2.1. Scouting Experiments
Scouting experiments showed that no polymeric substance was formed without TEA. A series of exploratory reactions was performed varying the ratios of DODT, TEA and H
2O
2 (
Table 1).
Table 1.
Scouting experiments.
Table 1.
Scouting experiments.
| Reaction | DODT | TEA | H2O2 | Initial |
|---|
| No. | mmol | mmol | mmol | Observations |
|---|
| E1 | 6.1 | 18.3 | 1.5 | No change |
| E2 | 6.1 | 18.3 | 3.1 | Viscous liquid |
| E3 | 6.1 | 18.3 | 6.1 | Sticky polymer |
| E4 | 6.1 | 12.2 | 1.5 | No change |
| E5 | 6.1 | 12.2 | 3.1 + 13.2 | Rubbery polymer |
| E6 | 6.1 | 12.2 | 6.1 | Sticky polymer |
| E7 | 6.1 | 9.2 | 1.5 | No change |
| E8 | 6.1 | 9.2 | 3.1 | Viscous liquid |
| E9 | 6.1 | 9.2 | 6.1 | Sticky polymer |
The vials with only 1.5 mmol of H2O2 (0.25 molar equivalent to DODT, E1, E4, E7) did not show any visible, permanent change in appearance. Translucent viscous liquids separated to the bottom of the vials in reactions E2 and E8 (0.5 molar equivalent to DODT). The volume of the translucent products was approximately 1 mL, which was the volume of DODT in the reaction mixture. Reactions E3, E6 and E9 (one molar equivalent to DODT) each formed a translucent, white globular polymeric product which separated to the bottom of the reaction mixture. The liquid reaction mixtures were decanted, and the remaining products were rinsed with fresh DI water followed by methanol. Products were soaked in acetone to extract residual water. When the products became translucent, the acetone was decanted and the products were dried overnight. The resulting products were clear, sticky polymeric substances that had spread to cover half of an aluminum pan (5.1 cm diameter). To reaction E5, an additional 15 mL of H2O2 was added (2.67 total molar equivalent to DODT) and after 2–5 min an opaque white, spherical mass with a layered surface and dense sponge-like texture had formed. After extraction in acetone and drying, the product was completely clear and colorless with a rubbery, tack-free texture. To verify the results from E5, an additional 12 mL aliquot of H2O2 was reacted with E1, E4, and E7 (2.00 total molar equivalent to DODT). All three reactions formed polymer products. The reaction in E7, which contained less than two molar equivalents of TEA relative to DODT, formed a gooey substance while the products of reactions E1 and E4 were solid, non-sticky polymers. These results demonstrated that a minimum of 1:2 molar ratio of DODT/TEA and DODT/H2O2 were needed to obtain non-sticky high molecular weight polymers. Based on the results of the scouting experiments, model reactions were carried out.
2.2. Two-Phase Model Reactions
The new dithiol polymerization is a two-phase system, with an organic phase (DODT + TEA) and an aqueous phase (H
2O/H
2O
2). In order to investigate the water phase a model reaction using D
2O was carried out. In the
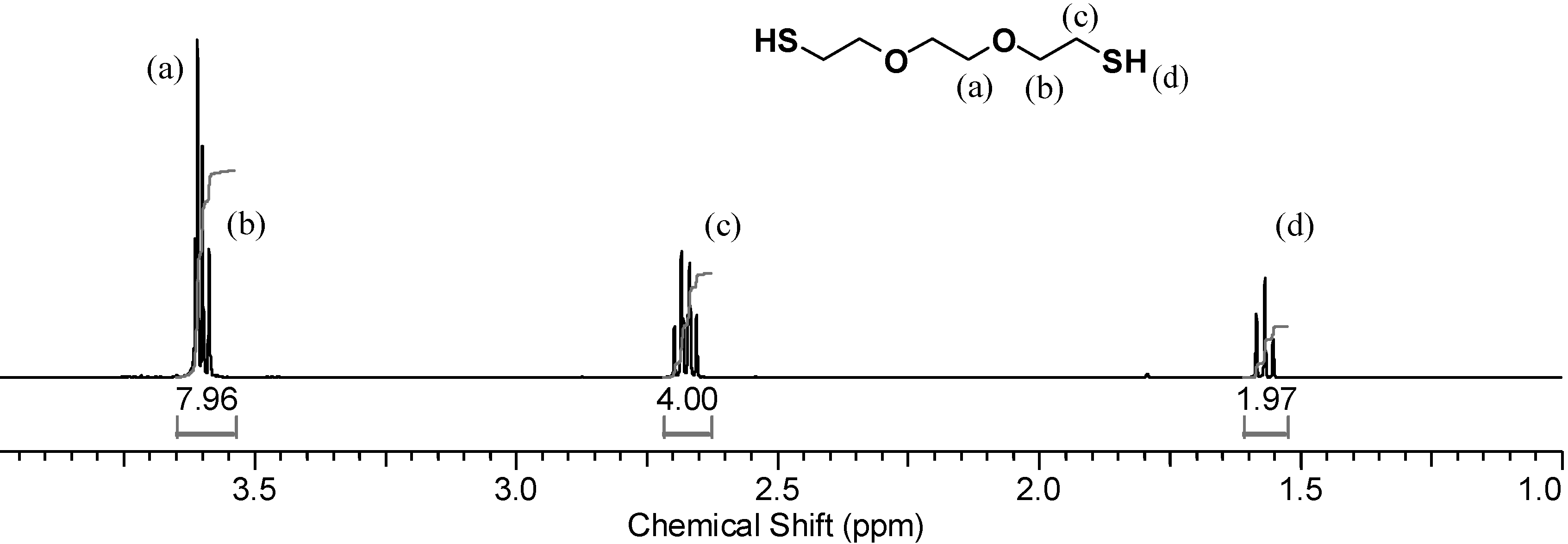
1H-NMR spectrum of the monomer dissolved in CDCl
3, shown in
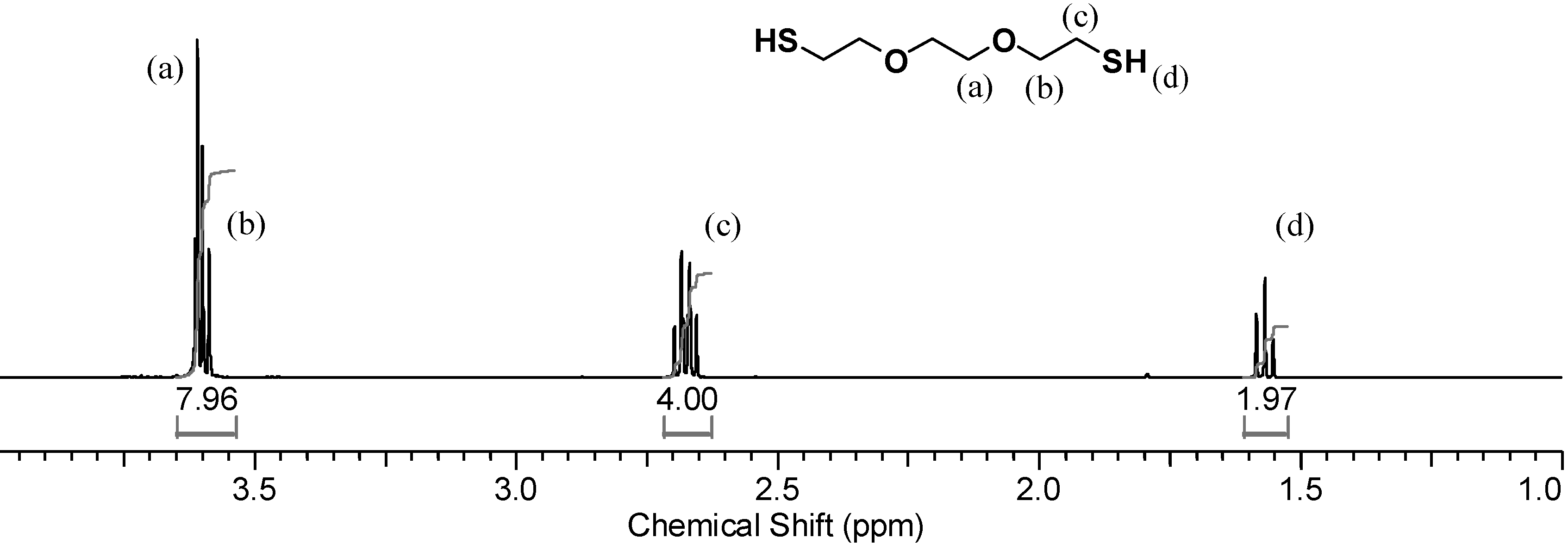
Figure 3, the thiol triplet of DODT appears at 1.58 ppm, the signal of the methylene protons (
c) appear as a quartet at 271 ppm and peaks from the oxygen adjacent methylene groups (a, b) overlap at about 3.62 ppm.
