3.1. General
Melting points were determined in open glass capillaries on a Gallenkamp melting point apparatus and are uncorrected. IR spectra (KBr discs) were recorded on Shimadzu FTIR-8201PC spectrophotometer (Giza, Egypt). 1H-NMR and 13C-NMR spectra were recorded on a Varian Mercury 300 MHz or Varian Gemini 200 MHz spectrometers (Giza, Egypt) using TMS as an internal standard and DMSO-d6 as solvent. Microwave reactions were performed with a Millstone Organic Synthesis Unit with touch control terminal (MicroSYNTH, Giza, Egypt) with a continuous focused microwave power delivery system in a pressure glass vessel (10 mL) sealed with a septum under magnetic stirring. The temperature of the reaction mixture was monitored using a calibrated infrared temperature control under the reaction vessel, and control of the pressure was performed with a pressure sensor connected to the septum of the vessel. Elemental analysis was carried out at the Microanalytical Center of Cairo University, Giza, Egypt.
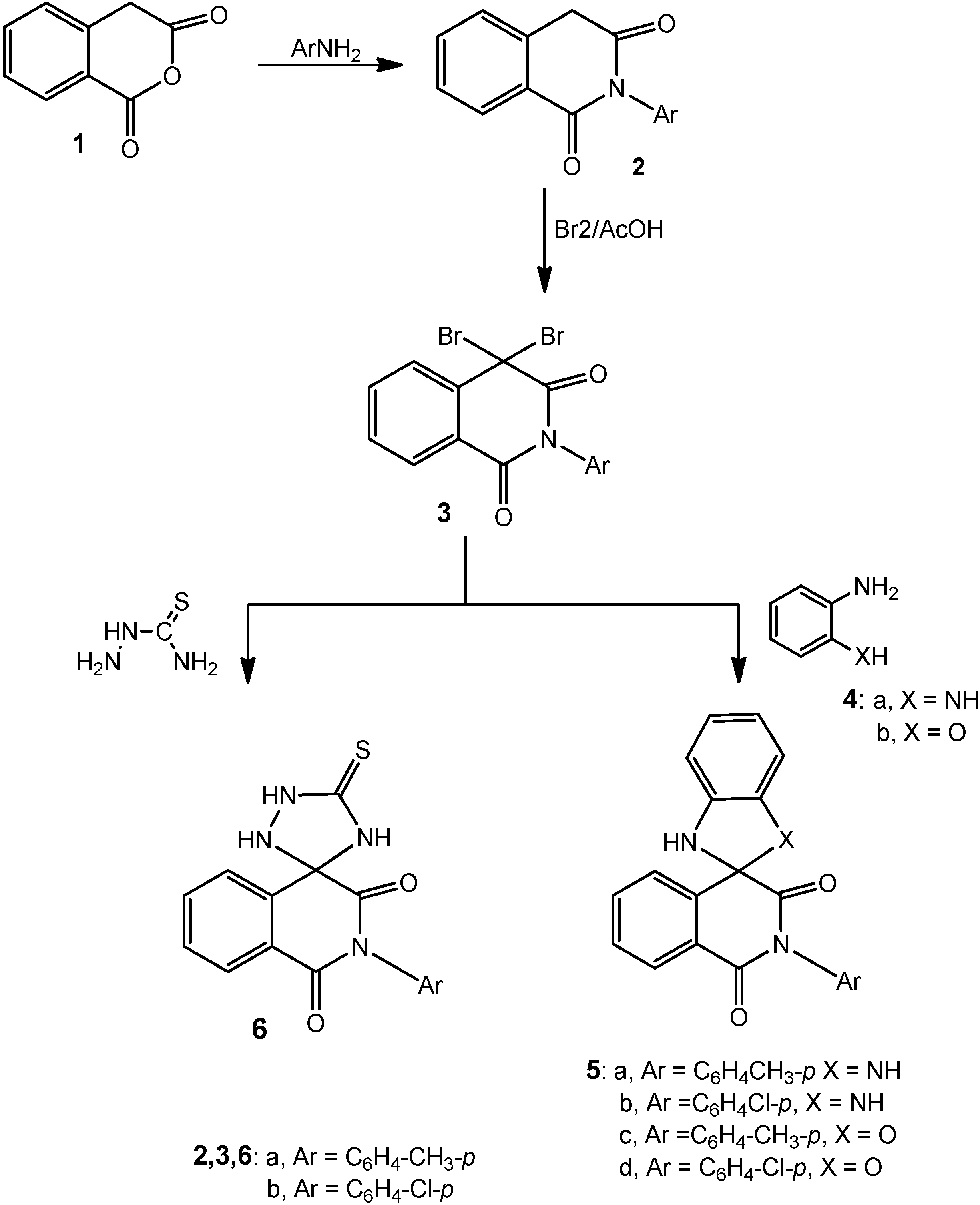
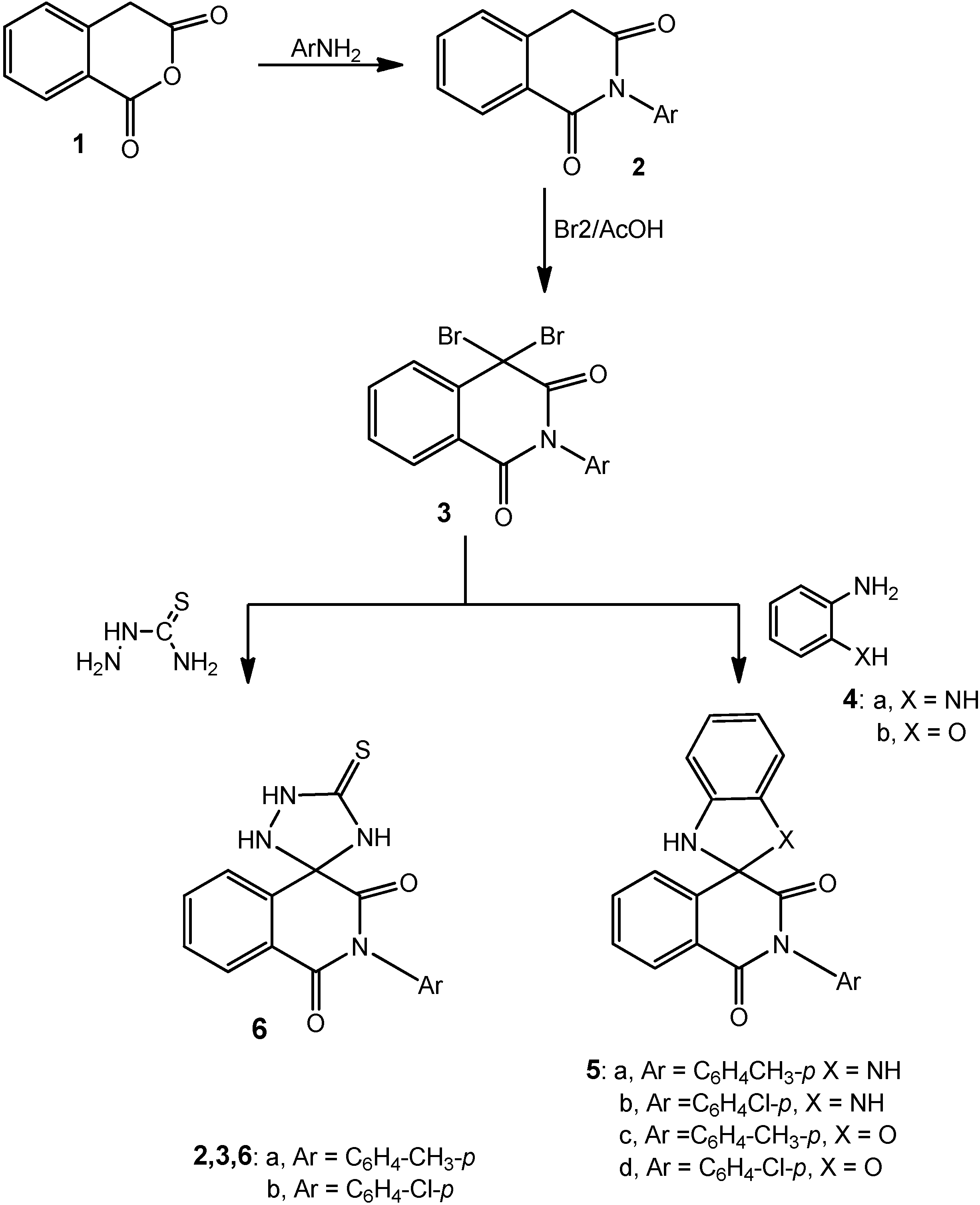
3.1.1. 2-Aryl-4,4-dibromoisoquinoline-1,3-(2H,4H)dione Derivatives 3a,b
A solution of either of 2a (2.37 g, 0.01 mol), 2b (2.51 g, 0.01 mol) or 2c (2.72 g, 0.01 mol) in glacial acetic acid (20 mL) was heated under reflux with bromine (1.1 mL, 3.0 g, 0.02 mole) for 2 h. After cooling, the reaction mixture was poured onto ice-water and the solid that precipitated was filtered off, dried and crystallized from the proper solvent.
4,4-Dibromo-2-p-tolylisoquinoline-1,3-(2H,4H)dione (3a): white crystals after crystallization from acetic acid then washing with ethanol; 66% yield; m.p. 236–238 °C; 1H-NMR: 2.60 (s, 3H, CH3), 7.10–8.30 (m, 8H, Ar-H); 13C-NMR: 25.3 (CH3), 80.2 (sp3 C-4), 118.2, 122.7, 125.3, 126.1, 128.7, 130.0, 131.5, 134.6, 135.2, 136.4 (aromatic C), 158.4, 167.6 (2 CO); IR ν: 3066 cm−1 (aromatic CH), 2970 (aliphatic CH), 1645 (broad, 2C=O), 1605, 1500 (aromatic C=C); MS: M+ m/z 407 (3.2%), 409 (6.7%), 411 (3.0%); Anal. Calcd. for C16H11Br2NO2 (407.07): C (46.98%), H (2.71%), Br (39.07%), N (3.42%); Found: C (46.7%), H (2.9%), Br (38.93%), N (3.1%).
