
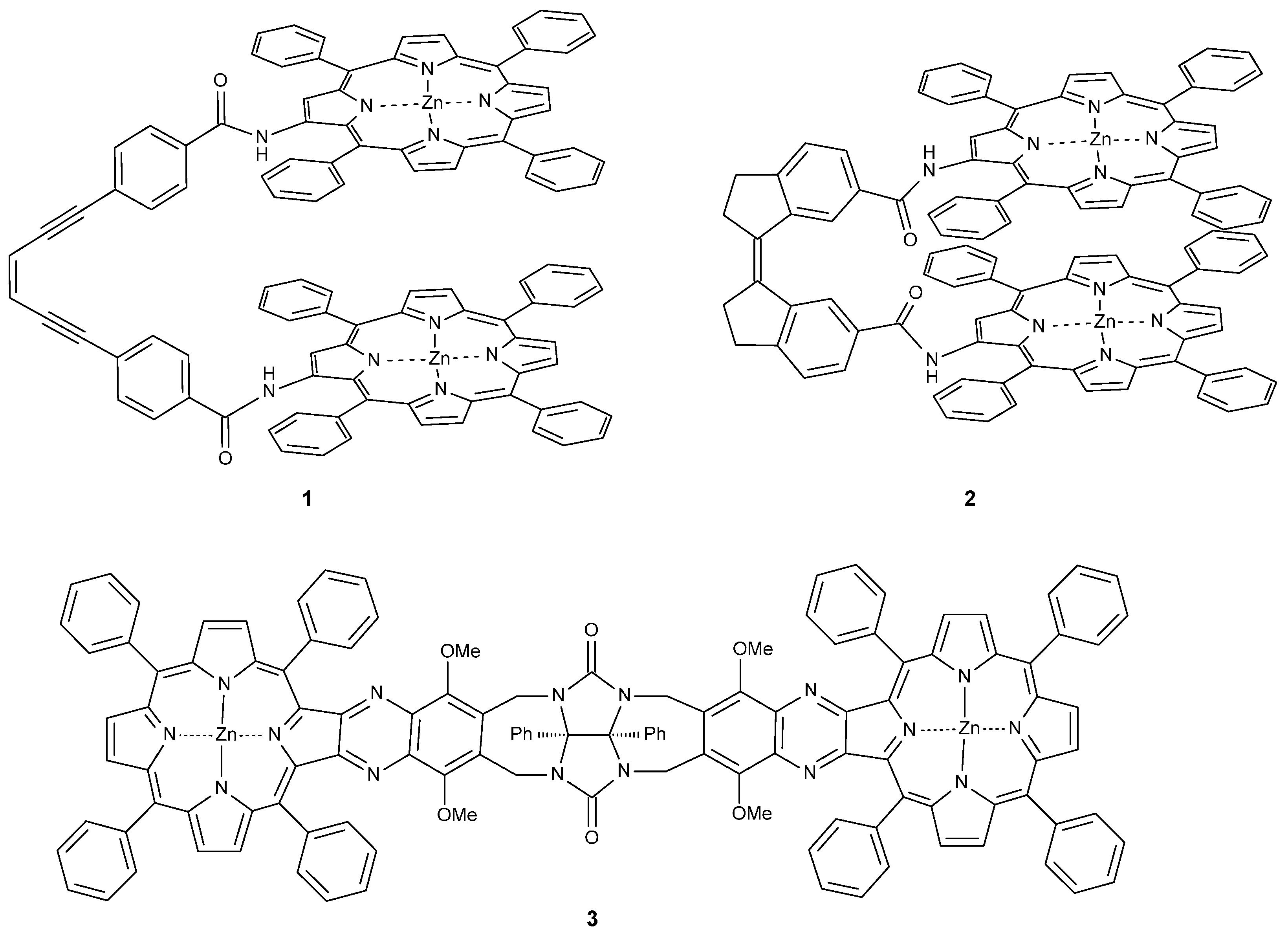
Synthesis and Properties of Bis-Porphyrin Molecular Tweezers: Effects of Spacer Flexibility on Binding and Supramolecular Chirogenesis


 ,
,
Abstract
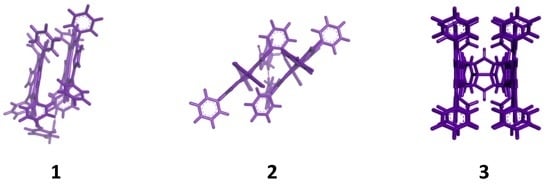
:
1. Introduction
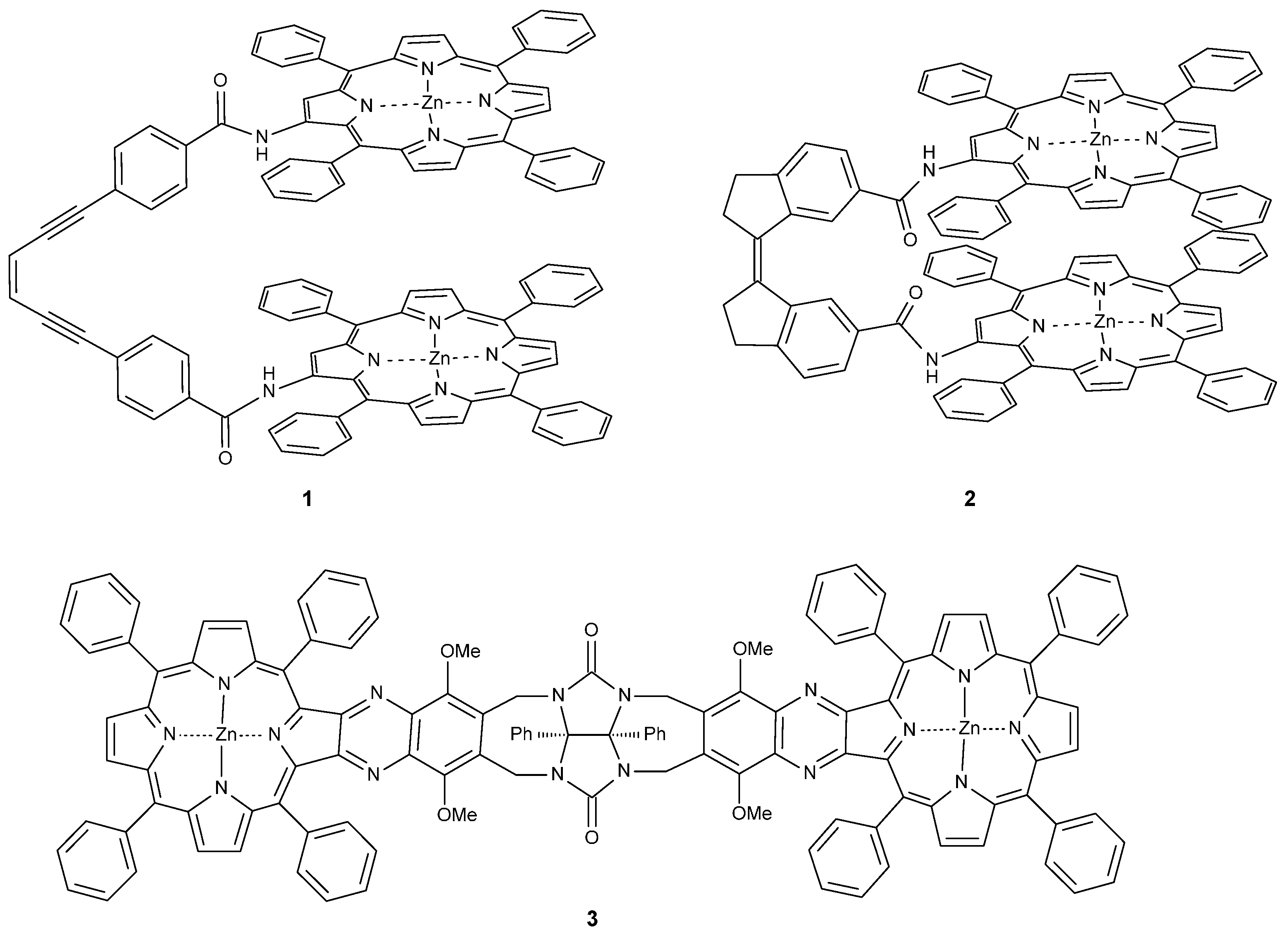

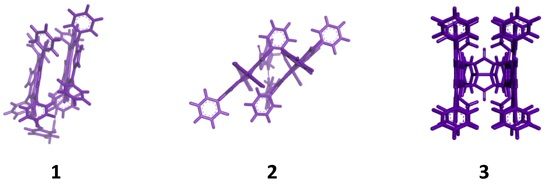{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
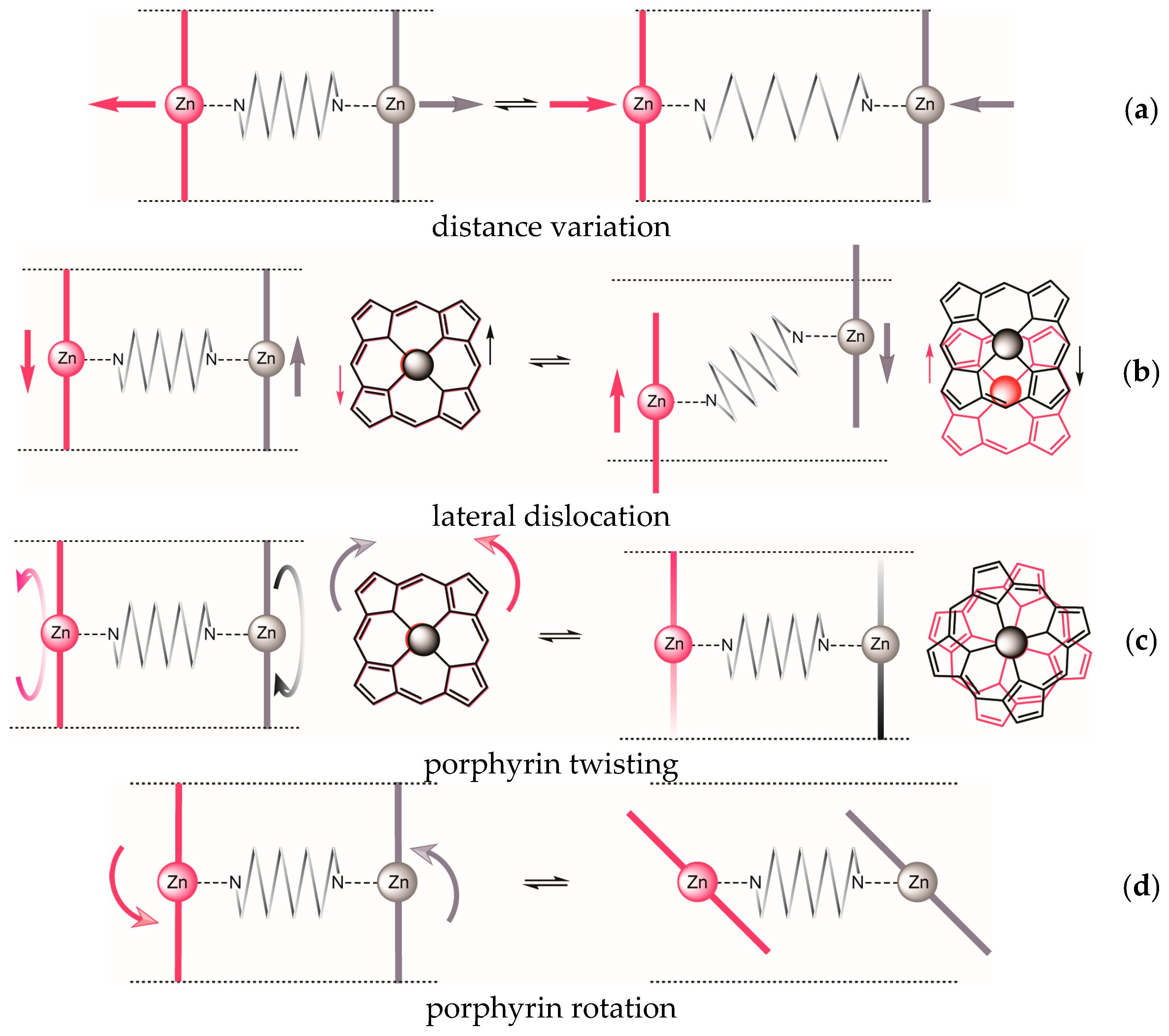
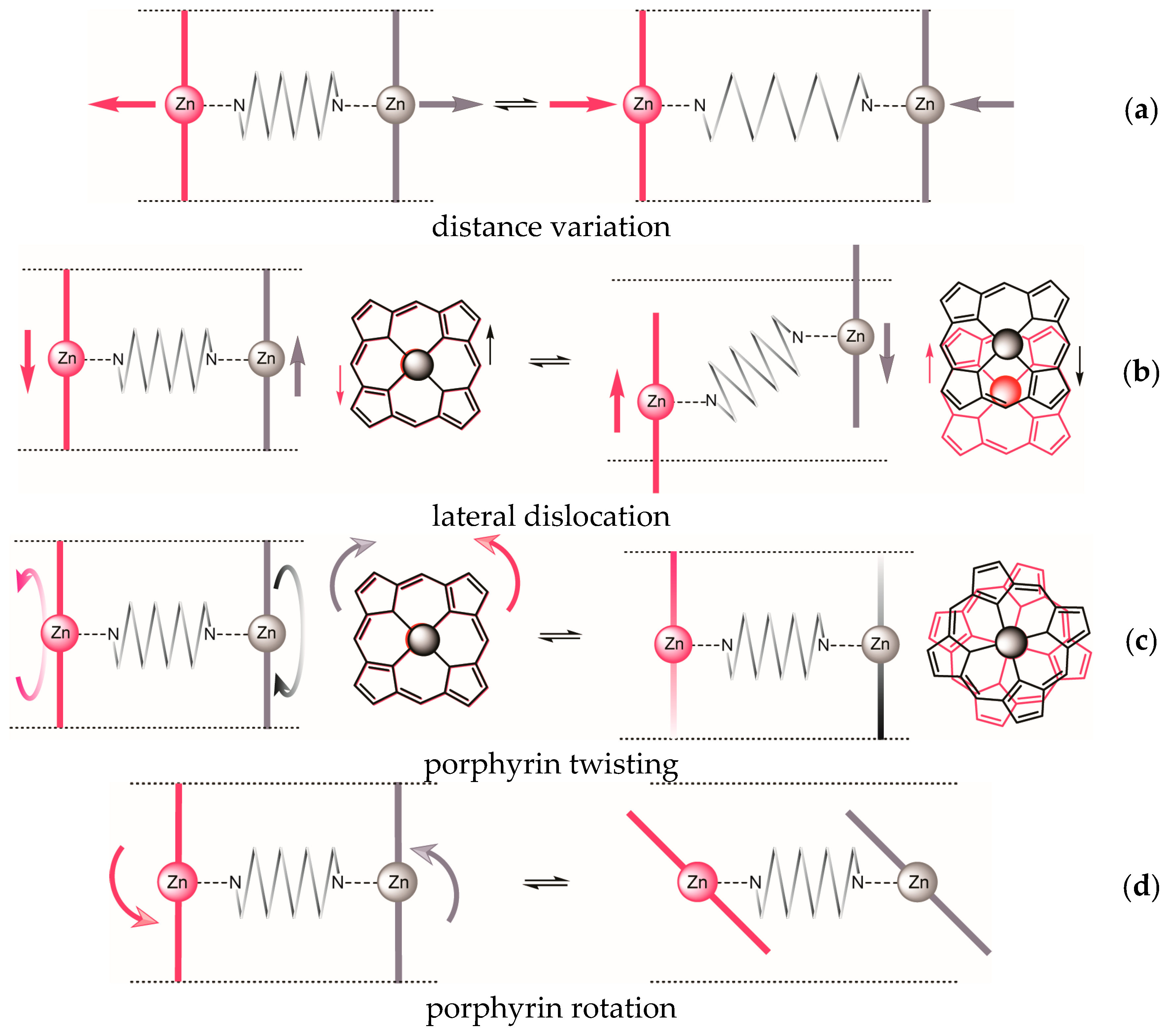
| Dislocation | 1 | 2 | 3 | |
|---|---|---|---|---|
| a | distance variation | + | (+) | + |
| b | lateral dislocation | + | inherent | − |
| c | porphyrin twisting | + | inherent | + |
| d | porphyrin rotation | + | + | − |
| helical complex achievable? | + | inherent | − | |
2. Results and Discussion
2.1. Synthesis
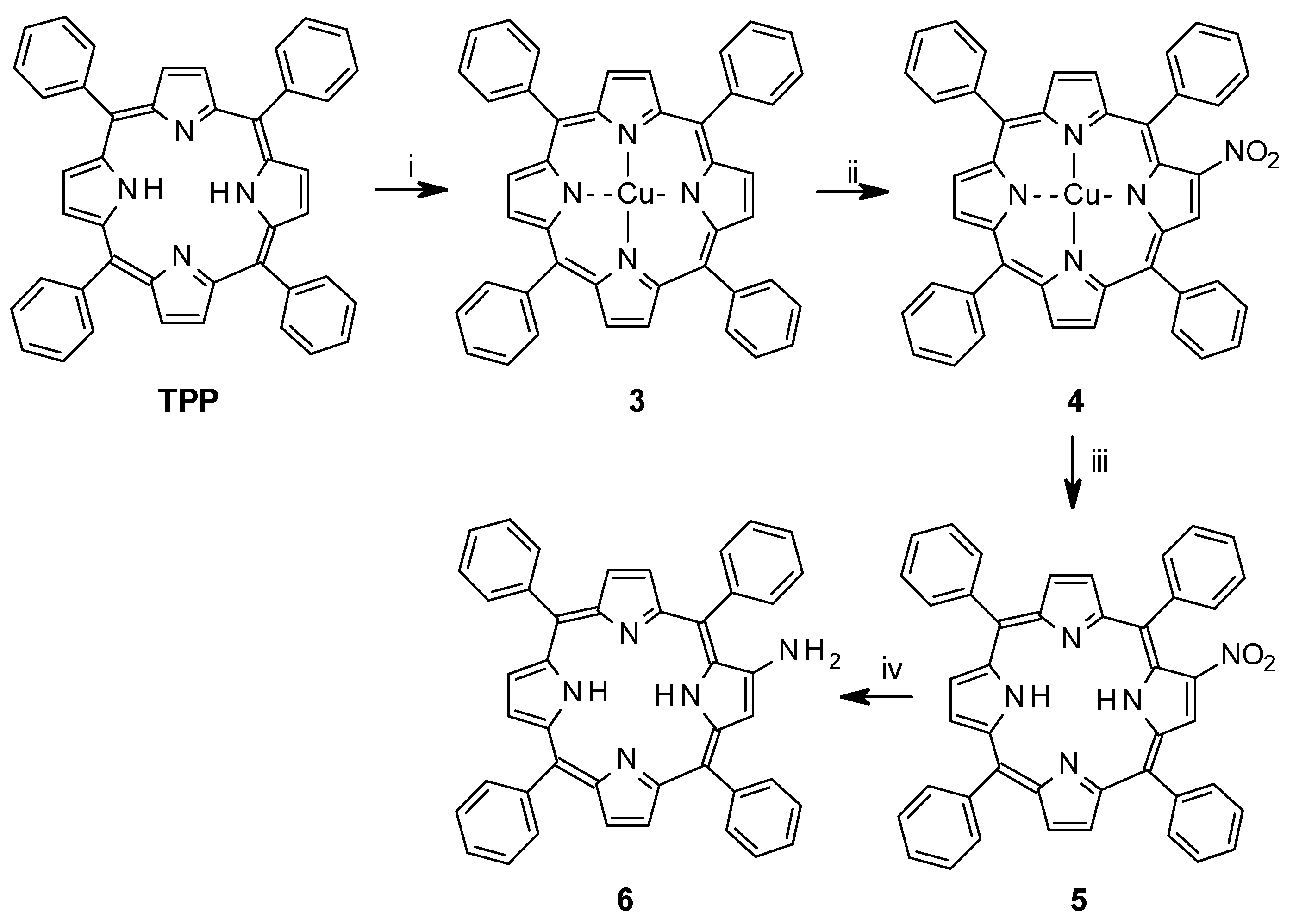
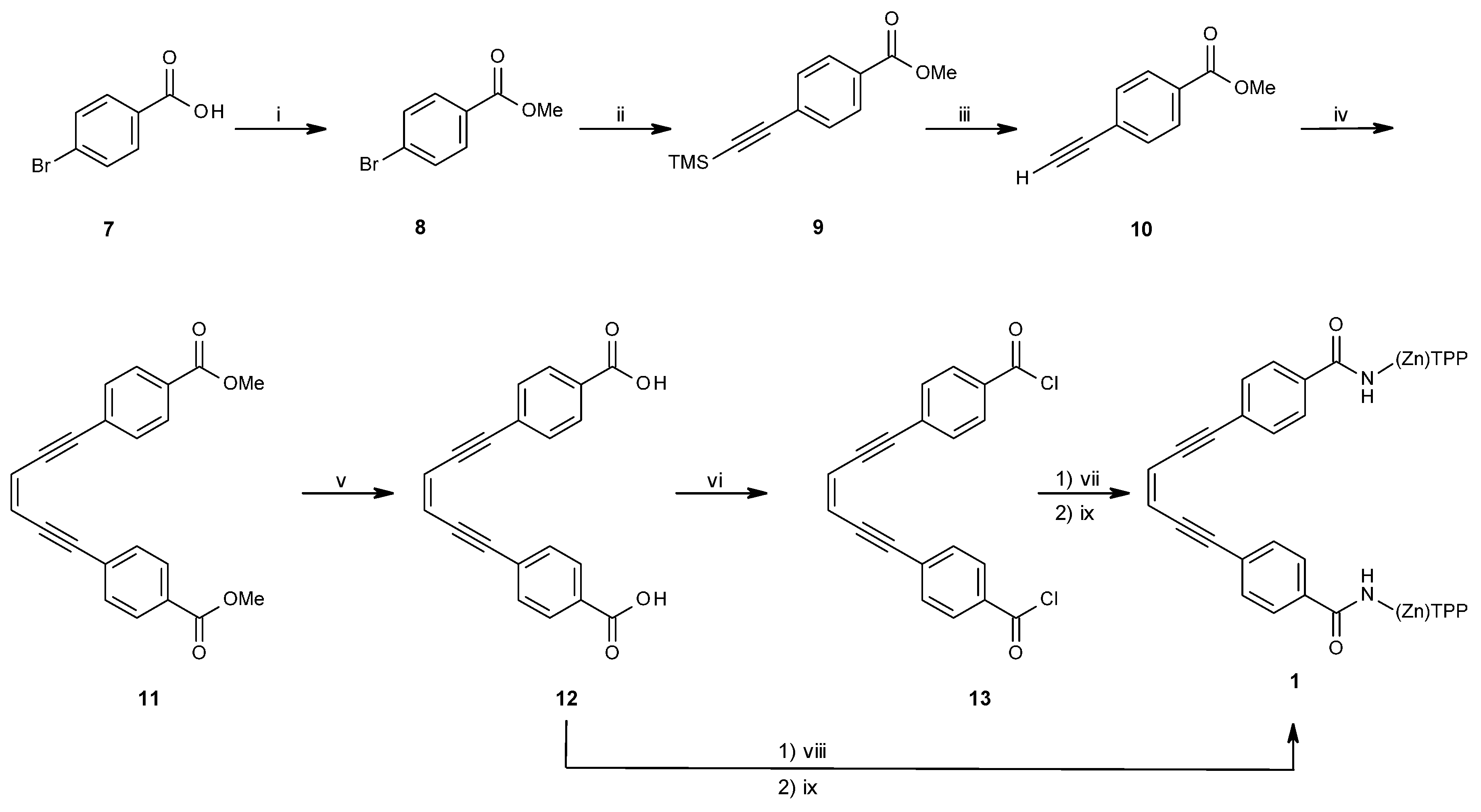
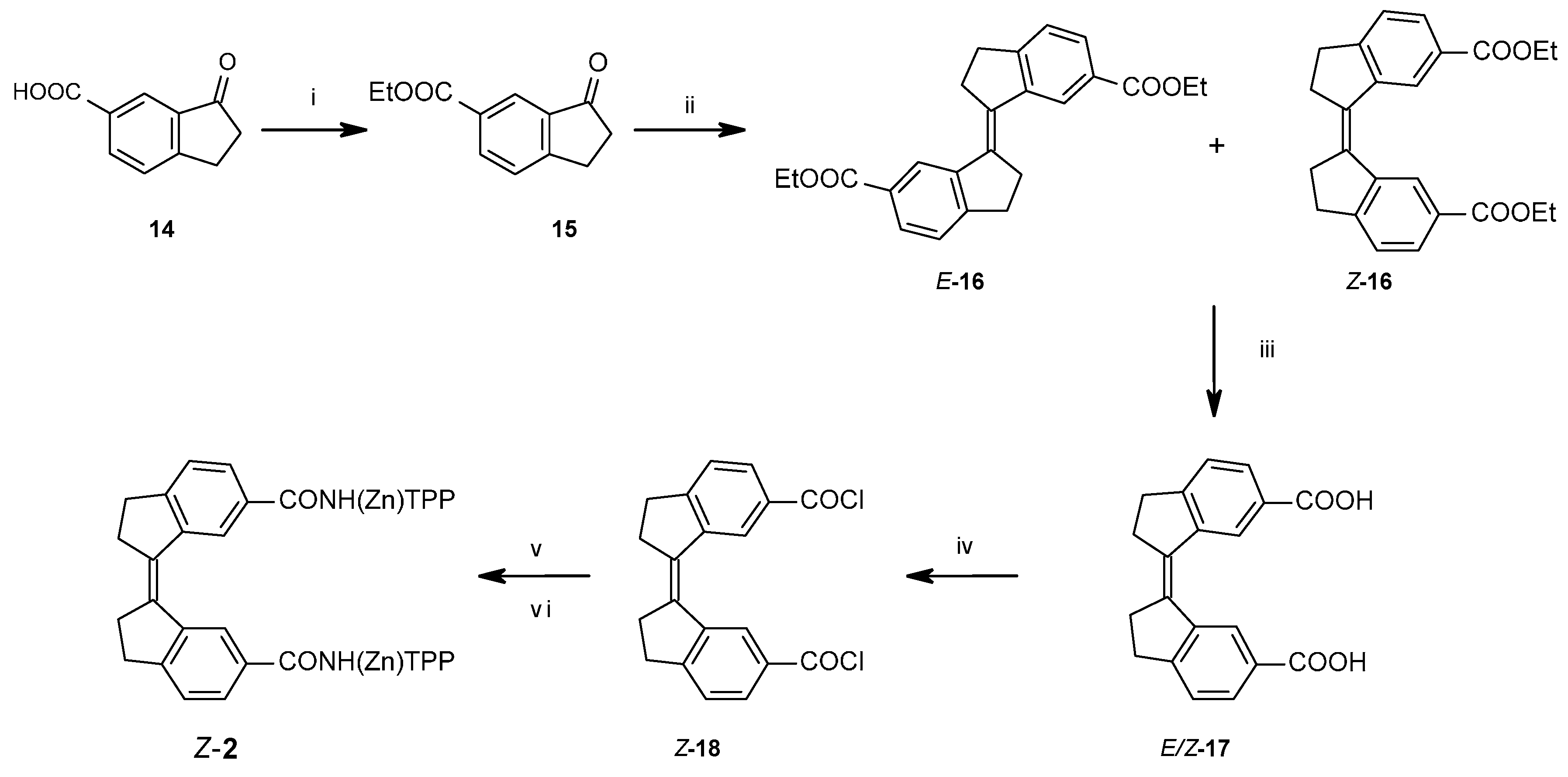
2.1.1. Synthesis of Porphyrin Units and Spacers
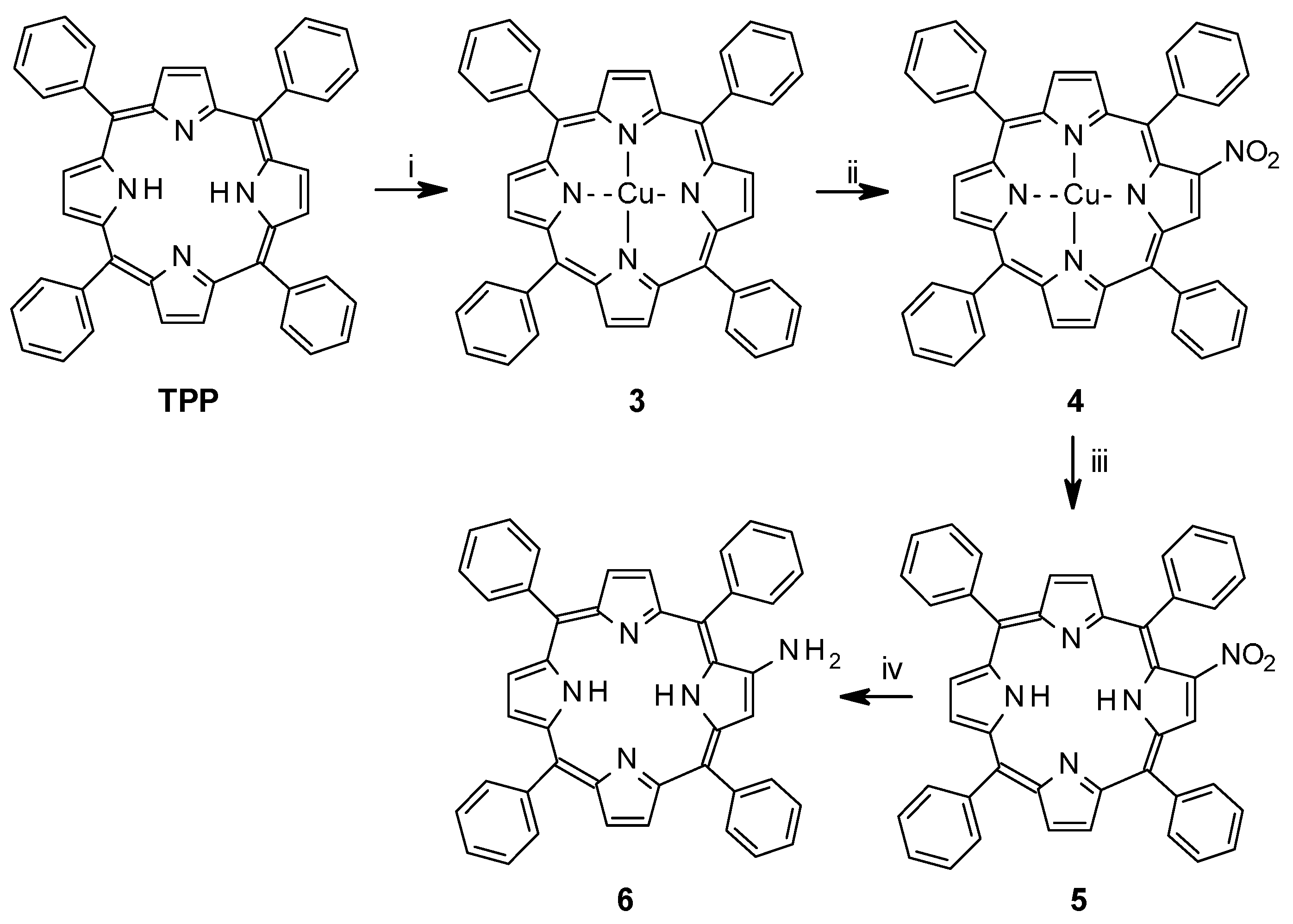
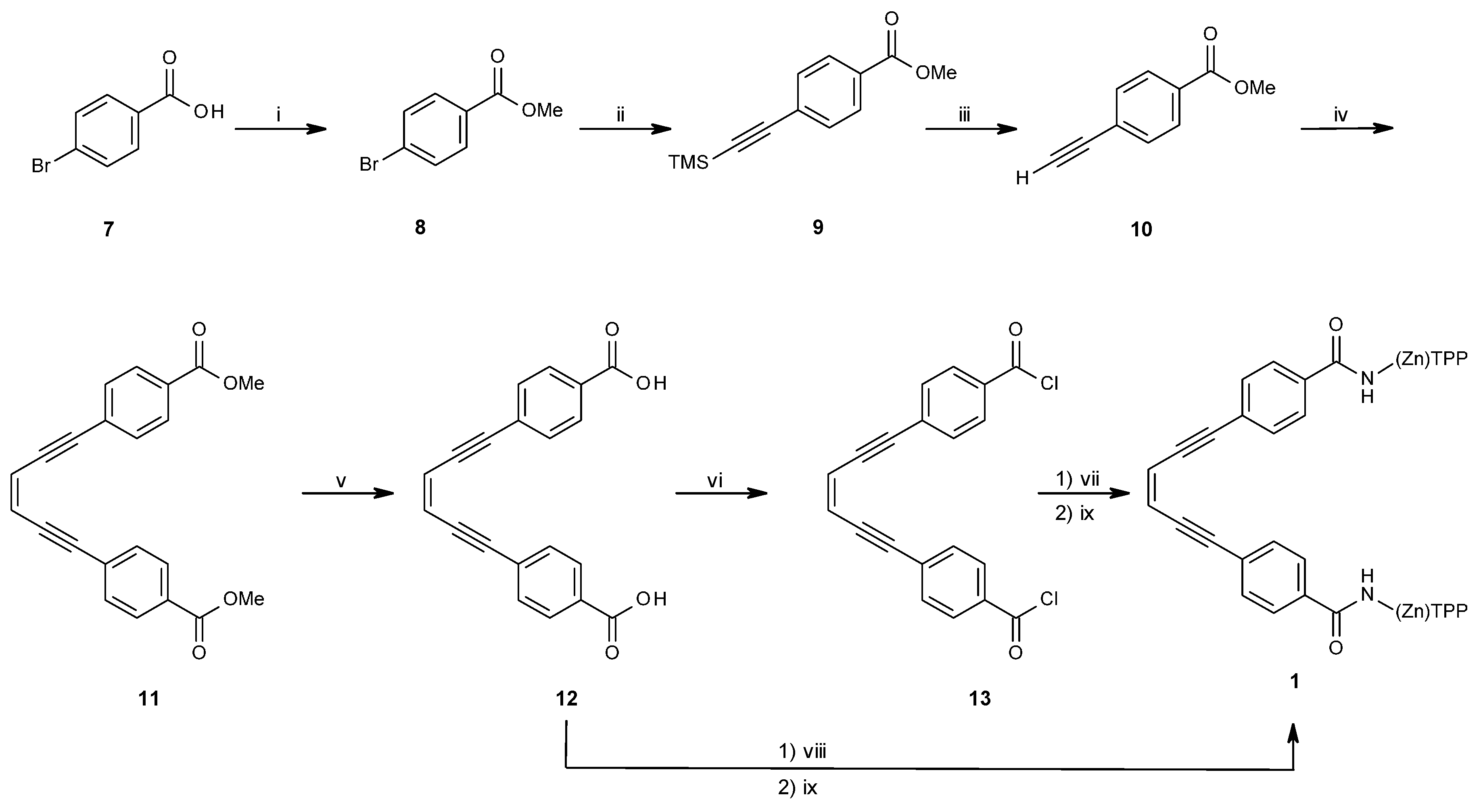
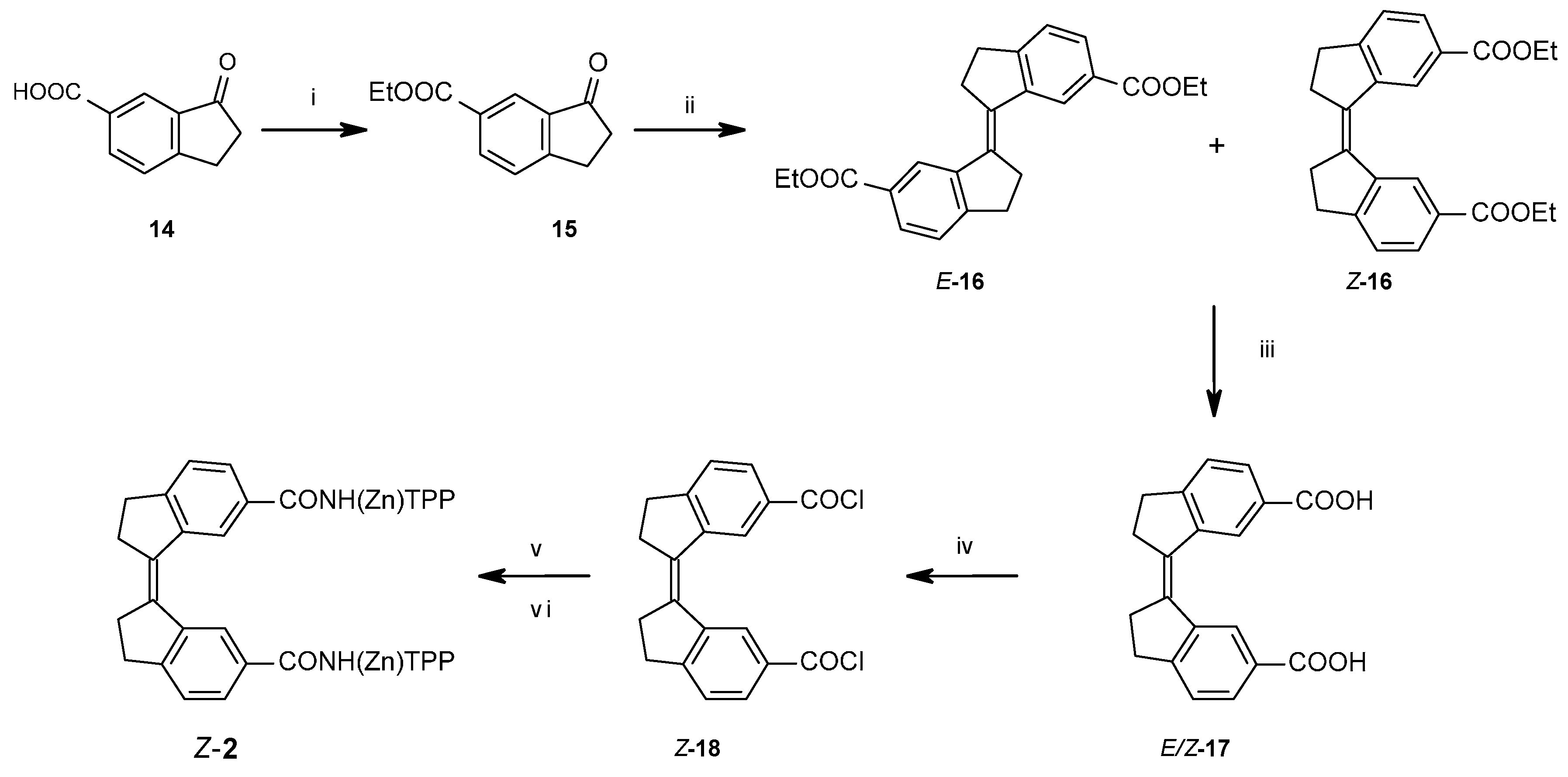
2.1.2. Coupling of Spacers to β-Monoaminotetraphenylporphyrin
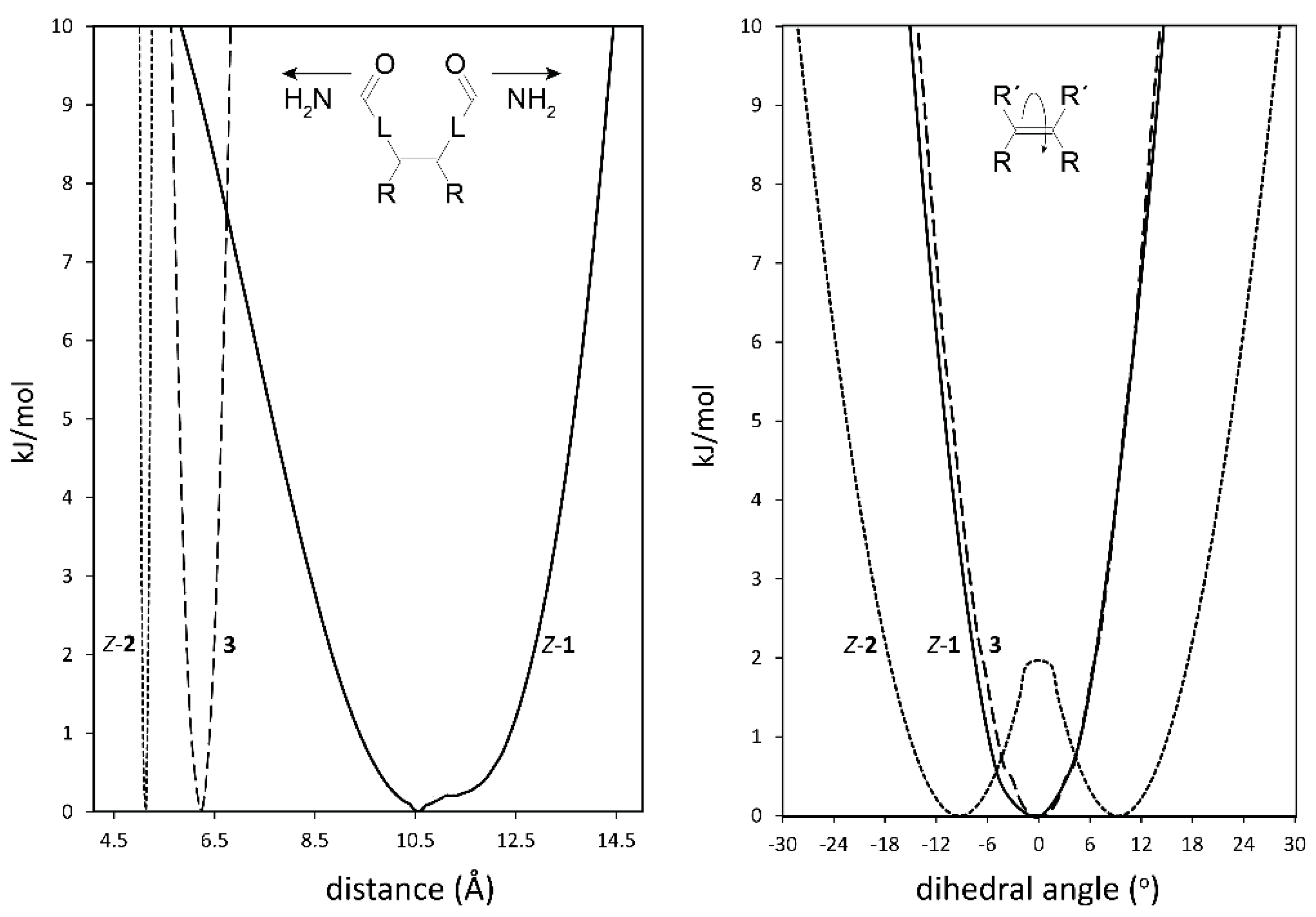
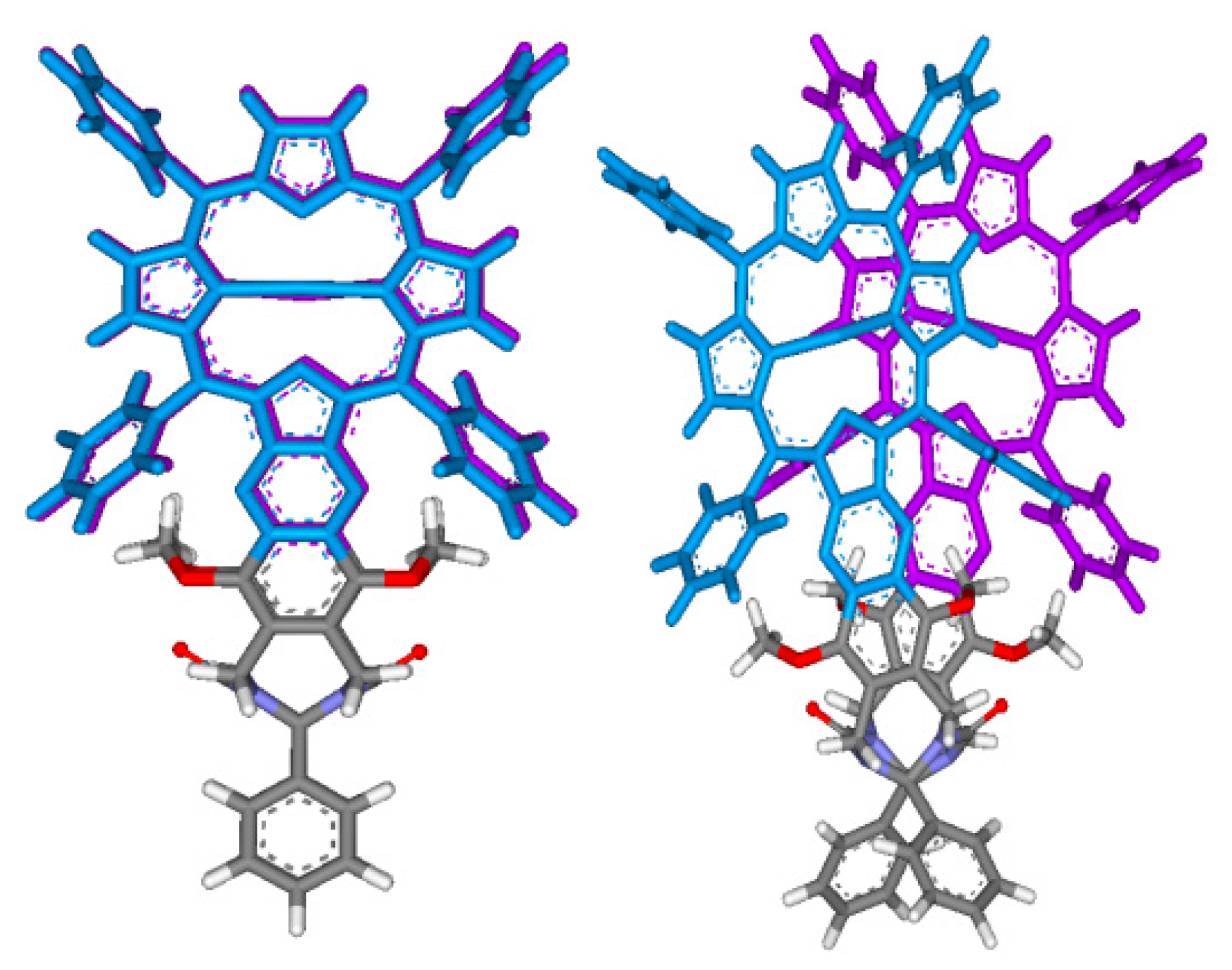
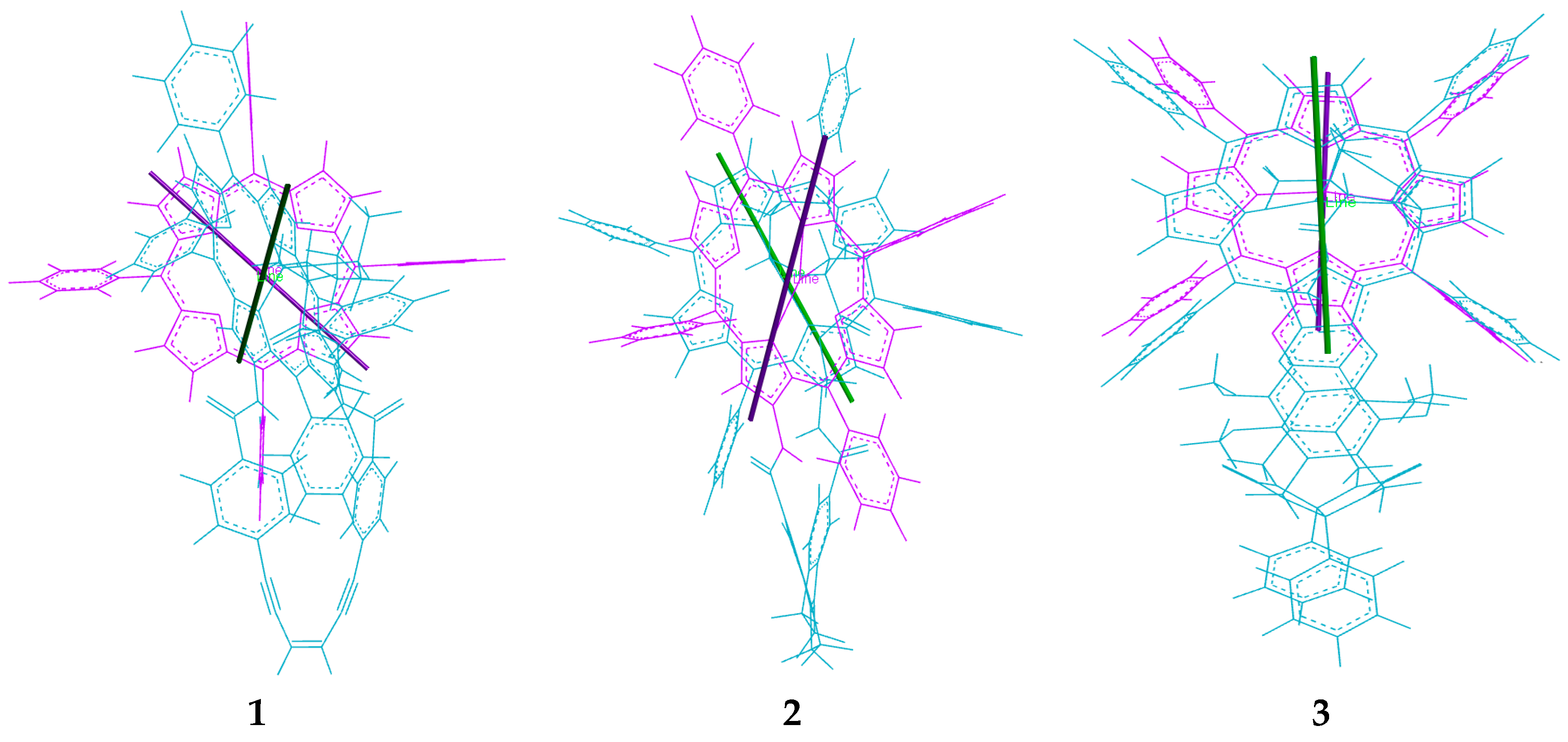
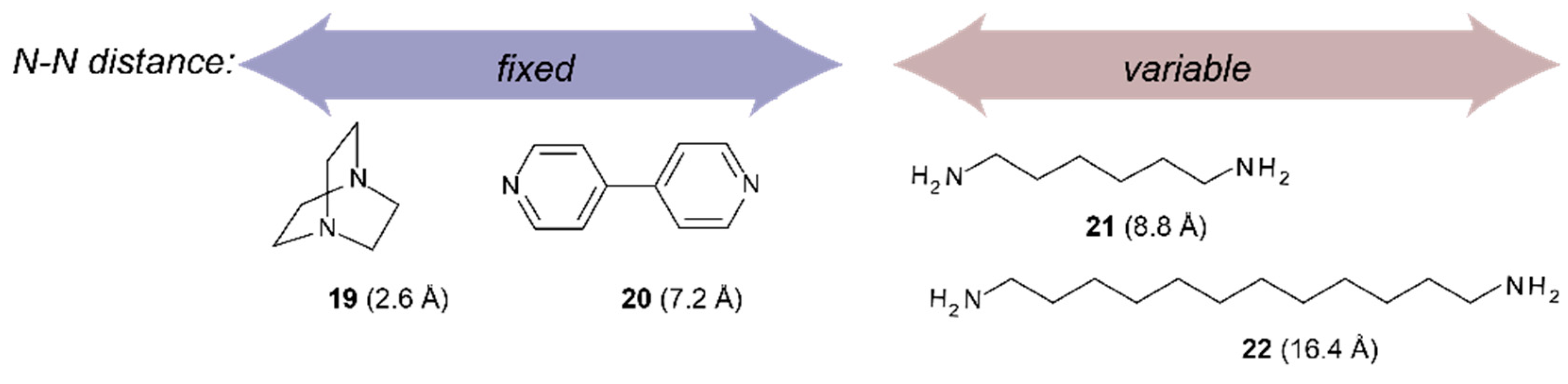
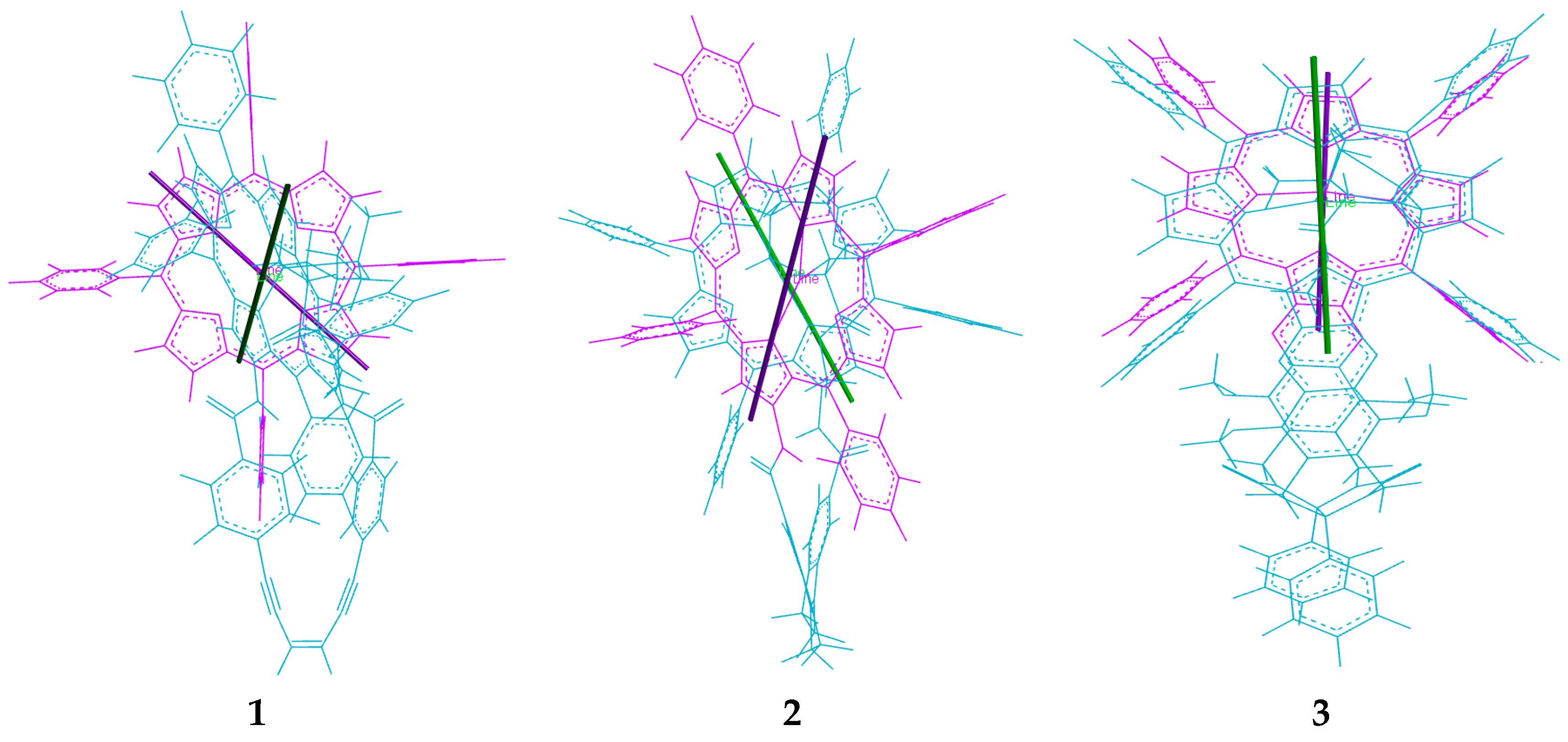
2.2. Conformational Analysis of Spacer and Tweezer Geometry


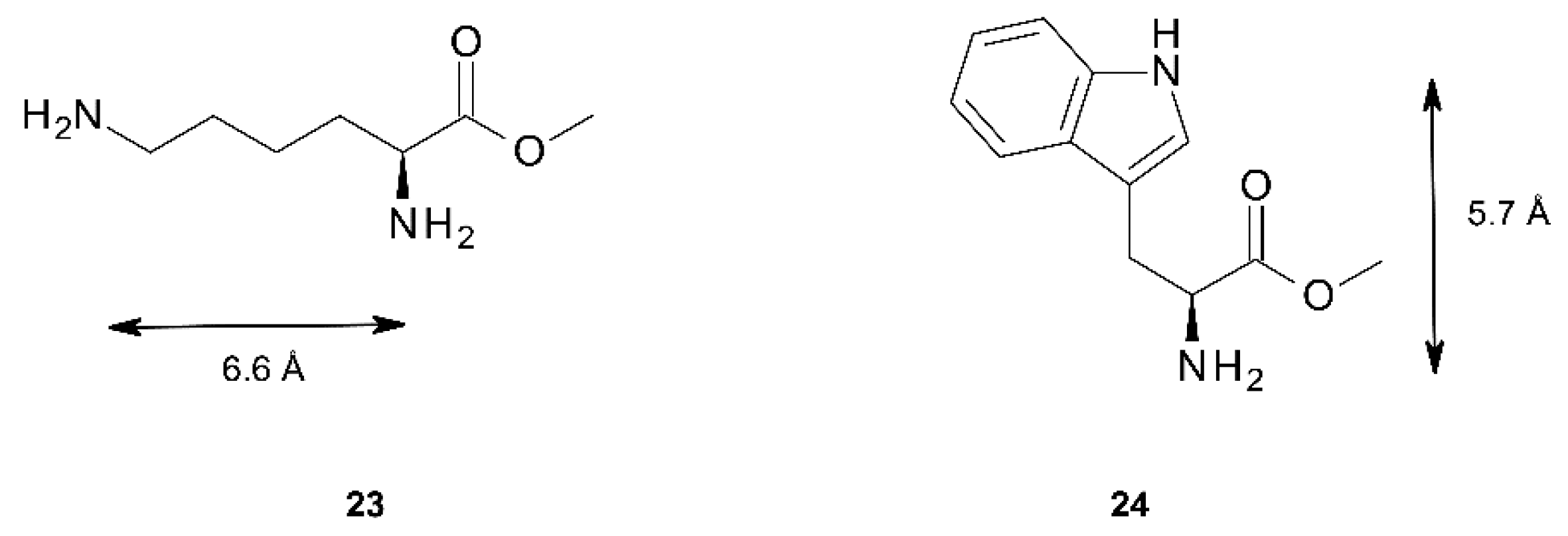
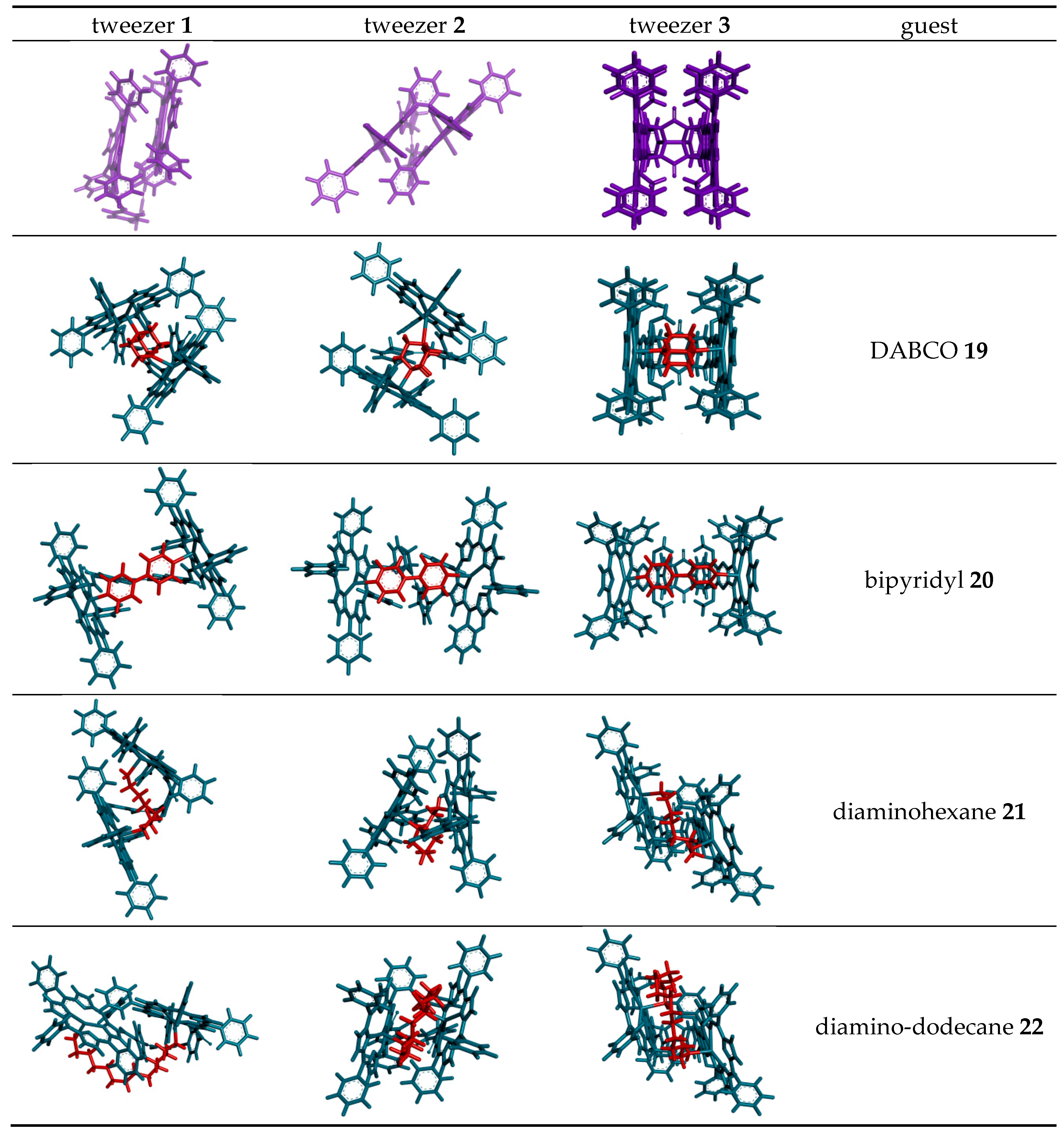
2.3. Conformational Analysis of Host-Guest Complex Geometry

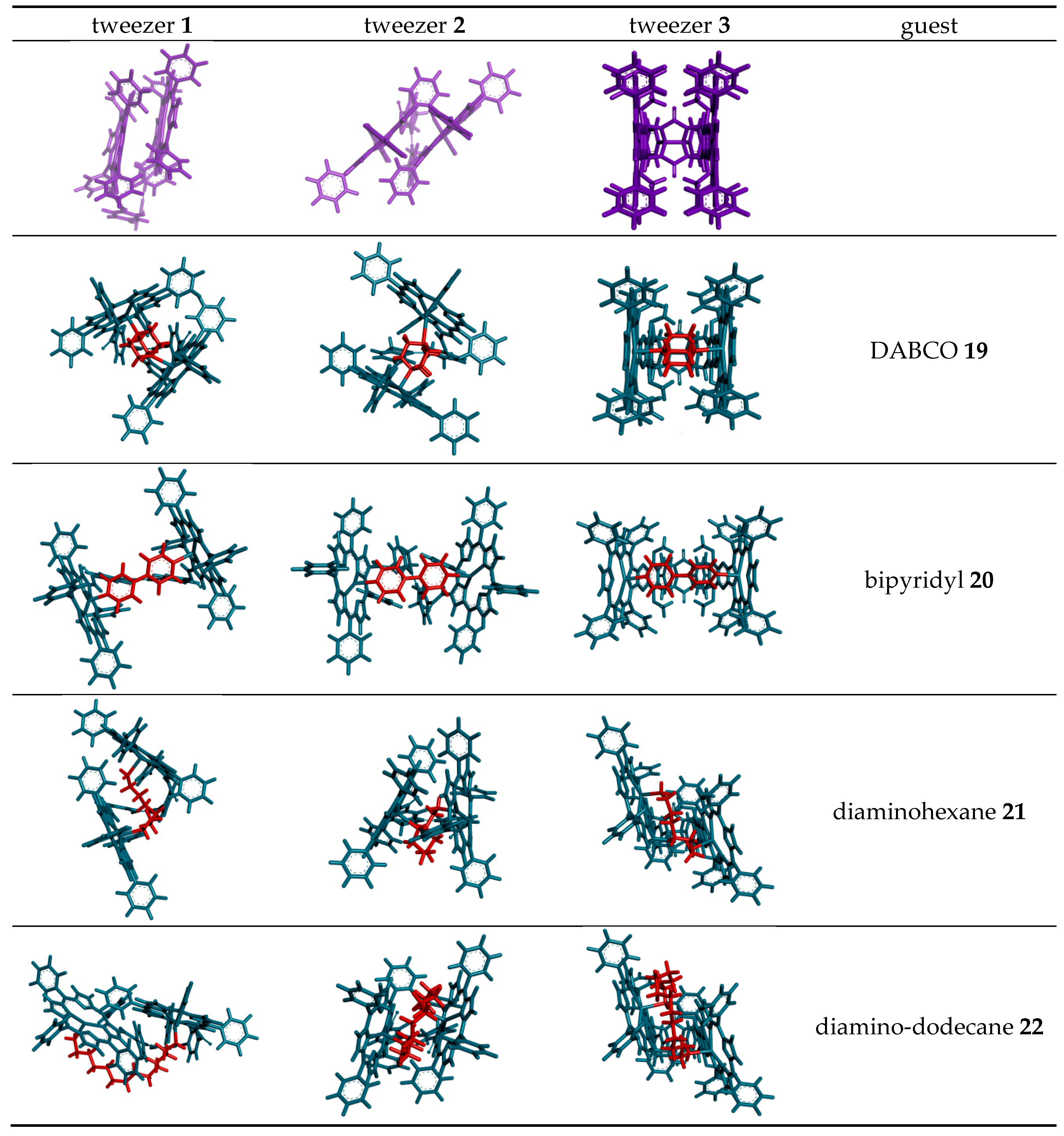
| Guest | Host | ||||||
|---|---|---|---|---|---|---|---|
| Z-1 | Z-2 | 3 | |||||
| Dihedral Angle a | CO–CO Distance | Zn–Zn Distance | Dihedral Angle a | CO–CO Distance | Zn–Zn Distance | Zn–Zn Distance | |
| (free tweezer) | 0° | 8.5 Å | 4.9 Å | 6.5° | 4.9 Å | 6.1 Å | 6.3 Å b |
| DABCO 19 | 0.2° | 7.9 Å | 7.0 Å | 11.2° | 5.4 Å | 7.3 Å | 7.3 Å |
| 4,4′-bipyridyl 20 | 0.1° | 10.6 Å | 11.3 Å | 11.5° | 5.9 Å | 11.6 Å | 11.5 Å |
| 1,6-diaminohexane 21 | 0.5° | 7.1 Å | 8.4 Å | 10.4° | 5.7 Å | 5.8 Å | 8.5 Å |
| 1,12-diaminododecane 22 | c | c | c | 10.5° | 4.8 Å | 6.1 Å | 8.2 Å |
| Lys methylester 23 | 0.3° | 6.9 Å | 9.9 Å | 8.5° | 5.3 Å | 8.5 Å | 11.3 Å |
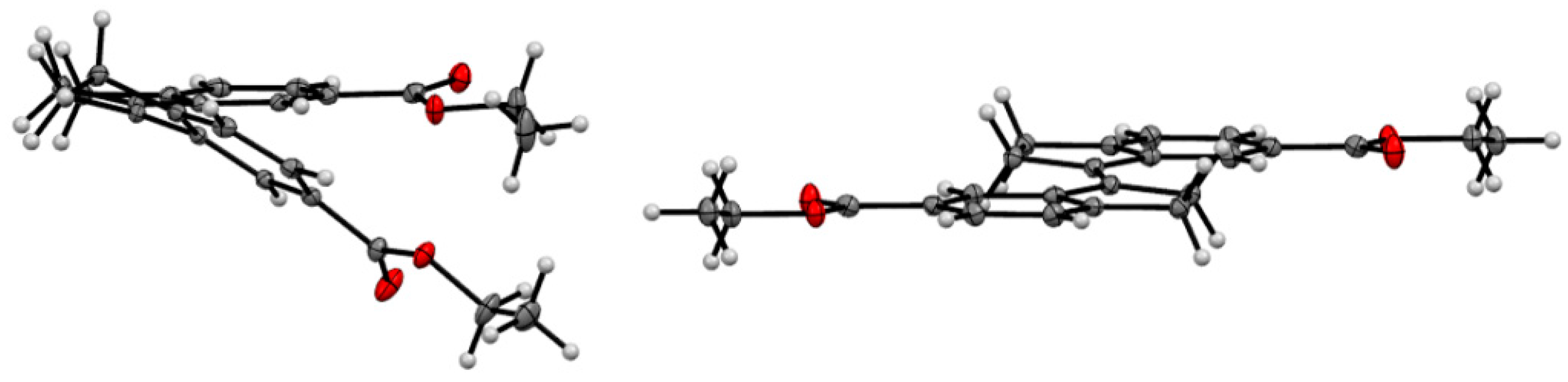