Figure 3.
1H-NMR spectrum of DODT in CDCl3. (500 MHz; CDCl3; 12 s relax; 128 trans).
Figure 3.
1H-NMR spectrum of DODT in CDCl3. (500 MHz; CDCl3; 12 s relax; 128 trans).
When DODT and D
2O are combined, the system partitions into two phases where the organic phase (
ρ(DODT) = 1.114 g/mL) settles below the D
2O aqueous phase. The
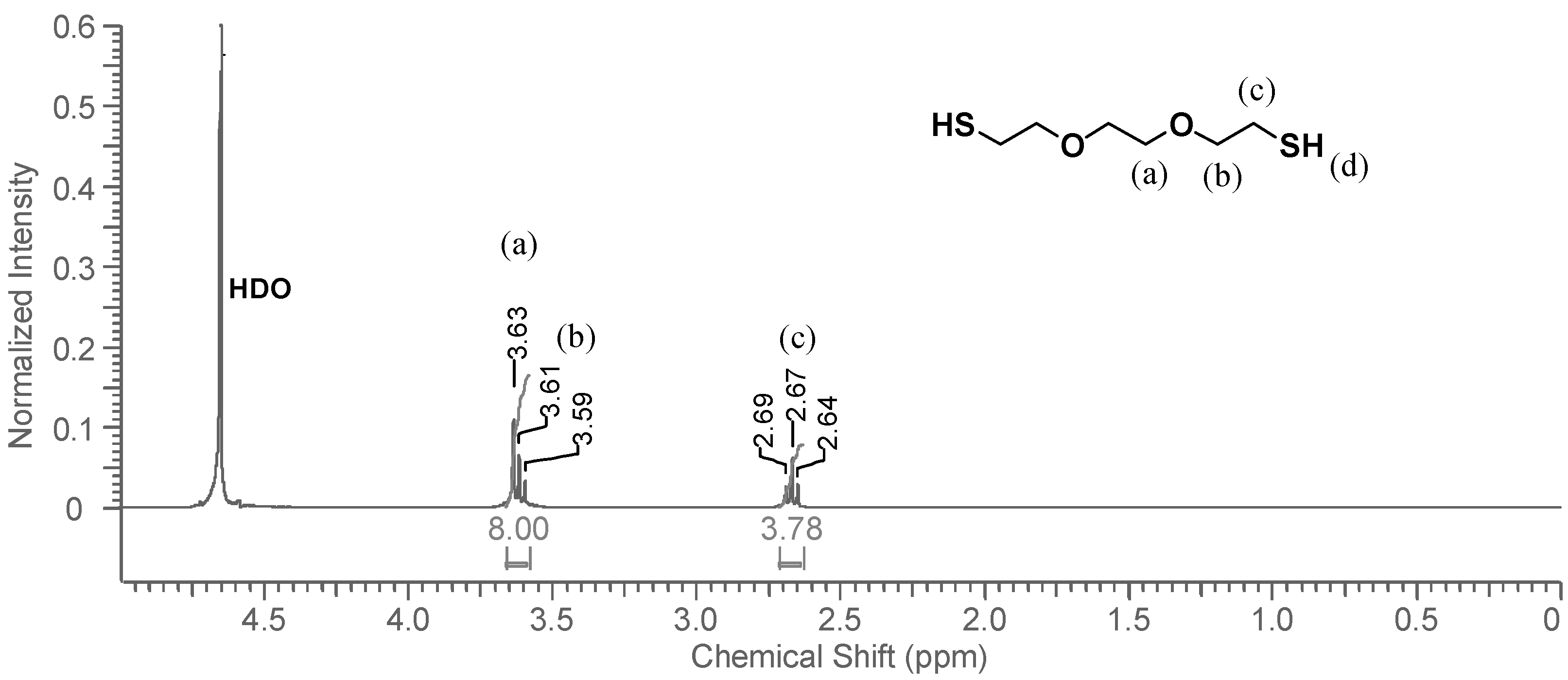
1H-NMR spectrum of the D
2O phase is shown in
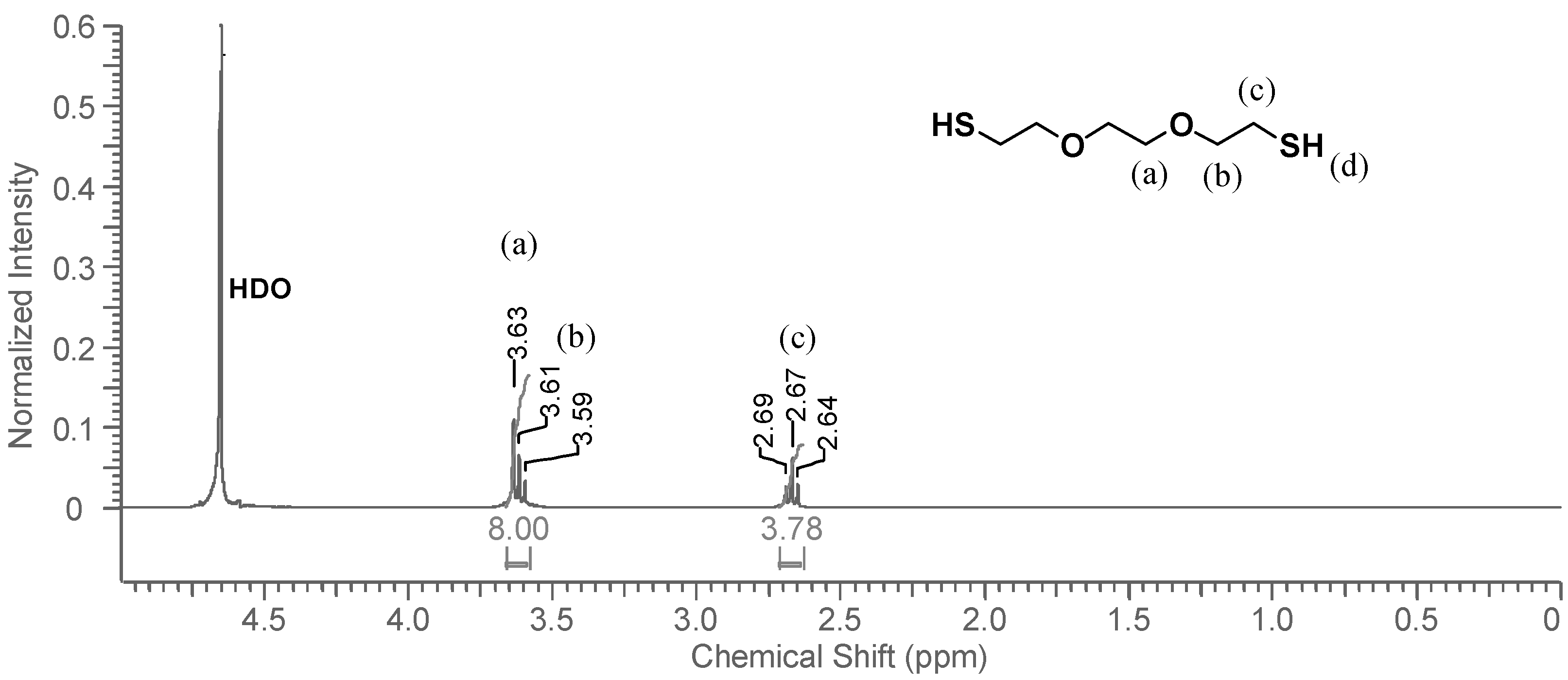
Figure 4. The thiol proton, which exchanges easily with deuterium, is not visible in the spectrum, and does not influence the neighboring methylene protons (
c), whose signal appears as a triplet. The signals representing protons
a and
b show the same overlapping pattern that they did in CDCl
3. The DODT signals are very small, and have a much lower intensity than the solvent residual peak, HDO. Visual observation indicates that DODT does not dissolve in the aqueous phase, however
1H-NMR analysis indicates that a small fraction of the DODT is soluble in water. Only traces of DODT are present in the D
2O phase.
Figure 4.
1H-NMR spectrum of DODT in D2O. (3 s relax; 64 scans; 300 MHz; D2O).
Figure 4.
1H-NMR spectrum of DODT in D2O. (3 s relax; 64 scans; 300 MHz; D2O).
Once TEA is added, the phases switch positions, with the organic phase moving above the aqueous phase.
