
Synthesis of 5,10-bis(Trifluoromethyl) Substituted β-Octamethylporphyrins and Central-Metal-Dependent Solvolysis of Their meso-Trifluoromethyl Groups


Abstract
:
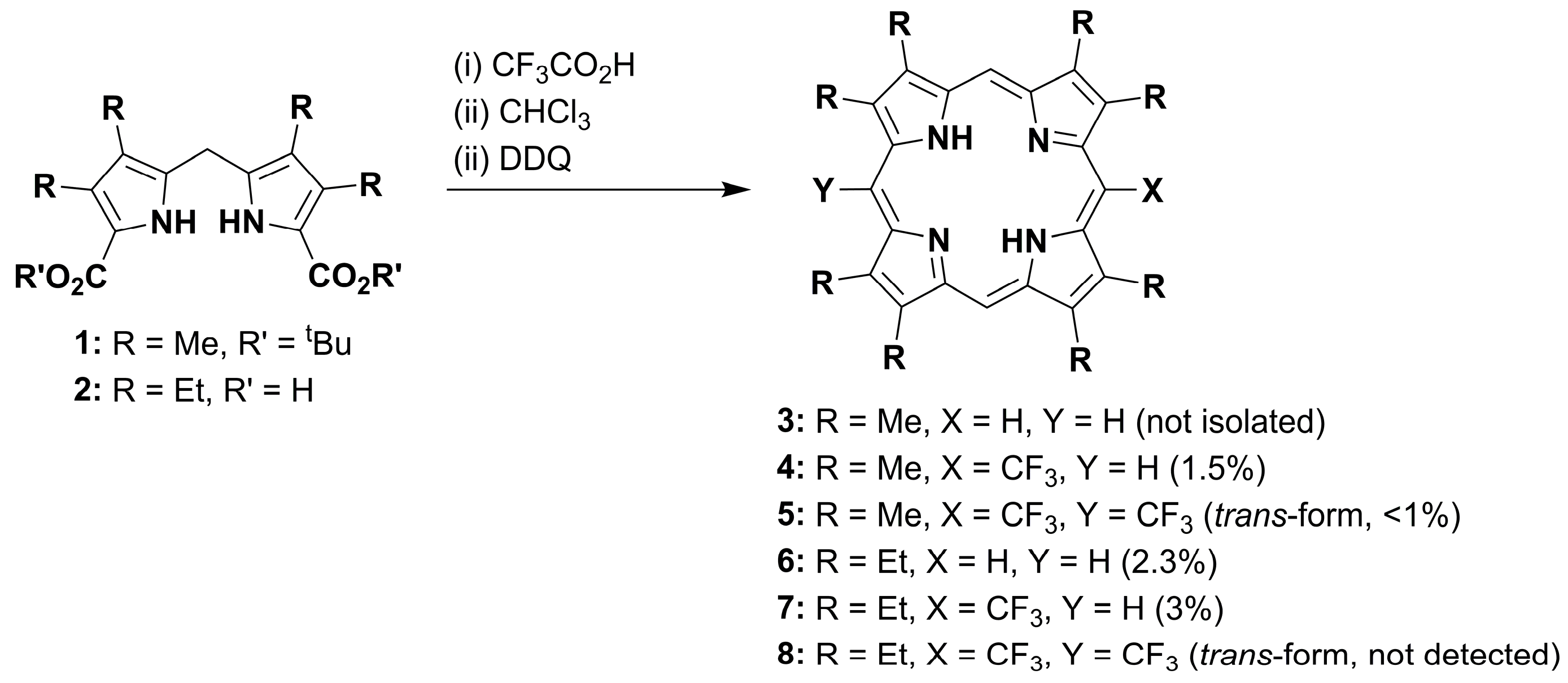
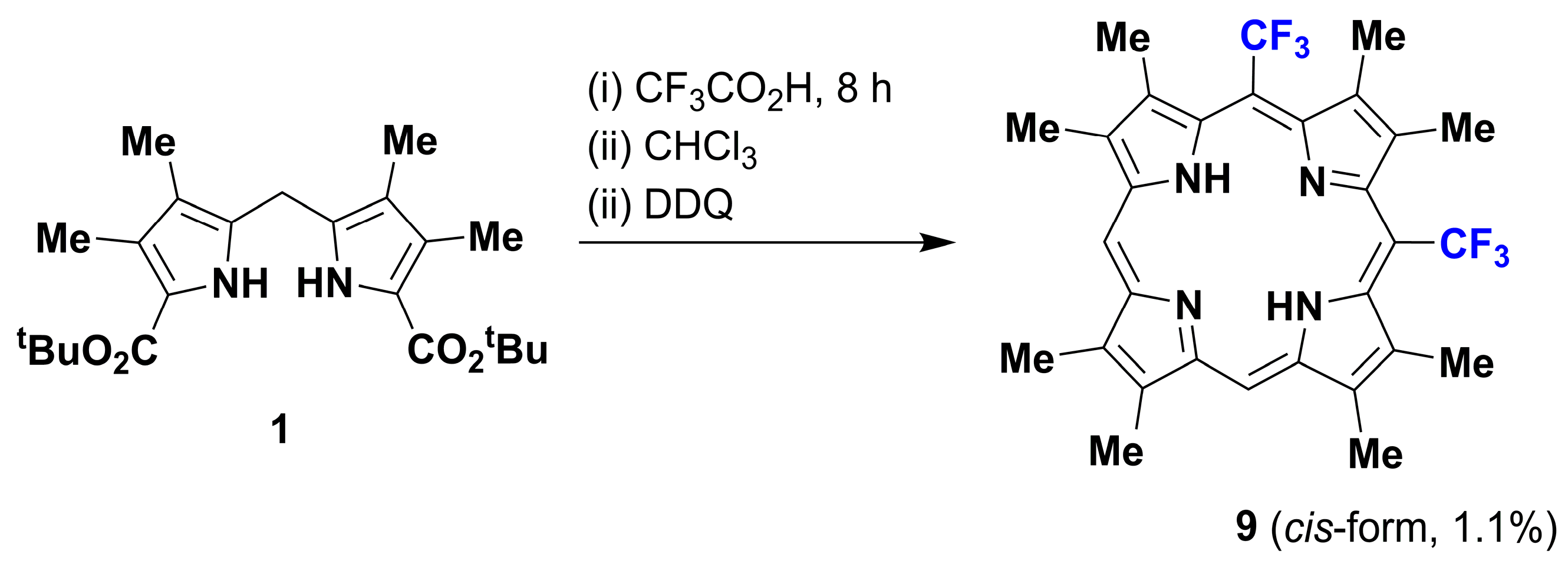
1. Introduction
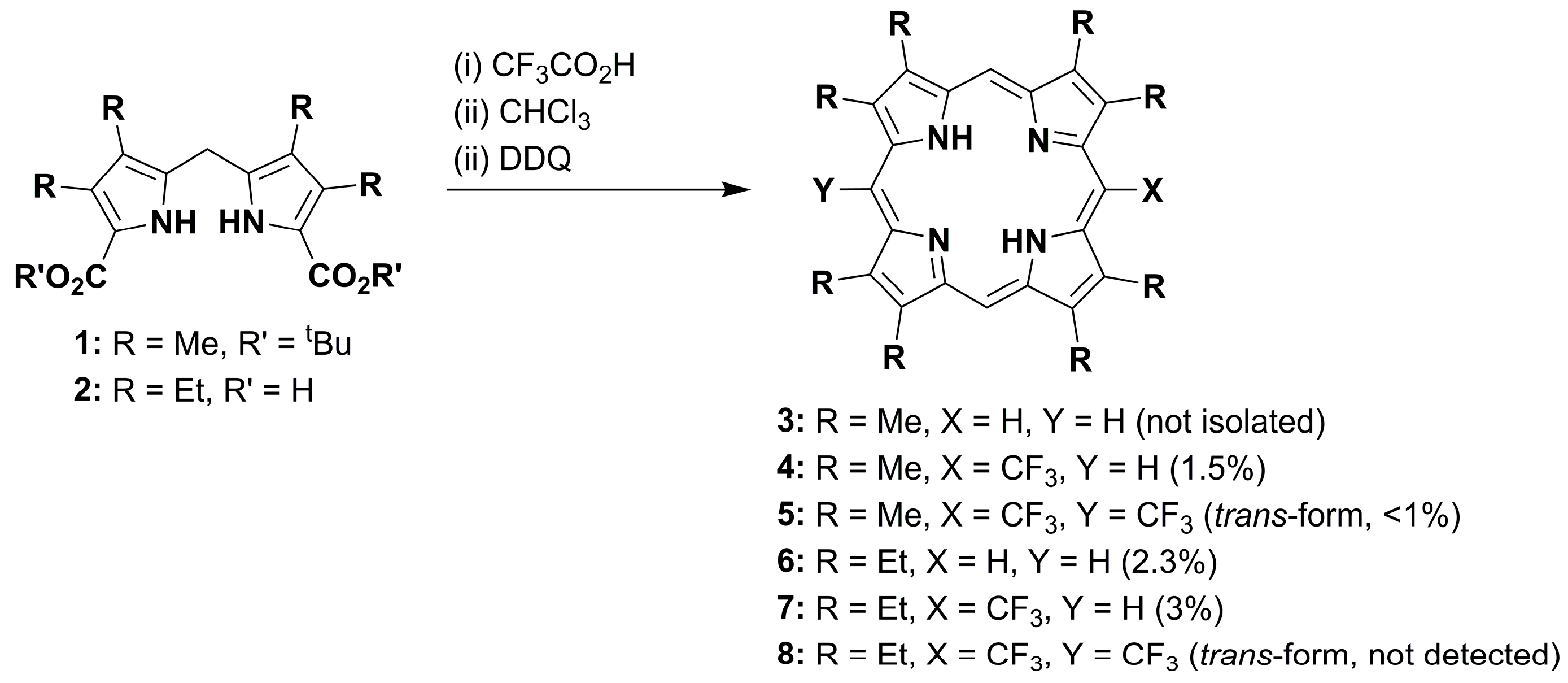
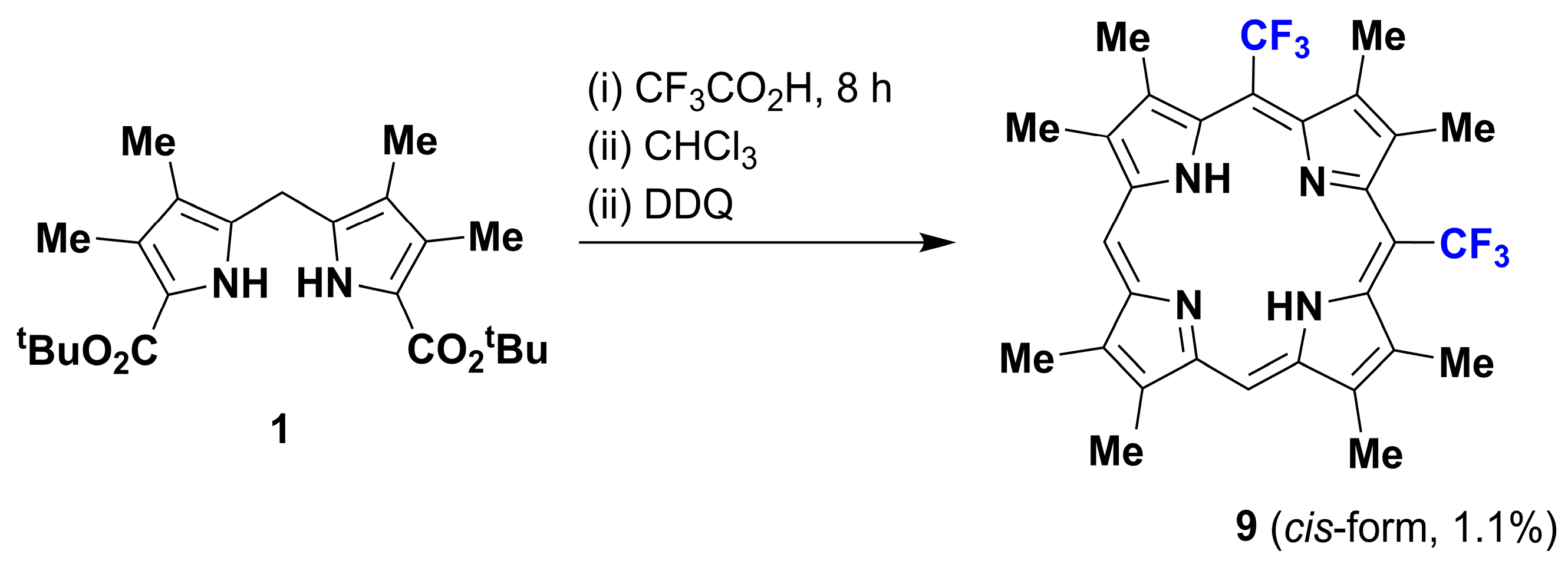
2. Results and Discussion
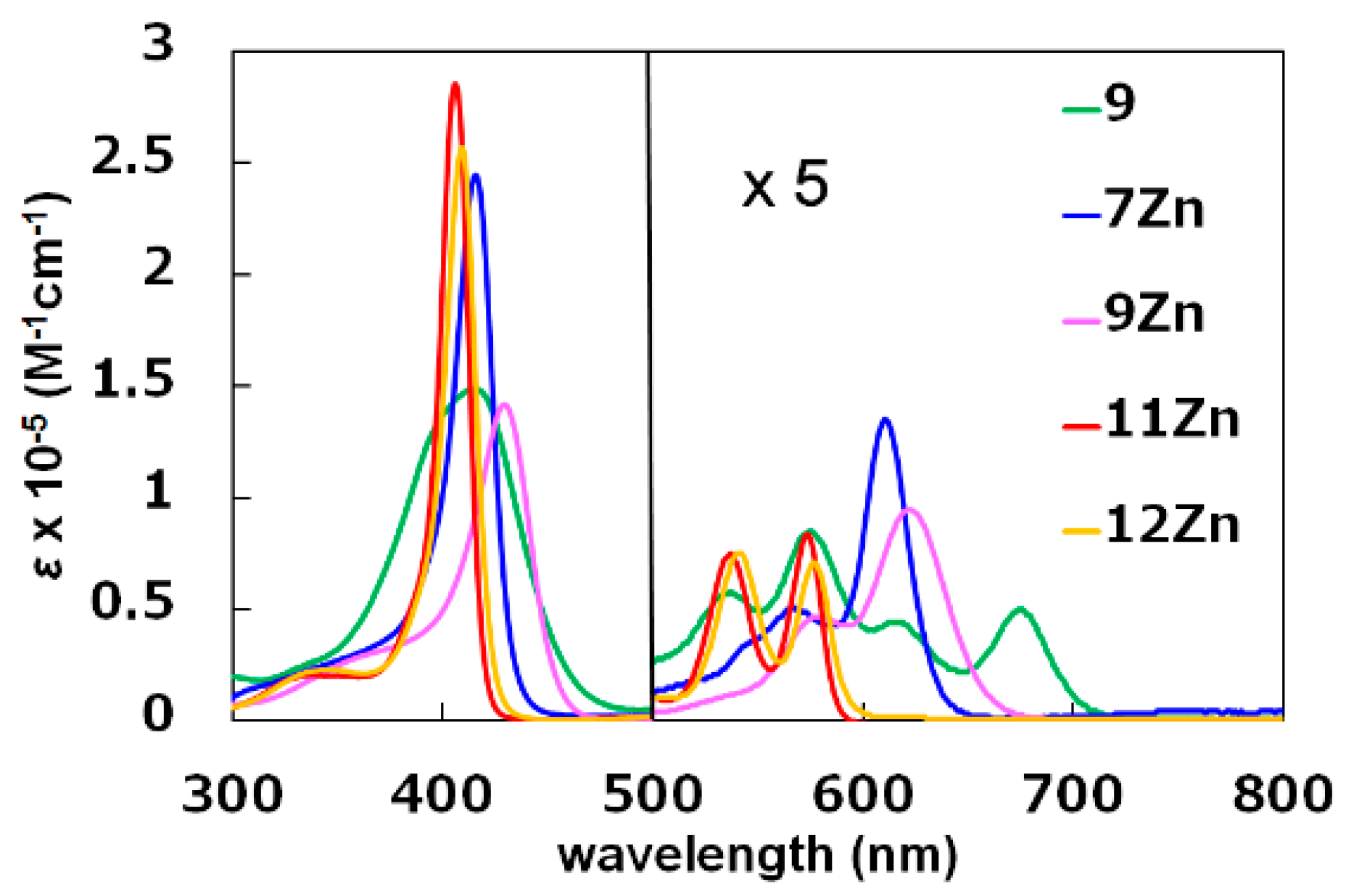
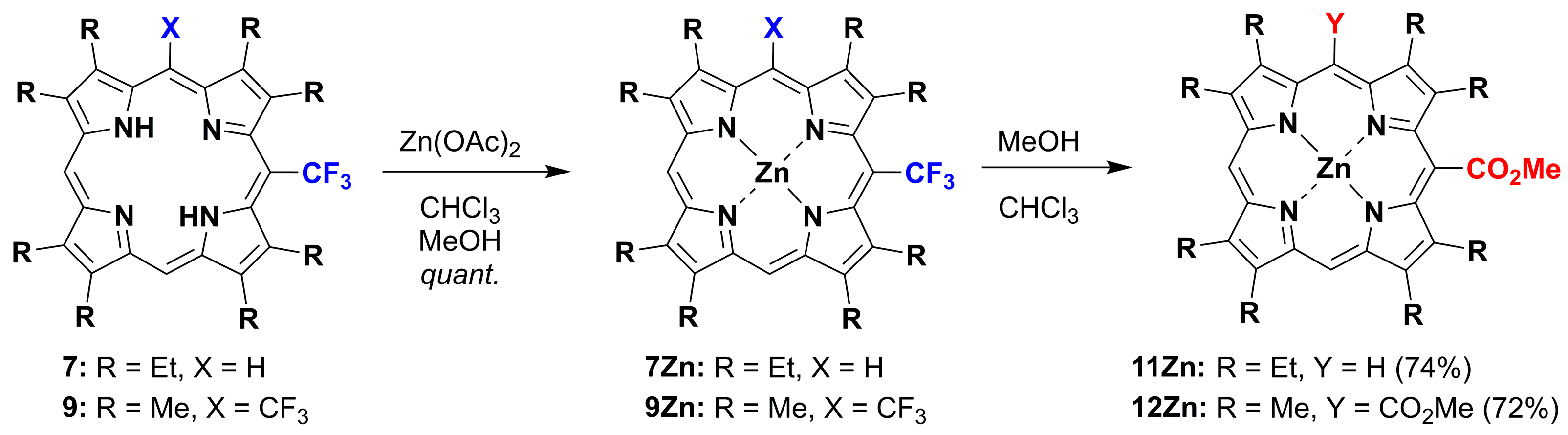
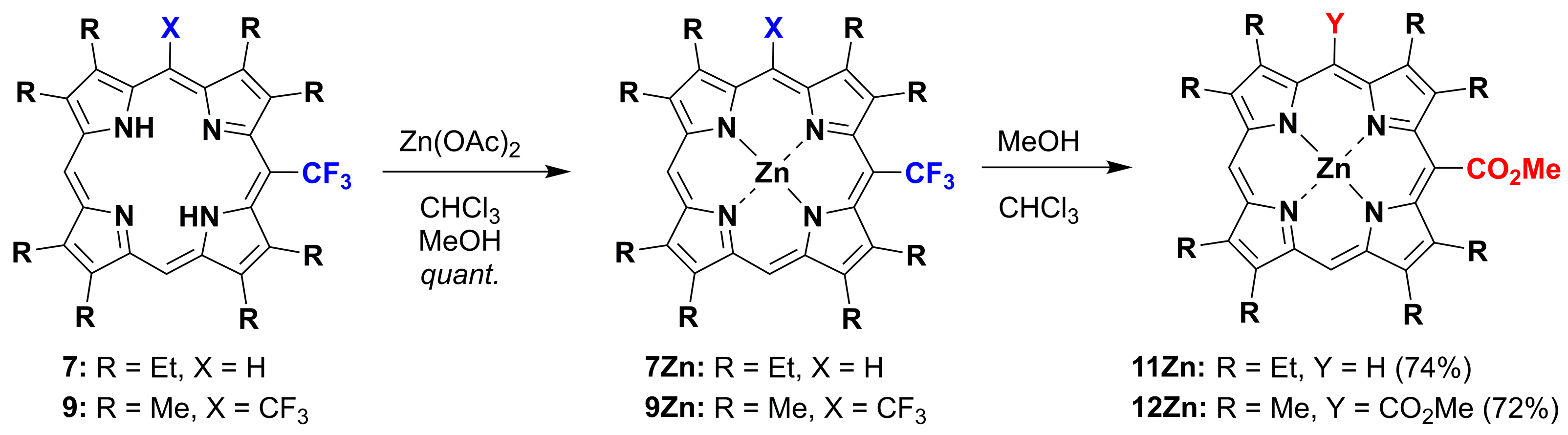
2.1. Metallation Reaction
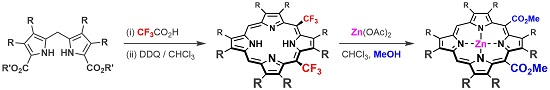
2.2. Formation of meso-CO2Me Porphyrins by Solvolysis of CF3 Substituents
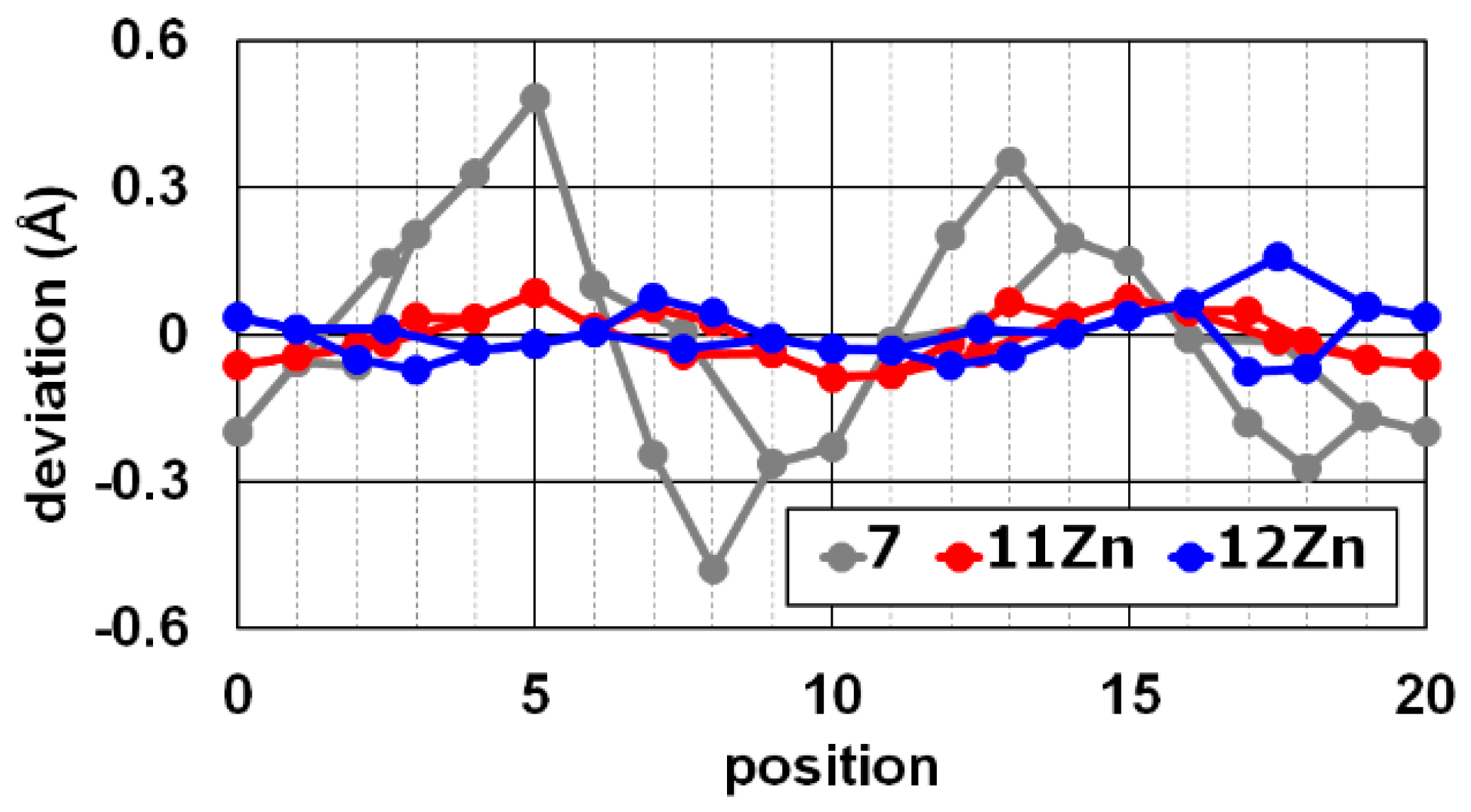
2.3. X-ray Crystal Structures
3. Experimental Section