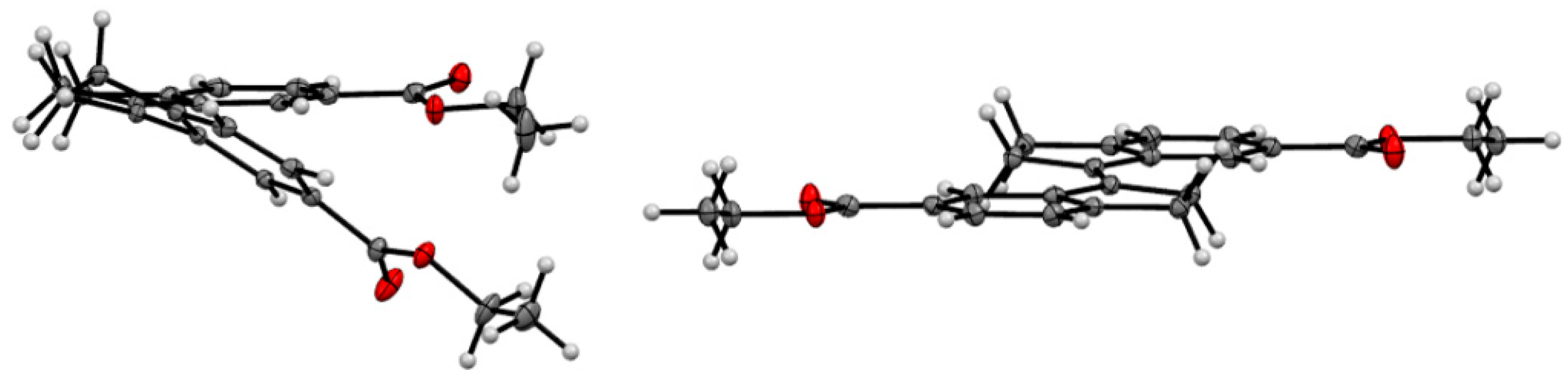
2.4. X-ray Crystallography of the Stiff Stilbene Spacer
2.5. Binding of Dinitrogen Guests
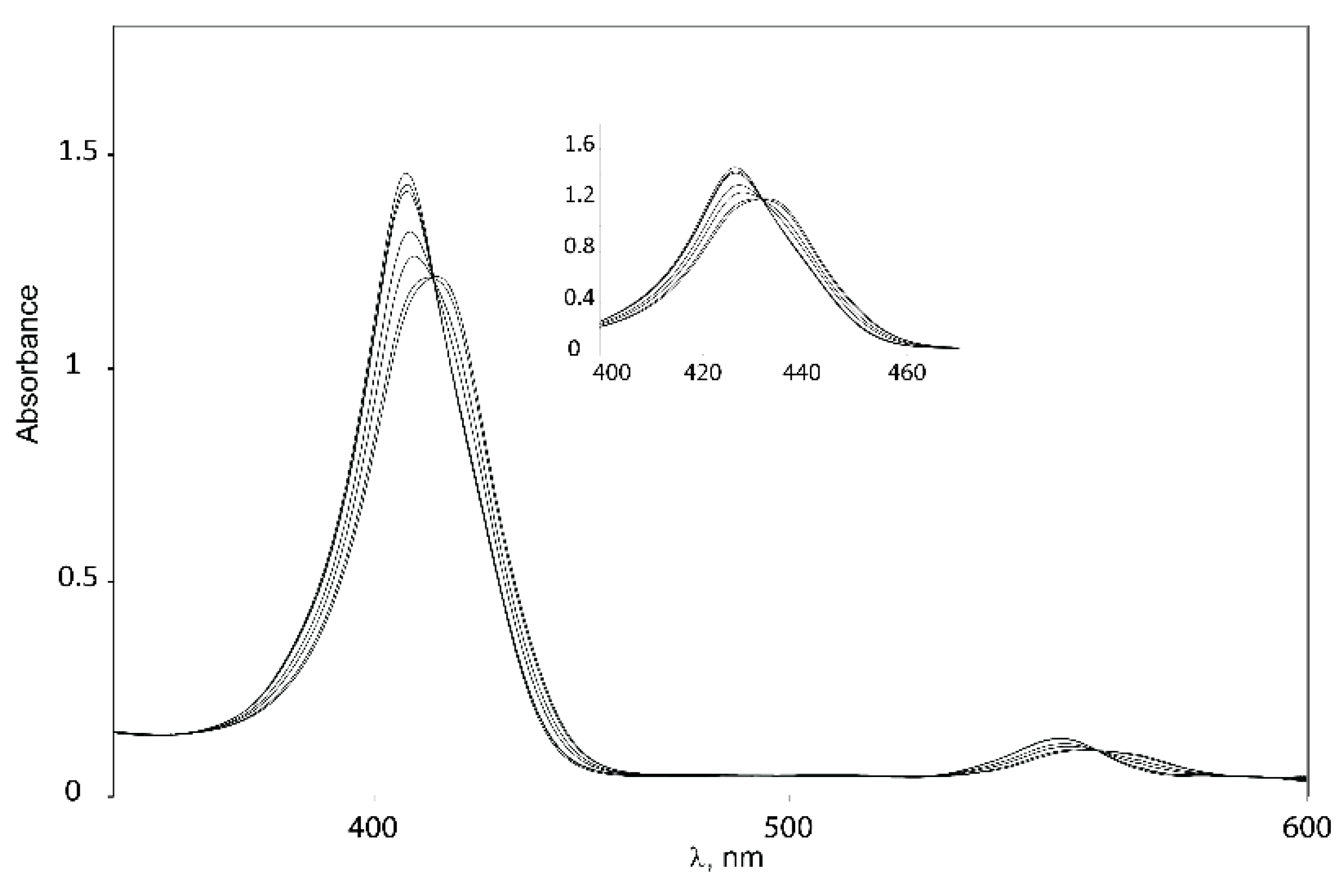
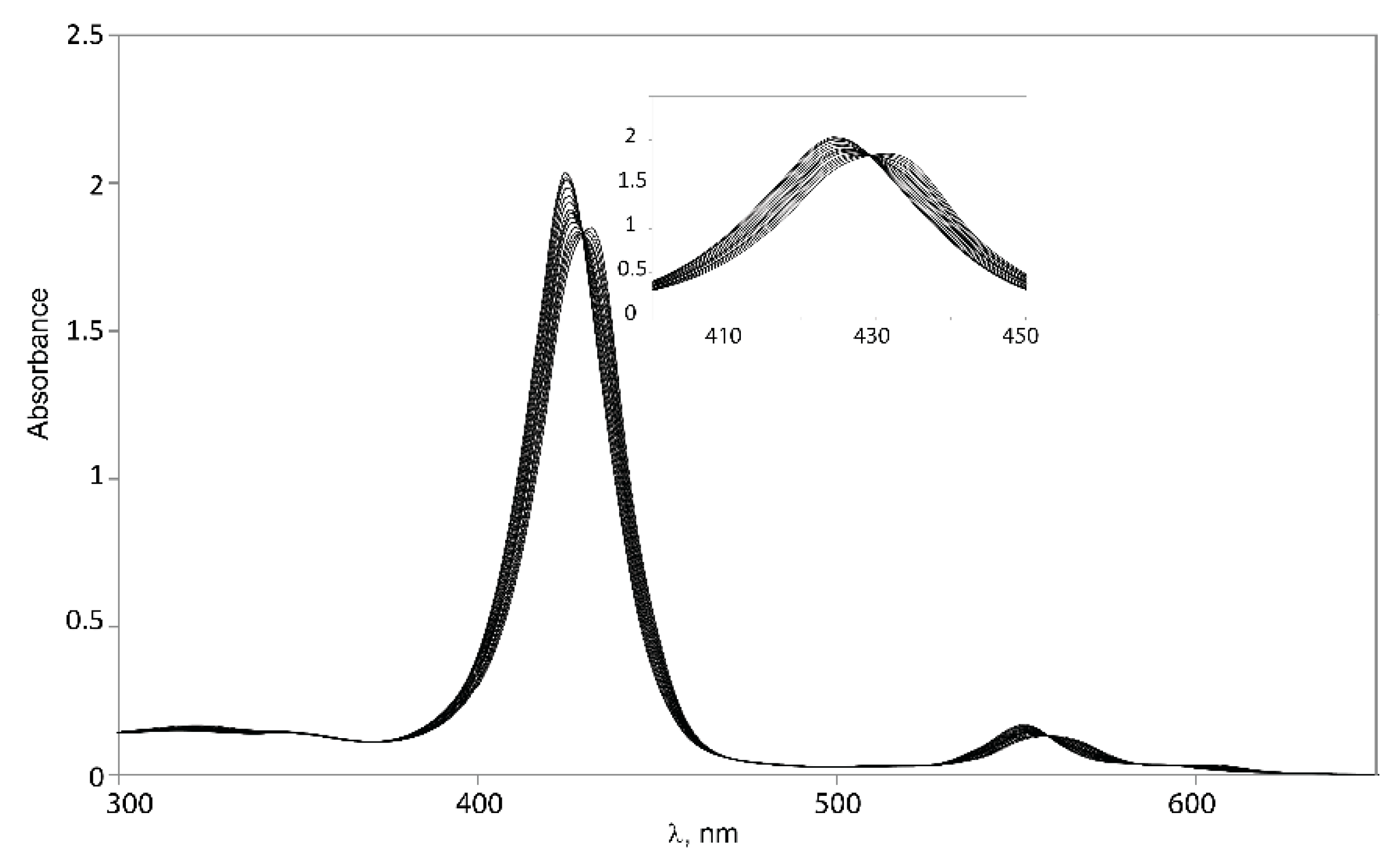
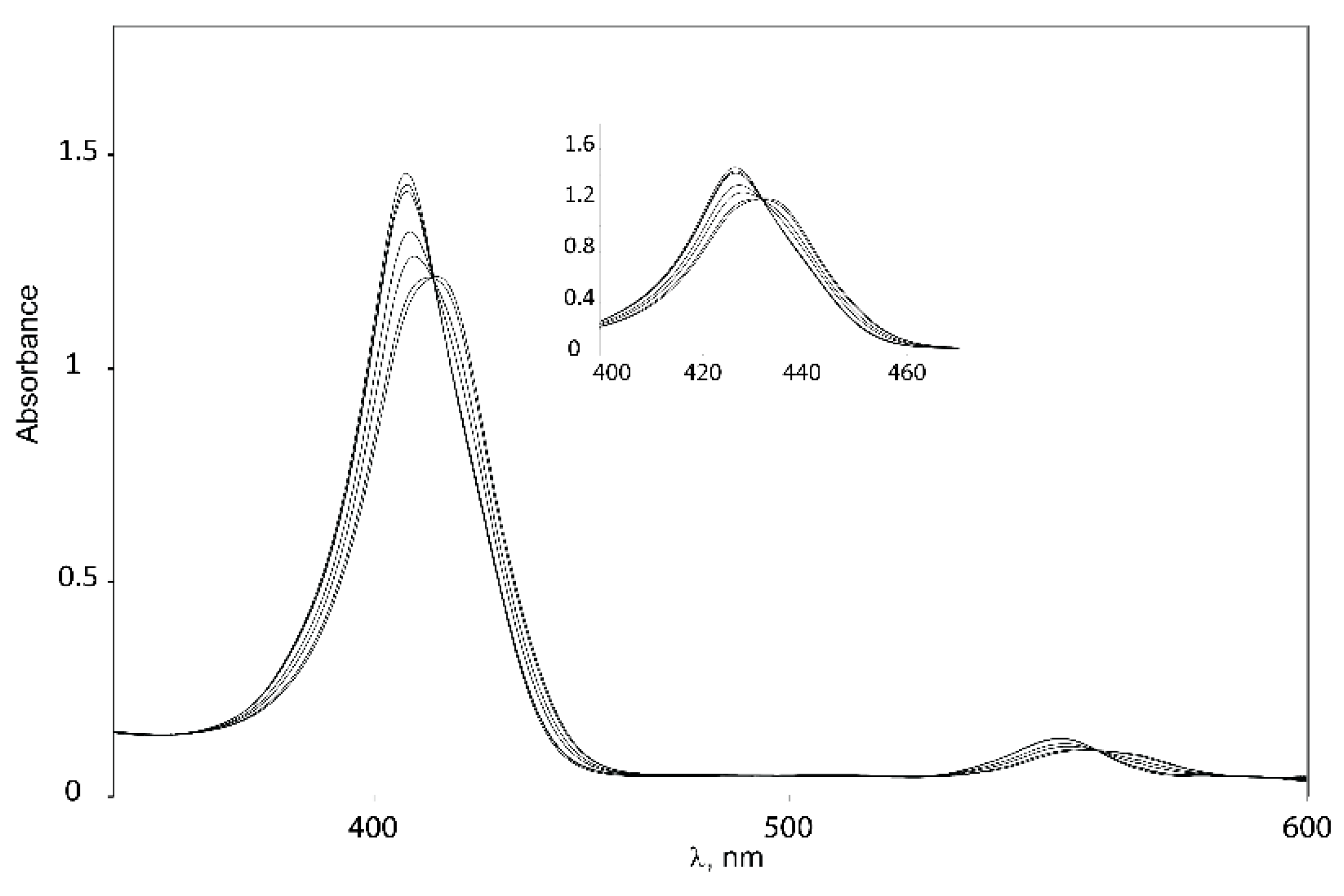
2.5.1. UV-Vis Spectroscopy
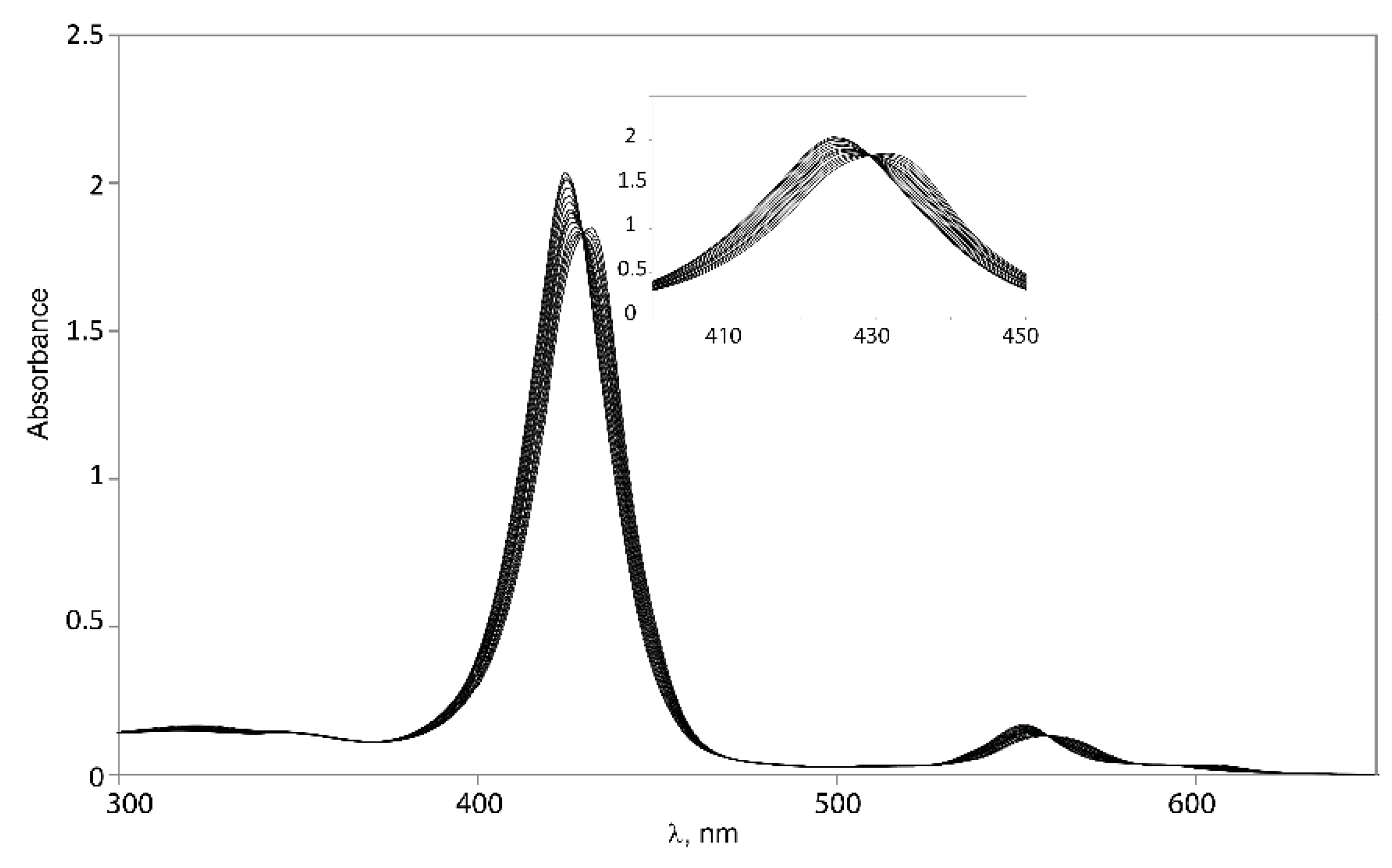
| Guest | (Δλ = λcomplex – λtweezer) Host | |||
|---|---|---|---|---|
| Z-1 | Z-2 | 3 | Zn-TPP | |
| DABCO 19 a | 5 | 5 | 4 b | 10 |
| 4,4′-bipyridyl 20 | 7 | 7 | 5 | 8 |
| 1,6-diaminohexane 21 | 9 | 9 | 10 b | 10 |
| 1,12-diaminododecane 22 | 10 | 10 | 10 | 10 |
| Guest | Host Ka/M−1 | |||
|---|---|---|---|---|
| Z-1 | Z-2 | 3 | Zn-TPP a | |
| DABCO 19 b | 2 × 104 | 4 × 105 | 3 × 106 c | 8 × 103 |
| 4,4′-bipyridyl 20 | 4 × 106 | 2 × 105 | 2 × 106 | - |
| 1,6-diaminohexane 21 | 6 × 106 | 5 × 105 | 2 × 106 c | 5 × 104 |
| 1,12-diaminododecane 22 | 8 × 105 | 2 × 105 | 2 × 105 | - |
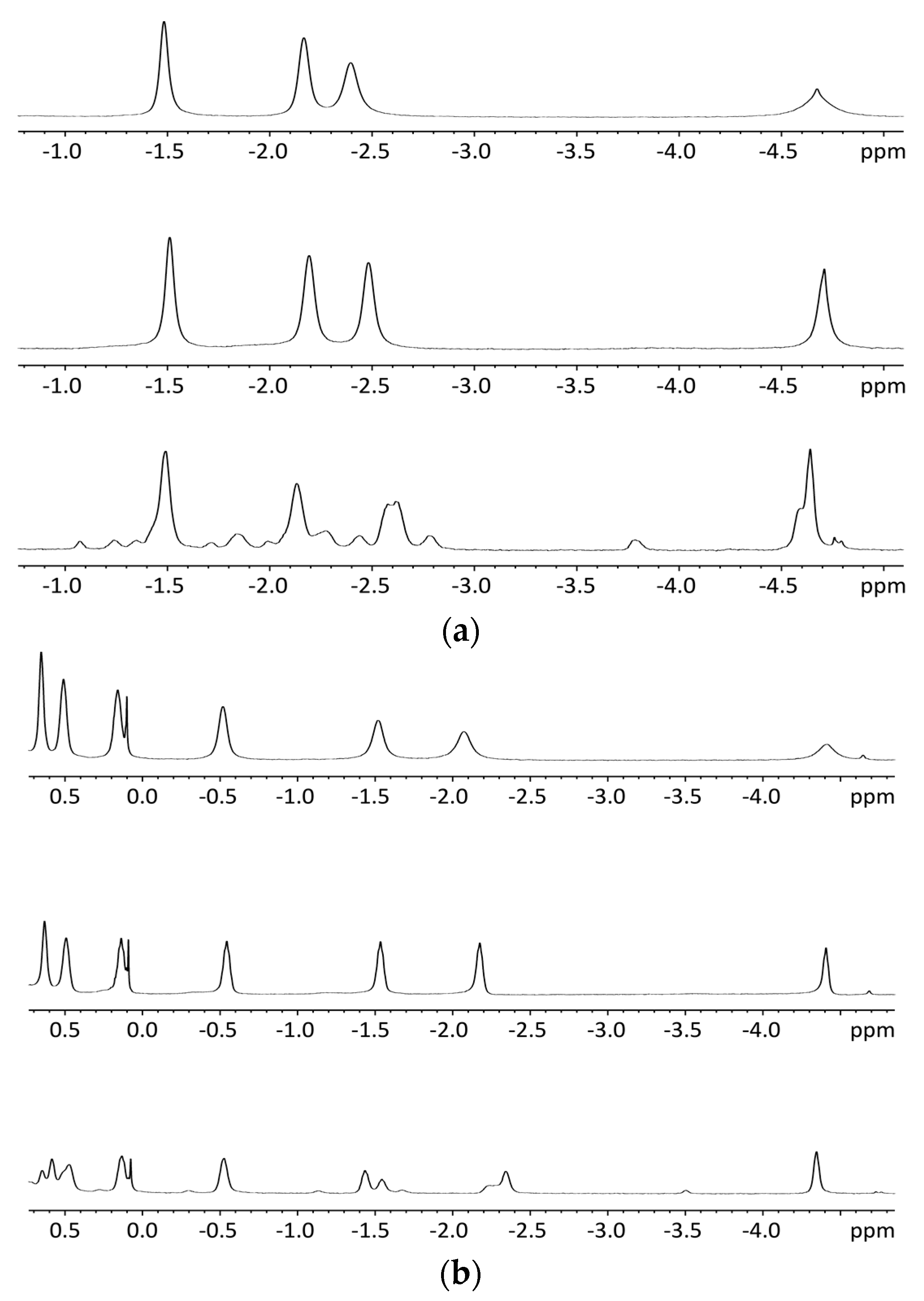
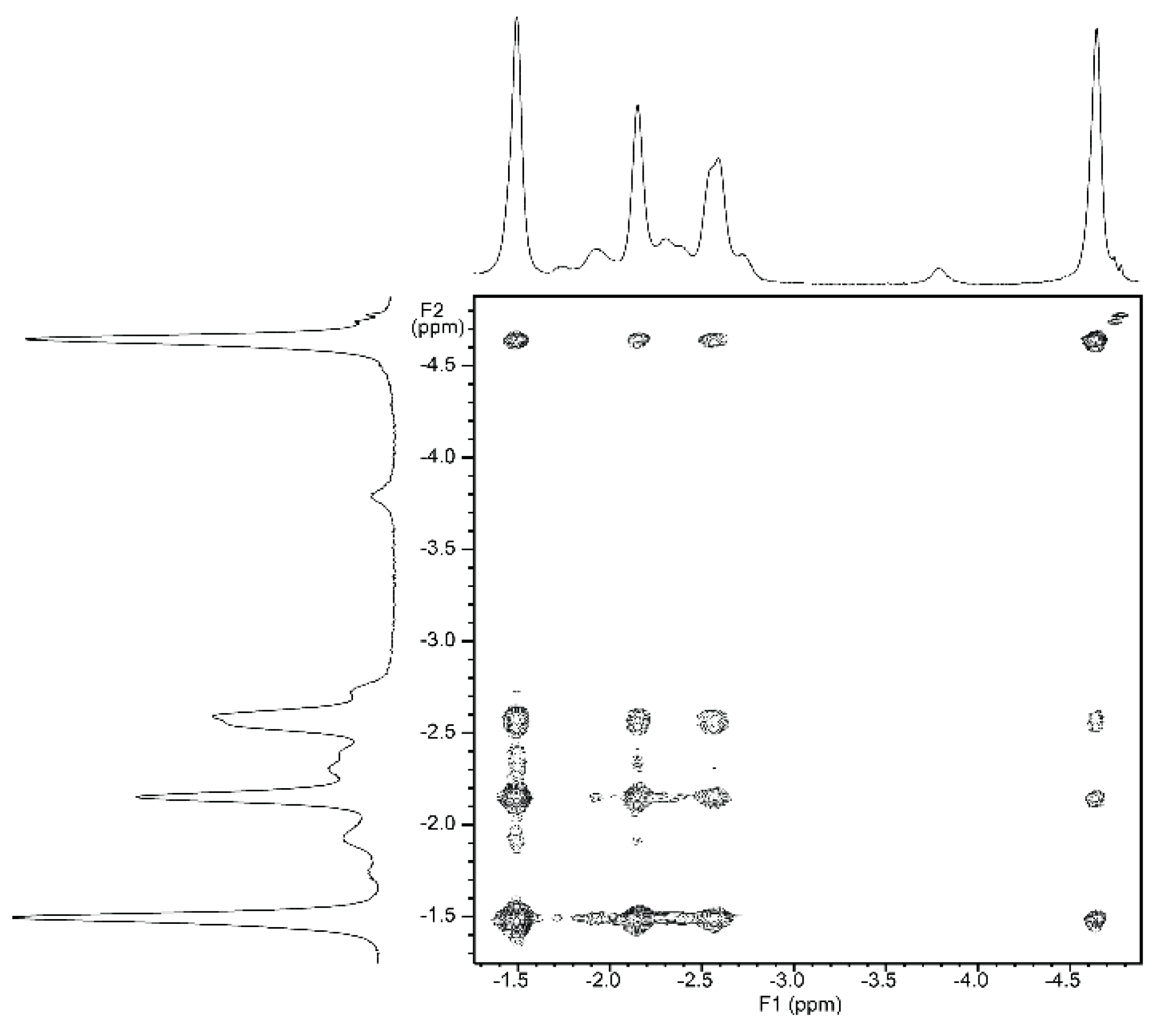
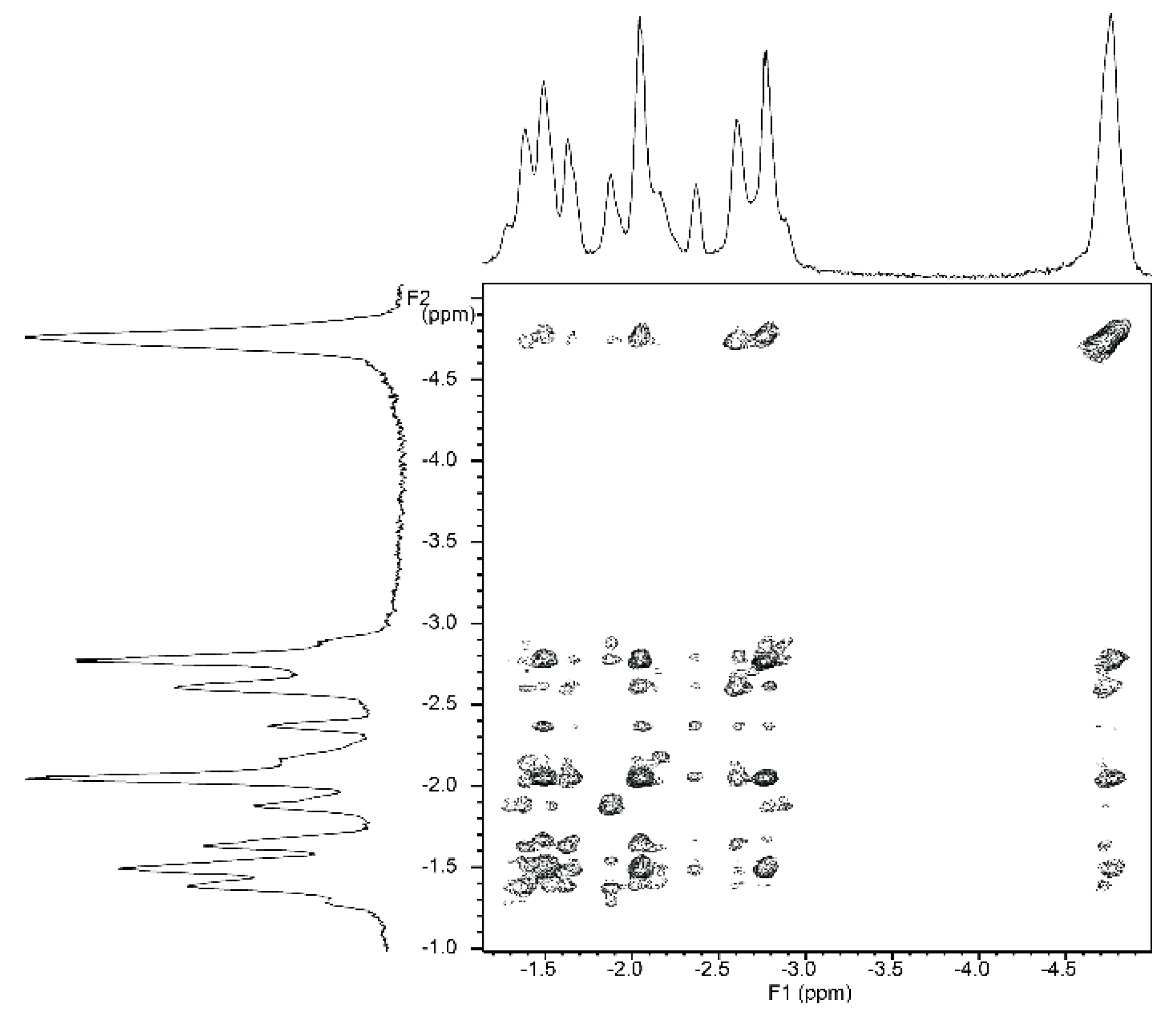
2.5.2. NMR Spectroscopy
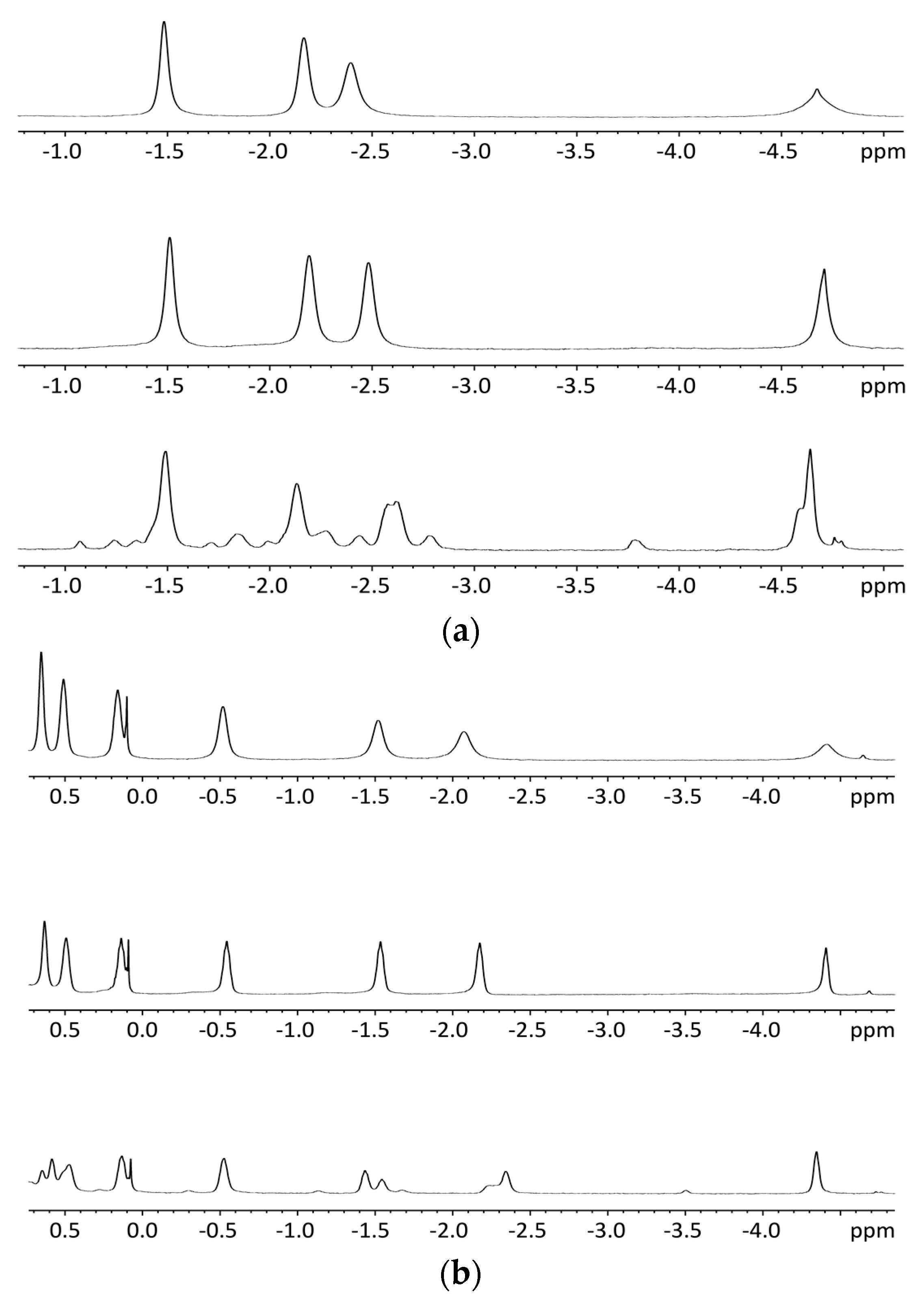
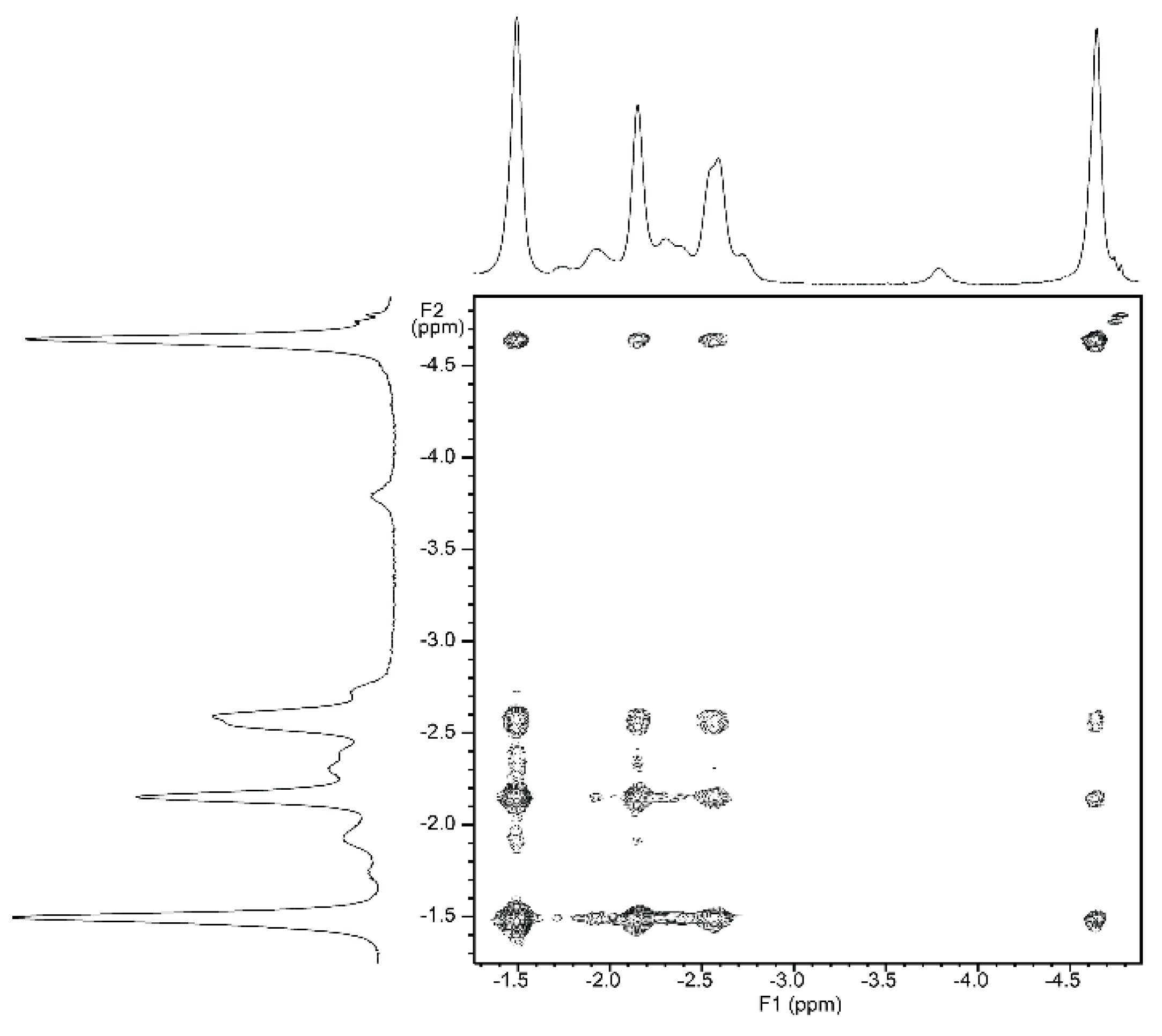
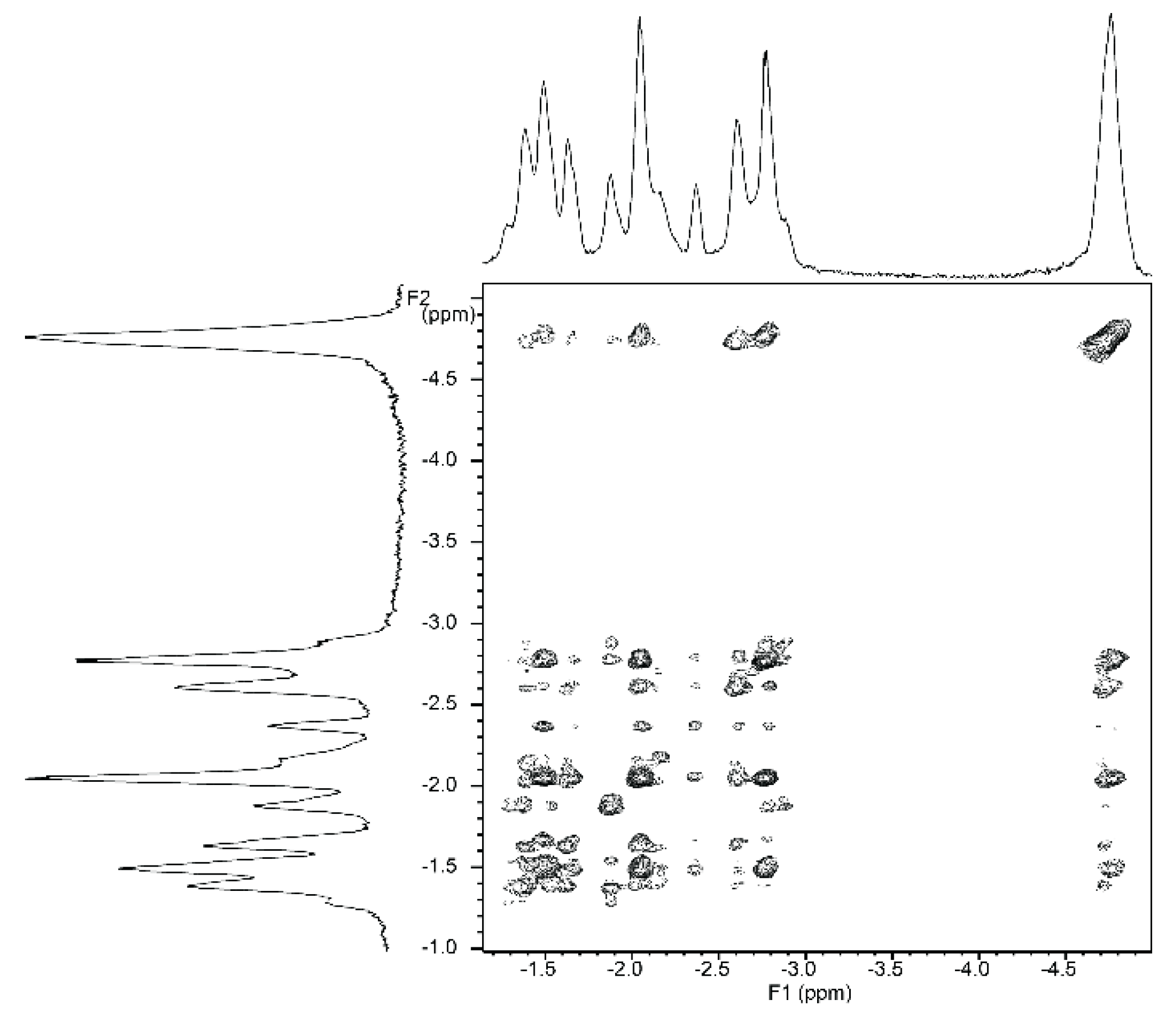
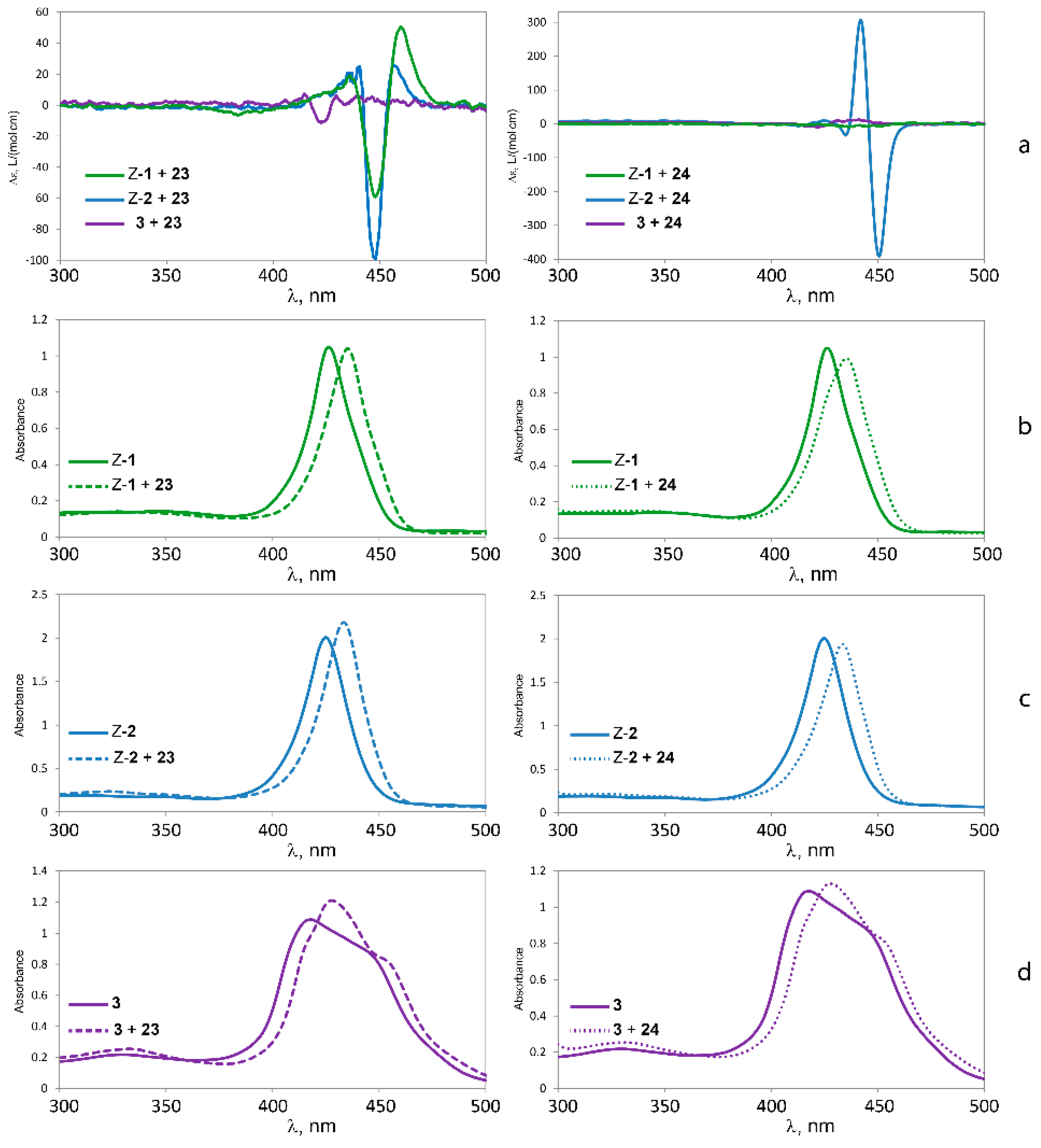
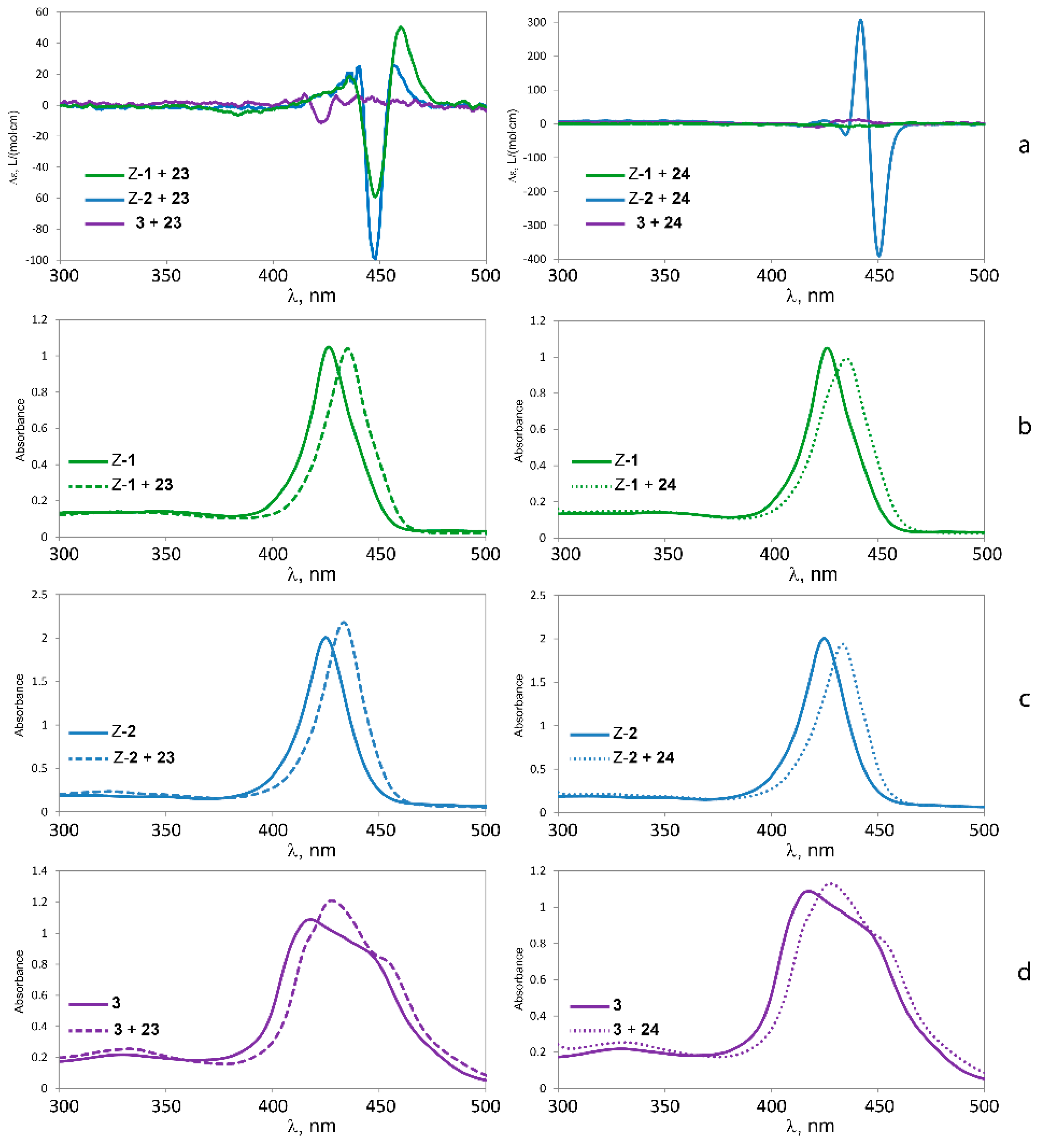
2.5.3. Circular Dichroism
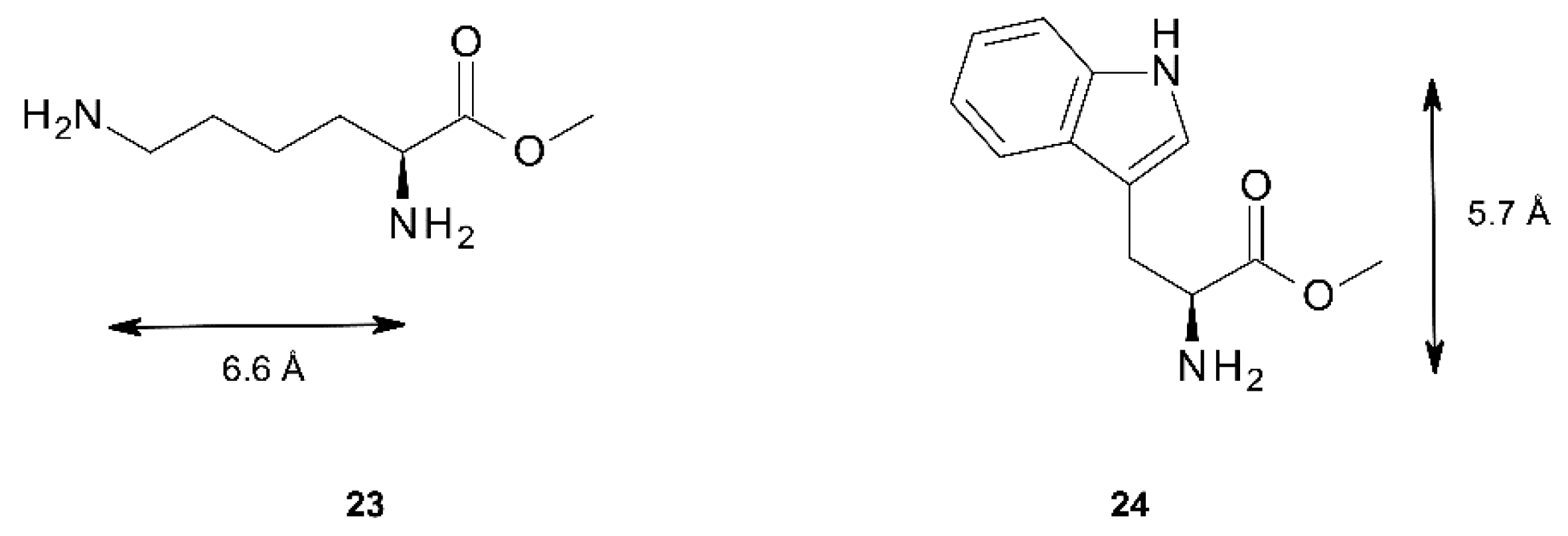
| Guest | Host | |||||
|---|---|---|---|---|---|---|
| Z-1 | Z-2 | 3 | ||||
| λ/Δε | ACD | λ/Δε | ACD | λ/Δε | ACD | |
| 23 | 460 nm (+50) | +109 | 457 nm (+26) | +125 | - | |
| 448 nm (−59) | 448 nm (−99) | |||||
| 24 | - | 451 nm (−391) | −697 | - | ||
| 442 nm (+306) | ||||||
3. Experimental Section
3.1. General
3.2. Syntheses
Alternative Route 1
Alternative Route 2



Photoisomerizations

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Valderreya, V.; Aragaya, G.; Ballester, P. Porphyrin tweezer receptors: Binding studies, conformational properties and applications. Coord. Chem. Rev. 2014, 258–259, 137–156. [Google Scholar] [CrossRef]
- Beletskaya, I.; Tyurin, V.S.; Tsivadze, A.Y.; Guilard, R.; Stern, C. Supramolecular chemistry of metalloporphyrins. Chem. Rev. 2009, 109, 1659–1713. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Yotsukura, M.; Noji, M.; Takanami, T. Bis(zinc porphyrin) as a CD-sensitive bidentate host molecule: Direct determination of absolute configuration of mono-alcohols. Chem. Commun. 