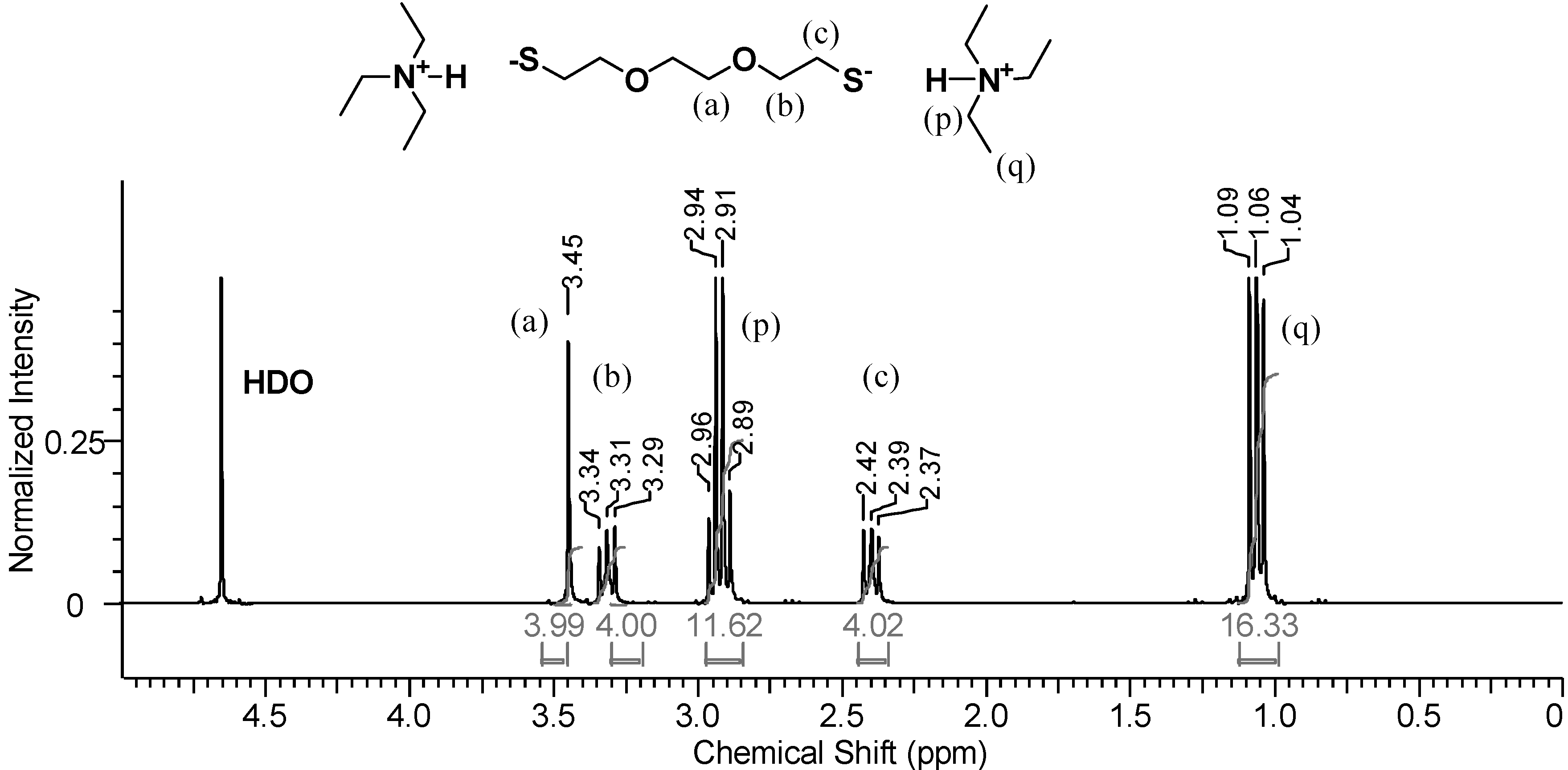
Figure 5 shows the
1H-NMR spectrum of the D
2O phase after the addition of
slightly more than two molar equivalents of TEA relative to DODT. The methyl proton signals from TEA are now the strongest signals in the spectrum. DODT proton signals are also stronger relative to their appearance in
Figure 4. The singlet peak representing DODT central methylene protons (
a) and the triplet peak representing oxygen adjacent protons (
b) are no longer overlapped and have shifted upfield. Change in chemical shifts from the DODT-only spectrum indicates that the thiol protons have been abstracted and that DODT is present in a new, ionic (ion triad) form.
Figure 5.
1H-NMR spectrum of the D2O phase after the addition of TEA to DODT/ D2O. (3 s relax; 64 scans; 300 MHz; D2O).
Figure 5.
1H-NMR spectrum of the D2O phase after the addition of TEA to DODT/ D2O. (3 s relax; 64 scans; 300 MHz; D2O).
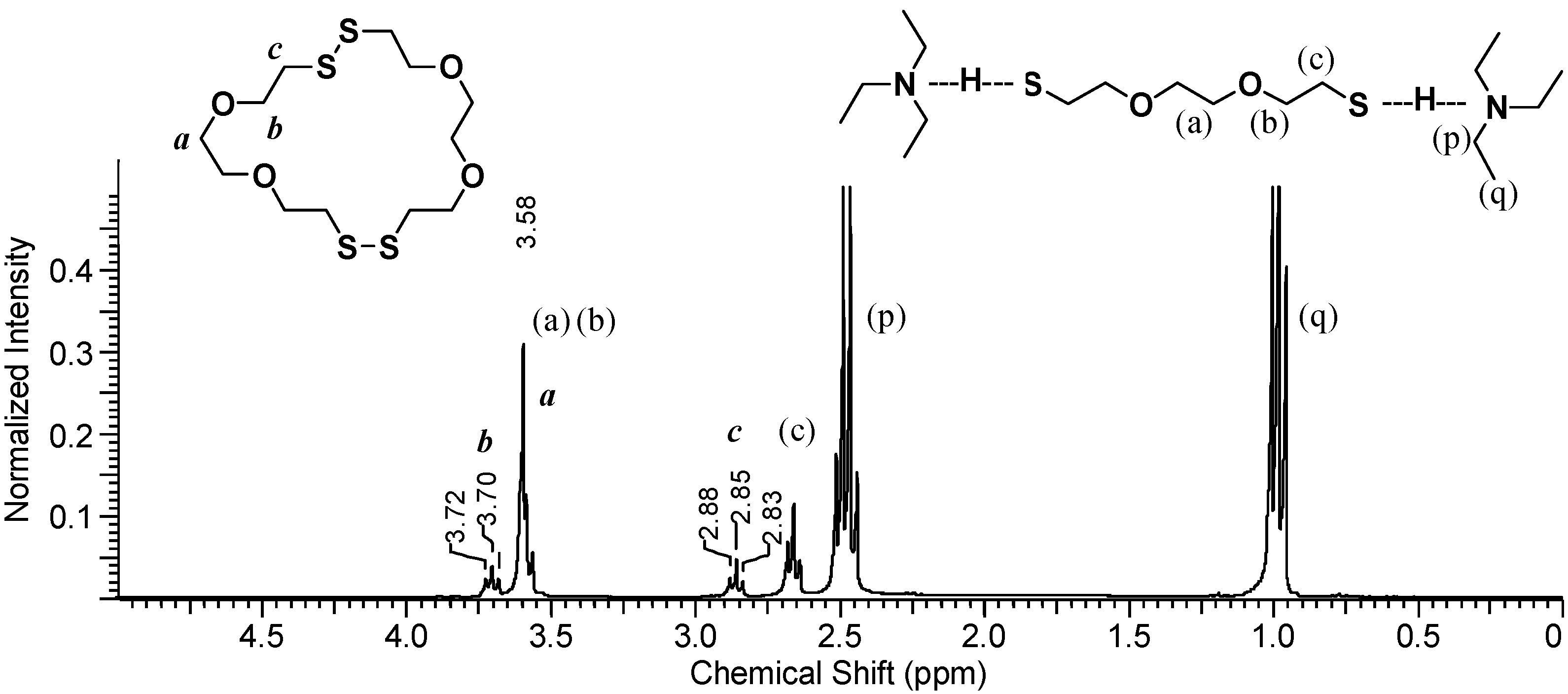
In addition to the DODT signals, a quartet at 2.94 ppm (p) and a triplet at 1.06 ppm (q) represent the ethyl substituents of the amine. Neat TEA is immiscible with water at room temperature, however it becomes miscible once ionized. Two moles of TEA for every one mol DODT was added to the DODT/D2O mixture. The signal intensity ratios (p/b and p/c = 12/4) show that there are two moles of TEA to every mole of DODT, while the ratios of q/b and q/c = 16/4 and are somewhat smaller than the theoretical of 18/4. This indicates that most DODT molecules are dianions which migrate into the aqueous phase accompanied by two TEA cations as an ionic triad. This is in agreement with the proposed mechanism.