3.1. General Information
3.2. Syntheses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TFA | Trifluoroacetic acid |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| TLC | solid thin layer chromatography |
References and Notes
- Chambron, J.-C.; Heitz, V.; Sauvage, J.-P.; Chou, J.-H.; Nalwa, H.S.; Kosal, M.E.; Rakow, N.A.; Suslick, K.S.; Aida, T.; Inoue, S.; et al. The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R, Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 6. [Google Scholar]
- Ali, H.; Lier, J.E.v.; Alonso, C.; Boyle, R.W.; Vicente, M.d.G.H.; Sibrian-Vazquez, M.; Ethirajan, M.; Patel, N.J.; Pandey, R.K.; Jux, N.; et al. Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R, Eds.; World Scientific: Singapore, 2010; Volume 4. [Google Scholar]
- Shelnutt, J.A.; Song, X.Z.; Ma, J.G.; Jia, S.L.; Jentzen, W.; Medforth, C.J. Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev. 1998, 27, 31–41. [Google Scholar] [CrossRef]
- Senge, M.O. Exercises in molecular gymnastics-bending, stretching and twisting porphyrins. Chem. Commun. 2006, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Di Magno, S.G.; Dussault, P.H.; Schultz, J.A. Fluorous biphasic singlet oxygenation with a perfluoroalkylated photosensitizer. J. Am. Chem. Soc. 1996, 118, 5312–5313. [Google Scholar] [CrossRef]
- Lahaye, D.; Muthukumaran, K.; Hung, C.