
Chemical Constituents from the Aerial Parts of Cyrtopodium paniculatum

 ,
,  , , ,
, , ,
Abstract
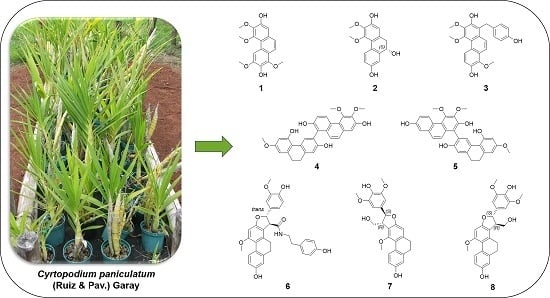
:
1. Introduction
2. Results and Discussion
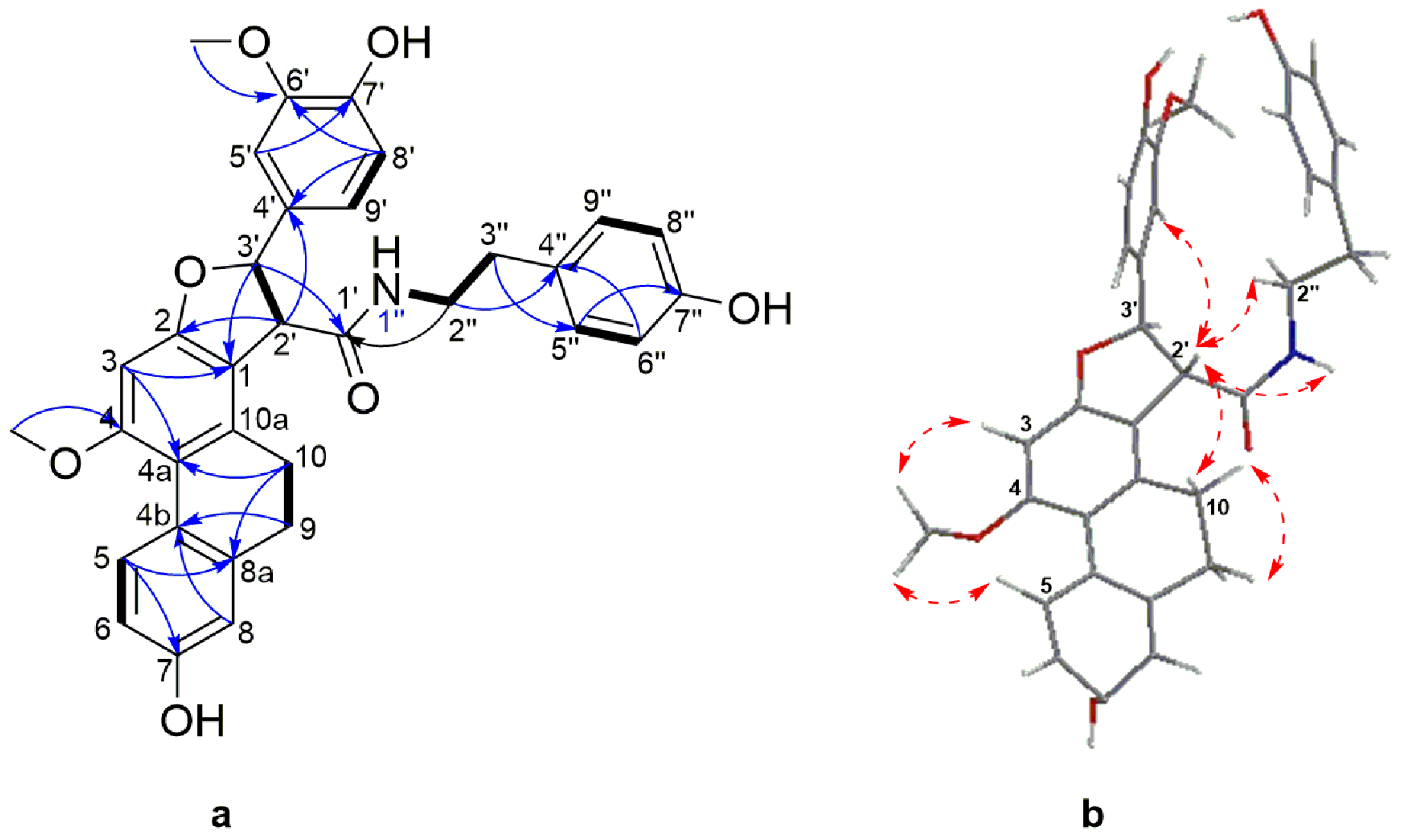
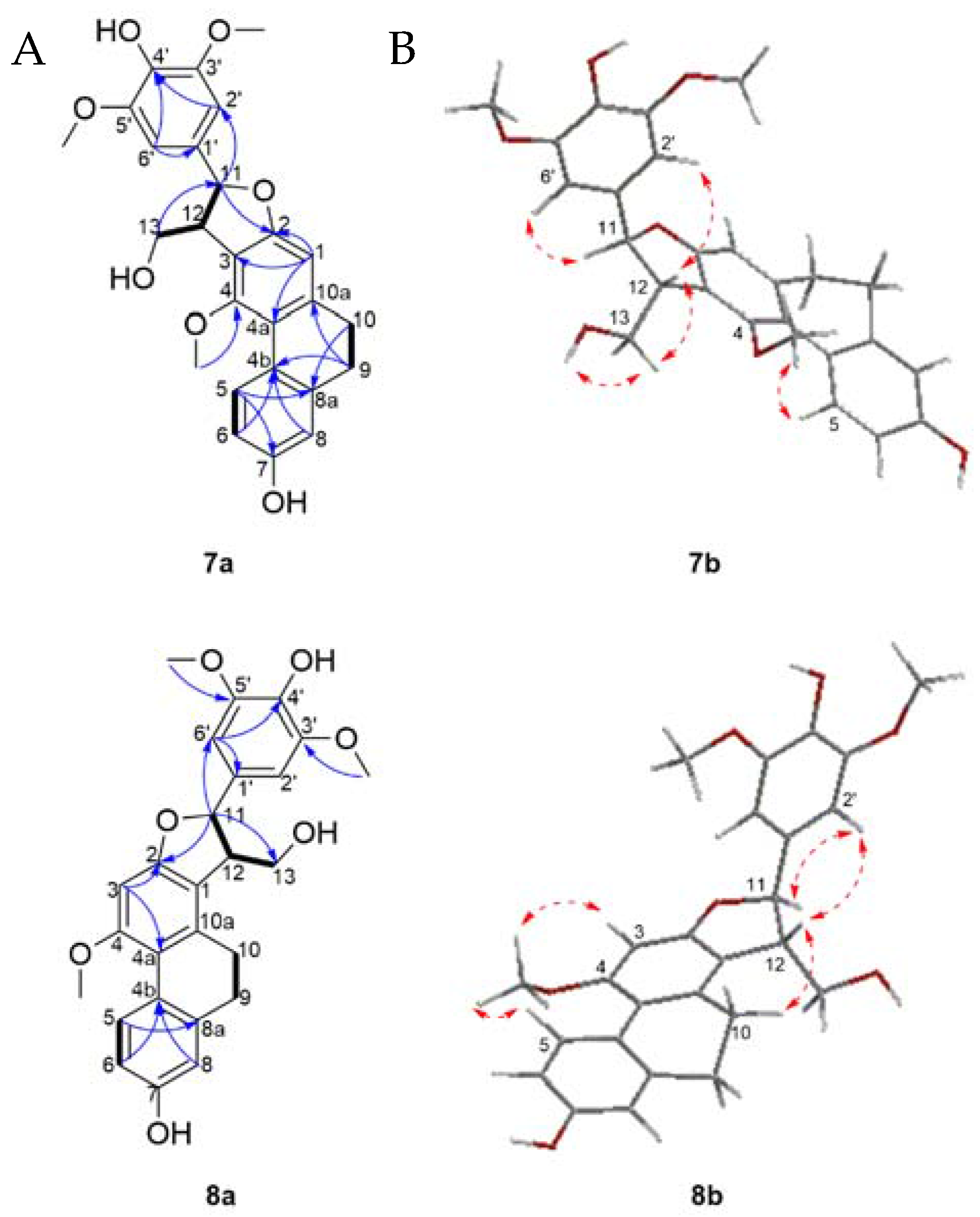
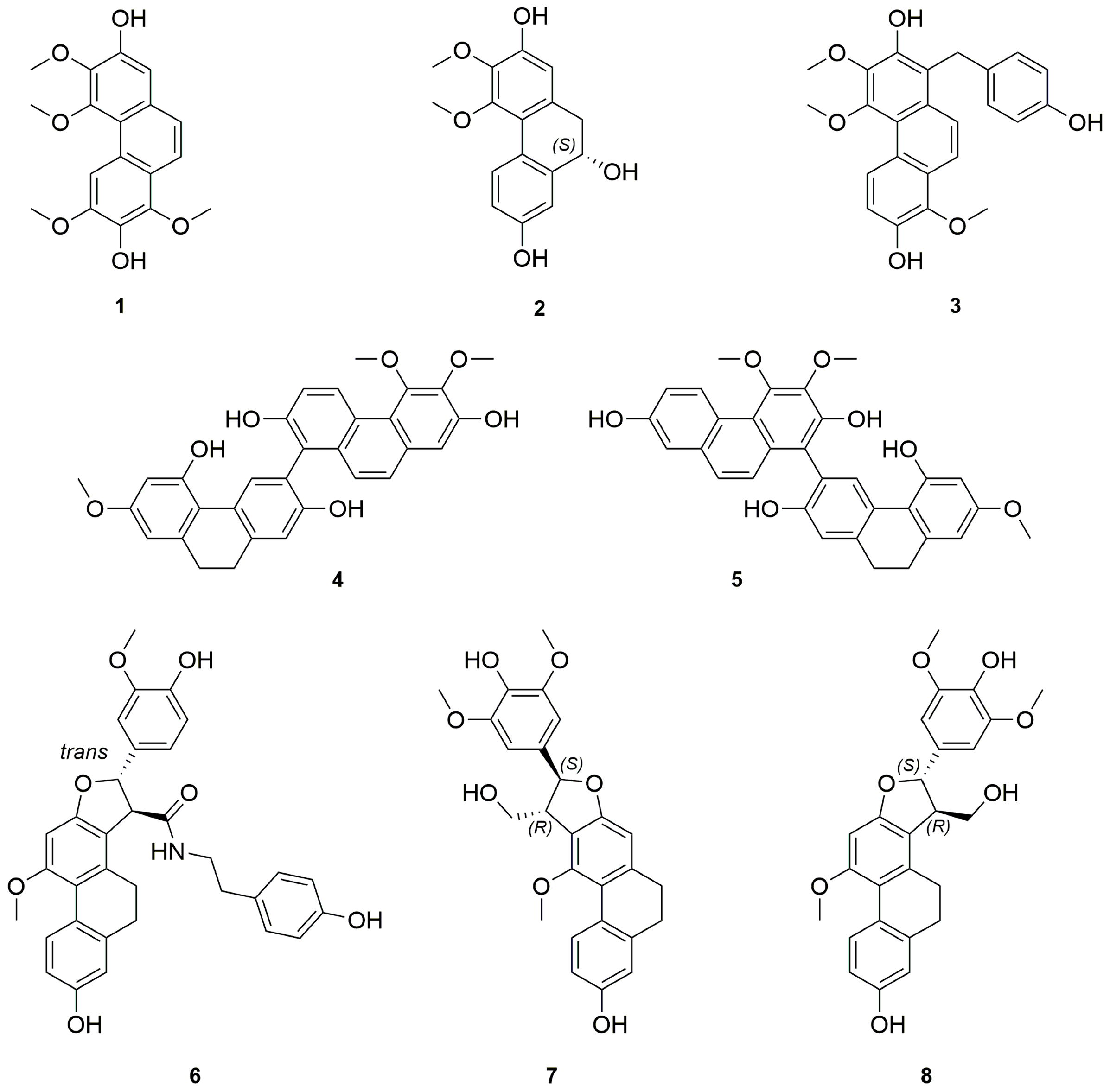
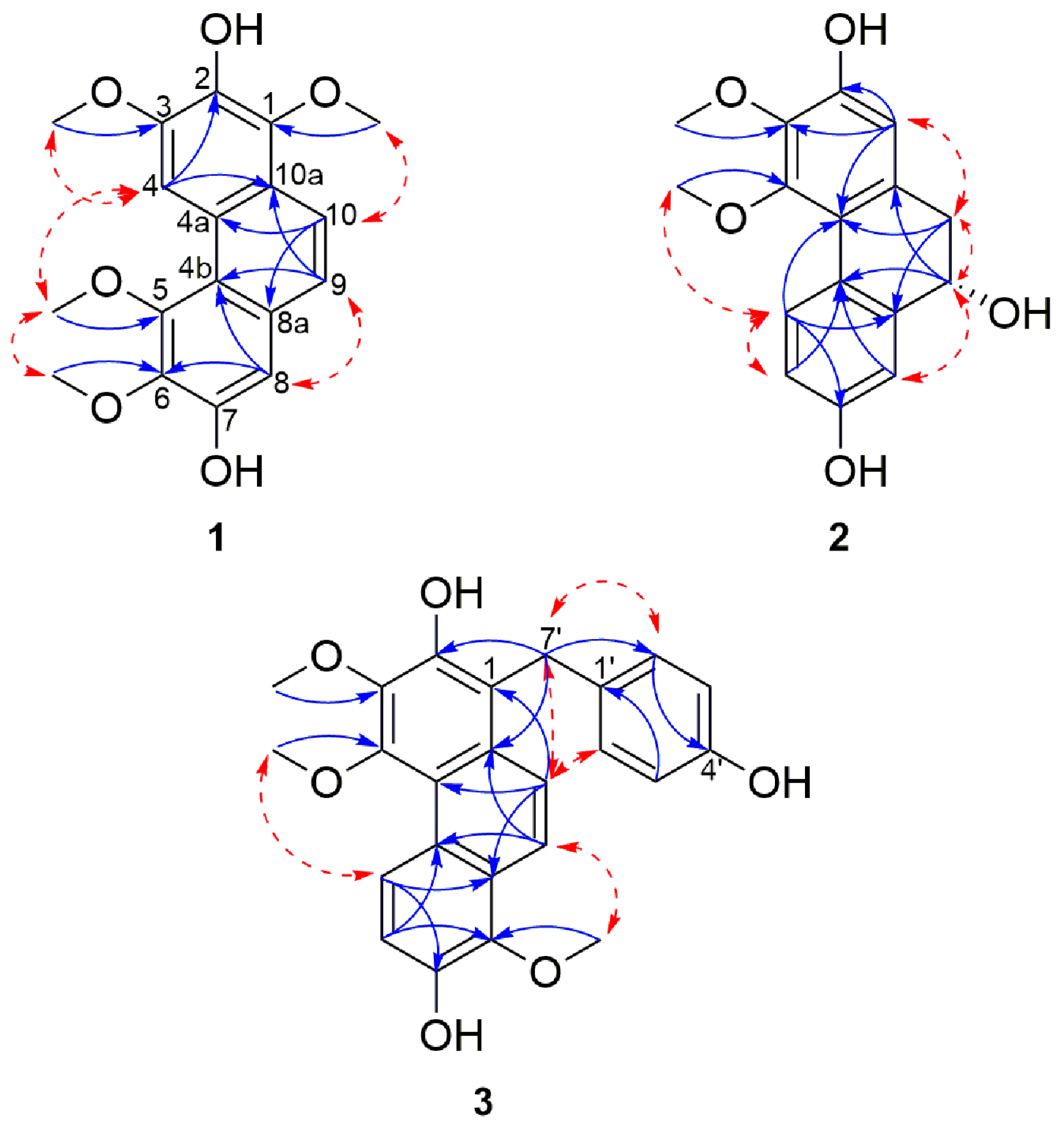
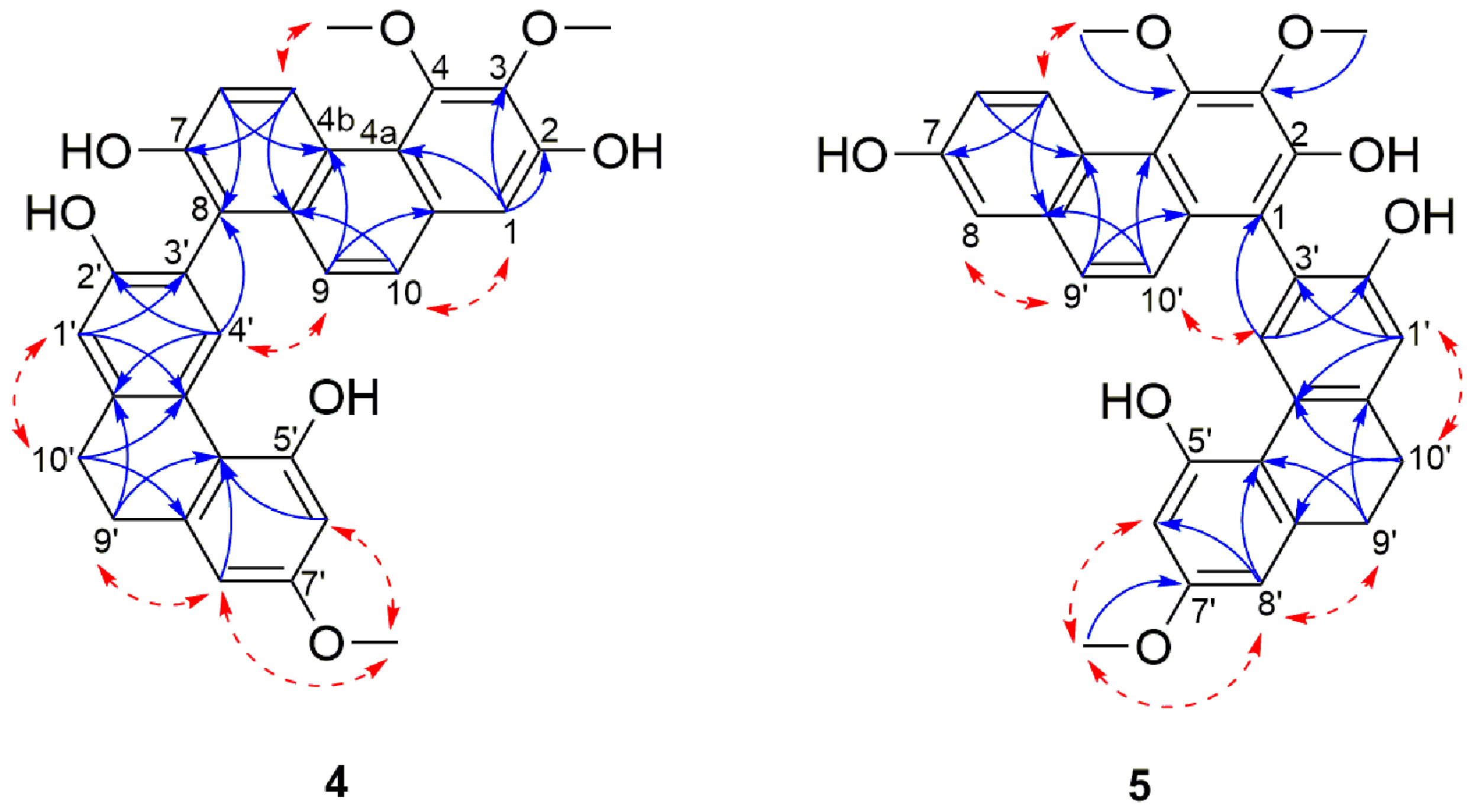
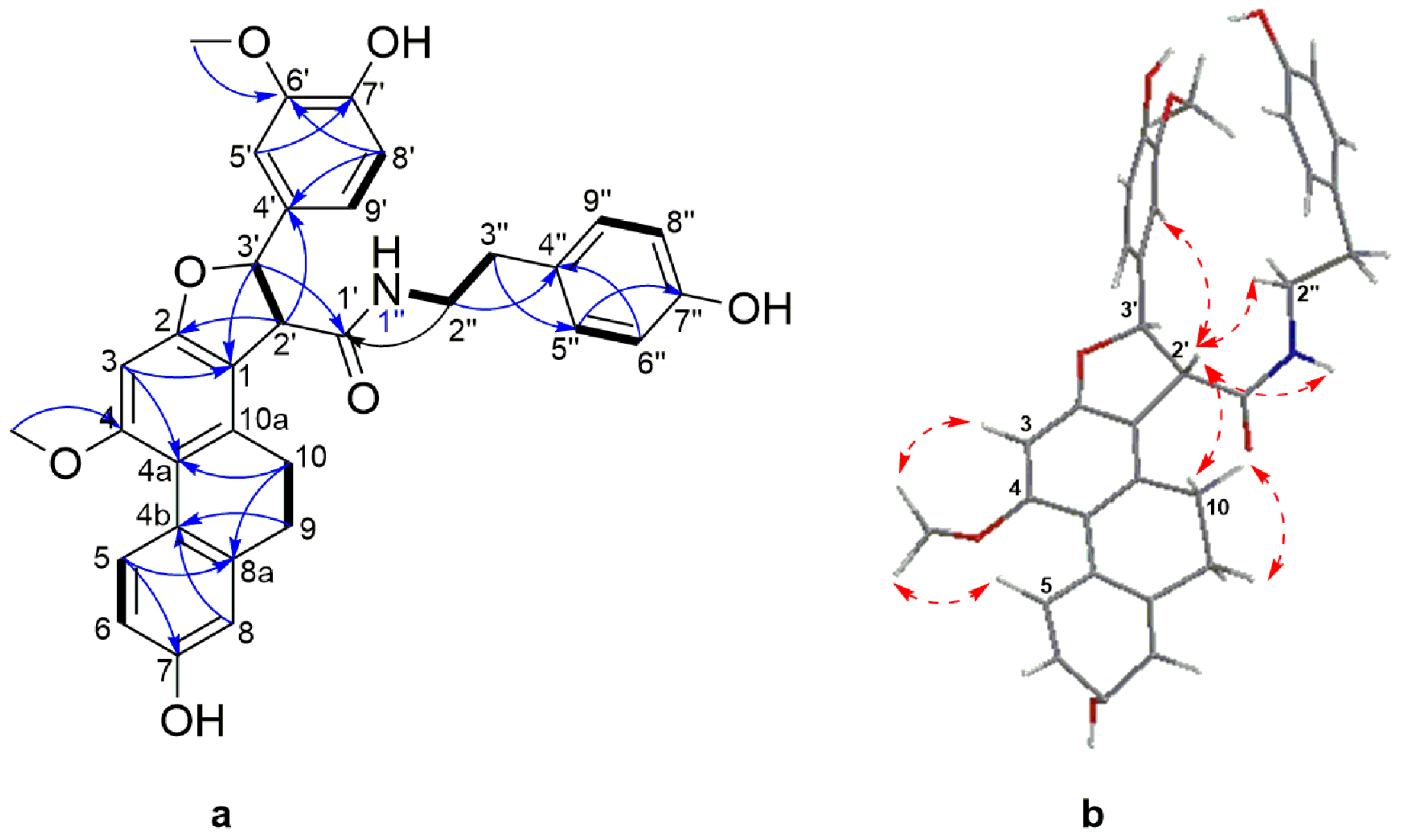
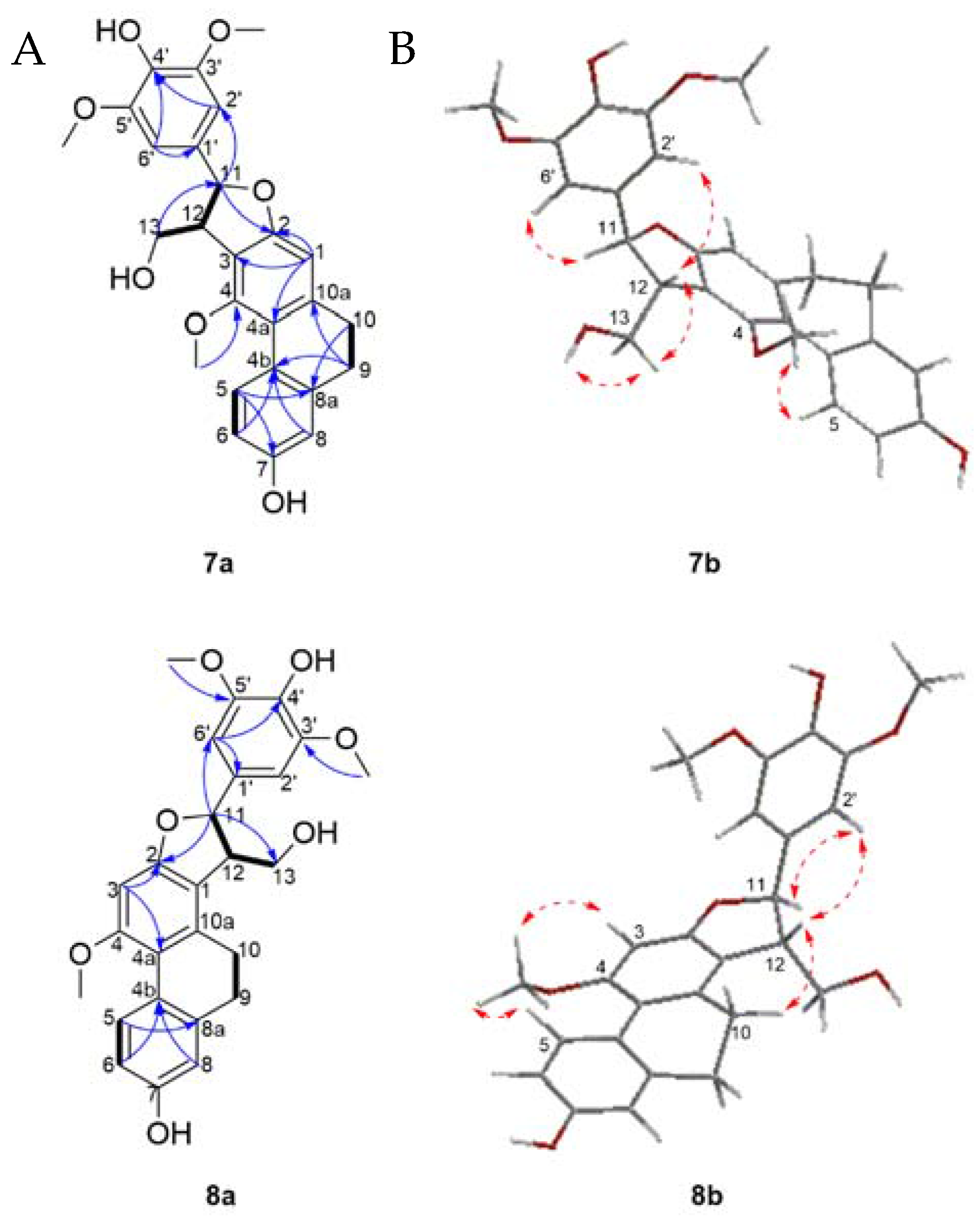
Structure Elucidation
3. Experimental Section
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Compound Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Estrada, S.; Rojas, A.; Mathison, Y.; Israel, A.; Mata, R. Nitric oxide/cGMP mediates the spasmolytic action of 3,4′-dihydroxy-5,5′-dimethoxybibenzyl from Scaphyglottis livida. Planta Med. 1999, 65, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Mata, R.; Rivero-Cruz, J.F. Bioactive secondary metabolites from selected mexican medicinal plants: Recent progress. In Bioactive Constituents from Natural Sources: Isolation, Characterization and Biological properties; Tringali, C., Ed.; Taylor & Francis Publishing: London, UK, 2001; pp. 129–158. [Google Scholar]
- Estrada, S.; Acevedo, L.; Rodriguez, M.; Toscano, R.A.; Mata, R. New triterpenoids from the orchids Scaphyglottis livida and Nidema boothii. Nat. Prod. Lett. 2002, 16, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Estrada, S.; Toscano, R.A.; Mata, R. New phenanthrene derivatives from Maxillaria densa. J. Nat. Prod. 1999, 62, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Islas, N.A.; Paul, R.N.; Shier, W.T.; Mata, R.; Abbas, H.K. Phytotoxicity and ultrastructural effects of gymnopusin from the orchid Maxillaria densa on duckweed (Lemna pausicostata) frond and root tissues. Phytochemistry 2002, 61, 141–148. [Google Scholar] [CrossRef]
- Deciga-Campos, M.; Palacios-Espinosa, J.F.; Reyes-Ramirez, A.; Mata, R. Antinociceptive and anti-inflammatory effects of compounds isolated from Scaphyglottis livida and Maxillaria densa. J. Ethnopharmacol. 2007, 114, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Rendon-Vallejo, P.; Hernandez-Abreu, O.; Vergara-Galicia, J.; Millan-Pacheco, C.; Mejia, A.; Ibarra-Barajas, M.; Estrada-Soto, S. Ex vivo study of the vasorelaxant activity induced by phenanthrene derivatives isolated from Maxillaria densa. J. Nat. Prod. 2012, 75, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.W.; Parente, J.P. Chemical properties and biological activity of a polysaccharide from Cyrtopodium cardiochilum. Carbohyd. Polym. 2006, 64, 287–291. [Google Scholar] [CrossRef]
- Morales-Sanchez, V.; Rivero-Cruz, I.; Laguna-Hernandez, G.; Salazar-Chavez, G.; Mata, R. Chemical composition, potential toxicity, and quality control. Procedures of the crude drug of Cyrtopodium macrobulbon. J. Ethnopharmacol. 2014, 154, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Parente, J.P.; Adao, C.R.; da Silva, B.P.; Tinoco, L.W. Structural characterization of an acetylated glucomannan with antiinflammatory activity and gastroprotective property from Cyrtopodium andersonii. Carbohyd. Res. 2014, 391, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Cogniaux, A. Orchidaceae. In Cyrtopodium in Flora Brasiliensis; Martius, C.F.P., Eichler, A.G.I., Eds.; R. Oldenbourg Verlag GmbH: Munich, Germany, 1898–1902; Volume 3, pp. 356–375. [Google Scholar]
