
Investigating the Dissolution Performance of Amorphous Solid Dispersions Using Magnetic Resonance Imaging and Proton NMR


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Solution-State Assays
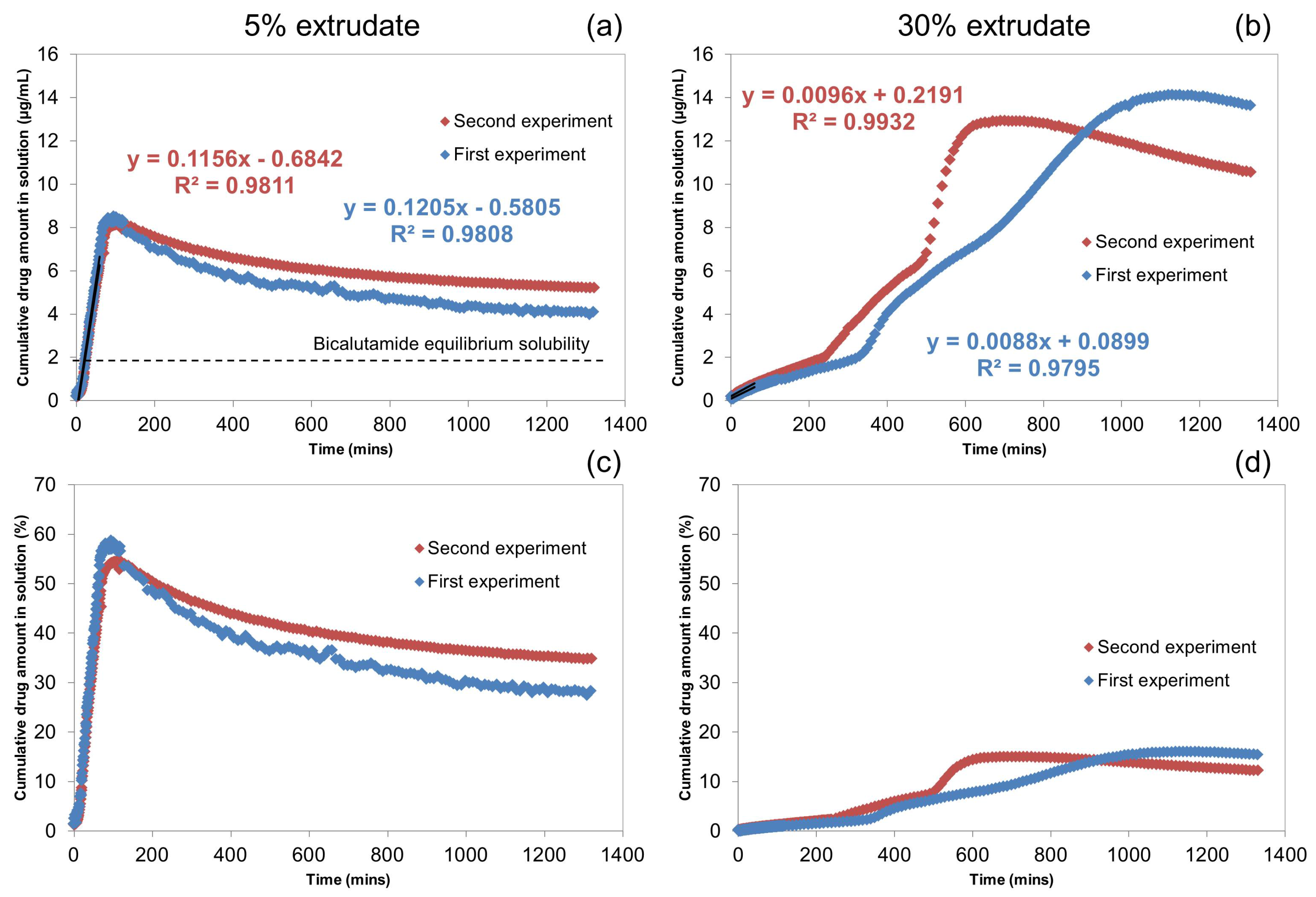
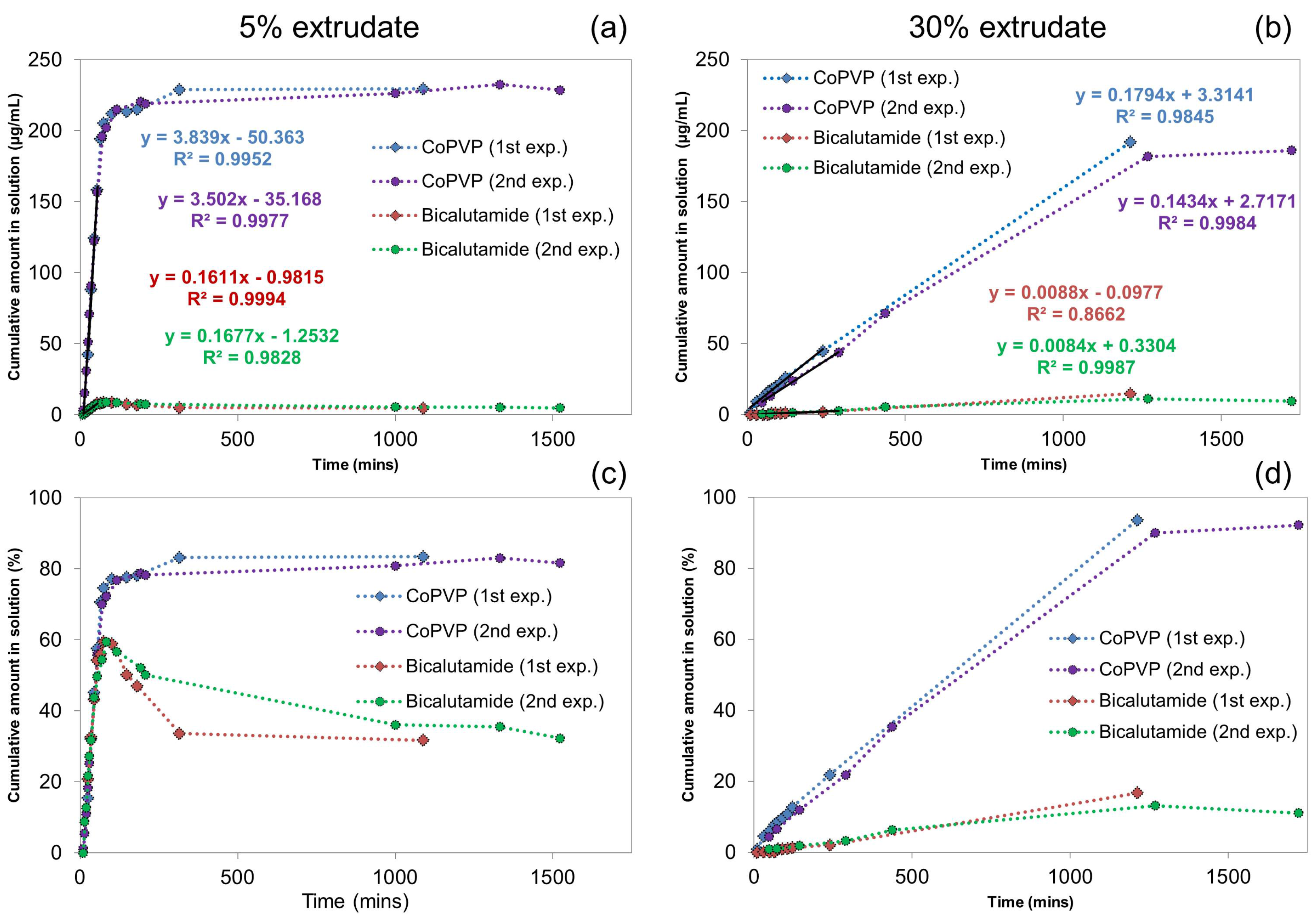
2.1.1. UV-Vis Flow Cell Experiments
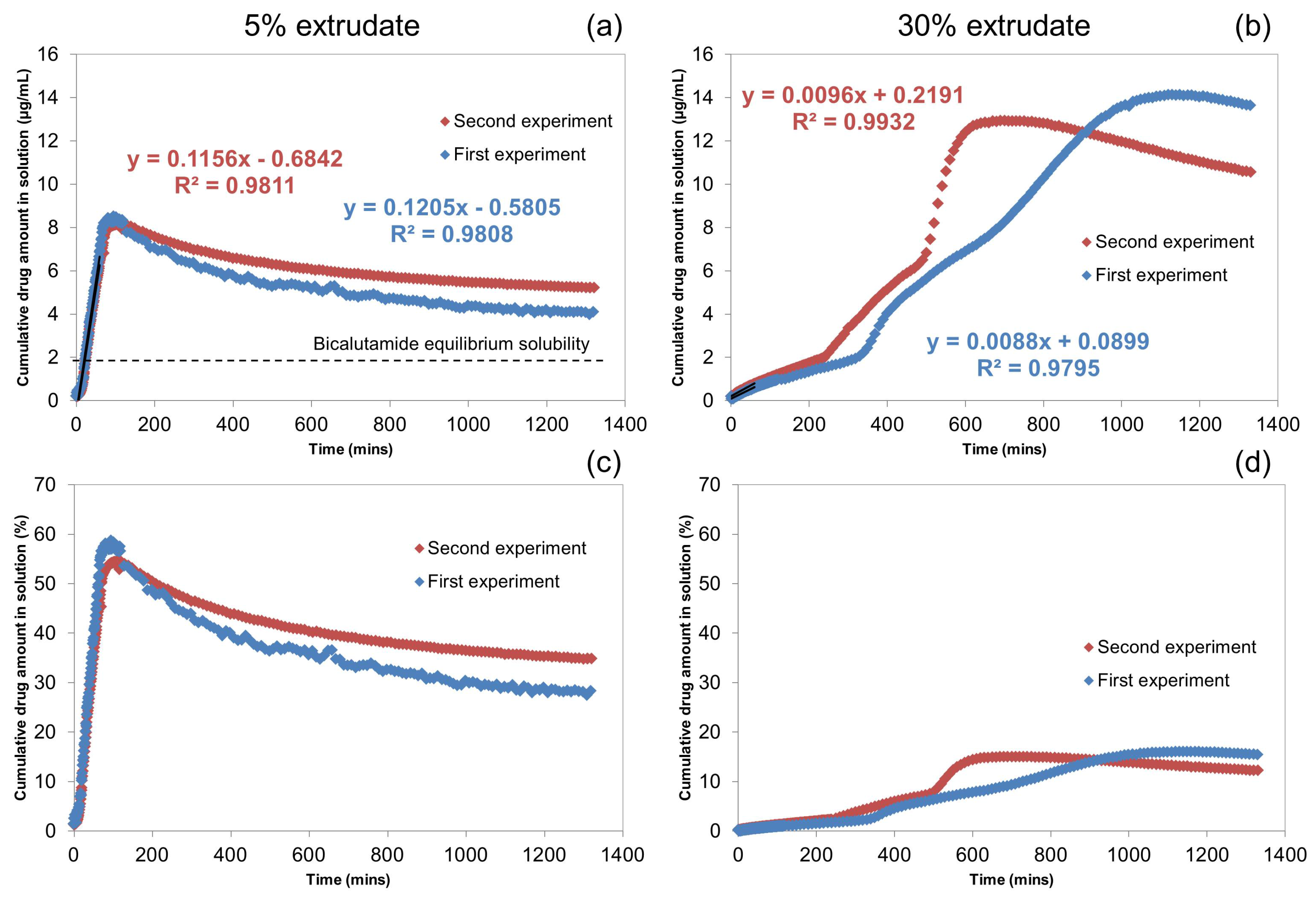
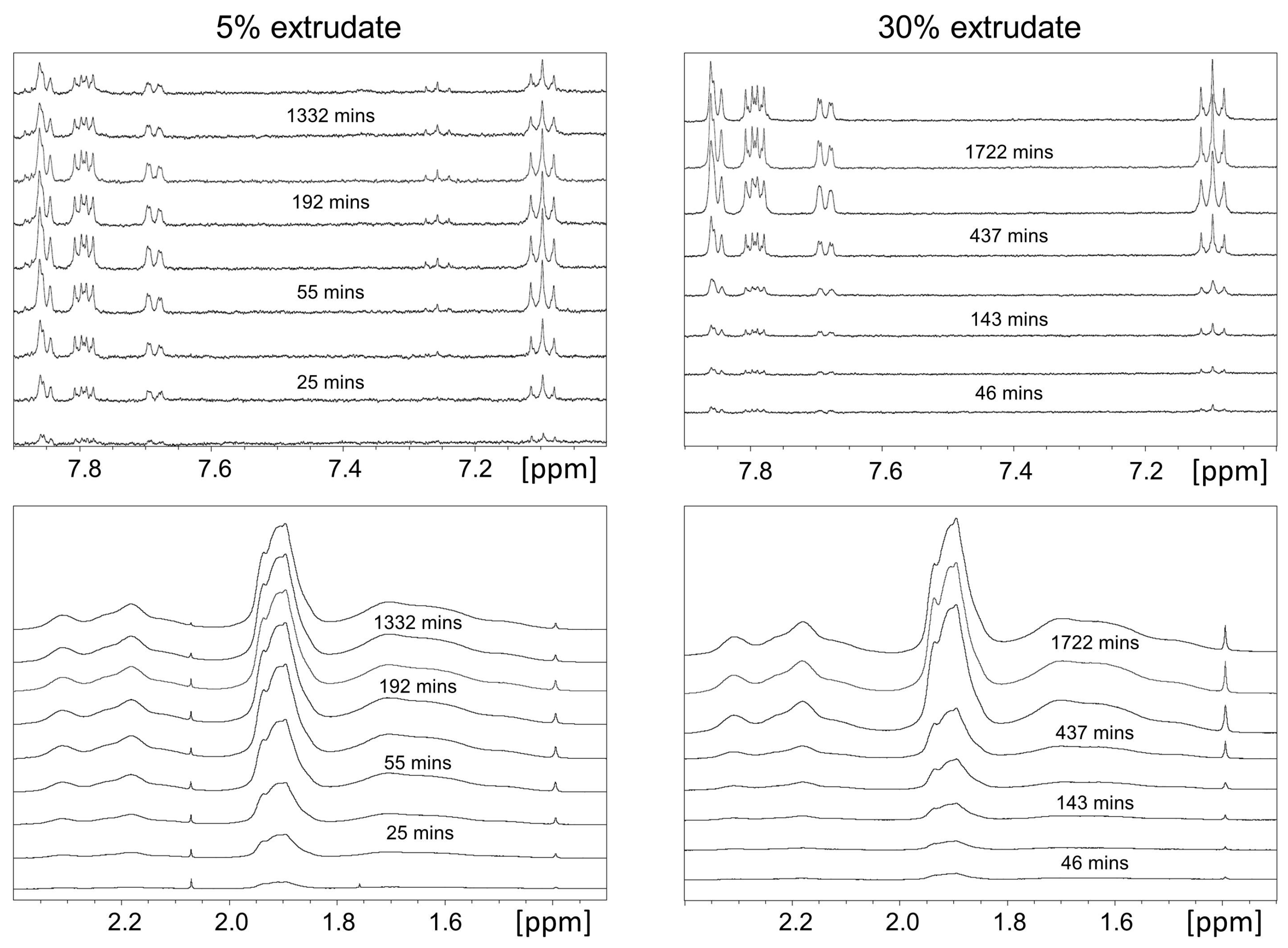
2.1.2. Off-Line H-NMR


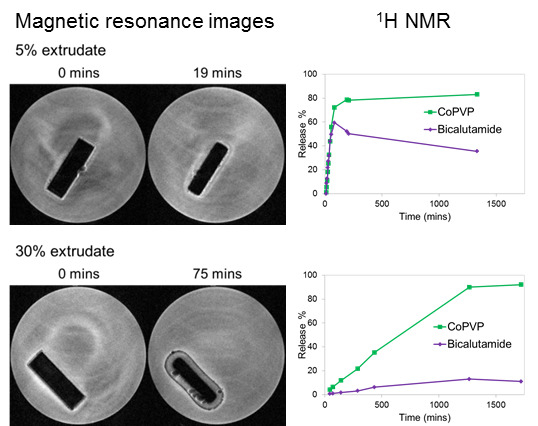
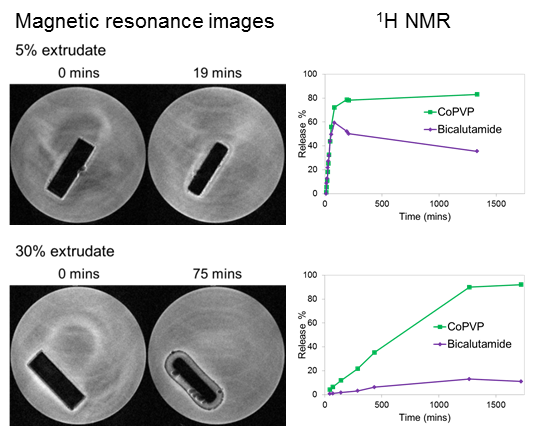
2.2. Solid-State Assays
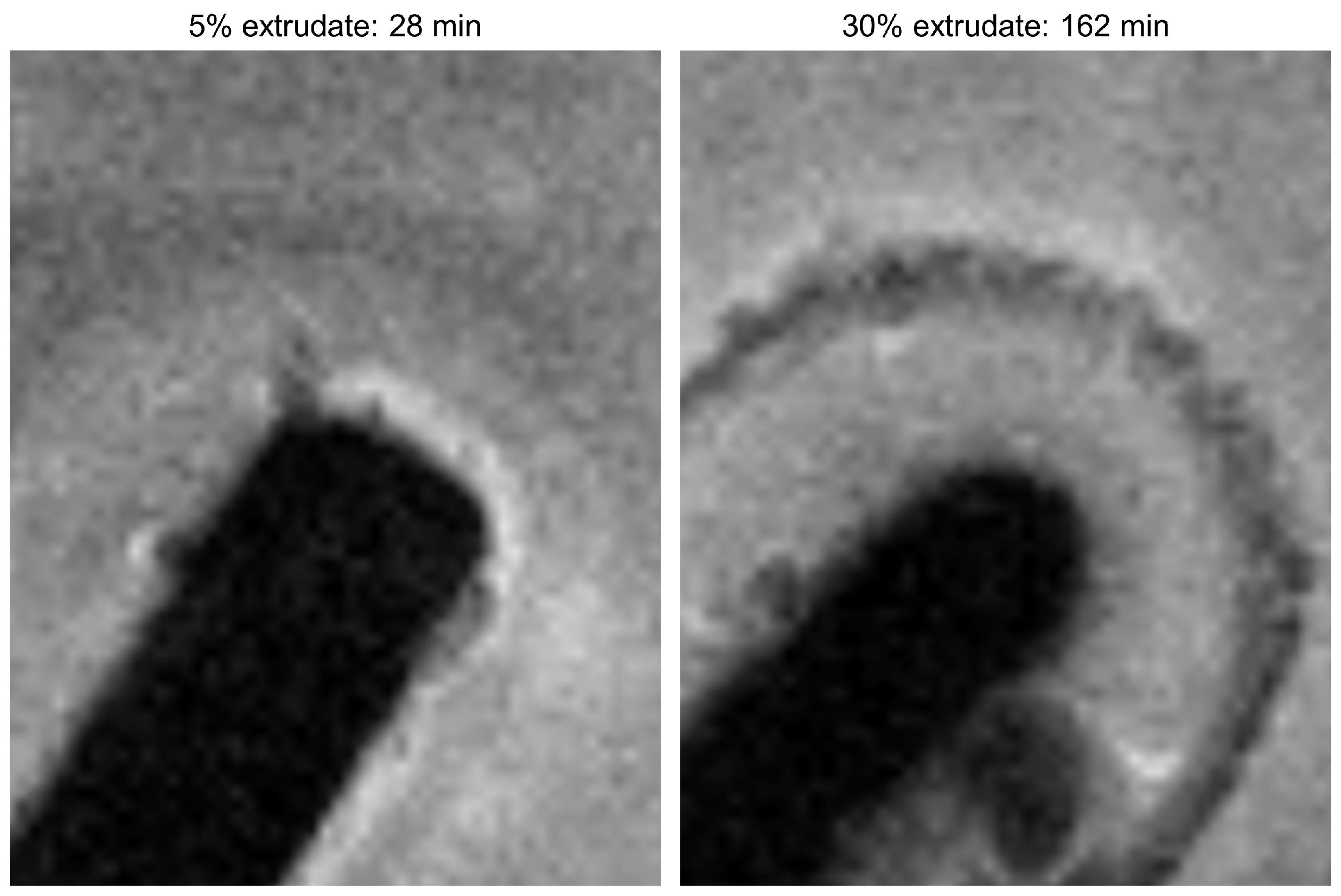
MRI Flow Cell Experiments
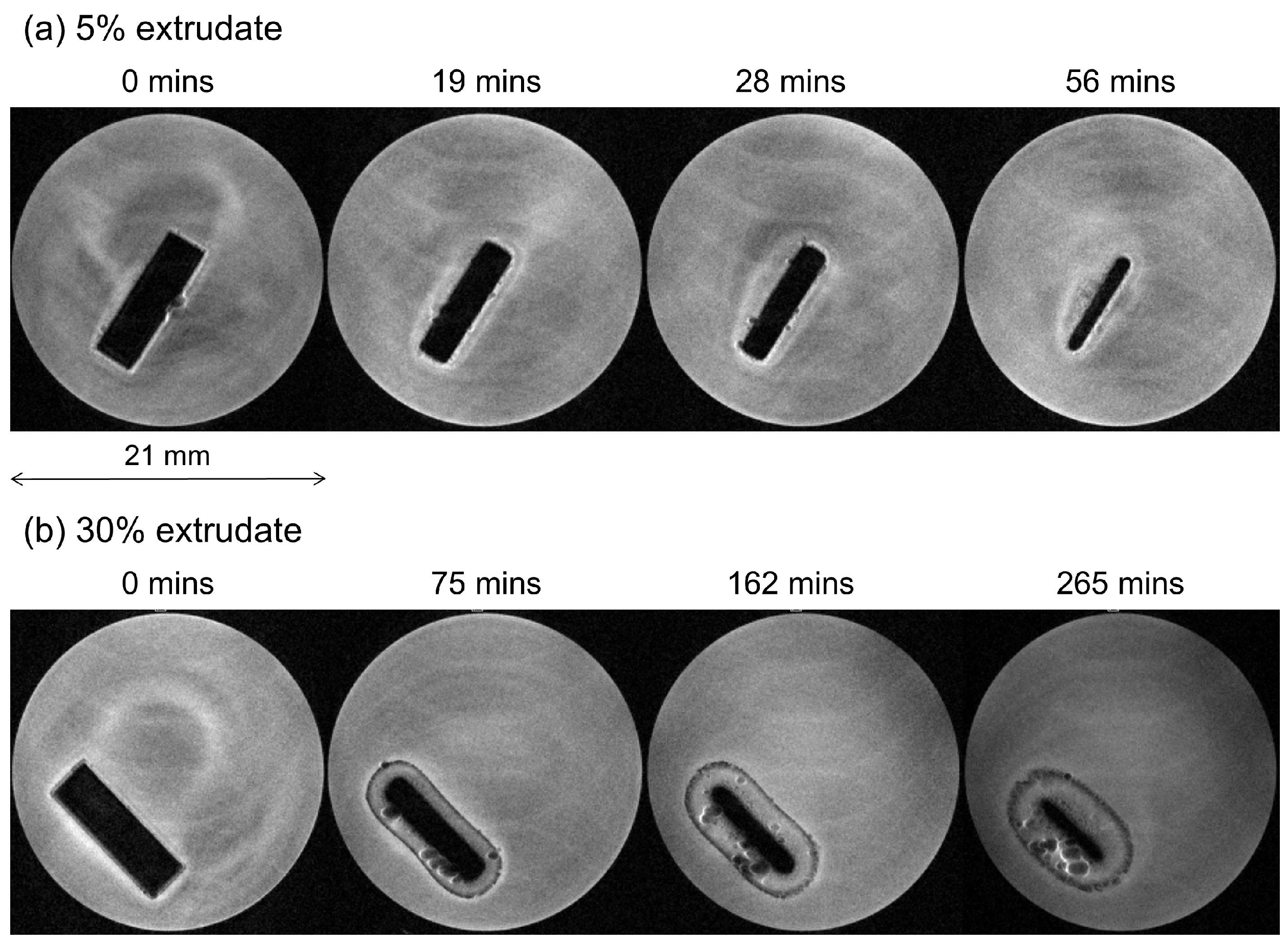
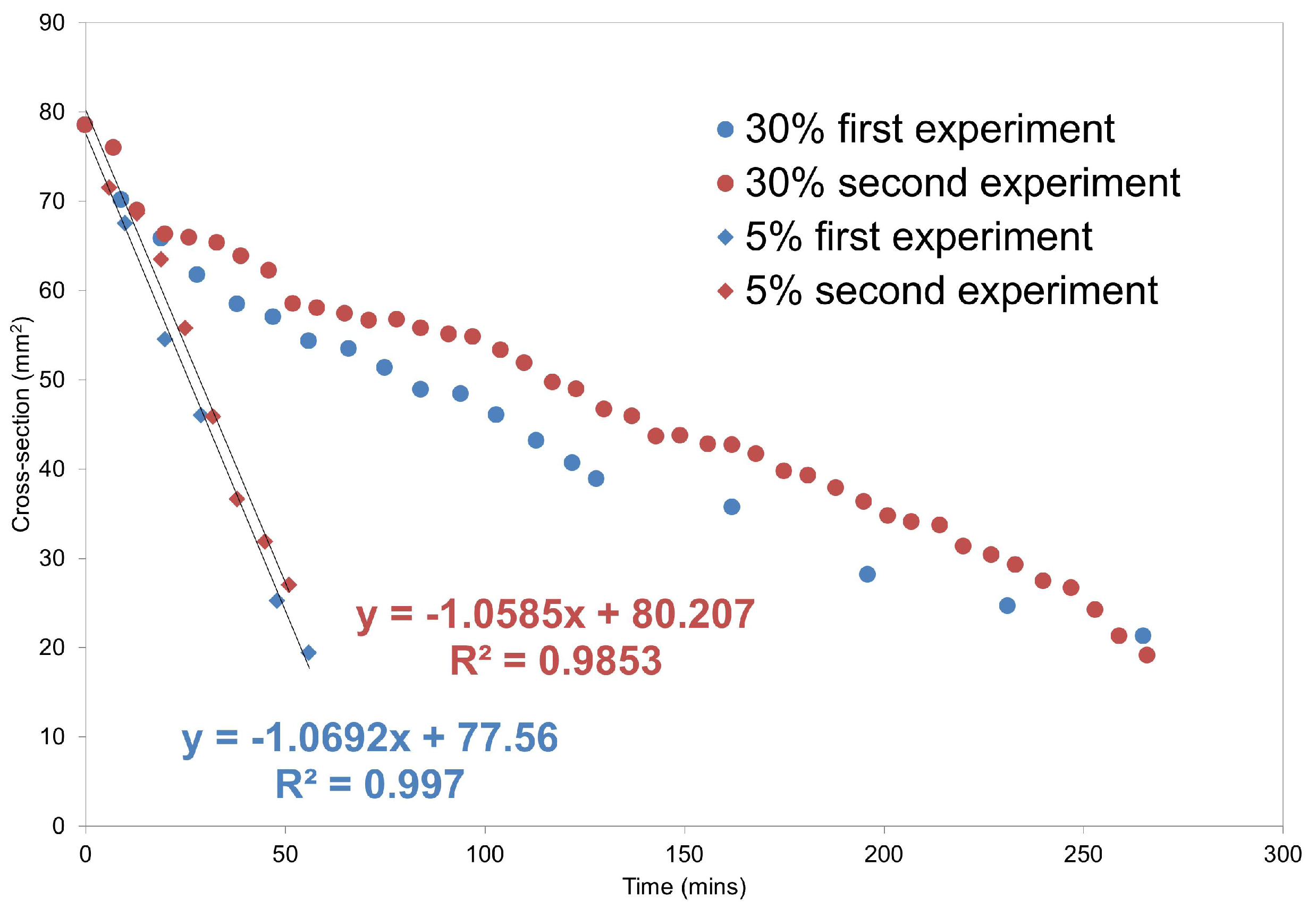
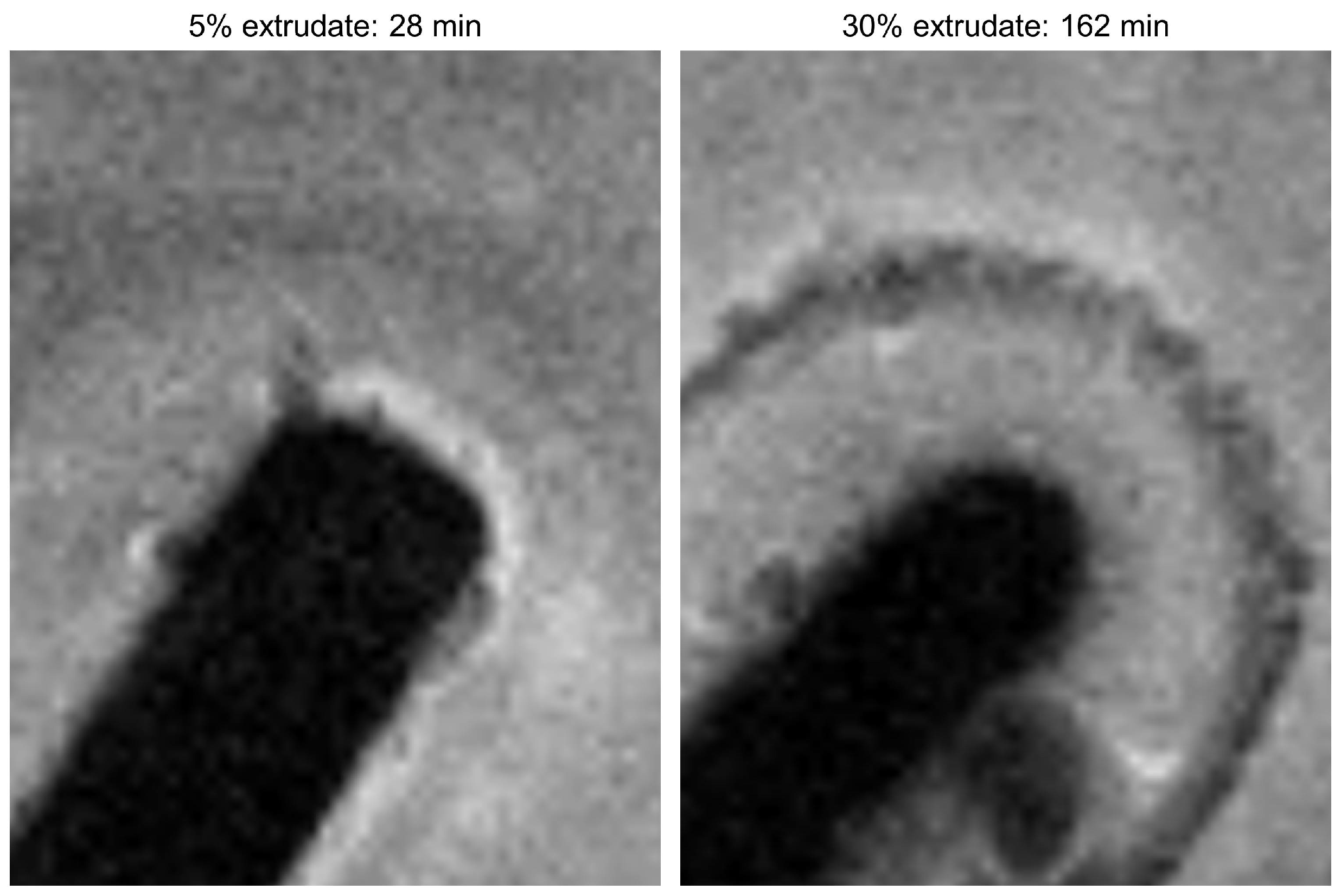
3. Experimental Section
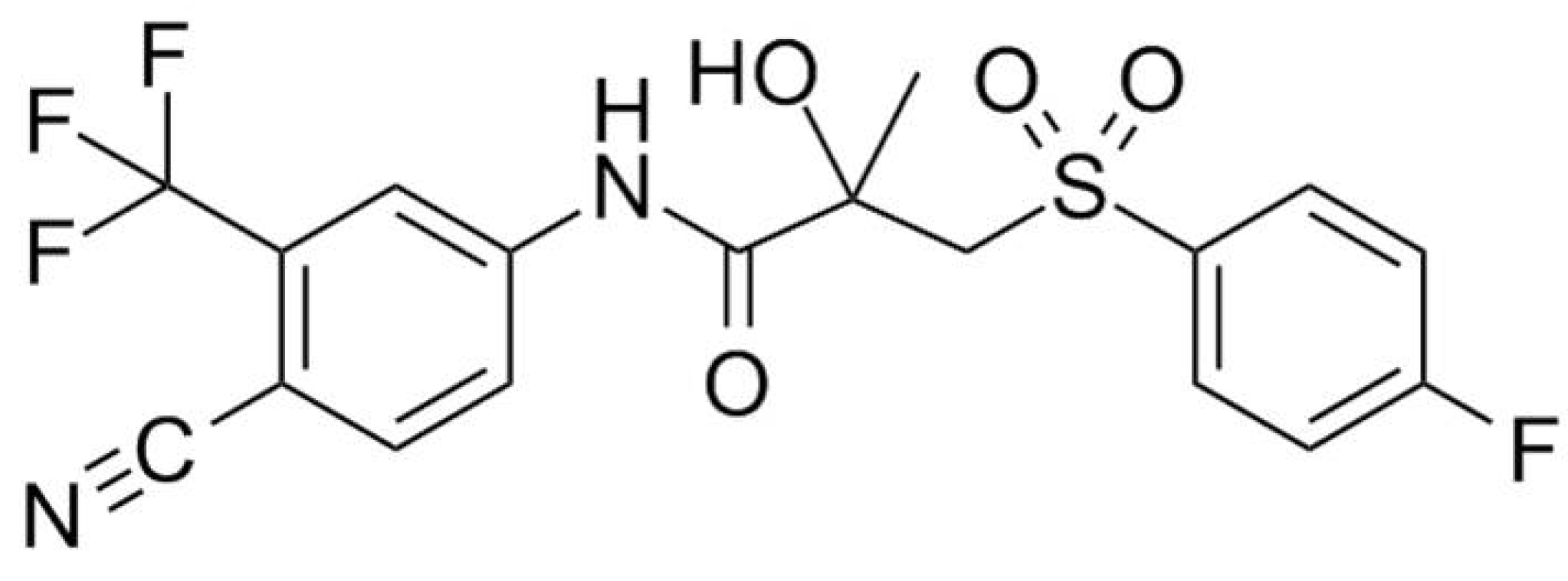
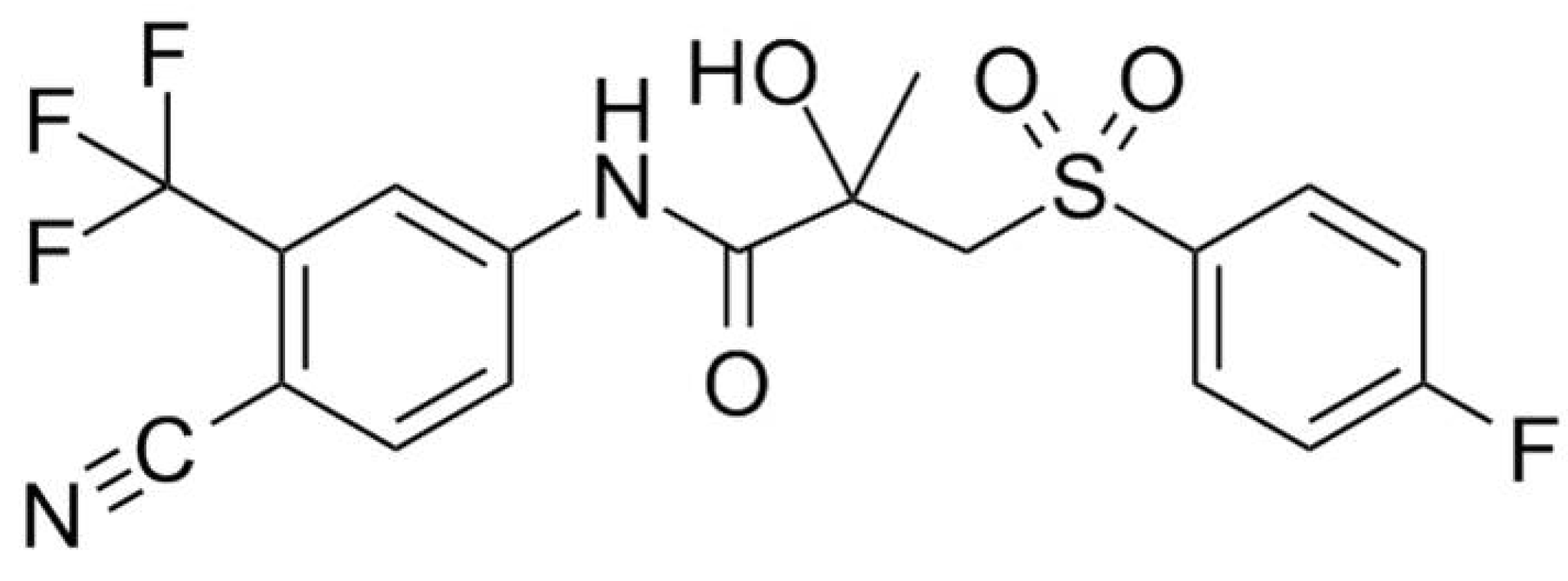
3.1. Materials
3.2. Sample Preparation