To determine what species are present in the organic phase
during polymerization, DODT, TEA and H
2O
2 were mixed in a vial and a sample of the organic layer was immediately taken and diluted in CDCl
3.
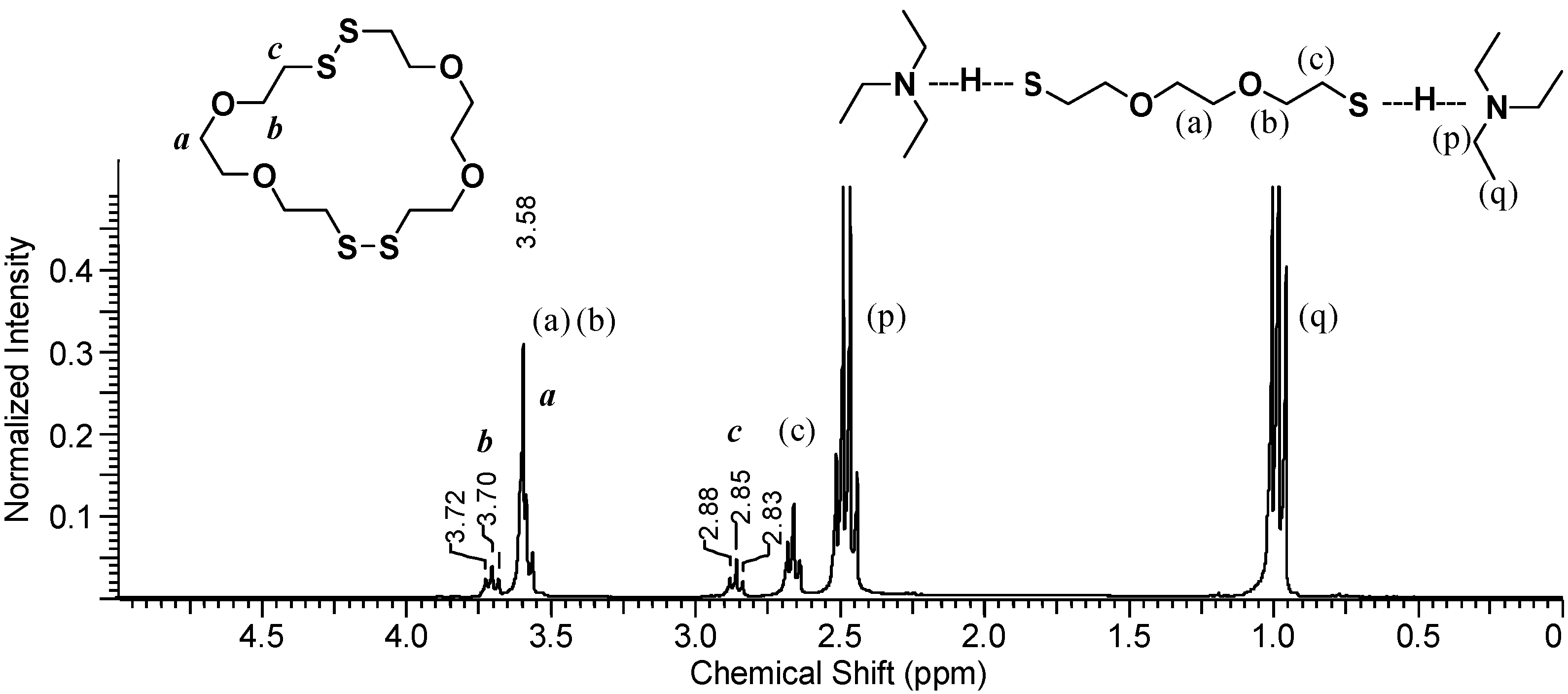
Figure 6 shows the
1H-NMR spectrum of the organic layer.
The TEA signals, (p) and (q), appear at about 1.0 ppm and 2.5 ppm, which represents a slight downfield shift from their signal shifts under neat conditions. Signals from non-ionic DODT [(a), (b) and (c)] appear at 3.6 ppm (singlet and triplet) and at 2.6 ppm (triplet), however the thiol triplet is not visualized either due to complexation with TEA or exchange with deuterium from CDCl3. New peaks with shifts identical to the those seen in poly(DODT) spectra also appear.
Figure 6.
1H-NMR spectrum of the organic phase after H2O2 addition shows a mixture of DODT species and TEA (3 s relax; 64 scans; 300 MHz; CDCl3).
Figure 6.
1H-NMR spectrum of the organic phase after H2O2 addition shows a mixture of DODT species and TEA (3 s relax; 64 scans; 300 MHz; CDCl3).
A triplet at 3.70 ppm, b, represents the oxygen-adjacent methylene protons, and a triplet at 2.85 ppm, c, is characteristic of disulfide-adjacent methylene protons. The singlet corresponding to the methylene protons central to the DODT unit (a) is hidden under the stronger peak at about 3.6 ppm from the dithiol monomer. Poly(DODT) is not soluble in water or TEA and precipitates from the polymerization mixture, so these new signals must arise from small molecules with disulfide bonds. No thiol proton signals (around 1.5 ppm) are seen for any species which is in agreement with the MALDI-ToF data published earlier. This supports a mechanism which involves cyclic species.