4,4-Dibromo-2-(4-chlorophenyl)isoquinoline-1,3(2H,4H)-dione (3b): white crystals after crystallization from acetic acid then washing with ethanol; 52% yield; m.p. 216–218 °C; 1H-NMR: 7.40–8.50 (m, 8H, Ar-H); 13C-NMR: 80.2 (sp3 C-4), 121.5, 123.9, 125.8, 127.1, 128.7, 130.0, 131.5, 134.6, 135.2, 136.4 (aromatic C), 158.2, 167.5 (2 CO); IR ν: 3060 cm−1 (aromatic CH), 1645 (broad, 2C=O), 1605, 1500 (aromatic C=C); MS: M+ m/z 427 (4.7%), 429 (10.3%), 431 (7.2%); Anal. Calcd.forC15H8Br2ClNO2 (429.49): C (41.95%), H (1.88%), Br (37.21%), Cl (8.25%), N (3.26%); Found: C (41.7%), H (1.7%), Br (37.1%), Cl (8.4%), N (3.1%).
3.1.2. Cyclocondensation of 3a with o-Phenylenediamine and o-Aminophenol; Formation of 5a–d
Method A: Compounds 3a,b (0.01 mol) were heated under reflux with either of o-phenylenediamine (1.08 g, 0.01 mol) or o-aminophenol (1.09 g, 0.01 mol) in absolute ethanol (25 mL) and few drops of piperidine for 5 h. The reaction mixture was then cooled, acidified with few drops of conc. hydrochloric acid and the solid that precipitated was filtered at the pump and crystallized from the appropriate solvent.
Method B: The same reactants of method A were heated in microwave oven at 500 W and 140 °C for 15 min. The reaction mixture was treated similar to method A to obtain compounds 5a–d.
2'-(4-Tolyl)-1,3-dihydro-1'H-spiro[benzimidazole-2,4'-isoquinoline]-1',3'(2'H)-dione (5a): grey crystals after crystallization from acetic acid; 42% yield (Method A) and 91% (Method B); 229–231 °C; 1H-NMR: 2.40 (s, 3H, CH3), 3.80 (s, 2H, 2NH, D2O exchangeable), 6.40–7.70 (m, 12H, Ar-H); 13C-NMR: 25.5 (CH3), 83.4 (sp3-spiro C), 113.0, 115.9, 120.2, 122.9, 127.3, 127.8, 128.3, 128.9, 131.2, 133.7, 135.1, 135.9, 136.8 (aromatic C), 155.4, 163.6 (2 CO); IR ν: 3180 cm−1 (broad, NH), 3065 (aromatic CH), 2970 (aliphatic CH), 1655, 1640 (2C=O), 1605, 1500 (aromatic C=C); MS: M+ m/z 355 (12.3%); Anal. Calcd. for C22H17N3O2 (355.39): C (74.35%), H (4.82%), N (11.82%); Found: C (74.1%), H (4.4%), N (12.1%).
