
Synthesis of Phenolic Compounds by Trapping Arynes with a Hydroxy Surrogate

Abstract
:
1. Introduction
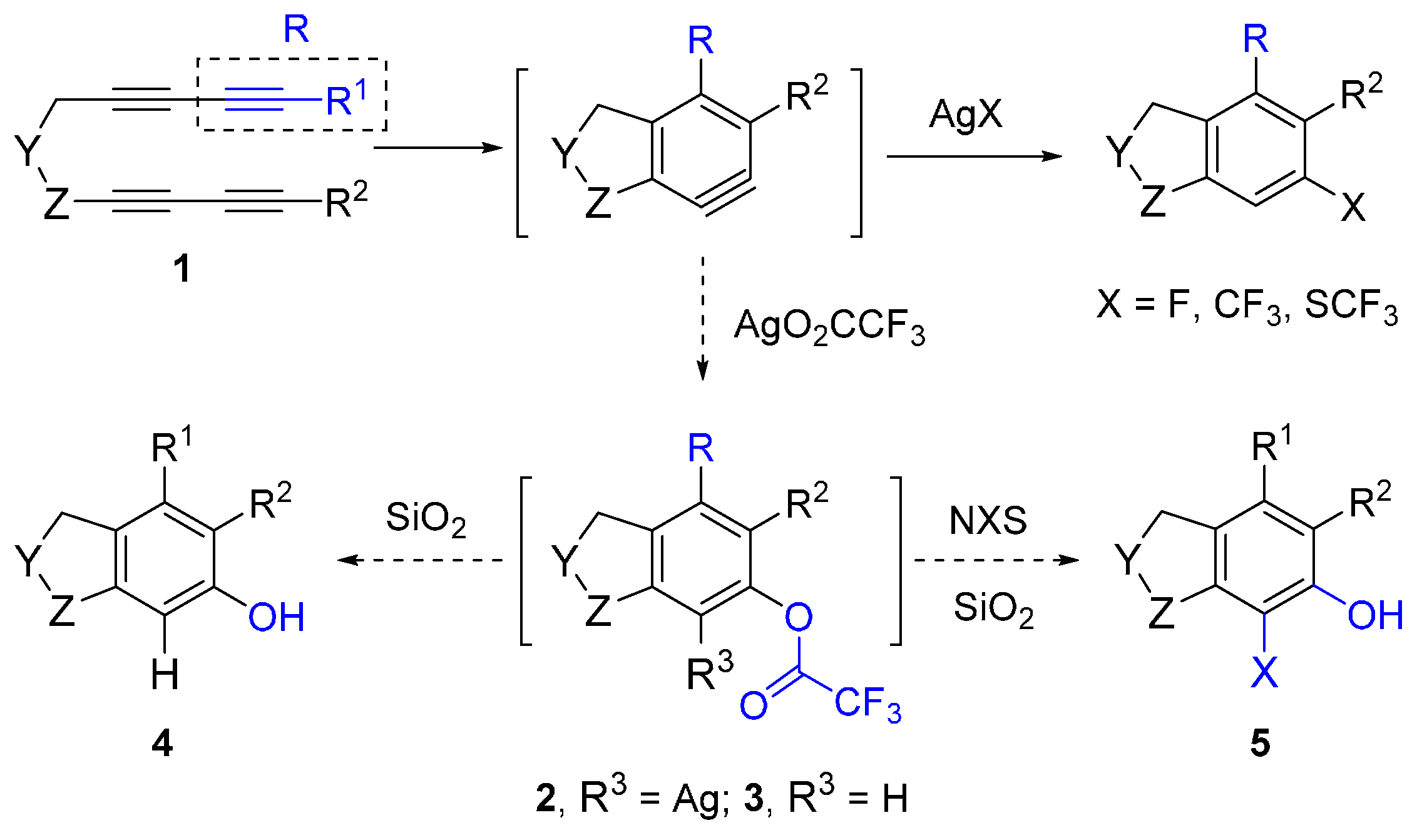
2. Results and Discussion


| Entry | Diyne | Products Ratio a | Yield (%) b | |
|---|---|---|---|---|
| 1 | 1a |  | 75 | |
| 2 | 1b |  | 87 | |
| 3 | 1c |  | 66 | |
| 4 | 1d |  | 66 | |
| 5 | 1e |  | 69 |

| Entry | Diyne | Products Ratio a | Yield (%) b | |
|---|---|---|---|---|
| 1 | 1a |  | 69 | |
| 2 | 67 | |||
| 3 | 1b |  | 63 | |
| 4 | 1f |  | 83 | |
| 5 | 88 | |||
| 6 | 1e |  | 36 |

{kind=link}
{kind=link}
| Entry | Diyne | Products Ratio a | Yield (%) b | |
|---|---|---|---|---|
| 1 | 1g |  | 82 | |
| 2 | 1c | 52 | ||
| 3 | 1c |  | 65 | |
| 4 | 1a |  | 82 | |
| 5 | 1f | 94 | ||
| 6 | 1f |  | 30 | |
| 7 | 76 |
3. Experimental Section
3.1. General Information
3.2. Experimental Details
3.2.1. General Procedure for the Mono-Functionalization (GPM)
3.2.2. General Procedure for the Bis-Functionalization (GPB)
3.2.3. Characterization Data of the Products


























4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Pellissier, H.; Santelli, M. The Use of Arynes in Organic Synthesis. Tetrahedron 2003, 59, 701–730. [Google Scholar] [CrossRef]
- Dyke, A.M.; Hester, A.J.; Lloyd-Jones, G.C. Organometallic Generation and Capture of ortho-Arynes. Synthesis 2006, 24, 4093–4112. [Google Scholar] [CrossRef]
- Sanz, R. Recent Applications of Aryne Chemistry to Organic Synthesis. A Review. Org. Prep. Proced. Int. 2008, 40, 215–291. [Google Scholar] [CrossRef]
- Chen, Y.; Larock, R.C. Arylation Reactions Involving the formation of arynes. In Modern Arylation Methods; Akermann, L., Ed.; WILEY-VCH: Weinheim, Germany, 2009; pp. 401–473. [Google Scholar]
- Kitamura, T. Synthetic Methods for the Generation and Preparative Application of Benzyne. Aust. J. Chem. 2010, 63, 987–1001. [Google Scholar] [CrossRef]
- Tadross, P.M.; Stoltz, B.M. A Comprehensive History of Arynes in Natural Product Total Synthesis. Chem. Rev. 2012, 112, 3550–3577. [Google Scholar] [CrossRef] [PubMed]
