | 133–135 |
| 3f | 4-FC6H4 | 7/61 | 3.5/43 | 2 | 98 | 118–120 |
| Entry | Reaction Temperature °C | Reaction Time (min) | Isolated Yield (%) |
|---|---|---|---|
| 1 | 80 | 5 | - |
| 2 | 100 | 4 | 32 |
| 3 | 120 | 4 | 53 |
| 4 | 150 | 2 | 90 |
| 5 | 180 | 5 | 90 |
| Compound | C15H17N2O2, H2O |
|---|---|
| Crystal shape, color | Polyhedron, yellow |
| Crystal size (mm) | 0.32 × 0.12 × 0.10 |
| Crystal system, Space group | Monoclinic, P21/n (N°14) |
| a (Å) | 10.704 (2) |
| b (Å) | 10.106 (2) |
| c (Å) | 13.735 (3) |
| β (°) | 95.35 (3) |
| V (Å3) | 1479.3 (5) |
| Z | 4 |
| Wavelength, Mo Kα (Å) | 0.71073 |
| T (K) | 297 (2) |
| F(000) | 580 |
| θ-range (°) | 2.50 < θ < 25.0 |
| hkl-range | −12:12, −12:11, −16:16 |
| μ (mm−1) | 0.086 |
| Reflections collected/Rint/Rσ | 10,360/0.099/0.0909 |
| Reflections unique/ parameters | 2588/ 412 |
| R1, wR2 [F2 > 2σ (F2)] | 0.0586, 0.0927 |
| Goodness-of-Fit on F2 (GooF = S) | 1.106 |
| Residual electron density ∆ρmax/∆ρmin (e Å−3) | 0.15/−0.11 |
| Compound/(µg/mL) | 100 | 50 | 10 | IC50 |
|---|---|---|---|---|
| 3a | 13.51 ± 1.5 | 8.10 ± 0.6 | 3.78 ± 0.3 | >100 |
| 3b | 29.19 ± 4.3 | 17.84 ± 3.4 | 11.09 ± 1.7 | >100 |
| 3c | 31.35 ± 2.6 | 16.49 ± 2.2 | 8.92 ± 1.0 | >100 |
| 3d | 25.89 ± 5.3 | 13.45 ± 2.8 | 3.05 ± 0.8 | >100 |
| 3e | 47.72 ± 4.4 | 21.32 ± 2.1 | 7.87 ± 1.2 | >100 |
| 3f | 85.41 ± 12.6 | 55.14 ± 9.7 | 19.46 ± 6.7 | 42.35 ± 4.3 |
| Ascorbic acid | - | - | - | 1 ± 0.3 |
| Compound/(µg/mL) | 100 | 75 | 50 | 25 | IC50 |
|---|---|---|---|---|---|
| 3a | 56.47 ± 12.2 | 35 ± 8.7 | 28.62 ± 5.3 | 0 | 87.94 ± 15.8 |
| 3b | 52.14 ± 10.5 | 37.23 ± 11.6 | 12.04 ± 8.1 | 5.08 ± 0.8 | 92.15 ± 18.3 |
| 3c | 46.88 ± 9.7 | 27.38 ± 9.9 | 19.67 ± 7.4 | 9.19 ± 1.7 | >100 |
| 3d | 21.34 ± 10.3 | 17.65 ± 8.7 | 12.10 ± 5.5 | 5.55 ± 2.4 | >100 |
| 3e | 49.24 ± 15.3 | 31.09 ± 9.0 | 3.87 ± 1.6 | 0 | >100 |
| 3f | 93.12 ± 17.5 | 88.37 ± 10.3 | 83.79 ± 8.5 | 53.65 ± 3.5 | 17.54 ± 4.8 |
| Ascorbic Acid | - | - | - | - | 35 ± 2.7 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polo, E.; Trilleras, J.; Ramos, J.; Galdámez, A.; Quiroga, J.; Gutierrez, M. Efficient MW-Assisted Synthesis, Spectroscopic Characterization, X-ray and Antioxidant Properties of Indazole Derivatives. Molecules 2016, 21, 903. https://doi.org/10.3390/molecules21070903
Polo E, Trilleras J, Ramos J, Galdámez A, Quiroga J, Gutierrez M. Efficient MW-Assisted Synthesis, Spectroscopic Characterization, X-ray and Antioxidant Properties of Indazole Derivatives. Molecules. 2016; 21(7):903. https://doi.org/10.3390/molecules21070903
Chicago/Turabian StylePolo, Efrain, Jorge Trilleras, Juan Ramos, Antonio Galdámez, Jairo Quiroga, and Margarita Gutierrez. 2016. "Efficient MW-Assisted Synthesis, Spectroscopic Characterization, X-ray and Antioxidant Properties of Indazole Derivatives" Molecules 21, no. 7: 903. https://doi.org/10.3390/molecules21070903
APA StylePolo, E., Trilleras, J., Ramos, J., Galdámez, A., Quiroga, J., & Gutierrez, M. (2016). Efficient MW-Assisted Synthesis, Spectroscopic Characterization, X-ray and Antioxidant Properties of Indazole Derivatives. Molecules, 21(7), 903. https://doi.org/10.3390/molecules21070903