- Romero-González, G.A.; Fernández-Concha, G.C. Notes on the species on Cyrtopodium (Cyrtopodinae, Orchidaceae) from Florida, the Greater Antilles, Mexico, Central and Northern America. Harv. Pap. Bot. 1999, 4, 327–341. [Google Scholar]
- Romero-González, G.A. Cyrtopodium. In Flora of North-America; Oxford Univ. Press New York: New York, NY, USA, 2002; Volume 26, pp. 642–643. [Google Scholar]
- Romero-González, G.A.; Batista, J.A.; Bianchetti, L.D.B. A synopsis of the genus Cyrtopodium (Catasetinae: Orchidaceae). Harv. Pap. Botany 2008, 13, 189–206. [Google Scholar] [CrossRef]
- Simmler, C.; Antheaume, C.; Lobstein, A. Antioxidant biomarkers from Vanda coerulea stems reduce irradiated HaCaT PGE-2 production as a result of COX-2 inhibition. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Simmler, C.; Antheaume, C.; Andre, P.; Bonte, F.; Lobstein, A. Glucosyloxybenzyl eucomate derivatives from Vanda teres stimulate HaCaT cytochrome c oxidase. J. Nat. Prod. 2011, 74, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Cakova, V.; Urbain, A.; Antheaume, C.; Rimlinger, N.; Wehrung, P.; Bonte, F.; Lobstein, A. Identification of phenanthrene derivatives in Aerides rosea (Orchidaceae) using the combined systems HPLC-ESI-HRMS/MS and HPLC-DAD-MS-SPE-UV-NMR. Phytochem. Anal. 2015, 26, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Barl, M.D.C.D.; Siegel, H.D.C.D.; Degner, D.D.C.D.; Mercker, H.J.D.C. Benzyl alcohols. DE3107147A1, 9 September 1982. [Google Scholar]
- Duan, J.; Li, W.; Hu, X.; Koike, K.; Fu, H. Chemical constituents of Silene rubicunda Franch. Zhong Cao Yao 2009, 40, 528–530. [Google Scholar]
- Letcher, R.M.; Nhamo, L.R.M. Chemical constituents of the Combretaceae. Iii. Substituted phenanthrenes, 9,10-dihydrophenanthrenes, and bibenzyls from the heartwood of Combretum psidioides. J. Chem. Soc. 1972, 2941–2946. [Google Scholar] [CrossRef]
- Majumder, P.L.; Kar, A. Confusarin and confusaridin, two phenanthrene derivatives of the orchid Eria confusa. Phytochemistry 1987, 26, 1127–1129. [Google Scholar] [CrossRef]
- Majumder, P.L.; Joardar, M. Erianthridin, a new 9,10-dihydrophenanthrene derivative from the orchids Eria carinata and Eria stricta. Ind. J. Chem. Sect. B 1985, 24B, 1192–1194. [Google Scholar]
- Bhandari, S.R.; Kapadi, A.H.; Mujumder, P.L.; Joardar, M.; Shoolery, J.N. Nudol, a phenanthrene of the orchids Eulophia nuda, Eria carinata and Eria stricta. Phytochemistry 1985, 24, 801–804. [Google Scholar] [CrossRef]
- Chang, C.-F.; Hsu, Y.-L.; Lee, C.-Y.; Wu, C.-H.; Wu, Y.-C.; Chuang, T.-H. Isolation and cytotoxicity evaluation of the chemical constituents from Cephalantheropsis gracilis. Int. J. Mol. Sci. 2015, 16, 3980–3989. [Google Scholar] [CrossRef] [PubMed]
- Juneja, R.K.; Sharma, S.C.; Tandon, J.S. A substituted 1,2-diarylethane from Cymbidium giganteum. Phytochemistry 1985, 24, 321–324. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hasegawa, K.; Kawarada, A. Batatasins: New dormancy-inducing substances of yam bulbils. Planta 1972, 108, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Tezuka, Y.; Hirano, H.; Kikuchi, T.; Xu, G. Constituents of orchidaceous plants. X. Constituents of Ephemerantha lonchophylla; isolation and structure elucidation of new phenolic compounds, ephemeranthol-a, ephemeranthol-b, and ephemeranthoquinone, and of a new diterpene glucoside, ephemeranthoside. Chem. Pharm. Bull. 1991, 39, 593–598. [Google Scholar] [CrossRef]