- Gampe, C.M.; Carreira, E.M. Arynes and Cyclohexyne in Natural Product Synthesis. Angew. Chem. Int. Ed. 2012, 51, 3766–3778. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Shi, F. A Closer Look at Aryne Chemistry: Details that Remain Mysterious. Asian J. Org. Chem. 2013, 2, 116–125. [Google Scholar] [CrossRef]
- Rodan, N.G.; Domelsmith, L.N.; Houk, K.N. The Relative Rates of Electron-Rich and Electron-Deficient Alkene Cycloadditions to Benzyne. Enhanced Electrophilicity as a Consequence of Alkyne Bending Distortion. Tetrahedron Lett. 1979, 20, 3237–3240. [Google Scholar] [CrossRef]
- Kitamura, T.; Yamane, M. (Phenyl)[o-(Trimethylsilyl)phenyl]iodonium Triflate. A New and Efficient Precursor of Benzyne. J. Chem. Soc. Chem. Commun. 1995, 983–984. [Google Scholar] [CrossRef]
- Himeshima, Y.; Sonoda, T.; Kobayashi, H. Fluoride-Induced 1,2-Elimination of o-Trimethylsilylphenyl Triflate to Benzyne Under Mild Conditions. Chem. Lett. 1983, 12, 1211–1214. [Google Scholar] [CrossRef]
- Campbell, C.D.; Rees, C.W. Reactive Intermediates. Part 1. Synthesis and Oxidation of 1- and 2-Aminobenzotriazole. J. Chem. Soc. C 1969, 742–747. [Google Scholar] [CrossRef]
- Matsumoto, T.; Hosoya, T.; Katsuki, M.; Suzuki, K. New Efficient Protocol for Aryne Generation. Selective Synthesis of Differentially Protected 1,4,5-Naphthalenetriols. Tetrahedron Lett. 1991, 32, 6735–6736. [Google Scholar] [CrossRef]
- Friedman, L.; Logullo, F.M. Reactions of Benzyne Intermediates in Non-basic Media. J. Am. Chem. Soc. 1963, 85, 1792–1797. [Google Scholar] [CrossRef]
- Hoffmann, R.W. Dehydrobenzene and Cycloalkynes; Verlag Chemie-Academic Press: New York, NY, USA, 1967. [Google Scholar]
- Wittig, G.; Hoffmann, R.W. 1,2,3-Benzothiadiazole 1,1-dioxide. Org. Synth. 1967, 47. [Google Scholar] [CrossRef]