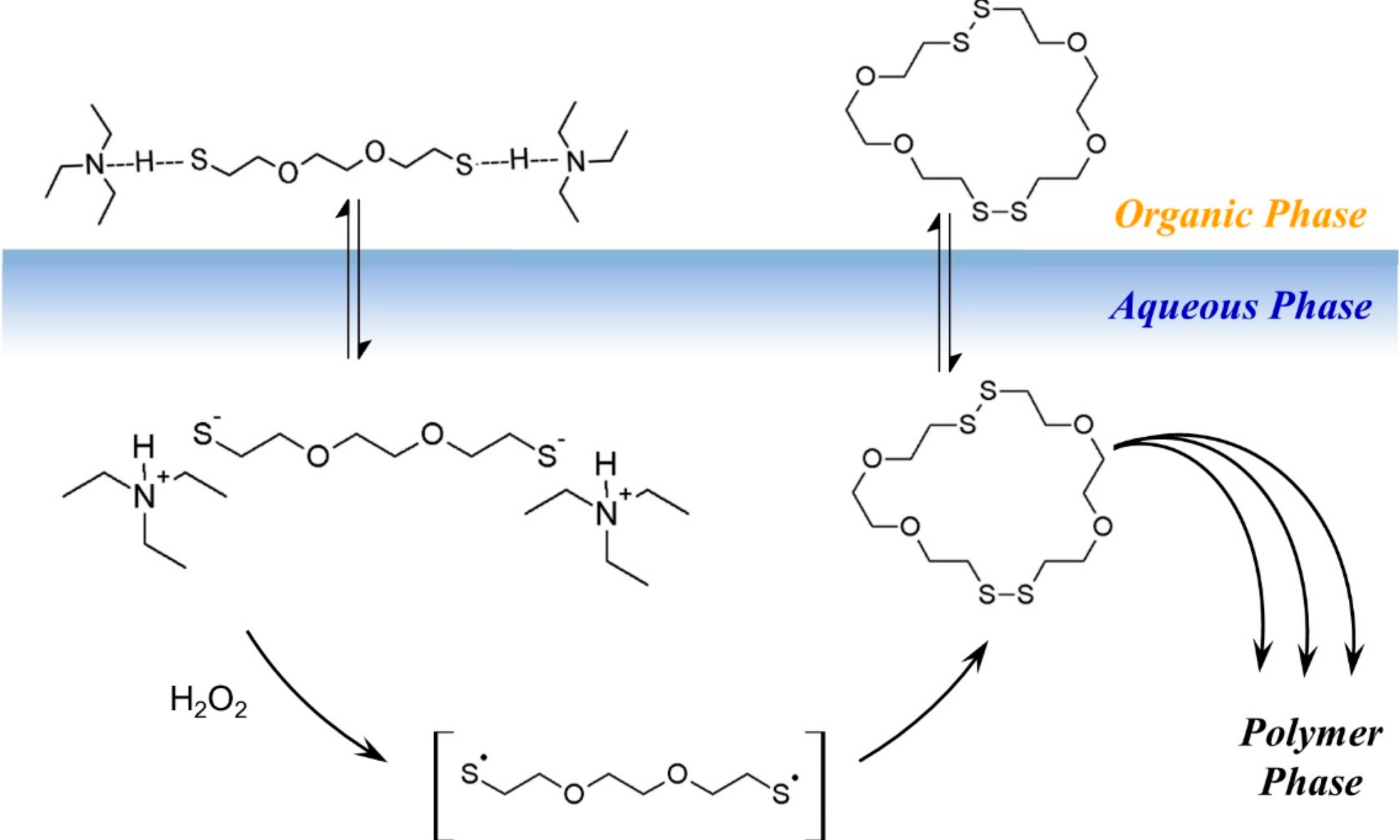
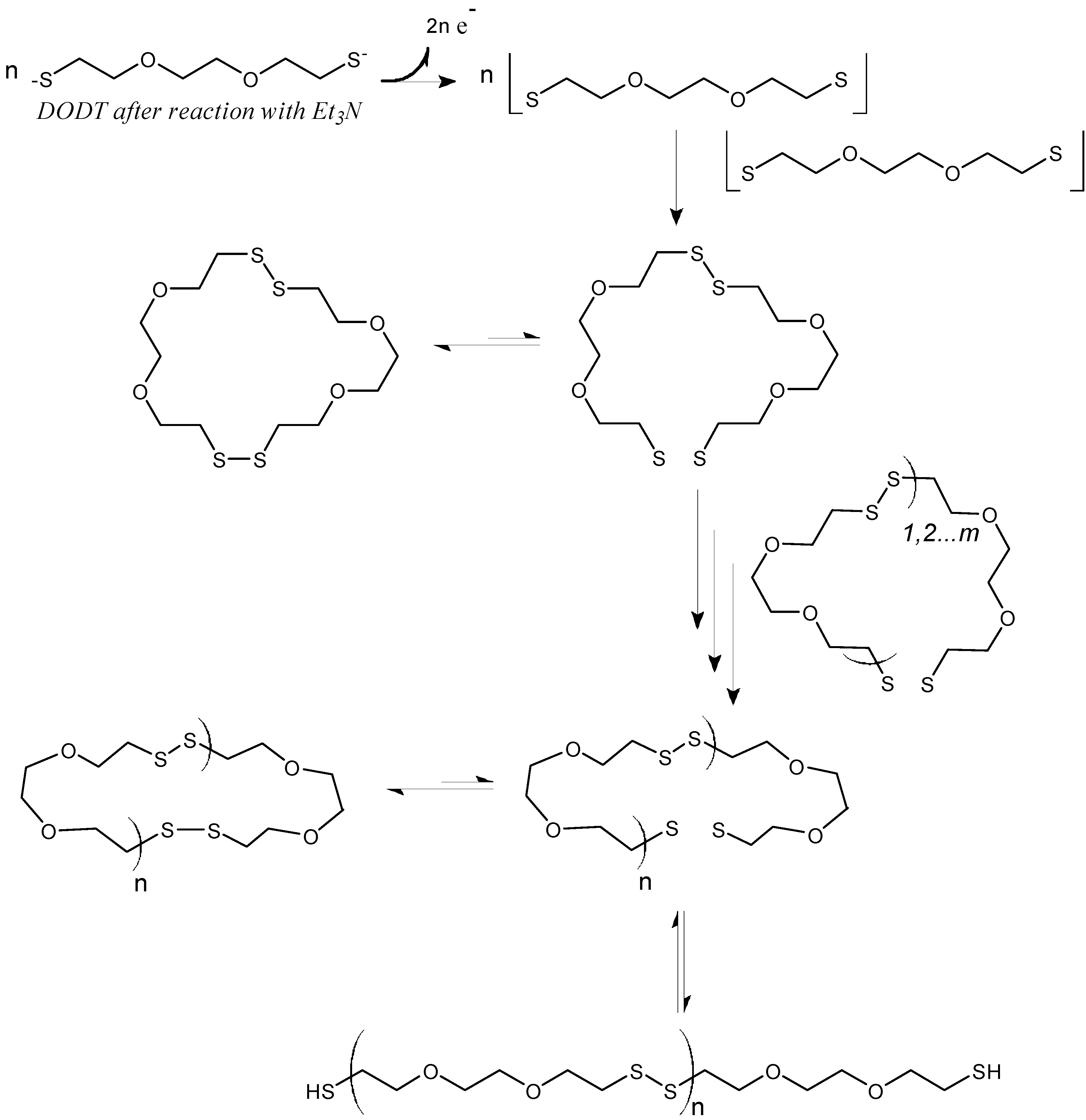
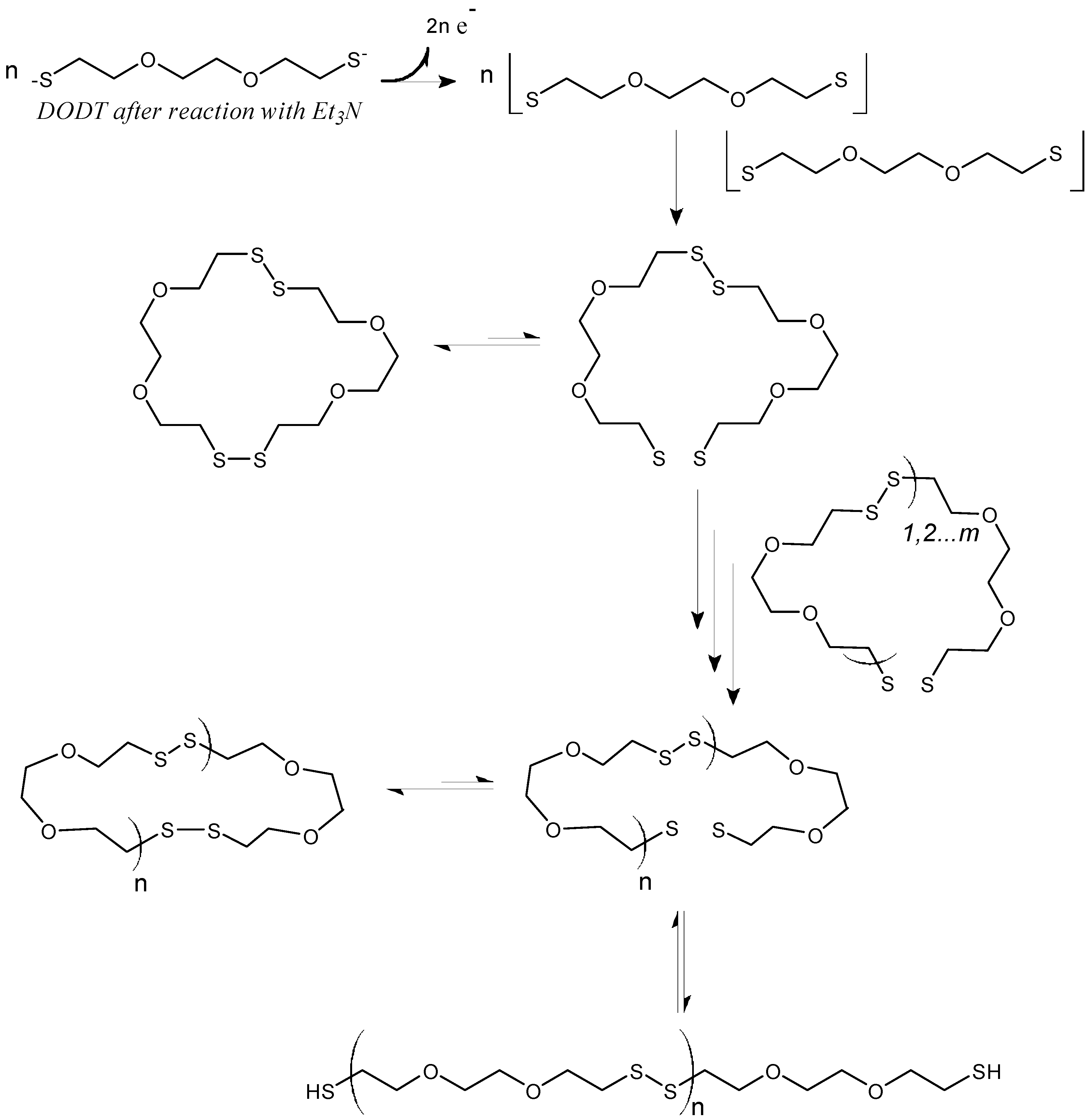
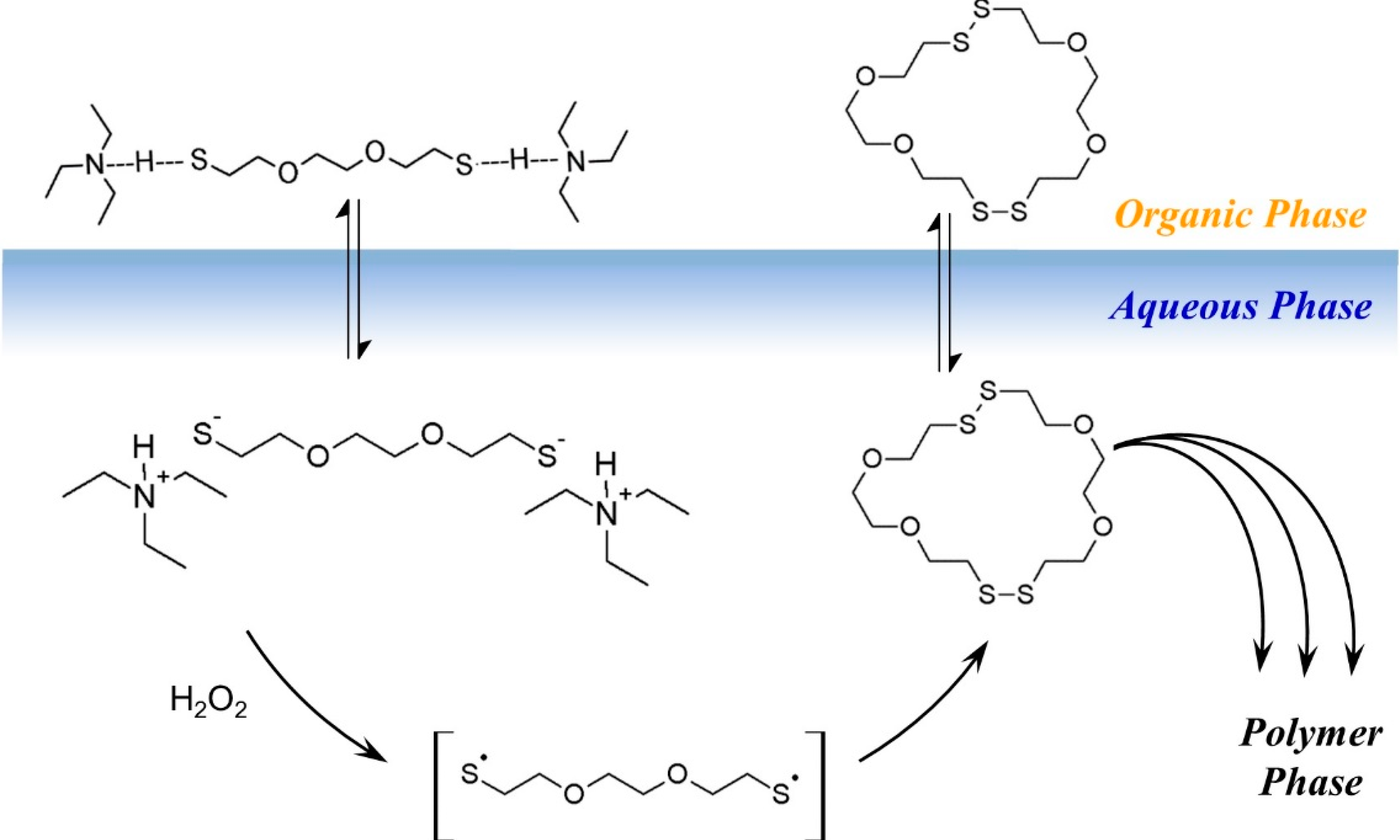
Based on the model experiments, a cartoon of phase-transfer events is presented in
Scheme 3. The DODT monomer is carried into the aqueous phase by TEA as the ionized trio, where H
2O
2 oxidizes it to form the thiyl diradical, which in turn recombines with another thiyl diradical to form a dimer. Intramolecular recombination of a single DODT unit would form a 10-membered ring which is highly prohibited by torsional strain.
Scheme 3.
Cartoon of the R3P phase transfer reaction.
Scheme 3.
Cartoon of the R3P phase transfer reaction.
This is confirmed by mass spectrometry which never showed the presence of cyclic monomers. Low molecular weight cyclic oligomers move back to the organic phase. Oligomers beyond the tetramer are too large to migrate into the organic phase or to remain in suspension. These oligomers begin to separate to the bottom of the reaction and rapidly add DODT units as they move through the oxidant phase. As the oligomers and polymers of DODT precipitate from the system, TEA returns to the organic phase. Under certain reaction conditions, the proton NMR of the high molecular weight polymers does not show the characteristic thiol triplet, suggesting that even the high molecular weight polymer may be cyclic.
2.3. Model Reactions Starting with a Single Aqueous Phase
A model reaction was performed by first dissolving DODT and TEA in DI H
2O to get 0.85 M DODT and 1.56 M TEA concentrations at less than a 1:2 ratio—at these concentrations there was no organic phase separation (