3.2.1. Preparation of the Amorphous Reference form of Bicalutamide
3.2.2. Preparation of the Bicalutamide: coPVP Extrudates
3.3. Analytical Methods
3.3.1. Raman Spectroscopy
3.3.2. X-ray Powder Diffraction (XRPD)
3.3.3. MRI UV-Vis Flow Cell System
3.3.4. H-NMR Spectroscopy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Di, L.; Fish, P.V.; Mano, T. Bridging solubility between drug discovery and development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, I. Physical stability studies of miscible amorphous solid dispersions. J. Pharm. Sci. 2010, 99, 4005–4012. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G.; Wuyts, M.; Blaton, N.; Busson, R.; Grobet, P.; Augustijns, P.; Kinget, R. Physical stabilisation of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur. J. Pharm. Sci. 2001, 12, 261–269. [Google Scholar] [CrossRef]
- Langham, Z.A.; Booth, J.; Hughes, L.P.; Reynolds, G.K.; Wren, S.A.C. Mechanistic insights into the dissolution of spray-dried amorphous solid dispersions. J. Pharm. Sci. 2012, 101, 2798–2810. [Google Scholar] [CrossRef] [PubMed]
- Tres, F.; Treacher, K.; Booth, J.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Real time Raman imaging to understand dissolution performance of amorphous solid dispersions. J. Control. Release 2014, 188, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.Q.M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Tres, F.; Patient, J.D.; Williams, P.M.; Treacher, K.; Booth, J.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Monitoring the dissolution mechanisms of amorphous bicalutamide solid dispersions via real-time raman mapping. Mol. Pharm. 