- Miyawaki, K.; Suzuki, R.; Kawano, T.; Ueda, I. Cycloaromatization of a Non-Conjugated Polyenyne System: Synthesis of 5H-Benzo[d]fluoreno[3,2-b]pyrans via Diradicals Generated from 1-[2-{4-(2-Alkoxymethylphenyl)butan-1,3-diynyl}]phenylpentan-2,4-diyn.l-ols and Trapping Evidence for the 1,2-Didehydrobenzene Diradical. Tetrahedron Lett. 1997, 38, 3943–3946. [Google Scholar]
- Bradley, A.Z.; Johnson, R.P. Thermolysis of 1,3,8-Nonatriyne: Evidence for Intramolecular [2 + 4] Cycloaromatization to a Benzyne Intermediate. J. Am. Chem. Soc. 1997, 119, 9917–9918. [Google Scholar] [CrossRef]
- Hoye, T.R.; Baire, B.; Niu, D.; Willoughby, P.H.; Woods, B.P. The Hexadehydro-Diels-Alder Reaction. Nature 2012, 490, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.Y.; Wang, K.P.; Lee, N.K.; Mamidipalli, P.; Lee, D. Alkane C–H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc. 2013, 135, 4668–4671. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, R.; Yun, S.Y.; Wang, K.P.; Lee, D. Regioselectivity in the Nucleophile Trapping of Arynes: The Electronic and Steric Effects of Nucleophiles and Substituents. Org. Lett. 2014, 16, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, P.H.; Niu, D.; Wang, T.; Haj, M.K.; Cramer, C.J.; Hoye, T.R. Mechanism of the Reactions of Alcohols with o-Benzynes. J. Am. Chem. Soc. 2014, 136, 13657–13665. [Google Scholar] [CrossRef] [PubMed]
- Whiting, D.A. Natural Phenolic Compounds 1900–2000: A Bird’s Eye View of a Century’s Chemistry. Nat. Prod. Rep. 2001, 18, 583–606. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Mussatto, S.I.; Martínez-Avila, G.; Montañez-Saenz, J.; Aguilar, C.N.; Teixeira, J.A. Bioactive Phenolic Compounds: Production and Extraction by Solid-State Fermentation. A Review. Biotechnol. Adv. 2011, 29, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.P.; Yun, S.Y.; Mamidipalli, P.; Lee, D. Silver-Mediated Fluorination, Trifluoromethylation and Trifluoromethylthiolation of Arynes. Chem. Sci. 2013, 4, 3205–3211. [Google Scholar] [CrossRef]
- The reaction needed overnight heating at 120 °C for complete consumption of the substrate.