2015, 51, 11068–11071. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Ikbal, S.A.; Dhamija, A.; Rath, S.P. Highly Enhanced Bisignate Circular Dichroism of Ferrocene-Bridged Zn(II) Bisporphyrin Tweezer with Extended Chiral Substrates due to Well-Matched Host-Guest System. Inorg. Chem. 2014, 53, 2381–2395. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Ikbal, S.A.; Rath, S.P. Synthesis, Structure, and Properties of a Series of Chiral Tweezer-Diamine Complexes Consisting of an Achiral Zinc(II) Bisporphyrin Host and Chiral Diamine Guest: Induction and Rationalization of Supramolecular Chirality. Inorg. Chem. 2014, 53, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Ikbal, S.A.; Brahma, S.; Rath, S.P. Transfer and control of molecular chirality in the 1:2 host-guest supramolecular complex consisting of Mg(II)bisporphyrin and chiral diols: The effect of H-bonding on the rationalization of chirality. Chem. Commun. 2014, 50, 14037–14040. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Ikbal, Sk.A.; Brahma, S.; Rath, S.P. Formation of exo–exo, exo–endo and tweezer conformation induced by axial ligand in a Zn(II) bisporphyrin: Synthesis, structure and properties. Polyhedron 2013, 52, 761–769. [Google Scholar] [CrossRef]
- Tanasova, M.; Borhan, B. Conformational Preference in Bis(porphyrin) Tweezer Complexes: A Versatile Chirality Sensor for α-Chiral Carboxylic Acids. Eur. J. Org. Chem. 2012, 2012, 3261–3269. [Google Scholar] [CrossRef]
- Petrovic, A.G.; Vantomme, G.; Negrón-Abril, Y.; Lubian, E.; Saielli, G.; Menegazzo, I.; Cordero, R.; Proni, G.; Nakanishi, K.; Carofiglio, T.; et al. Bulky Melamine-Based Zn-Porphyrin Tweezer as a CD Probe of Molecular Chirality. Chirality 2011, 23, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Nakanishi, K.; Berova, N. Porphyrins and metalloporphyrins: Versatile circular dichroic reporter groups for structural studies. Chirality 2000, 12, 237–255. [Google Scholar] [CrossRef]