2'-(4-Chlorophenyl)-1,3-dihydro-1'H-spiro[benzimidazole-2,4'-isoquinoline]-1',3'(2'H)-dione (5b): grey crystals after crystallization from acetic acid; 48% yield (Method A) and 89% (Method B); m.p. 215–217 °C; 1H-NMR: 3.90 (s, 2H, 2NH, D2O exchangeable), 6.90–8.10 (m, 12H, Ar-H); 13C-NMR: 83.4 (sp3-spiro C), 115.0, 118.1, 123.6, 125.2, 127.3, 128.8, 129.6, 130.9, 131.8, 133.7, 135.1, 135.9, 136.8 (aromatic C), 155.4, 163.6 (2 CO); IR ν: 3180 cm−1 (broad, NH), 3065 (aromatic CH), 1655, 1640 (2C=O), 1605, 1500 (aromatic C=C); MS: M+ m/z 375 (11.5%), 377 (4.1%); Anal. Calcd. for C21H14ClN3O2 (375.81): C (67.12%), H (3.75%), Cl (9.43%), N (11.18%); Found: C (76.0%), H (3.5%), Cl (9.3%), N (11.3%).
2'-p-Tolyl-1,3-dihydro-1'H-spiro[benzo[d]imidazole-2,4'-isoquinoline]-1',3'(2'H)-dione (5c): grey crystals after crystallization from acetic acid; 47% yield (Method A) and 82% (Method B); m.p. 240–242 °C; 1H-NMR: 2.60 (s, 3H, CH3), 3.90 (s, 1H, NH, D2O exchangeable), 6.70–7.90 (m, 12H, Ar-H); 13C-NMR: 83.4 (sp3-spiro C), 115.4, 118.8, 122.7, 126.2, 127.3, 128.8, 129.6, 130.9, 131.8, 133.2, 136.8, 137.9, 142.8 (aromatic C), 155.4, 163.6 (2 CO); IR ν: 3190 cm−1 (broad, NH), 3065 (aromatic CH), 2975 (aliphatic CH), 1655,1645 (2C=O), 1605, 1500 (aromatic C=C); MS: M+ m/z 356 (10.3%); Anal. Calcd. for C22H16N2O3 (356.37): C (74.15%), H (4.53%), N (7.86%); Found: C (74.05%), H (4.43%), N (7.75%).
2'-(4-Chlorophenyl)-1'H,3H-spiro[benzo[d]oxazole-2,4'-isoquinoline]-1',3'(2'H)-dione (5d): grey crystals after crystallization from acetic acid; 53% yield (Method A) and 92% (Method B); m.p. 220–222 °C; 1H-NMR: 3.90 (s, 1H, NH, D2O exchangeable), 6.70–8.00 (m, 12H, Ar-H); 13C-NMR: 93.8 (sp3-spiro C), 119.0, 122.1, 124.9, 125.8, 127.5, 128.9, 129.6, 131.2, 132.9, 134.7, 137.1, 139.9, 146.8 (aromatic C), 155.4, 163.6 (2 CO); IR ν: 3150 cm−1 (broad, NH), 3065 (aromatic CH), 1655,1645 (2C=O), 1605, 1500 (aromatic C=C); MS: M+ m/z 376 (8.5%), 378 (2.7%); Anal. Calcd. for C21H13ClN2O3 (376.79): C (66.94%), H (3.48%), Cl (9.41%), N (7.43%); Found: C (66.6%), H (3.6%), Cl (9.1%), N (7.3%).
3.1.3. Cyclocondensation of 3a,b with Thiosemicarbazide; Formation of 6a,b
Method A: Each of compounds 3a,b (0.01 mol), was heated under reflux with thiosemicarbazide (0.91 g, 0.01 mol), absolute ethanol (25 mL) and few drops of piperidine for 4 h. The reaction mixture was then cooled, acidified with few drops of conc. hydrochloric acid and the solid that precipitated was filtered at the pump and crystallized from the appropriate solvent.
Method B: The same reactants of method A were heated in microwave oven at 500 W and 140 °C for 10 min. The reaction mixture was treated similar to method A to obtain compounds 6a,b.
5'-Thioxo-2-p-tolyl-1H-spiro[isoquinoline-4,3'-[1,2,4]triazolidine]-1,3(2H)-dione (6a): white crystals after crystallization from acetic acid and washing with ethanol; 55% yield (Method A) and 90% (Method B); m.p. 156–158 °C; 1H-NMR: 2.40 (s, 3H, CH3), 3.10 (s, 1H, NH, D2O exchangeable), 3.60 (s, 1H, NH, D2O exchangeable), 4.00 (s, 1H, NH, D2O exchangeable), 7.20–7.90 (m, 8H, Ar-H); 13C-NMR: 23.2 (CH3), 91.7 (sp3 spiro C), 121.1, 123.4, 127.6, 128.4, 128.8, 129.8, 132.1, 134.0, 135.2, 136.8 (aromatic C), 154.9, 159.5 (2 CO), 176.9 (CS); IR ν: 3220, 3185, 3150 cm−1 (NH), 3060 (aromatic CH), 2970 (aliphatic CH), 1670,1650 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 338 (10.3%); Anal. Calcd. for C17H14N4O2S (338.36): C (60.34%), H (4.17%), N (16.56%), S(9.48%); Found: C (60.0%), H (3.9%), N (16.8%), S (9.7%).