- Majumder, P.; Laha, S.; Datta, N. Coelonin, a 9,10-dihydrophenanthrene from the orchids Coelogyne ochracea and C. elata. Phytochemistry 1982, 21, 478–480. [Google Scholar] [CrossRef]
- Yamaki, M.; Honda, C. The stilbenoids from Dendrobium plicatile. Phytochemistry 1996, 43, 207–208. [Google Scholar] [CrossRef]
- Majumder, P.L.; Lahiri, S. Lusianthrin and lusianthridin, two stilbenoids from the orchid lusia indivisa. Phytochemistry 1990, 29, 621–624. [Google Scholar] [CrossRef]
- Fan, C.Q.; Zhao, W.M.; Qin, G.W. New bibenzyl and phenanthrenedione from Dendrobium densiflorum. Chin. Chem. Lett. 2000, 11, 705–706. [Google Scholar]
- Zhang, G.-N.; Zhong, L.-Y.; Annie Bligh, S.W.; Guo, Y.-L.; Zhang, C.-F.; Zhang, M.; Wang, Z.-T.; Xu, L.-S. Bi-bicyclic and bi-tricyclic compounds from Dendrobium thyrsiflorum. Phytochemistry 2005, 66, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Hwang, T.-L.; Chen, F.-A.; Huang, C.-H.; Hung, H.-Y.; Wu, T.-S. Chemical constituents of the rhizomes of Bletilla formosana and their potential anti-inflammatory activity. J. Nat. Prod. 2016, 79, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.O.; Varshney, I.P.; Rahman, W.; Gangwar, P.C. The synthesis of chrysoeriol (3′-methyl ether of luteolin) and homoeriodictyol (5,7,4′-trihydroxy-3′-methoxyflavanone). Naturwissenschaften 1959, 46, 76. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, Y.H.; Park, S.M.; Lee, K.E.; Lee, J.J.; Lee, B.C.; Pyo, H.B.; Song, K.S.; Park, H.D.; Yun, Y.P. Antioxidants and inhibitor of matrix metalloproteinase-1 expression from leaves of Zostera marina L. Arch. Pharm. Res. 2004, 27, 177–183. [Google Scholar] [CrossRef]
- Menke, A.E.; Bentley, W.B. Preliminary note on some new derivatives of vanillin. J. Am. Chem. Soc. 1898, 20, 316–317. [Google Scholar] [CrossRef]
- Li, S.; Lundquist, K. A new method for the analysis of phenolic groups in lignins by proton NMR spectrometry. Nord. Pulp Pap. Res. J. 1994, 9, 191–195. [Google Scholar] [CrossRef]
- Cai, J.-Y.; Zhao, L.; Zhang, D.-Z. Chemical constituents from Bletilla ochracea Schltr. Chem. Res. Chin. Univ. 2007, 23, 705–707. [Google Scholar] [CrossRef]
- Takagi, S.; Yamaki, M.; Inoue, K. Antimicrobial agents from Bletilla striata. Phytochemistry 1983, 22, 1011–1015. [Google Scholar] [CrossRef]
- Bai, L.; Yamaki, M.; Takagi, S. Stilbenoids from Pleione bulbocodioides. Phytochemistry 1996, 42, 853–856. [Google Scholar] [CrossRef]
- Yamaki, M.; Bai, L.; Inoue, K.; Takagi, S. Biphenanthrenes from Bletilla striata. Phytochemistry 1989, 28, 3503–3505. [Google Scholar] [CrossRef]
- Das, K.C.; Farmer, W.J.; Weinstein, B. Phytochemical studies. IX. A new flavone, velutin. J. Org. Chem. 1970, 35, 3989–3990. [Google Scholar] [CrossRef]
- Lodhi, M.A.; Choudhary, M.I.; Malik, A.; Ahmad, S. Α-chymotrypsin inhibition studies on the lignans from Vitex negundo Linn. J. Enzyme Inhib. Med. Chem. 2008, 23, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Haruna, M.; Koube, T.; Ito, K.; Murata, H. Balanophonin, a new neo-lignan from Balanophora japonica Makino. Chem. Pharm. Bull. 1982, 30, 1525–1527. [Google Scholar] [CrossRef]
- Hussain, S.F.; Goezler, B.; Shamma, M.; Goezler, T. Feruloyltyramine from Hypecoum. Phytochemistry 1982, 21, 2979–2980. [Google Scholar] [CrossRef]