- Huang, X.; Fujioka, N.; Pescitelli, G.; Koehn, F.E.; Williamson, R.T.; Nakanishi, K.; Berova, N. Absolute Configurational Assignments of Secondary Amines by CD-Sensitive Dimeric Zinc Porphyrin Host. J. Am. Chem. Soc. 2002, 124, 10320–10335. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Borhan, B.; Rickman, B.H.; Nakanishi, K.; Berova, N. Zinc Porphyrin Tweezer in Host ± Guest Complexation: Determination of Absolute Configurations of Primary Monoamines by Circular Dichroism. Chem. Eur. J. 2000, 6, 216–224. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. Origin, Control, and Application of Supramolecular Chirogenesis in Bisporphyrin-Based Systems. Acc. Chem. Res. 2004, 37, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Borovkov, V.V.; Lintuluoto, J.M.; Hembury, G.A.; Sugiura, M.; Arakawa, R.; Inoue, Y. Supramolecular Chirogenesis in Zinc Porphyrins: Interaction with Bidentate Ligands, Formation of Tweezer Structures, and the Origin of Enhanced Optical Activity. J. Org. Chem. 2003, 68, 7176–7192. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Nonoguchi, M.; Aya, T.; Ogoshi, H. Molecular recognition of α,ω-diamines by metalloporphyrin dimer. Tetrahedron Lett. 1997, 38, 1603–1606. [Google Scholar] [CrossRef]
- Norrehed, S.; Polavarapu, P.; Yang, W.; Gogoll, A.; Grennberg, H. Conformational restriction of flexible molecules in solution by a semirigid bis-porphyrin molecular tweezer. Tetrahedron 2013, 69, 7131–7138. [Google Scholar] [CrossRef]
- Norrehed, S.; Johansson, H.; Grennberg, H.; Gogoll, A. Improved Stereochemical Analysis of Conformationally Flexible Diamines by Binding to a Bisporphyrin Molecular Clip. Chem. Eur. J. 2013, 19, 14631–14638. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, A.; Kunimune, T.; Ikeda, Y.; Moriguchi, T.; Araki, K. Binding properties of calixarene-based cofacial bisporphyrins. Chem. Lett. 2010, 39, 1155–1157. [Google Scholar] [CrossRef]
- Wahadoszamen, M.; Yamamura, T.; Momotake, A.; Nishimura, Y.; Arai, T. High Binding Affinity of DABCO with Porphyrin in a Porphyrin-cis-Stilbene-Porphyrin Triad. Heterocycles 2009, 79, 331–337. [Google Scholar]
- Fathalla, J.M. Jayawickramarajah. Configurational Isomers of a Stilbene-Linked Bis(porphyrin) Tweezer: Synthesis and Fullerene-Binding Studies. Eur. J. Org. Chem. 2009, 6095–6099. [Google Scholar] [CrossRef]
- Tsuge, A.; Ikeda, Y.; Moriguchi, T.; Araki, K. Preparation of cyclophanes having cofacial bisporphyrins and their binding properties. J. Porphyr. Phthalocyanines 2012, 16, 250–254. [Google Scholar] [CrossRef]