2-(4-Chlorophenyl)-5'-thioxo-1H-spiro[isoquinoline-4,3'-[1,2,4]triazolidine]-1,3(2H)-dione (6b): white crystals after crystallization from dilute acetic acid and washing with ethanol; 60% yield (Method A) and 94% (Method B); m.p. 188–190 °C; 1H-NMR: 3.10 (s, 1H, NH, D2O exchangeable), 3.50 (s, 1H, NH, D2O exchangeable), 4.00 (s, 1H, NH, D2O exchangeable), 7.20–7.90 (m, 8H, Ar-H); 13C-NMR: 92.7 (sp3 spiro C), 127.1, 127.9, 132.6, 133.4, 134.8, 135.6, 136.1, 137.0, 137.9, 138.8 (aromatic C), 156.9, 160.7 (2CO), 180.1 (CS); IR ν: 3220, 3185, 3150 cm−1 (NH), 3060 (aromatic CH), 1670,1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 358 (12.7%) and 360 (4.3%); Anal. Calcd. for C16H11N4O2S (358.80): C (53.56%), H (3.09%), Cl (9.88%), N (15. 61%), S (8.94%); Found: C (53.4%), H (2.90%), Cl (9.6%), N (15.5%), S (8.7%).
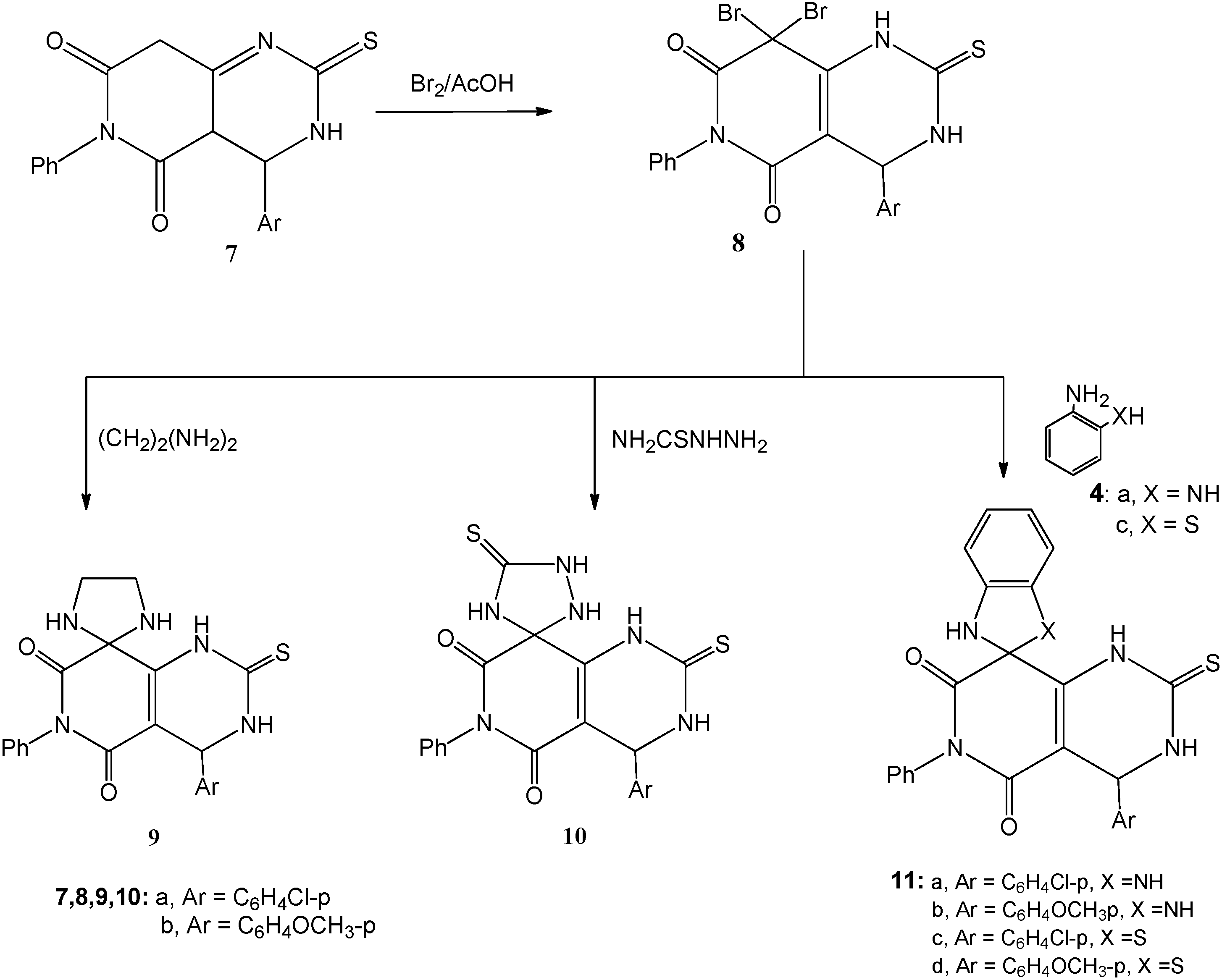
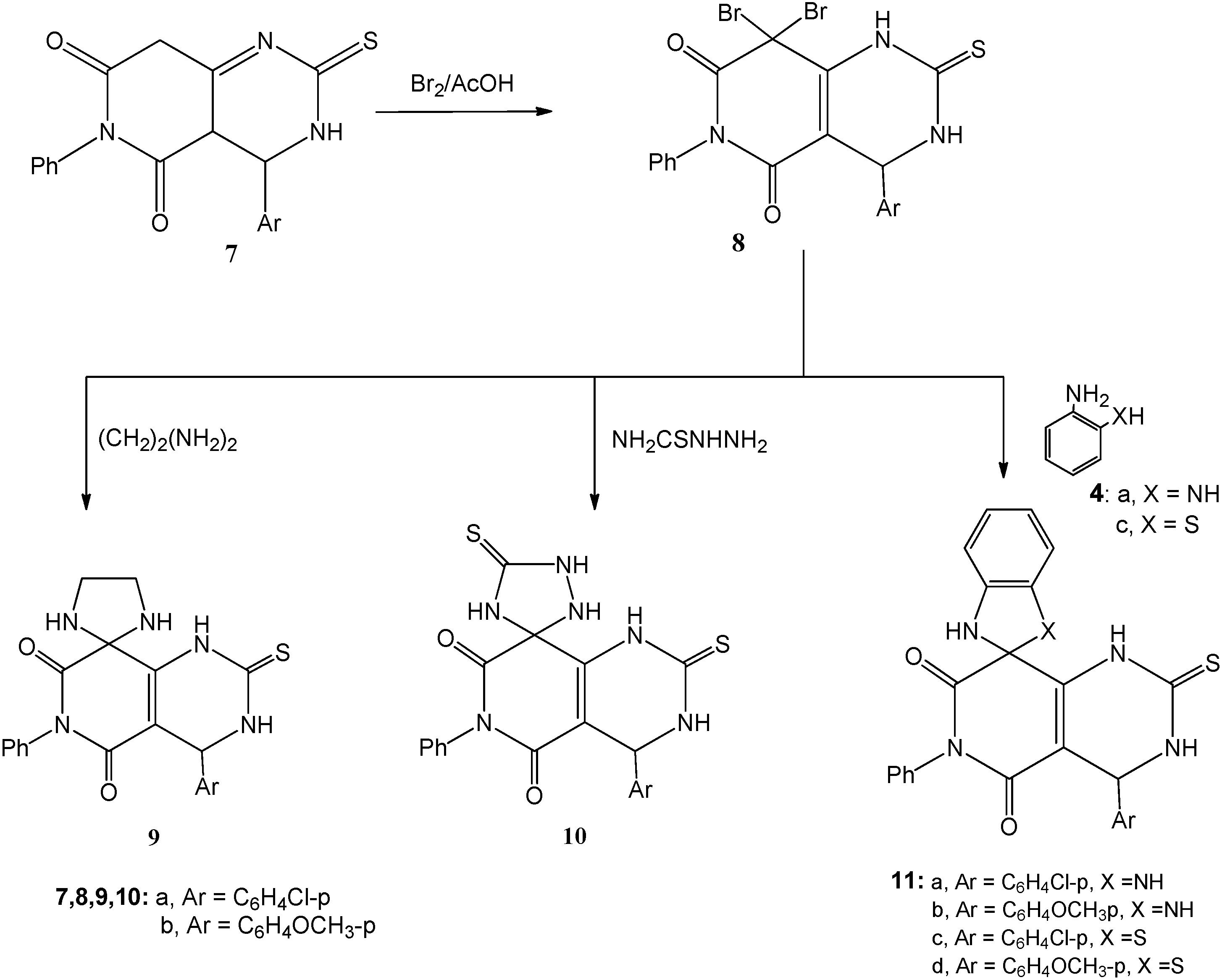
3.1.4. Bromination of 7a,b; Formation of 8a,b
A solution of either of 7a or 7b (0.01 mol) in ethanol (20 mL) was heated under reflux with bromine (1.1 mL, 3.0 g, 0.02 mol) for 2 h. After cooling, the reaction mixture was poured onto ice-water and the solid that precipitated was filtered off, dried and crystallized from the proper solvent.
8,8-Dibromo-4-(4-chlorophenyl)-6-phenyl-2-thioxo-1,2,3,4-tetrahydropyrido[4,3-d]pyrimidine-5,7(6H,8H)-dione (8a): white crystals after crystallization from absolute ethanol; 55% yield; m.p. 255–257 °C; 1H-NMR: 3.00 (s, 1H, NH, D2O exchangeable), 5.40 (s, 1H, CH), 7.20–7.70 (m, 9H, Ar-H), 11.30 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 127.1, 127.9, 132.6, 133.4, 134.8, 135.6, 136.1, 137.0, 139.9, 150.8 (aromatic C), 156.9, 160.7 (2CO), 178.0 (CS); IR ν: 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 539 (4.7%), 541 (10.7%) and 543 (7.1%); Anal. Calcd. for C19H12Br2ClN3O2S (541.64): C (42.13%), H (2.23%), Br (29.50%), Cl (6.55%), N (7.76%), S (5.92%); Found: C (42.0%), H (2.3%), Br (29.4%), Cl (6.7%), N (7.9%), S (5.8%).