- Majumder, P.L.; Basak, M. Cirrhopetalin, a phenanthrene derivative from Cirrhopetalum andersonii. Phytochemistry 1990, 29, 1002–1004. [Google Scholar] [CrossRef]
- Majumder, P.L.; Sen, R.C. Pendulin, a polyoxygenated phenanthrene derivative from the orchid Cymbidium pendulum. Phytochemistry 1991, 30, 2432–2434. [Google Scholar] [CrossRef]
- Honda, C.; Yamaki, M. Phenanthrenes from Dendrobium plicatile. Phytochemistry 2000, 53, 987–990. [Google Scholar] [CrossRef]
- Majumder, P.L.; Pal, S. Rotundatin, a new 9,10-dihydrophenanthrene derivative from Dendrobium rotundatum. Phytochemistry 1992, 31, 3225–3228. [Google Scholar] [CrossRef]
- Majumder, P.L.; Sen, S.; Majumder, S. Phenanthrene derivatives from the orchid Coelogyne cristata. Phytochemistry 2001, 58, 581–586. [Google Scholar] [CrossRef]
- Resnick, S.M.; Gibson, D.T. Regio- and stereospecific oxidation of 9,10-dihydroanthracene and 9,10-dihydrophenanthrene by naphthalene dioxygenase: Structure and absolute stereochemistry of metabolites. Appl. Environ. Microbiol. 1996, 62, 3355–3359. [Google Scholar] [PubMed]
- Lin, Y.; Wang, F.; Yang, L.-J.; Chun, Z.; Bao, J.-K.; Zhang, G.-L. Anti-inflammatory phenanthrene derivatives from stems of Dendrobium denneanum. Phytochemistry 2013, 95, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.L.; Pal, A.; Joardar, M. Cirrhopetalanthrin, a dimeric phenanthrene derivative from the orchid Cirrhopetalum maculosum. Phytochemistry 1990, 29, 271–274. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, S.-H.; Meng, Y.-H.; Zhang, Y.-B.; Cheng, C.-R.; Shi, Y.-Y.; Feng, R.-H.; Zeng, F.; Wu, Z.-Y.; Zhang, J.-X.; et al. Phenanthrenes, 9,10-dihydrophenanthrenes, bibenzyls with their derivatives, and malate or tartrate benzyl ester glucosides from tubers of Cremastra appendiculata. Phytochemistry 2013, 94, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Leong, Y.-W.; Harrison, L.J. A biphenanthrene and a phenanthro[4,3-b]furan from the orchid Bulbophyllum vaginatum. J. Nat. Prod. 2004, 67, 1601–1603. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Tang, C.-P.; Li, X.-Q.; Ye, Y. Phochinenins a–f, dimeric 9,10-dihydrophenanthrene derivatives, from Pholidota chinensis. Helv. Chim. Acta 2008, 91, 2122–2129. [Google Scholar] [CrossRef]
- Ali, Z.; Tanaka, T.; Iliya, I.; Iinuma, M.; Furusawa, M.; Ito, T.; Nakaya, K.; Murata, J.; Darnaedi, D. Phenolic constituents of Gnetum klossii. J. Nat. Prod. 2003, 66, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Antus, S.; Kurtan, T.; Juhasz, L.; Kiss, L.; Hollosi, M.; Majer, Z. Chiroptical properties of 2,3-dihydrobenzo[b]furan and chromane chromophores in naturally occurring O-heterocycles. Chirality 2001, 13, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, C.; Li, Y.; Guo, S.; Yang, J. Complete assignments of 1H and 13C-NMR data of three new dihydrophenanthrofurans from Pleione yunnanensis. Magn. Reson. Chem. 2010, 48, 256–260. [Google Scholar] [PubMed]
- Fisch, M.H.; Flick, B.H.; Arditti, J. Orchid phytoalexins. I. Structure and antifungal activity of hircinol, loroglossol, and orchinol. Phytochemistry 1973, 12, 437–441. [Google Scholar] [CrossRef]
- Ward, E.W.B.; Unwin, C.H.; Stoessl, A. Loroglossol. Orchid phytoalexin. Phytopathology 1975, 65, 632–633. [Google Scholar] [CrossRef]