- Collman, J.P.; Wagenknecht, P.S.; Hutchinson, J.H. Molecular catalysis for multielectron redox reactions of small molecules: The “cofacial metallodiphorphyrin approach”. Angew. Chem. Int. Ed. Engl. 1994, 33, 1537–1553. [Google Scholar] [CrossRef]
- Merkasa, S.; Bouatraa, S.; Reina, R.; Piantanidab, I.; Zinicb, M.; Solladié, N. Pre-organized dinucleosides with pendant porphyrins for the formation of sandwich type complexes with DABCO with high association constants. J. Porphyr. Phthalocyanines 2015, 19, 535–546. [Google Scholar] [CrossRef]
- Solladié, N.; Aziat, F.; Bouatra, S.; Rein, R. Bis-porphyrin tweezers: Rigid or flexible linkers for better adjustment of the cavity to bidentate bases of various size. J. Porphyr. Phthalocyanines 2008, 12, 1250–1260. [Google Scholar] [CrossRef]
- Pescitelli, G.; Gabriel, S.; Wang, Y.; Fleischhauer, J.; Woody, R.W.; Berova, N. Theoretical Analysis of the Porphyrin-Porphyrin Exciton Interaction in Circular Dichroism Spectra of Dimeric Tetraarylporphyrins. J. Am. Chem. Soc. 2003, 125, 7613–7628. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Ikbal, S.A.; Dey, S.; Rath, S.P. Induction of supramolecular chirality in di-zinc(II) bisporphyrin via tweezer formation: Synthesis, structure and rationalization of chirality. Chem. Commun. 2012, 48, 4070–4072. [Google Scholar] [CrossRef] [PubMed]
- Promarak, V.; Burn, P.L. A new synthetic approach to porphyrin-α-diones and a -2,3,12,13-tetraone: Building blocks for laterally conjugated porphyrin arrays. J. Chem. Soc. Perkin Trans. I 2001, 14–20. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Crossley, M.J.; DeBernardis, J. Efficient peripheral functionalization of porphyrins. Tetrahedron 1982, 38, 685–692. [Google Scholar] [CrossRef]
- Shine, H.J.; Padilla, A.G.; Wu, S.-M. Ion radicals. 45. Reactions of zinc tetraphenylporphyrin cation radical perchlorate with nucleophiles. J. Org. Chem. 1979, 44, 4069–4075. [Google Scholar] [CrossRef]
- Fanning, J.C.; Mandel, F.S.; Gray, T.L. The reaction of metalloporphyrins with nitrogen dioxide. Tetrahedron 1979, 33, 1251–1255. [Google Scholar] [CrossRef]
- Giraudeau, A.; Callot, H.J.; Jordan, J.; Ezhar, I.; Gross, M. Substituent effects in the electroreduction of porphyrins and metalloporphyrins. J. Am. Chem. Soc. 1979, 101, 3857–3862. [Google Scholar] [CrossRef]