-H.; Gryko, D.; Reboucas, J.S.; Spasojevic, I.; Batinic-Haberle, I.; Lindsey, J.S. Design and synthesis of manganese porphyrins with tailored lipophilicity: Investigation of redox properties and superoxide dismutase activity. Bioorg. Med. Chem. 2007, 15, 7066–7086. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.-M.; Chen, L.; Yin, J.-J.; Zhou, J.-M.; Guo, C.-C.; Chen, Q.-Y. Rational synthesis of meso- or β-fluoroalkylporphyrin derivatives via halo-fluoroalkylporphyrin precursors: Electronic and steric effects on regioselective electrophilic substitution in 5-fluoroalkyl-10,20-diarylporphyrins. J. Org. Chem. 2006, 71, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, A.; Jaquinod, L.; Nurco, D.J.; Smith, K.M. Investigations on the directive effects of a single meso-substituent via nitration of 5,12,13,17,18-pentasubstituted porphyrins: Syntheses of conjugated β-nitroporphyrins. Tetrahedron 2001, 57, 4261–4269. [Google Scholar] [CrossRef]
- Smith, K.M. Synthesis and organic chemistry. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R, Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 1–44. [Google Scholar]
- Senge, M.O. Highly substituted porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R, Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 239–348. [Google Scholar]
- Suzuki, M.; Ishii, S.; Hoshino, T.; Neya, S. Syntheses of Highly Distorted meso-Trifluoromethyl-Substituted β-Octaalkylporphyrins. Chem. Lett. 2014, 43, 1563–1565. [Google Scholar] [CrossRef]