- Fritzemeier, K.H.; Kindl, H. 9,10-dihydrophenanthrenes as phytoalexins of Orchidaceae. Biosynthetic studies in vitro and in vivo proving the route from l-phenylalanine to dihydro-m-coumaric acid, dihydrostilbene and dihydrophenanthrenes. Eur. J. Biochem. 1983, 133, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Hills, K.A.; Stoessl, A.; Oliva, A.P.; Arditti, J. Effects of orchinol, loroglossol, dehydroorchinol, batatasin III, and 3,4′-dihydroxy-5-methoxydihydrostilbene on orchid seedlings. Bot. Gaz. 1984, 145, 298–301. [Google Scholar] [CrossRef]
- Reinecke, T.; Kindl, H. Characterization of bibenzyl synthase catalyzing the biosynthesis of phytoalexins of orchids. Phytochemistry 1994, 35, 63–66. [Google Scholar] [CrossRef]
- Coxon, D.T.; Ogundana, S.K.; Dennis, C. Antifungal phenanthrenes in yam tubers. Phytochemistry 1982, 21, 1389–1392. [Google Scholar] [CrossRef]
- Chong, J.; Poutaraud, A.; Hugueney, P. Metabolism and roles of stilbenes in plants. Plant. Sci. 2009, 177, 143–155. [Google Scholar] [CrossRef]
- Majumder, P.L.; Ghosal, S. Two stilbenoids from the orchid Arundina bambusifolia. Phytochemistry 1993, 32, 439–444. [Google Scholar] [CrossRef]
- Liu, M.-F.; Han, Y.; Xing, D.-M.; Shi, Y.; Xu, L.-Z.; Du, L.-J.; Ding, Y. A new stilbenoid from Arundina graminifolia. J. Asian Nat. Prod. Res. 2004, 6, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.; Chen, W.-P.; Macabalang, A.D. Dihydrophenanthrenes from Bletilla formosana. Chem. Pharm. Bull. 2005, 53, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tang, C.; Zhao, P.; Shu, G.; Mei, Z. Antimicrobial constituents from the tubers of Bletilla ochracea. Planta Med. 2012, 78, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Kato, T.; Inoue, K.; Yamakai, M.; Takagi, S. Nonpolar constituents from Bletilla striata. Part 6. Blestrianol A, B and C, biphenanthrenes from Bletilla striata. Phytochemistry 1991, 30, 2733–2735. [Google Scholar] [CrossRef]
- Feng, J.-Q.; Zhang, R.-J.; Zhao, W.-M. Novel bibenzyl derivatives from the tubers of Bletilla striata. Helv. Chim. Acta 2008, 91, 520–525. [Google Scholar] [CrossRef]
- Liu, X.-Q.; Yuan, Q.-Y.; Guo, Y.-Q. Two new stilbenoids from Pleione bulbocodioides. J. Asian Nat. Prod. Res. 2009, 11, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Han, S.-W.; Cui, B.-S.; Wang, X.-J.; Li, S. Chemical constituents from Pleione bulbocodioides. Zhongguo Zhongyao Zazhi 2014, 39, 442–447. [Google Scholar] [PubMed]
- Bai, L.; Yamaki, M.; Takagi, S. Flavan-3-ols and dihydrophenanthropyrans from Pleione bulbocodioides. Phytochemistry 1998, 47, 1125–1129. [Google Scholar] [CrossRef]
- Liu, X.Q.; Gao, W.Y.; Guo, Y.Q.; Zhang, T.J.; Yan, L.L. A new phenanthro[2,3-b]furan from Pleione bulbocodioides. Chin. Chem. Lett. 2007, 18, 1089–1091. [Google Scholar] [CrossRef]
- Liu, X.-Q.; Guo, Y.-Q.; Gao, W.-Y.; Zhang, T.-J.; Yan, L.-L. Two new phenanthrofurans from Pleione bulbocodioides. J. Asian Nat. Prod. Res. 2008, 10, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.-D.; Jiang, F.-S.; Yu, H.-S.; Shen, Y.; Fu, Y.-H.; Cheng, D.-Q.; Gan, L.-S.; Ding, Z.-S. Antibacterial biphenanthrenes from the fibrous roots of Bletilla striata. J. Nat. Prod. 2015, 78, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Liu, J.; Pang, S.; Lin, J.; Xu, R. Two novel phenanthraquinones with anti-cancer activity isolated from Bletilla striata. Bioorg. Med. Chem. Lett. 2016, 26, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, J.; Xu, C.; Lv, J.; Wang, C.; Song, P. Antimicrobial stilbenoids from Bletilla yunnanensis. Chem. Nat. Compd. 2016, 52, 19–22. [Google Scholar] [CrossRef]