- Crossley, M.J.; King, L.G.; Newsom, I.A.; Sheehan, G.S. Investigation of a “reverse” approach to extended porphyrin systems. Synthesis of a 2,3-diaminoporphyrin and its reactions with α-diones. J. Chem. Soc. Perkin Trans. 1 1996, 2675–2684. [Google Scholar] [CrossRef]
- Erdélyi, M.; Gogoll, A. Rapid Homogeneous-Phase Sonogashira Coupling Reactions Using Controlled Microwave Heating. J. Org. Chem. 2001, 66, 4165–4169. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Rukavishnikov, A.V.; Petukhov, P.A.; Zaikova, T.O.; Jin, C.; Keana, J.F.W. Nanoscale Tripodal 1,3,5,7-Tetrasubstituted Adamantanes for AFM Applications. J. Org. Chem. 2003, 68, 4862–4869. [Google Scholar] [CrossRef] [PubMed]
- Kosinki, C.; Hirsch, A.; Heineman, F.W.; Hampel, F. An Iterative Approach to cis-Oligodiacetylenes. Eur. J. Org. Chem. 2001, 2001, 3879–3890. [Google Scholar] [CrossRef]
- McMurry, J.E. Carbonyl-coupling reactions using low-valent titanium. Chem. Rev. 1989, 89, 1513–1524. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Frank, R.; Lemmen, P.; Lenoir, D.; Lex, J.; Inoue, Y. Synthesis, structural characterization and photoisomerization of cyclic stilbenes. Tetrahedron 2012, 68, 4048–4056. [Google Scholar] [CrossRef]
- Quick, M.; Berndt, F.; Dobryakov, A.L.; Ioffe, I.N.; Granovsky, A.A.; Knie, C.; Mahrwald, R.; Lenoir, D.; Ernsting, N.P.; Kovalenko, S.A. Photoisomerization Dynamics of Stiff-Stilbene in Solution. J. Phys. Chem. B 2014, 118, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, M.; Tanatani, A.; Kaneko, T.; Azumaya, I.; Masu, H.; Hashizume, D.; Kagechika, H.; Muranaka, A.; Uchiyama, M. Synthesis of porphyrinylamide and observation of N-methylation-induced trans-cis amide conformational alteration. Tetrahedron 2013, 69, 10927–10932. [Google Scholar] [CrossRef]
- Schaefer, W.P.; Abulu, J. An Indanyl Precursor to a Chiral Spiro Compound. Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 1995, 51, 2364–2366. [Google Scholar] [CrossRef]
- Ogawa, K.; Harada, J.; Tomoda, S. Unusually short ethylene bond and large amplitude torsional motion of (E)-stilbenes in crystals. X-ray crystallographic study of “stiff” stilbenes. Acta Crystallogr. Sect. B Struct. Sci. 1995, 51, 240–248. [Google Scholar] [CrossRef]
- Jovanovic, J.; Elling, W.; Schurmann, M.; Preut, H.; Spiteller, M. (Z)-2,3,2′,3′-Tetra-hydro-[1,1′]biindenylidene. Acta Crystallogr. Sect. E: Struct. Rep. Online 2002, 58, o35–o36. [Google Scholar] [CrossRef]