8,8-Dibromo-4-(4-methoxyphenyl)-6-phenyl-2-thioxo-1,2,3,4-tetrahydropyrido[4,3-d]pyrimidine-5,7(6H,8H)-dione (8b): white crystals after crystallization from absolute ethanol; 53% yield; m.p. 232–234 °C; 1H-NMR: 3.00 (s, 1H, NH, D2O exchangeable), 3.80 (s, 3H, OCH3), 5.40 (s, 1H, CH), 6.90–7.60 (m, 9H, Ar-H), 11.30 (s, 1H, NH, D2O exchangeable); 13C-NMR: 53.9 (OCH3), 55.5 (pyrimidine C-4), 83.1 (pyridine C-8), 127.4, 128.4, 132.7, 133.8, 134.9, 136.6, 137.1, 138.5, 148.3, 150.8 (aromatic C), 156.9, 160.7 (2CO), 182.0 (CS); IR ν: 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2880 (aliphatic CH), 1665, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 535 (6.5%), 537 (13.7%) and 539 (6.1%); Anal. Calcd. for C20H15Br2N3O3S (537.22): C (44.71%), H (2.81%), Br (29.75%), N (7.82%), S (5.97%); Found: C (44.8%), H (2.8%), Br (29.9%), N (7.7%), S (6.1%).
3.1.5. Cyclocondensation of 8a,b with Ethylene Diamine and Thiosemicarbazide; Formation of 9a and 10a
Method A: Each of compounds 8a,b (0.01 mol) was heated under reflux with either of ethylene diamine (0.67 mL, 0.01 mol) or thiosemicarbazide (0.91 g, 0.01 mol) in absolute ethanol (25 mL)] and few drops of piperidine for 2 h. The reaction mixture was then cooled, acidified with few drops of conc. hydrochloric acid and the solid that precipitated was filtered at the pump and crystallized from the appropriate solvent to give 9a,b and 10a,b.
Method B: The same reactants of method A were heated in microwave oven at 500 W and 140 °C for 5 min. The reaction mixture was treated similar to method A to obtain compounds 9a,b and 10a,b.
4'-(4-Chlorophenyl)-6'-phenyl-2'-thioxo-3',4'-dihydro-1'H-spiro[imidazolidine-2,8'-pyrido[4,3-d]pyrimidine]-5',7'(2'H,6'H)-dione (9a): white crystals after crystallization from absolute ethanol; 65% yield (Method A) and 93% (Method B); m.p. 247–249 °C; 1H-NMR: 2.60 (s, 4H, 2CH2-imidazolidine), 3.10 (s, 1H, NH, D2O exchangeable), 3.80 (s, 2H, 2NH, D2O exchangeable), 5.30 (s, 1H, CH), 7.20–7.70 (m, 9H, Ar-H), 11.30 (s, 1H, NH, D2O exchangeable); 13C-NMR: 51.2 (imidazolidine 2CH2), 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 127.1, 127.9, 132.6, 133.4, 134.8, 135.6, 136.1, 137.0, 138.5, 147.3 (aromatic C), 156.9, 160.4 (2CO), 181.0 (CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 1670, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 439 (8.7%) and 441 (3.2%). Anal. Calcd. for C21H18ClN5O2S (439.92): C (57.33%), H (4.12%), Cl (8.06%), N (15.92%), S (7.29%); Found: C (57.1%), H (4. 2%), Cl (7.9%), N (16.1%), S (7.1%).