2015, 12, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Alonzo, D.; Taylor, L. Influence of particle size on the ultraviolet spectrum of particulate-containing solutions: Implications for in-situ concentration monitoring using UV/vis fiber-optic probes. Pharm. Res. 2011, 28, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Boetker, J.P.; Savolainen, M.; Koradia, V.; Tian, F.; Rades, T.; Müllertz, A.; Cornett, C.; Rantanen, J.; Østergaard, J. Insights into the early dissolution events of amlodipine using UV imaging and raman spectroscopy. Mol. Pharm. 2011, 8, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.H.; Gordon, S.; Pajander, J.P.; Østergaard, J.; Rades, T.; Müllertz, A. Biorelevant characterisation of amorphous furosemide salt exhibits conversion to a furosemide hydrate during dissolution. Int. J. Pharm. 2013, 457, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.A.; Kazarian, S.G. FTIR spectroscopic imaging of dissolution of a solid dispersion of nifedipine in poly(ethylene glycol). Mol. Pharm. 2004, 1, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Wray, P.S.; Clarke, G.S.; Kazarian, S.G. Dissolution of tablet-in-tablet formulations studied with ATR-FTIR spectroscopic imaging. Eur. J. Pharm. Sci. 2013, 48, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Hughes, L.P.; Gladden, L.F.; Mantle, M.D. Quantitative ultra-fast MRI of HPMC swelling and dissolution. J. Pharm. Sci. 2010, 99, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Coombes, S.R.; Hughes, L.P.; Phillips, A.R.; Wren, S.A.C. Proton NMR: A new tool for understanding dissolution. Anal. Chem. 2014, 86, 2474–2480. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Wren, S.; Reynolds, G. Linking dissolution to disintegration in immediate release tablets using image analysis and a population balance