- Harada, J.; Ogawa, K.; Tomoda, S. “Stiff” cis-Stilbenes. (Z)-6,6′-Dimethyl-1,1′-biindanylidene and (Z)-4,4′,7,7′-Tetramethyl-1,1′-biindanylidene. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1995, 51, 2125–2127. [Google Scholar] [CrossRef]
- Shimasaki, T.; Kato, S.; Shinmyozu, T. Synthesis, Structural, Spectral, and Photoswitchable Properties of cis- and trans-2,2,2′,2′-Tetramethyl-1,1′-indanylindanes. J. Org. Chem. 2007, 72, 6251–6254. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Fischer, E. Isosbestic points. J. Chem. Soc. 1962, 3044–3052. [Google Scholar] [CrossRef]
- Mauzerall, D. Spectra of Molecular Complexes of Porphyrins in Aqueous Solution. Biochemistry 1965, 4, 1801–1810. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A.; Marsol, C.; Pescitelli, G.; di Bari, L.; Salvadori, P.; Huang, X.; Fujioka, N.; Berova, N.; Cao, X.; Freedman, T.B.; et al. (R)-(+)- and (S)-(−)-1-(9-Phenanthryl)ethylamine: Assignment of Absolute Configuration by CD Tweezer and VCD Methods, and Difficulties Encountered with the CD Exciton Chirality Method. Eur. J. Org. Chem. 2002, 2002, 1788–1796. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M.; Stone, A.J. Exciton coupling in porphyrin dimers. Chem. Phys. 1989, 133, 395–404. [Google Scholar] [CrossRef]
- Hunter, C.A.; Meah, M.N.; Sanders, J.K.M. DABCO-Metalloporphyrin Binding: Ternary Complexes, Host-Guest Chemistry, and the Measurement of T-T Interactions. J. Am. Chem. Soc. 1990, 112, 5773–5780. [Google Scholar] [CrossRef]
- Anderson, H.L.; Hunter, C.A.; Meah, M.N.; Sanders, J.K.M. Thermodynamics of induced-fit binding inside polymacrocyclic porphyrin hosts. J. Am. Chem. Soc. 1990, 112, 5780–5789. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Yamamoto, N.; Lintuluoto, J.M.; Tanaka, T.; Inoue, Y. Supramolecular Chirality Induction in Bis(Zinc Porphyrin) by Amino Acid Derivatives: Rationalization and Applications of the Ligand Bulkiness Effect. Chirality 2001, 13, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Wokaun, A.; Ernst, R.R. Selective detection of multiple quantum transitions in NMR by two-dimensional spectroscopy. Chem. Phys. Lett. 1977, 52, 407–412. [Google Scholar] [CrossRef]