- The absorption bands of zinc octaethylporphyrin were observed at λmax (log ε) = 331 (4.28), 402 (5.63), 490 (3.51), 532 (4.21), and 569 (4.39) nm in CH2Cl2. These data also support the opinion discussed here. Gong, L.-C.; Dolphin, D. Nitrooctaethylporphyrins: Synthesis, optical and redox properties. Can. J. Chem. 1985, 63, 401–405. [Google Scholar]
- Crystal data for 11Zn: C38H46N4O2Zn = 656, triclinic, space group P-1 (No. 2), a = 10.6883(2) Å, b = 13.0924(2) Å, c = 24.3733(4) Å, α = 73.7710(10), β = 89.9650(10), γ = 89.9610(10), V = 3274.78(10) Å3, Z = 4, Dcalcd. = 1.331 g/cm3, T = −180 °C, R1 = 0.0946 (I > 2σ(I)), RW = 0.2274 (all data), GOF= 0.859, CCDC-1444312. Crystal data for 12Zn: C33H33Cl3N4O4Zn = 721, triclinic, space group P-1 (No. 2), a = 9.5307(2) Å, b = 12.0063(3) Å, c = 14.4714(3) Å, α = 78.7230(10), β = 72.5150(10), γ = 85.381(2), V = 1548.52(6) Å3, Z = 2, Dcalcd. = 1.547 g/cm3, T = −180 °C, R1 = 0.0921 (I > 2σ(I)), RW = 0.2943 (all data), GOF= 1.126, CCDC-1444313. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (accessed on 19 February 2016).
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXS-97, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- CCP14, Sir97. Available online: http://www.ccp14.ac.uk/tutorial/sir97/ (accessed on 19 February 2016).
- Yadokari-XG HomePage, Yadokari-XG Version 2005.9.19 with OpenGL. Available online: http://www.hat.hi-ho.ne.jp/k-wakita/yadokari/ (accessed on 19 February 2016).