4'-(4-Methoxyphenyl)-6'-phenyl-2'-thioxo-3',4'-dihydro-1'H-spiro[imidazolidine-2,8'-pyrido[4,3-d]pyrimidine]-5',7'(2'H,6'H)-dione (9b): white crystals after crystallization from absolute ethanol; 67% yield (Method A) and 91% (Method B); m.p. 215–217 °C; 1H-NMR: 2.60 (s, 4H, 2CH2-imidazolidine), 3.00 (s, 1H, NH, D2O exchangeable), 3.60 (s, 2H, 2NH, D2O exchangeable), 3.80 (s, 3H, OCH3), 5.40 (s, 1H, CH), 6.90–7.60 (m, 9H, Ar-H), 11.30 (s, 1H, NH, D2O exchangeable); 13C-NMR: 51.0 (imidazolidine 2CH2), 53.9 (OCH3), 55.5 (pyrimidine C-4), 83.1 (pyridine C-8), 116.4, 118.5, 125.7, 128.0, 134.9, 136.6, 137.1, 138.5, 148.3, 150.8 (aromatic C), 156.9, 160.7 (2CO), 182.0 (CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2880 (aliphatic CH), 1670, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 435 (7.1%); Anal. Calcd. for C22H21N5O3S (435.50): C (60.67%), H (4.86%), N (16.08%), S (7.36%); Found: C (60.7%), H (4.9%), N (15.9%), S (7.1%).
4-(4-Chlorophenyl)-6-phenyl-2,5'-dithioxo-3,4-dihydro-1H-spiro[pyrido[4,3-d]pyrimidine-8,3'-[1,2,4]triazolidine]-5,7(2H,6H)-dione (10a): white crystals after crystallization from absolute ethanol; 61% yield (Method A) and 91% (Method B); m.p. 225–227 °C; 1H-NMR: 3.00 (s, 1H, NH, D2O exchangeable), 3.10 (s, 1H, NH, D2O exchangeable), 3.40 (s, 1H, NH, D2O exchangeable), 5.30 (s, 1H, CH), 7.20–7.70 (m, 9H, Ar-H), 8.10 (s, 1H, NH, D2O exchangeable), 11.00 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 127.1, 127.9, 132.6, 133.4, 134.8, 135.6, 136.1, 137.0, 140.5, 152.3 (aromatic C), 156.9, 160.4 (2CO), 177.3, 181.0 (2CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 1675, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 470 (11.3%) and 441 (3.9%). Anal. Calcd. for C20H15ClN6O2S2 (470.96): C (51.01%), H (3.21%), Cl (7.53%), N (13.62%), S (7.29%); Found: C (50.9%), H (3.1%), Cl (7.3%), N (13.4%), S (7.4%).
4-(4-Methoxyphenyl)-6-phenyl-2,5'-dithioxo-3,4-dihydro-1H-spiro[pyrido[4,3-d]pyrimidine-8,3'-[1,2,4]triazolidine]-5,7(2H,6H)-dione (10b): white crystals after crystallization from absolute ethanol; 58% yield (Method A) and 89% (Method B); m.p. 206–208 °C; 1H-NMR: 3.00 (s, 1H, NH, D2O exchangeable), 3.10 (s, 1H, NH, D2O exchangeable), 3.60 (s, 1H, NH, D2O exchangeable), 3.80 (s, 3H, OCH3), 5.40 (s, 1H, CH), 6.90–7.60 (m, 9H, Ar-H), 8.00 (s, 1H, NH, D2O exchangeable), 11.30 (s, 1H, NH, D2O exchangeable); 13C-NMR: 53.3 (OCH3), 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 114.3, 115.9, 122.6, 126.4, 131.8, 135.6, 136.1, 137.0, 142.5, 150.3 (aromatic C), 160.9, 164.0 (2CO), 173.4, 181.0 (2CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 1675, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 466 (8.3%). Anal. calcd. for C21H18N6O3S2 (466.54): C (54.06%), H (3.89%), N (18.01%), S (13.75%); Found: C (53.9%), H (3.7%), N (17.9%), S (13.6%).
3.1.6. Cyclocondensation of 8a,b with o-Phenylenediamine (4a) and o-Aminothiophenol (4c); Formation of 11a–d
Method A: Each of compounds 8a,b (0.01 mol) was heated under reflux with either of o-phenylenediamine (4a; 1.08 g, 0.01 mol) or o-aminothiophenol (4c; 1.25 mL, 0.01 mol) in absolute ethanol (25 mL) and few drops of piperidine for 2 h. The reaction mixture was then cooled, acidified with few drops of conc. hydrochloric acid and the solid that precipitated was filtered at the pump and crystallized from the appropriate solvent.
Method B: The same reactants of method A were heated in microwave oven at 500 W and 140 °C for 5 min. The reaction mixture was treated similar to method A to obtain compounds 11a–d.
4'-(4-Chlorophenyl)-6'-phenyl-2'-thioxo-1,3,3',4'-tetrahydro-1'H-spiro[benzo[d]imidazole-2,8'-pyrido[4,3-d]pyrimidine]-5',7'(2'H,6'H)-dione (11a): white crystals after crystallization from absolute ethanol; 47% yield (Method A) and 84% (Method B); m.p. 258–260 °C; 1H-NMR: 3.10 (s, 1H, NH, D2O exchangeable), 4.40 (s, 2H, 2NH, D2O exchangeable), 5.30 (s, 1H, CH), 6.60–7.50 (m, 13H, Ar-H), 12.10 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 116.8, 119.2, 127.1, 127.9, 132.6, 133.3, 134.6, 135.6, 136.1, 137.0, 138.5, 145.3, 151.4 (aromatic C), 160.9, 164.4 (2CO), 175.1 (CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3040 (aromatic CH), 2830 (aliphatic CH), 1665, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 487 (6.3%) and 489 (2.2%). Anal. Calcd. for C25H18ClN5O2S (487.96): C (61.54%), H (3.72%), Cl (7.27%), N (14.35%), S (6.57%); Found: C (61.3%), H (3.6%), Cl (7.1%), N (14.3%), S (6.4%).