- Majumder, P.L.; Pal, S.; Majumder, S. Dimeric phenanthrenes from the orchid Bulbophyllum reptans. Phytochemistry 1999, 50, 891–897. [Google Scholar] [CrossRef]
- Majumder, P.L.; Bandyopadhyay, S.; Pal, S. Rigidanthrin, a new dimeric phenanthrene derivative of the orchid Bulbophyllum rigidum. J. Indian Chem. Soc. 2008, 85, 1116–1123. [Google Scholar]
- Xu, J.; Yu, H.; Qing, C.; Zhang, Y.; Liu, Y.; Chen, Y. Two new biphenanthrenes with cytotoxic activity from Bulbophyllum odoratissimum. Fitoterapia 2009, 80, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Li, S.; Wang, S.; Wang, Y.; Yang, Y.; Shi, J.; He, L. Mono-, bi-, and triphenanthrenes from the tubers of Cremastra appendiculata. J. Nat. Prod. 2006, 69, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Q.; Li, X.-P.; Yuan, Q.-Y. A new biphenanthrene glucoside from Cremastra appendiculata. Chem. Nat. Compd. 2015, 51, 1035–1037. [Google Scholar] [CrossRef]
- Yuan, Q.-Y.; Liu, X.-Q. Chemical constituents from Cremastra appendiculata. Zhongyaocai 2015, 38, 298–301. [Google Scholar] [PubMed]
- Liu, X.-Q.; Li, X.-P.; Yuan, W.-K.; Yuan, Q.-Y.; Qin, B.-H. Two new phenanthrene glucosides from Cremastra appendiculata and their cytotoxic activities. Nat. Prod. Commun. 2016, 11, 477–479. [Google Scholar] [PubMed]
- Liu, L.; Zeng, K.-W.; Jiang, Y.; Tu, P.-F.; Liu, L.; Liu, L.; Li, J. Five new biphenanthrenes from Cremastra appendiculata. Molecules 2016, 21, 1089. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.; Cohen, T. Some biogenetic aspects of phenol oxidation. Festschr. Arthur. Stoll. 1957, 117–143. [Google Scholar]
- Stadler, R.; Zenk, M.H. The purification and characterization of a unique cytochrome P-450 enzyme from Berberis stolonifera plant cell cultures. J. Biol. Chem. 1993, 268, 823–831. [Google Scholar] [PubMed]
- Cottyn, B.; Kollmann, A.; Waffo-Teguo, P.; Ducrot, P.H. Rationalization and in vitro modeling of the chemical mechanisms of the enzymatic oxidation of phenolic compounds in planta: From flavonols and stilbenoids to lignins. Chem. Eur. J. 2011, 17, 7282–7287. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δH | δC | HMBC | NOESY | δH | δC | HMBC | NOESY | δH | δC | HMBC | NOESY | |
| 1 | 142.5 | 6.59 (s) | 112.7 | 2, 3, 4, 4a, 10 | 10, 10′ | 119.7 | ||||||
| 2 | 138.5 | 149.9 | 147.9 | |||||||||
| 3 | 149.5 | 141.1 | 142.3 | |||||||||
| 4 | 8.90 (s) | 103.9 | 2, 3, 4b, 10a | 3/5-OCH3 | 152.1 | 150.6 | ||||||
| 4a | 124.2 | 120.2 | 127.8 | |||||||||
| 4b | 118.8 | 124.3 | 125.5 | |||||||||
| 5 | 152.3 | 8.09 (d, 8.6) | 129.2 | 4a, 7, 8, 8a, 9 | 4-OCH3, 6 | 9.21 (d, 9.4) | 124.4 | 4a, 4b, 7 | 4-OCH3, 6 | |||
| 6 | 142.7 | 6.76 (dd, 8.6, 2.7) | 114.6 | 4b, 7, 8 | 5 | 7.24 (d, 9.4) | 118.2 | 4b, 7, 8 | 5 | |||
| 7 | 150.2 | 157.1 | 147.2 | |||||||||
| 8 | 7.17 (s) | 109.7 | 4b, 5, 6, 7, 9 | 7-OH, 9 | 7.11 (d, 2.7) | 112.8 | 4b, 6, 7, 9 | 9 | 142.1 | |||
| 8a | 131.0 | 143.3 | 128.8 | |||||||||
| 9 | 7.50 (d, 8.9) | 125.3 | 4b, 8, 10a | 8, 10 | 4.63 (dd, 10.6, 4.6) | 68.9 | 4b, 8, 8a, 10a, 10 | 8, 10, 10′ | 7.87 (d, 9.5) | 120.3 | 8, 4b, 10a | 8-OCH3 |