Table 2). After polymerization was initiated by the addition of H
2O
2, the system became two-phased. As the polymerization progressed and polymer precipitated, the organic (TEA) phase grew. This was compared with a two-phase reaction where DODT and TEA were mixed first, then H
2O
2 was added to form the two-phase system and polymer precipitate.
Table 2.
Concentration of reagents in two DODT polymerizations.
Table 2.
Concentration of reagents in two DODT polymerizations.
| Starting Mixture | [DODT] (mol/L) | [Et3N] (mol/L) | [H2O2] (mol/L) |
|---|
| DODT + TEA + H2O | 0.39 | 0.77 | 0.74 |
| DODT + TEA | 0.37 | 0.74 | 0.74 |
After 5 min reaction time, the polymer precipitates were removed from both systems and the hazy aqueous reaction liquids were analyzed by ESI-MS.
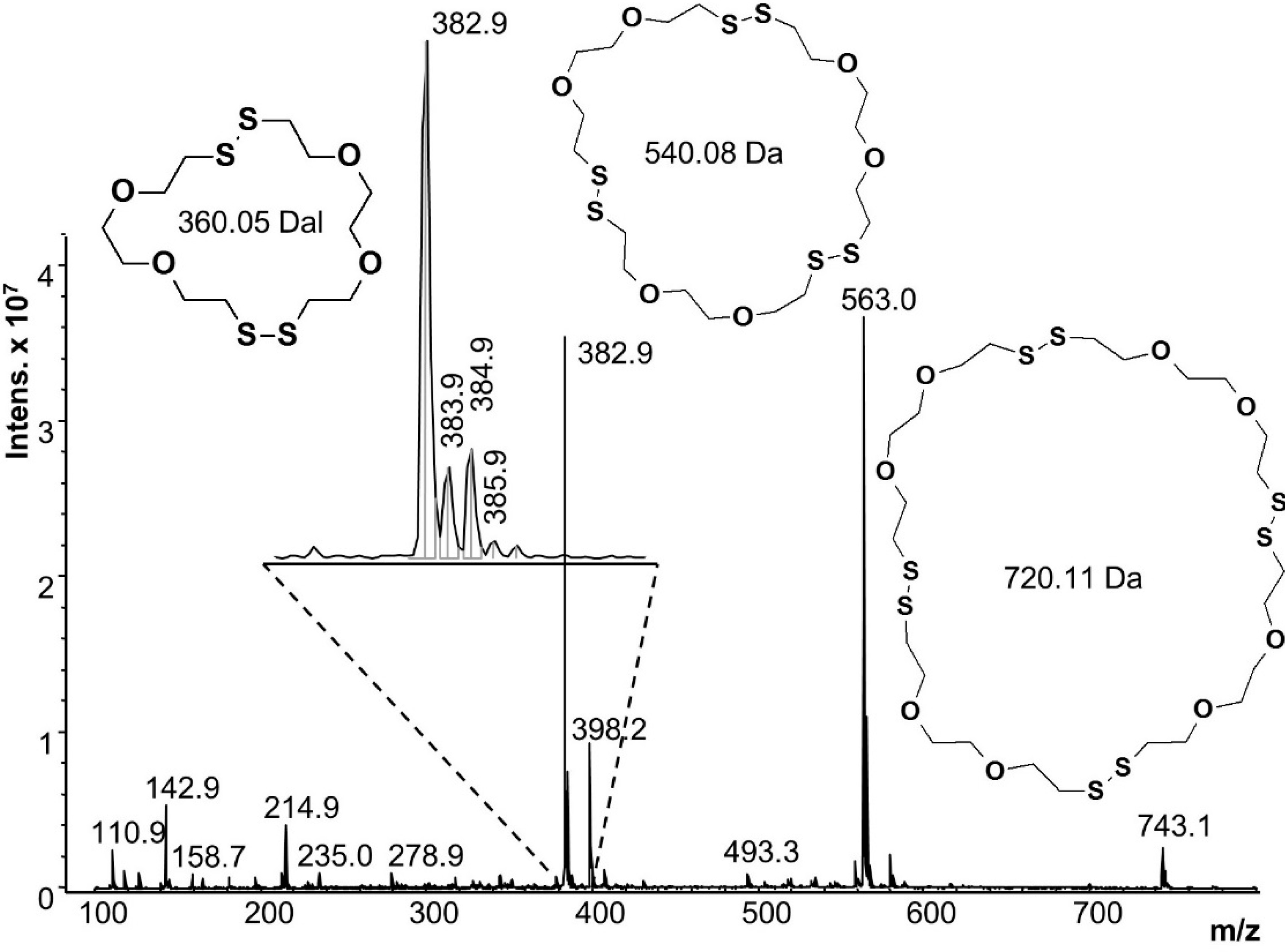
Figure 7 shows the ESI-MS of the first reaction, starting from a single phase. Dimer, trimer and tetramer cycles were identified, with the most intense peak at 382.9
m/
z corresponding to the mass of the monoisotopic peak of the cyclic dimer (360.05 Da) plus the mass of the sodium counter ion (22.9 Da). Isotope peaks corresponding to linear dimer species should appear at 388
m/
z or 389
m/
z but no signals were observed at these
m/
z values. The trimer and tetramer signals (563 and 743
m/
z) also verify cyclic structures. All other peaks (
i.e., 142.9, 214.9 and 398.2
m/
z) were also present in the solvent blank, or are fragments of the oligomeric species. Oligomers beyond the tetramer were not seen, but they are near or beyond the detection limits of ESI-MS (pentamer = 972 g/mol).