4'-(4-Methoxyphenyl)-6'-phenyl-2'-thioxo-1,3,3',4'-tetrahydro-1'H-spiro[benzo[d]imidazole-2,8'-pyrido[4,3-d]pyrimidine]-5',7'(2'H,6'H)-dione (11b): white crystals after crystallization from absolute ethanol; 50% yield (Method A) and 91% (Method B); m.p. 226–228 °C; 1H-NMR: 3.10 (s, 1H, NH, D2O exchangeable), 3.80 (s, 3H, OCH3), 4.10 (s, 2H, 2NH, D2O exchangeable), 5.40 (s, 1H, CH), 6.60–7.50 (m, 13H, Ar-H), 12.20 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.3 (OCH3), 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 112.8, 116.2, 123.1, 124.9, 132.6, 133.3, 134.6, 135.6, 136.1, 137.0, 138.5, 145.4, 150.6 (aromatic C), 160.9, 164.4 (2CO), 175.1 (CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3040 (aromatic CH), 2830 (aliphatic CH), 1665, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 483 (6.8%). Anal. Calcd. for C26H21N5O3S (483.54): C (64.58%), H (4.38%), N (14.48%), S (6.63%); Found: C (64.5%), H (4.1%), N (14.6%), S (6.5%).
4'-(4-Chlorophenyl)-6'-phenyl-2'-thioxo-3',4'-dihydro-1'H,3H-spiro[benzo[d]thiazole-2,8'-pyrido[4,3-d]pyrimidine]-5',7'(2'H,6'H)-dione (11c): white crystals after crystallization from absolute ethanol; m.p. 230–232 °C, in 52% yield (Method A) and 88% (Method B); 1H-NMR: 3.10 (s, 1H, NH, D2O exchangeable), 4.50 (s, 1H, NH, D2O exchangeable), 5.30 (s, 1H, CH), 6.60–7.50 (m, 13H, Ar-H), 11.80 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.7 (pyrimidine C-4), 82.0 (pyridine C-8), 116.0, 119.6, 121.3, 125.2, 127.4, 128.5, 132.6, 133.3, 134.6, 135.6, 136.1, 137.0, 138.5, 140.3 145.3, 151.4 (aromatic C), 160.9, 164.4 (2CO), 175.1 (CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3040 (aromatic CH), 2830 (aliphatic CH), 1665, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 504 (9.8%) and 489 (3.7%). Anal. Calcd. for C25H17ClN4O2S2 (505.01): C (59.46%), H (3.39%), Cl (7.02%), N (11.09%), S (12.70%); Found: C (59.5%), H (3.2%), Cl (6.9%), N (11.2%), S (12.6%).
4'-(4-Methoxyphenyl)-6'-phenyl-2'-thioxo-3',4'-dihydro-1'H,3H-spiro[benzo[d]thiazole-2,8'-pyrido[4,3-d]pyrimidine]-5',7'(2'H,6'H)-dione (11d): white crystals after crystallization from absolute ethanol; 45% yield (Method A) and 80% (Method B); m.p. 210–212 °C; 1H-NMR: 3.10 (s, 1H, NH, D2O exchangeable), 3.80 (s, 3H, OCH3), 4.00 (s, 1H, NH, D2O exchangeable), 5.40 (s, 1H, CH), 6.60–7.50 (m, 13H, Ar-H), 12.20 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.3 (OCH3), 55.7 (pyrimidine C-4), 80.9 (pyridine C-8), 116.4, 118.2, 121.2, 125.6, 127.1, 128.5, 132.6, 133.3, 134.6, 135.6, 136.1, 137.0, 138.5, 140.3, 145.1, 151.3 (aromatic C), 160.9, 164.4 (2CO), 175.1 (CS); IR ν: 3280, 3220, 3180 cm−1 (NH), 3040 (aromatic CH), 2830 (aliphatic CH), 1665, 1645 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 500 (11.3%). Anal. Calcd. for C26H20N4O3S2 (500.59): C (62.38%), H (4.03%), N (11.19%), S (12.81%); Found: C (62.4%), H (3.9%), N (11.0%), S (12.6%).