| 10 | 7.88 (d, 8.9) | 120.1 | 9, 1-OCH3 | 2.70 (dd, 14.3, 10.6) | 40.1 | 1, 4a, 8a, 9, 10a | 1, 9, 10 | 7.90 (d, 9.5) | 124.3 | 1, 4a, 8a | 2′/6′, 7′ | |
| 10 | 2.83 (dd, 14.3, 4.6) | 40.1 | 1, 4a, 8a, 9, 10a | 1, 9, 10′ | ||||||||
| 10a | 122.6 | 1, 4a, 8a | 132.1 | 129.2 | ||||||||
| 1-OCH3 | 3.99 (s) | 61.1 | 1 | 2-OH, 10 | ||||||||
| 3-OCH3 | 4.06 (s) | 56.3 | 3 | 2-OH, 4 | 3.85 (s) | 61.1 | 3 | 4.06 (s) | 61.6 | 3 | ||
| 4-OCH3 | 3.72 (s) | 60.3 | 4 | 3.95 (s) | 60.1 | 4 | ||||||
| 5-OCH3 | 4.03 (s) | 60.5 | 5 | 4 | ||||||||
| 6-OCH3 | 4.02 (s) | 61.4 | 6 | 7-OH | ||||||||
| 8-OCH3 | 3.90 (s) | 61.5 | 8 | |||||||||
| 2-OH | 7.91 (s) | 1, 2, 3 | 1/3-OCH3 | |||||||||
| 7-OH | 8.39 (s) | 6, 7, 8 | 6-OCH3, 8 | |||||||||
| 9-OH | 4.32 (brs) | |||||||||||
| 1′ | 133.0 | |||||||||||
| 2′/6′ | 7.08 (d, 8.6) | 130.1 | 3′/5′, 4′ | 3′/5′, 7′, 10 | ||||||||
| 3′/5′ | 6.68 (d, 8.6) | 115.9 | 2′/6′, 1′ | 2′/6′ | ||||||||
| 4′/ | 156.3 | |||||||||||
| 7′ | 4.41 (s) | 30.9 | 1, 10a, 1′, 2′/6′ | 10 | ||||||||
| No. | 4 | 5 | ||||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | HMBC | NOESY | δH | δC | HMBC | NOESY | |
| 1 | 7.11 (s) | 109.5 | 2, 3, 4a, 10 | 10 | 118.4 | |||
| 2 | 150.0 | 148.1 | ||||||
| 3 | 142.9 | 142.6 | ||||||
| 4 | 152.4 | 151.4 | ||||||
| 4a | 119.4 | 119.4 | ||||||
| 4b | 124.8 | 124.5 | ||||||
| 5 | 9.42 (d, 9.2) | 128.1 | 4a, 7, 8a | 4-OCH3, 6 | 9.42 (d, 9.2) | 129.3 | 4a, 7, 8b | 4-OCH3, 6 |
| 6 | 7.31 (d, 9.2) | 117.7 | 4b, 8 | 5 | 7.20 (dd, 9.2, 2.7) | 117.6 | 4b, 7, 8 | 5 |
| 7 | 153.5 | 156.1 | ||||||
| 8 | 121.0 | 7.21 (d, 2.7) | 112.2 | 4b, 6, 9 | 9 | |||
| 8a | 133.5 | 134.5 | ||||||
| 9 | 7.43 (s) | 125.6 | 4b, 8, 10a | 10, 4′ | 7.41 (d, 9.0) | 126.6 | 4b, 8, 10a | 8, 10 |
| 10 | 7.43 (s) | 127.2 | 1, 4a, 8a | 1, 9 | 7.44 (d, 9.0) | 126.0 | 1, 4a, 8a | 9, 4′ |
| 10a | 130.1 | 129.8 | ||||||
| 3-OCH3 | 4.01 (s) | 61.4 | 3 | 4.06 (s) | 61.3 | 3 | ||
| 4-OCH3 | 4.00 (s) | 60.2 | 4 | 5 | 4.00 (s) | 60.1 | 4 | 5 |
| 1′ | 6.92 (s) | 116.1 | 2′, 3′, 4a′, 10′ | 10′ | 6.89 (s) | 115.7 | 2′, 3′, 4a′, 10′ | 10′ |
| 2′ | 154.6 | 154.3 | ||||||
| 3′ | 120.6 | 121.1 | ||||||
| 4′ | 8.28 (s) | 133.2 | 8, 2′, 4b′, 10a′ | 9 | 8.27 (s) | 133.0 | 1, 2′, 4b′, 10a′ | 10 |
| 4a′ | 126.2 | 126.1 | ||||||
| 4b′ | 115.8 | 115.8 | ||||||
| 5′ | 156.0 | 155.9 | ||||||
| 6′ | 6.39 (d, 2.6) | 101.6 | 4b′, 7′, 8′ | 7′-OCH3 | 6.38 (d, 2.6) | 101.6 | 4b′, 7′, 8′ | 7′-OCH3 |
| 7′ | 159.5 | 159.4 | ||||||
| 8′ | 6.42 (d, 2.6) | 106.1 | 4b′, 6′, 7′, 9′ | 7′-OCH3, 9′ | 6.42 (d, 2.6) | 106.1 | 4b′, 6′, 7′, 9′ | 7′-OCH3, 9′ |
| 8a′ | 141.5 | 141.5 | ||||||
| 9′ | 2.80 (brs) | 30.7 | 4b′, 8′, 10a′ | 8′ | 2.80 (brs) | 30.7 | 4b′, 8′, 10a′ | 8′ |
| 10′ | 2.80 (brs) | 31.6 | 1′, 4a′, 8a′′ | 1′ | 2.80 (brs) | 31.7 | 1′, 4a′, 8a′′ | 1′ |
| 10a′ | 139.6 | 139.5 | ||||||
| 7′-OCH3 | 3.73 (s) | 55.4 | 7′ | 6′, 8′ | 3.74 (s) | 55.4 | 7′ | 6′, 8′ |
| No. | 6 | |||
|---|---|---|---|---|
| δH (J, Hz) | δC | HMBC | NOESY | |
| 1 | 116.7 | |||
| 2 | 160.4 | |||
| 3 | 6.54 (s) | 93.6 | 1, 2, 4, 4a | 4-OCH3 |
| 4 | 159.2 | |||
| 4a | 117.6 | |||
| 4b | 125.7 | |||
| 5 | 8.04 (d, 9.4) | 130.0 | 4a, 8a, 7 | 4-OCH3, 6 |
| 6 | 6.68 (dd, 9.4, 2.5) | 113.6 | 4b, 8 | 5 |
| 7 | 156.2 | |||
| 8 | 6.68 (d, 2.5) | 115.0 | 4b, 6, 9 | 9 |
| 8a | 139.8 | |||
| 9 | 2.55, 2.60 (m) | 30.7 | 8 | 8, 10 |
| 10 | 2.44, 2.55 (m) | 27.5 | 8a | 9, 2′ |
| 10a | 137.2 | |||
| 4-OCH3 | 3.88 (s) | 56.1 | 4 | 3, 5 |