The ESI-MS spectra of the second reaction (not shown) was nearly identical, except in the relative composition of oligomers.
Table 3 shows the oligomer distribution; it can be seen that the reaction starting with a single aqueous phase had more trimers.
Figure 7.
Mass spectrum of the reaction starting with a homogeneous solution of DODT, TEA and water.
Figure 7.
Mass spectrum of the reaction starting with a homogeneous solution of DODT, TEA and water.
Table 3.
Relative amount of oligomers by ESI-MS.
Table 3.
Relative amount of oligomers by ESI-MS.
| Starting Mixture | % Dimer | % Trimer | % Tetramer |
|---|
| DODT+TEA+H2O | 44.04 | 52.88 | 3.08 |
| DODT+TEA | 56.33 | 39.61 | 4.06 |
The first reaction starting from a single aqueous phase reached 94.8% conversion in 5 min, while the second reaction reached 80.1% conversion. Despite the lower conversion, the DODT/TEA system showed nearly twice the M
n of the DODT/TEA/H
2O system. The results of SEC analysis are given in
Table 4.
Table 4.
Results from SEC analysis of poly(DODT) samples.
Table 4.
Results from SEC analysis of poly(DODT) samples.
| Starting Mixture | Mn (g/mol) | Mw (g/mol) | PDI | Rgz (nm) | Rhw (nm) | [η]w (mL/g) |
|---|
| DODT+TEA+H2O | 55,000 | 96,000 | 1.76 | 15.0 | 9.2 | 59.1 |
| DODT+TEA | 92,000 | 192,000 | 2.09 | 27.2 | 12.7 | 79.8 |
An experiment was also carried out below 18.7 °C where TEA and water are miscible. DODT and TEA were mixed in 1:2 molar ratio at 0 °C, and chilled H
2O
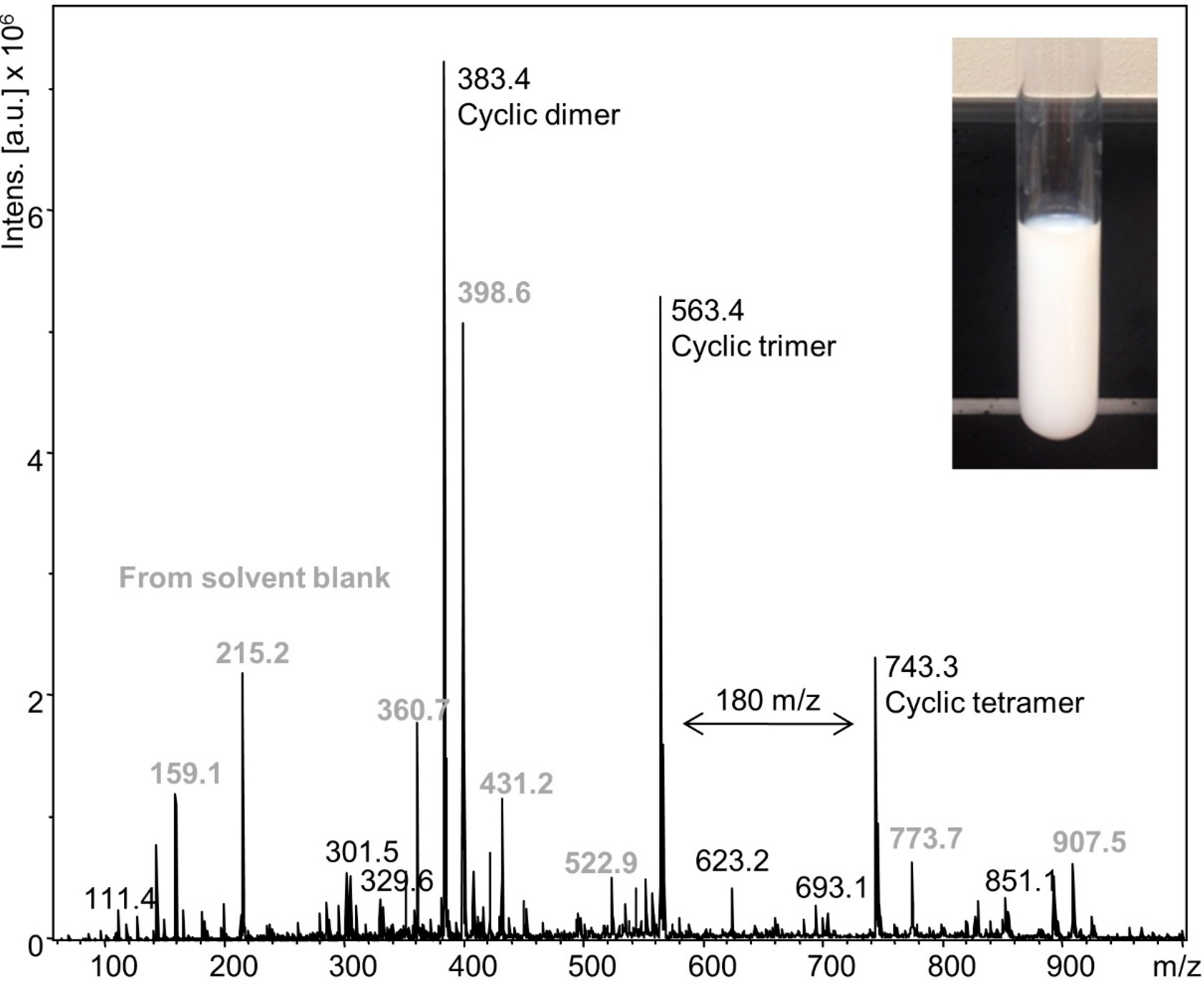
2 (2 °C) was slowly added over 2 min to prevent the exothermic oxidation reaction from exceeding 15 °C (2.15 mol per 1 mol DODT). A stable, milky emulsion was formed (
Figure 8 inset) which persisted at room temperature for over 1 week. ESI‑MS analysis of the emulsion (
Figure 8) showed cyclic dimers (48.6%), trimers (35.7%) and tetramers (15.7%), but no linear DODT species nor DODT monomer.
Figure 8.
ESI-MS spectrum of emulsion from the low temperature (15 °C) reaction.
Figure 8.
ESI-MS spectrum of emulsion from the low temperature (15 °C) reaction.
The model reactions which started with a single aqueous phase demonstrate the necessity of a two-phase system in order to produce high molecular weight polymers. The most dramatic example of this was seen in the chilled experiment where a single phase was maintained throughout the reaction and formed only cyclic oligomers. The two reactions performed at room temperature also demonstrate the effect of the two-phase system on the molecular weights of the polymers. While the DODT/TEA system always presents two phases, the DODT/TEA/H2O system only develops an organic phase when the ionic DODT polymerizes and TEA is released to form the organic phase. In this way, the two polymerizations were most different during the first seconds of the reaction. A lower concentration of initiating species leads to higher molecular weights in most chain-growth polymerization mechanisms. So the true initiating species, radicals of short DODT oligomers, may preferentially migrate to the organic phase, thereby limiting the number of available chain-initiating species in the oxidative phase. The mechanistic role of the two-phase system and initiating species merits further investigation.