modelling approach. Pharm. Res. 2012, 29, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Brillante, A.; Bilotti, I.; Della Valle, R.G.; Venuti, E.; Girlando, A. Probing polymorphs of organic semiconductors by lattice phonon Raman microscopy. CrystEngComm 2008, 10, 937–946. [Google Scholar] [CrossRef]
- Al-Dulaimi, S.; Aina, A.; Burley, J.C. Rapid polymorph screening on milligram quantities of pharmaceutical material using phonon-mode Raman spectroscopy. CrystEngComm 2010, 12, 1038–1040. [Google Scholar] [CrossRef]
- Fussell, A.; Garbacik, E.; Offerhaus, H.; Kleinebudde, P.; Strachan, C. In situ dissolution analysis using coherent anti-Stokes Raman scattering (CARS) and hyperspectral CARS microscopy. Eur. J. Pharm. Biopharm. 2013, 85, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Roy, R. Quantitative H-NMR spectroscopy. Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Dalitz, F.; Cudaj, M.; Maiwald, M.; Guthausen, G. Process and reaction monitoring by low-field NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Abu-Diak, O.A.; Jones, D.S.; Andrews, G.P. Understanding the performance of melt-extruded poly(ethylene oxide)-bicalutamide solid dispersions: Characterisation of microstructural properties using thermal, spectroscopic and drug release methods. J. Pharm. Sci. 2012, 101, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.P.; AbuDiak, O.A.; Jones, D.S. Physicochemical characterization of hot melt extruded bicalutamide-polyvinylpyrrolidone solid dispersions. J. Pharm. Sci. 2010, 99, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds used in this study are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tres, F.; Coombes, S.R.; Phillips, A.R.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Investigating the Dissolution Performance of Amorphous Solid Dispersions Using Magnetic Resonance Imaging and Proton NMR. Molecules 2015, 20, 16404-16418. https://doi.org/10.3390/molecules200916404
Tres F, Coombes SR, Phillips AR, Hughes LP, Wren SAC, Aylott JW, Burley JC. Investigating the Dissolution Performance of Amorphous Solid Dispersions Using Magnetic Resonance Imaging and Proton NMR. Molecules. 2015; 20(9):16404-16418. https://doi.org/10.3390/molecules200916404
Chicago/Turabian StyleTres, Francesco, Steven R. Coombes, Andrew R. Phillips, Leslie P. Hughes, Stephen A. C. Wren, Jonathan W. Aylott, and Jonathan C. Burley. 2015. "Investigating the Dissolution Performance of Amorphous Solid Dispersions Using Magnetic Resonance Imaging and Proton NMR" Molecules 20, no. 9: 16404-16418. https://doi.org/10.3390/molecules200916404