- Shaka, A.J.; Freeman, R. Simplification of NMR spectra by filtration through multiple-quantum coherence. J. Magn. Reson. 1983, 51, 169–173. [Google Scholar] [CrossRef]
- Mueller, L.P. E.COSY, a simple alternative to E.COSY. J. Magn. Reson. 1987, 72, 191–196. [Google Scholar]
- Davis, A.L.; Keeler, J.; Laue, E.D.; Moskau, D. Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. J. Magn. Reson. 1992, 98, 207–216. [Google Scholar] [CrossRef]
- Hurd, R.E.; John, B.K. Gradient-enhanced proton-detected heteronuclear multiple-quantum coherence spectroscopy. J. Magn. Reson. 1991, 91, 648–653. [Google Scholar] [CrossRef]
- Wagner, R.; Berger, S. Gradient-Selected NOESY—A Fourfold Reduction of the Measurement Time for the NOESY Experiment. J. Magn. Reson. A 1996, 123, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Bax, A.; Davis, D.G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. [Google Scholar] [CrossRef]
- Braunschweiler, L.; Ernst, R.R. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. [Google Scholar] [CrossRef]
- Thordarsson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blom, M.; Norrehed, S.; Andersson, C.-H.; Huang, H.; Light, M.E.; Bergquist, J.; Grennberg, H.; Gogoll, A. Synthesis and Properties of Bis-Porphyrin Molecular Tweezers: Effects of Spacer Flexibility on Binding and Supramolecular Chirogenesis. Molecules 2016, 21, 16. https://doi.org/10.3390/molecules21010016
Blom M, Norrehed S, Andersson C-H, Huang H, Light ME, Bergquist J, Grennberg H, Gogoll A. Synthesis and Properties of Bis-Porphyrin Molecular Tweezers: Effects of Spacer Flexibility on Binding and Supramolecular Chirogenesis. Molecules. 2016; 21(1):16. https://doi.org/10.3390/molecules21010016
Chicago/Turabian StyleBlom, Magnus, Sara Norrehed, Claes-Henrik Andersson, Hao Huang, Mark E. Light, Jonas Bergquist, Helena Grennberg, and Adolf Gogoll. 2016. "Synthesis and Properties of Bis-Porphyrin Molecular Tweezers: Effects of Spacer Flexibility on Binding and Supramolecular Chirogenesis" Molecules 21, no. 1: 16. https://doi.org/10.3390/molecules21010016
APA StyleBlom, M., Norrehed, S., Andersson, C.-H., Huang, H., Light, M. E., Bergquist, J., Grennberg, H., & Gogoll, A. (2016). Synthesis and Properties of Bis-Porphyrin Molecular Tweezers: Effects of Spacer Flexibility on Binding and Supramolecular Chirogenesis. Molecules, 21(1), 16. https://doi.org/10.3390/molecules21010016