2.4. Model Reactions with DODT/TEA
DODT and TEA were dissolved in acetone in a 2:1 ratio (rather than the normal 1:2 ratio). Because a small volume of TEA was used, acetone solution was needed to prevent the dense DODT
monomer from separating to the bottom of the reaction flask upon the addition of the aqueous H
2O
2. H
2O
2 was then added to the acetone solution. Final concentrations of the reagents were 0.99 M [DODT], 0.51 M [TEA] and 0.23 M [H
2O
2]. Using less than equimolar amounts of TEA and H
2O
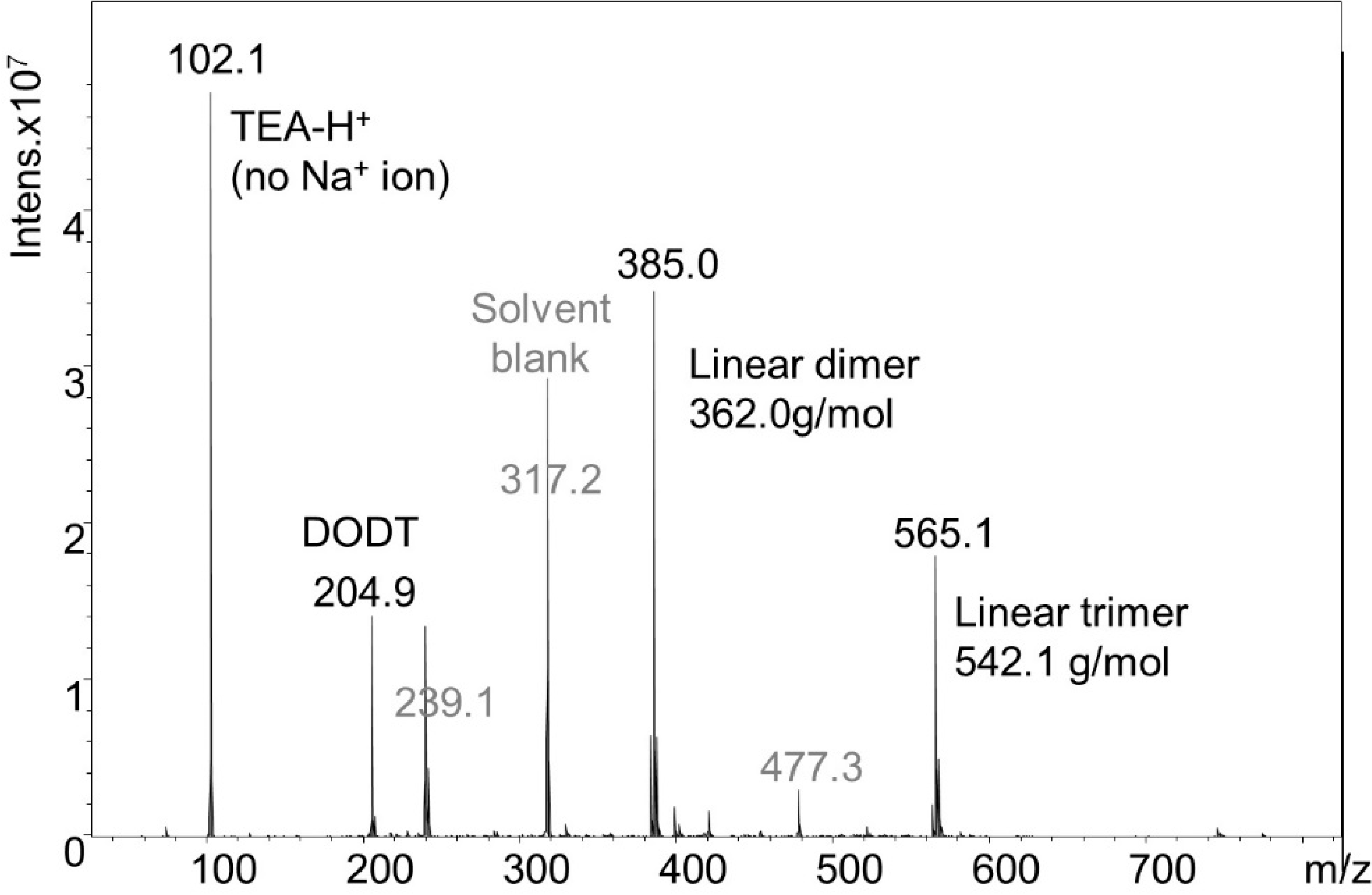
2 relative to DODT did not produce high molecular weight polymer. The resulting species were analyzed by ESI-MS (
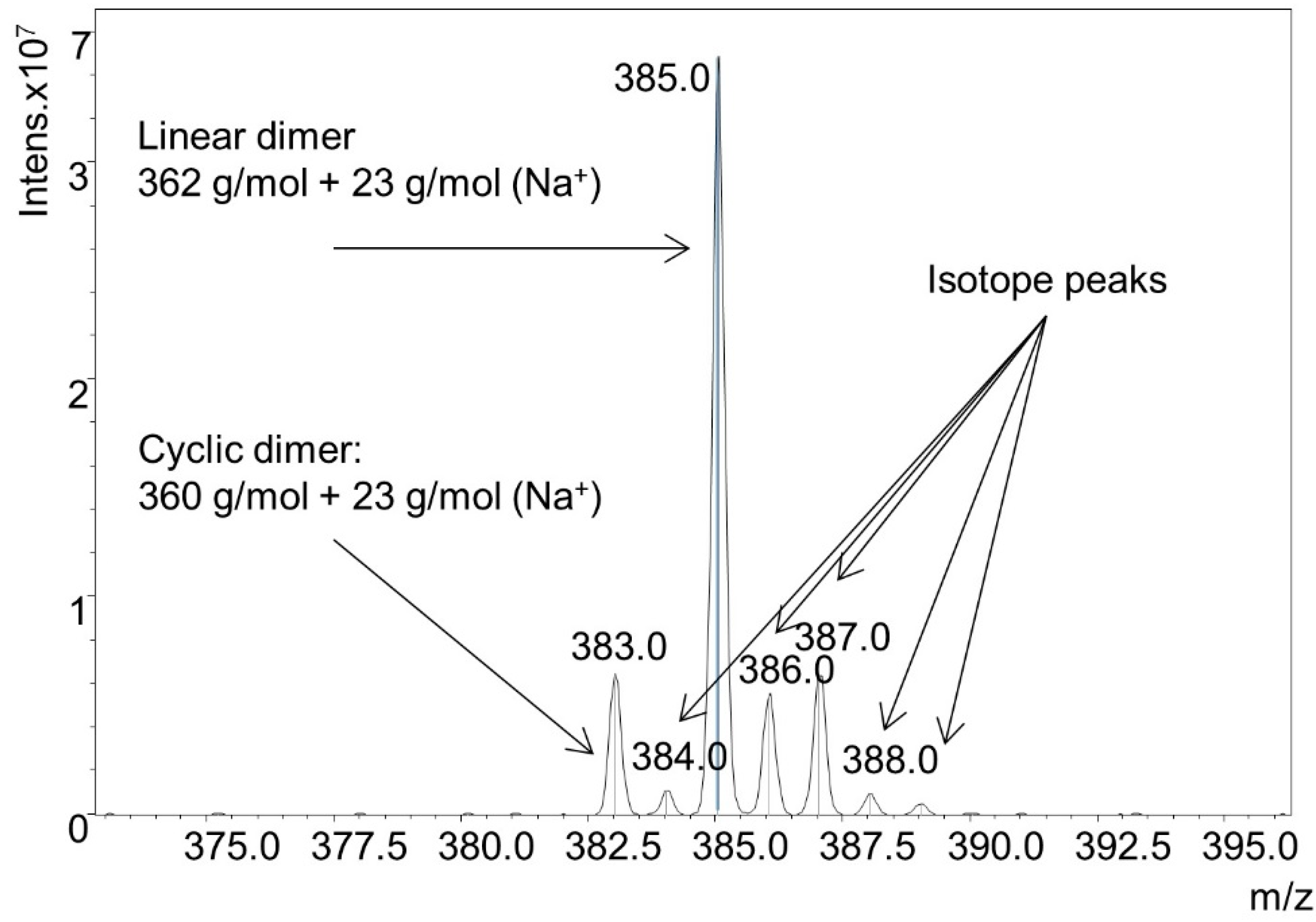
Figure 9). Monomer (≈20.8%), dimers (≈51.8%), trimers (≈26.7%), and trace amounts of tetramer (≈0.5%) were detected. The monomer was only found in linear form, but the dimer and trimer were present in both cyclic and linear form. The ratio of linear species to cyclic species was approximately 5:1. A detail of the dimer mass spectrum is shown in
Figure 10.
Figure 9.
ESI-MS spectrum from the solution synthesis of cyclic species.
Figure 9.
ESI-MS spectrum from the solution synthesis of cyclic species.
Figure 10.
Detail of ESI-MS spectrum from solution synthesis of cyclic species.
Figure 10.
Detail of ESI-MS spectrum from solution synthesis of cyclic species.
Thus under less activating, and less oxidizing conditions there is an equilibrium between cyclic disulfide and linear dithiol species. However, under conditions discussed in
Section 2.3, only cyclic disulfides were detected.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}