- Bronner, S.M.; Goetz, A.E.; Garg, N.K. Overturning Indolyne Regioselectivities and Synthesis of Indolactam V. J. Am. Chem. Soc. 2011, 133, 3832–3835. [Google Scholar] [CrossRef] [PubMed]
- Cheong, P.H.Y.; Paton, R.S.; Bronner, S.M.; Im, G.Y.J.; Garg, N.K.; Houk, K.N. Indolyne and Aryne Distortions and Nucleophilic Regioselectivites. J. Am. Chem. Soc. 2010, 132, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Bronner, S.M.; Mackey, J.L.; Houk, K.N.; Garg, N.K. Steric Effects Compete with Aryne Distortion to Control Regioselectivities of Nucleophilic Additions to 3-Silylarynes. J. Am. Chem. Soc. 2012, 134, 13966–13969. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.M.; Mackey, J.L.; Garg, N.K.; Houk, K.N. The Role of Aryne Distortions, Steric Effects, and Charges in Regioselectivities of Aryne Reactions. J. Am. Chem. Soc. 2014, 136, 15798–15805. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. Arynes in a Three-Component Coupling Reaction: Straightforward Synthesis of Benzoannulated Iminofurans. Angew. Chem. Int. Ed. 2004, 43, 3935–3938. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. Straightforward Access to 2-Iminoisoindolines via Three-Component Coupling of Arynes, Isocyanides and Imines. Tetrahedron Lett. 2004, 45, 8659–8662. [Google Scholar] [CrossRef]
- Yoshida, H.; Fukushima, H.; Morishita, T.; Ohshita, J.; Kunai, A. Three-Component Coupling using Arynes and Isocyanides: Straightforward Access to Benzo-Annulated Nitrogen or Oxygen Heterocycles. Tetrahedron 2007, 63, 4793–4805. [Google Scholar] [CrossRef]
- Allan, K.M.; Gilmore, C.D.; Stoltz, B.M. Benzannulated Bicycles by Three-Component Aryne Reactions. Angew. Chem. Int. Ed. 2011, 50, 4488–4491. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Takaki, K. Multicomponent Coupling Reaction of Arynes for Construction of Heterocyclic Skeletons. Heterocycles 2012, 85, 1333–1349. [Google Scholar] [CrossRef]
- Li, J.; Noyori, S.; Iwasaki, M.; Nakajima, K.; Nishihara, Y. A Novel Three-Component Coupling Reaction of Arynes, Isocyanides, and Cyanoformates: A Straightforward Access to Cyano-Substituted Iminoisobenzofurans. Heterocycles 2012, 86, 933–940. [Google Scholar] [CrossRef]
- Li, J.; Noyori, S.; Nakajima, K.; Nishihara, Y. New Entry to the Synthesis of α-Iminonitriles by Lewis Acid Mediated Isomerization of Cyano-Substituted Iminoisobenzofurans Prepared by Palladium-Catalyzed Three-Component Coupling of Arynes, Isocyanides, and Cyanoformates. Organometallics 2014, 33, 3500–3507. [Google Scholar] [CrossRef]
- The observed regioselectivity for the reactions of 1d and 1e can be further confirmed by the Mulliken Population analysis (but less accurately by NBO analysis) of aryne intermediates. See Supplementary Materials for the calculations.
- Regiochemistry of the products were predicted on the basis of product distribution in a reaction of 1f with AgOTf in the absence of any electrophile. The product was formed in 4.4:1 ratio favoring the ortho-addition of triflate.
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karmakar, R.; Ghorai, S.; Xia, Y.; Lee, D. Synthesis of Phenolic Compounds by Trapping Arynes with a Hydroxy Surrogate. Molecules 2015, 20, 15862-15880. https://doi.org/10.3390/molecules200915862
Karmakar R, Ghorai S, Xia Y, Lee D. Synthesis of Phenolic Compounds by Trapping Arynes with a Hydroxy Surrogate. Molecules. 2015; 20(9):15862-15880. https://doi.org/10.3390/molecules200915862
Chicago/Turabian StyleKarmakar, Rajdip, Sourav Ghorai, Yuanzhi Xia, and Daesung Lee. 2015. "Synthesis of Phenolic Compounds by Trapping Arynes with a Hydroxy Surrogate" Molecules 20, no. 9: 15862-15880. https://doi.org/10.3390/molecules200915862