| 1′ | 172.3 | |||
| 2′ | 4.11 (d, 6.6) | 58.6 | 1, 2, 1′, 3′, 4′ | 10, 3′, 5′, 9′, 1′′-NH |
| 3′ | 5.67 (d, 6.6) | 89.8 | 2, 1′, 2′, 4′, 5′, 9′ | 2′, 5′, 9′, 1′′-NH |
| 4′ | 133.8 | |||
| 5′ | 6.82 (dd, 8.3, 1.5) | 119.5 | 3′, 7′, 9′ | 2′, 3′ |
| 6′ | 6.82 (d, 8.3) | 115.9 | 4′, 8′ | |
| 7′ | 147.6 | |||
| 8′ | 148.5 | |||
| 9′ | 6.99 (d, 1.5) | 110.3 | 3′, 5′, 7′ | 2′, 3′, 8′-OCH3 |
| 8′-OCH3 | 3.82 (s) | 56.4 | 8′ | 9′ |
| 2′′ | 3.46 (td, 7.1, 5.5) | 41.9 | 1′, 3′′, 4′′ | 1′′-NH, 3′′ |
| 3′′ | 2.72 (brt, 7.1) | 35.5 | 2′′, 5′′/9′′ | 2′′ |
| 4′′ | 130.9 | |||
| 5′′/9′′ | 7.01 (d, 8.4) | 130.6 | 3′′, 9′′/5′′, 6′′ | 6′′/8′′ |
| 6′′/8′′ | 6.73 (d, 8.4) | 116.1 | 4′′, 8′′/6′′ | 5′′/9′′ |
| 7′′ | 156.8 | |||
| 1′′-NH | 7.28 (t, 5.5) | 2′, 3′, 2′′ | ||
| No. | 7 | 8 | ||||||
|---|---|---|---|---|---|---|---|---|
| δH (J, Hz) | δC | HMBC | NOESY | δH (J, Hz) | δC | HMBC | NOESY | |
| 1 | 6.56 (s) | 105.9 | 2, 3, 4a, 10 | 117.4 | ||||
| 2 | 160.5 | 160.3 | ||||||
| 3 | 118.7 | 6.56 (s) | 93.5 | 2, 4a | 4-OCH3 | |||
| 4 | 156.0 | 158.9 | ||||||
| 4a | 120.9 | 116.8 | ||||||
| 4b | 125.4 | 125.8 | ||||||
| 5 | 8.03 (d, 9.2) | 128.9 | 7, 8a | 4-OCH3, 6 | 8.04 (d, 9.3) | 130.0 | 7, 8a | 4-OCH3, 6 |
| 6 | 6.73 (dd, 9.2, 2.8) | 114.2 | 5, 7-OH | 6.68 (dd, 9.3, 2.7) | 113.6 | 4b, 8 | 5 | |
| 7 | 156.7 | 156.1 | ||||||
| 8 | 6.73 (d, 2.8) | 115.2 | 9, 7-OH | 6.69 overlapped | 115.0 | 4b, 6, 9 | 9 | |
| 8a | 140.2 | 139.8 | ||||||
| 9 | 2.69 (m) | 30.8 | 4b, 10, 10a | 8 | 2.59-2.64 (m) | 30,4 | 8 | |
| 10 | 2.67 (m) | 31.5 | 8a, 9, 1 | 1 | 2.59-2.71 (m) | 27.5 | 12 | |
| 10a | 142.1 | 137.1 | ||||||
| 4-OCH3 | 3.60 | 60.0 | 4 | 5, 12 | 3.87 (s) | 56.0 | 4 | 3, 5 |
| 7-OH | 8.29 (brs) | 6, 7, 8 | 6, 8 | 8.22 (brs) | 11, 12 | |||
| 11 | 5.66 (d, 5.4) | 88.4 | 2, 13, 2′/ 6′ | 12, 13, 2′/6′ | 5.71 (d, 3.4) | 88.2 | 2, 13, 2′ | 10, 11, 13, 2′/6′ |
| 12 | 3.73 (m) | 54.3 | 11, 13, 2′/6′ | 3.54 (dt, 8.7, 3,4) | 54.5 | 12 | ||
| 13 | 4.08 (ddd, 10.4, 4.7, 4.2) | 63.5 | 12 | 3.63 (m) | 64.6 | 12 | ||
| 3.83 (m) | 11, 12 | 3.90 (m) | - | |||||
| 13-OH | 4,17 (dd, 6.1, 4.7) | 13 | 4.22 (brt 5.3) | - | ||||
| 1′ | 133.9 | 134.4 | ||||||
| 2′/6′ | 6.75 (s) | 104.3 | 11, 1′, 4′, 6′/2′, 3′/5′ | 11, 12, 3′/5′-OCH3 | 6.69 (s) | 103.9 | 11, 1′, 4′, 6′/2′, 3′/5′ | 11, 12, 3′/5′-OCH3 |
| 3′/5′ | 148.8 | 148.8 | ||||||
| 4′ | 136.6 | 136.4 | ||||||
| 3′/5′-OCH3 | 3.80 (s) | 56.7 | 3′/5′ | 2′/6′ | 3.78 (s) | 56.7 | 3′/5′ | 2′/6′ |
| 4′-OH | 7.24 (brs) | 3′/5′, 4′ | 7.23 (brs) | 3′/5′ | ||||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auberon, F.; Olatunji, O.J.; Herbette, G.; Raminoson, D.; Antheaume, C.; Soengas, B.; Bonté, F.; Lobstein, A. Chemical Constituents from the Aerial Parts of Cyrtopodium paniculatum. Molecules 2016, 21, 1418. https://doi.org/10.3390/molecules21101418
Auberon F, Olatunji OJ, Herbette G, Raminoson D, Antheaume C, Soengas B, Bonté F, Lobstein A. Chemical Constituents from the Aerial Parts of Cyrtopodium paniculatum. Molecules. 2016; 21(10):1418. https://doi.org/10.3390/molecules21101418
Chicago/Turabian StyleAuberon, Florence, Opeyemi Joshua Olatunji, Gaëtan Herbette, Diamondra Raminoson, Cyril Antheaume, Beatriz Soengas, Frédéric Bonté, and Annelise Lobstein. 2016. "Chemical Constituents from the Aerial Parts of Cyrtopodium paniculatum" Molecules 21, no. 10: 1418. https://doi.org/10.3390/molecules21101418