- Yadokari-XG 2009 Project, Yadokari-XG 2009 Revision 835. Available online: http://www.xray.chem.tohoku.ac.jp/en/index.html (accessed on 19 February 2016).
- Neya, S.; Quan, J.; Hata, M.; Hoshino, T.; Funasaki, N. A novel and efficient synthesis of porphine. Tetrahedron Lett. 2006, 47, 8731–8732. [Google Scholar] [CrossRef]
- Yao, Z.; Bhaumik, J.; Dhanalekshmi, S.; Ptaszek, M.; Rodriguez, P.A.; Lindsey, J.S. Synthesis of porphyrins bearing 1–4 hydroxymethyl groups and other one-carbon oxygenic substituents in distinct patterns. Tetrahedron 2007, 63, 10657–10670. [Google Scholar] [CrossRef] [PubMed]
- Takanami, T.; Wakita, A.; Matsumoto, J.; Sekine, S.; Suda, K. An efficient one-pot procedure for asymmetric bifunctionalization of 5,15-disubstituted porphyrins: A simple preparation of meso acyl-, alkoxycarbonyl-, and carbamoyl-substituted meso-formylporphyrins. Chem. Commun. 2009, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, J.; Leone, S.A.; Sullivan, W.F.; Bennett, O.F. Facile hydrolysis of the trifluoromethyl group in the presence of base. Some trifluoromethylated indoles. J. Am. Chem. Soc. 1957, 79, 1745–1748. [Google Scholar] [CrossRef]
- Jones, R.G. Ortho and para substituted derivatives of benzotrifluoride. J. Am. Chem. Soc. 1947, 69, 2346–2350. [Google Scholar] [CrossRef] [PubMed]
- Fave, G.M.L. Some reactions of the trifluoromethyl group in the benzotrifluoride series. I. Hydrolysis. J. Am. Chem. Soc. 1949, 71, 4148–4149. [Google Scholar] [CrossRef]
- Mechoulam, R.; Cohen, S.; Kaluszyner, A. Basic alcoholysis of the trifluoromethyl group in 1,1,1-trifluoro-2,2-diarylethanes. J. Org. Chem. 1956, 21, 801–803. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds of 7, 9, 7Zn, 9Zn, 11Zn, and 12Zn are available from the authors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Soret Band (log ε)/nm | Q-Band (log ε)/nm |
|---|---|---|
| 5 | 397 | 575, 671 1 |
| 9 | 415 (5.17) | 537 (4.07), 575 (4.23), 616 (3.95), 675 (4.00) |
| 7Zn | 416 (5.39) | 567 (4.01), 611 (4.43) |
| 9Zn | 429 (5.15) | 578 (3.97), 623 (4.28) |
| 11Zn | 406 (5.46) | 537 (4.18), 574 (4.22) |
| 12Zn | 409 (5.41) | 541 (4.18), 577 (4.15) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, M.; Neya, S.; Nishigaichi, Y. Synthesis of 5,10-bis(Trifluoromethyl) Substituted β-Octamethylporphyrins and Central-Metal-Dependent Solvolysis of Their meso-Trifluoromethyl Groups. Molecules 2016, 21, 252. https://doi.org/10.3390/molecules21030252
Suzuki M, Neya S, Nishigaichi Y. Synthesis of 5,10-bis(Trifluoromethyl) Substituted β-Octamethylporphyrins and Central-Metal-Dependent Solvolysis of Their meso-Trifluoromethyl Groups. Molecules. 2016; 21(3):252. https://doi.org/10.3390/molecules21030252
Chicago/Turabian StyleSuzuki, Masaaki, Saburo Neya, and Yutaka Nishigaichi. 2016. "Synthesis of 5,10-bis(Trifluoromethyl) Substituted β-Octamethylporphyrins and Central-Metal-Dependent Solvolysis of Their meso-Trifluoromethyl Groups" Molecules 21, no. 3: 252. https://doi.org/10.3390/molecules21030252
APA StyleSuzuki, M., Neya, S., & Nishigaichi, Y. (2016). Synthesis of 5,10-bis(Trifluoromethyl) Substituted β-Octamethylporphyrins and Central-Metal-Dependent Solvolysis of Their meso-Trifluoromethyl Groups. Molecules, 21(3), 252. https://doi.org/10.3390/molecules21030252