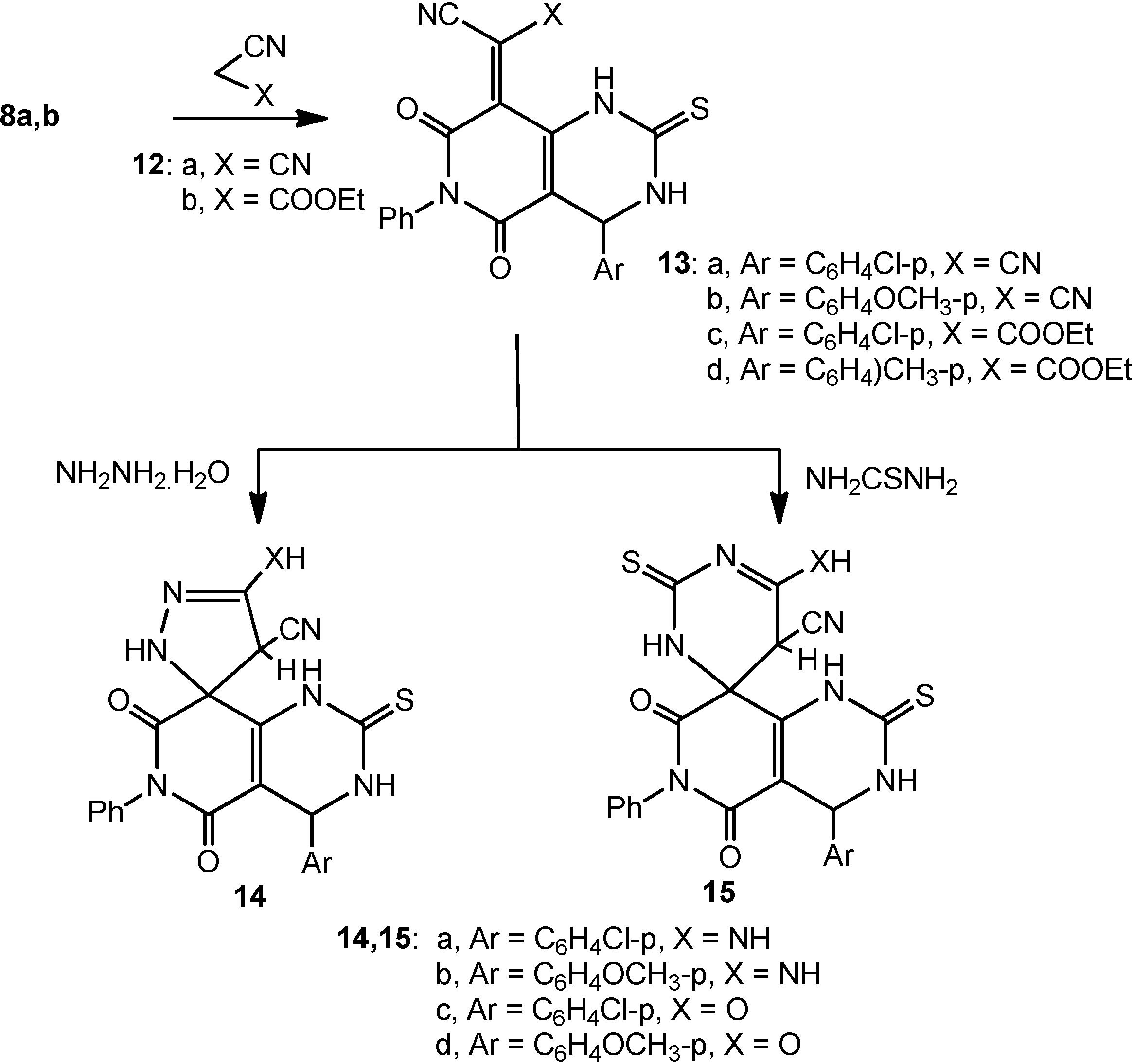
3.1.7. Reactions of 8a,b with Malononitrile (12a) and Ethyl cyanoacetate (12b): Formation of 13a–d
To a solution of each of compounds 8a,b (0.01 mol) in absolute ethanol (30 mL) containing a catalytic amount of piperidine was added either of malononitrile (12a; 0.66 g, 0.01 mol) or ethyl cyanoacetate (12b; 1.13 mL, 0.01 mol). The reaction mixture was heated under reflux for 3 h, under TLC monitoring, then cooled and poured onto ice-cold water. The solid product that separated was filtered off, dried and crystallized from ethanol.
2-(4-(4-Chlorophenyl)-5,7-dioxo-6-phenyl-2-thioxo-1,2,3,4,6,7-hexahydropyrido[4,3-d]pyrimidin-8(5H)-ylidene)malononitrile (13a): pale yellow crystals after crystallization from absolute ethanol; 52% yield; m.p. 223–225 °C; 1H-NMR: 3.20 (s, 1H, NH, D2O exchangeable), 4.80 (s, 1H, CH), 7.20–7.50 (m, 9H, Ar-H), 12.60 (s, 1H, NH, D2O exchangeable); 13C-NMR: 55.5 (pyrimidine C-4), 81.9 (methylidine C), 107.1 (CN), 112.1, 127.1, 127.9, 132.1, 132.9, 133.8, 134.7, 135.1, 137.3, 150.8, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0 (CS); IR ν: 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2210 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 445 (12.2%) and 447 (4.7%). Anal. Calcd. for C22H12ClN5O2S (445.88): C (59.26%), H (2.71%), Cl (7.95%), N (15.71%), S (7.19%); Found: C (59.2%), H (2.5%), Cl (7.8%), N (15.6%), S (7.0%).
2-(4-(4-Methoxyphenyl)-5,7-dioxo-6-phenyl-2-thioxo-1,2,3,4,6,7-hexahydropyrido[4,3-d]pyrimidin-8(5H)-ylidene)malononitrile (13b): yellow crystals after crystallization from absolute ethanol; 50% yield; m.p. 214–216 °C; 1H-NMR: 3.10 (s, 1H, NH, D2O exchangeable), 3.80 (s, 1H, OCH3), 4.70 (s, 1H, CH), 6.90–7.50 (m, 9H, Ar-H), 12.60 (s, 1H, NH, D2O exchangeable); 13C-NMR: 54.7 (OCH3), 55.5 (pyrimidine C-4), 81.8 (methylidine C), 107.1 (CN), 112.1, 114.8, 127.3, 132.1, 132.9, 133.8, 134.6, 135.1, 137.3, 150.8, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.1 (CS); IR ν: 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2210 (CN), 1670, 1640 (2C=O), 1600, 1500 (aromatic C=C); MS (70 eV): M+ m/z 441 (15.1%); Anal. Calcd. for C23H15N5O3S (441.46): C (62.58%), H (3.42%), N (15.86%), S (7.26%); Found: C (62.6%), H (3.5%), N (15.7%), S (7.0%).
Ethyl 2-(4-(4-chlorophenyl)-5,7-dioxo-6-phenyl-2-thioxo-1,2,3,4,6,7-hexahydropyrido[4,3-d]-pyrimidin-8(5H)-ylidene)-2-cyanoacetate (13c): white crystals after crystallization from absolute ethanol; 42% yield; m.p. 188–190 °C; 1H-NMR: 1.20 (t, 3h, CH3), 3.00 (s, 1H, NH, D2O exchangeable), 4.30 (q, 2H, CH2), 5.10 (s, 1H, CH), 7.20–7.60 (m, 9H, Ar-H), 12.50 (s, 1H, NH, D2O exchangeable); 13C-NMR: 16.7 (CH3), 55.5 (pyrimidine C-4), 59.1 (CH2), 93.9 (methylidine C), 108.1 (CN), 123.4, 127.1, 127.9, 132.1, 132.9, 133.8, 134.6, 135.1, 137.3, 151.8, 157.1 (sp2 + aromatic C), 156.9, 160.7, 165.0 (3CO), 178.0 (CS); IR ν: 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2230 (CN), 1710, 1670, 1640 (3C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 492 (14.2%) and 494 (5.0%). Anal. Calcd. for C24H17ClN4O4S (492.93): C (58.48%), H (3.48%), Cl (7.19%), N (11.37%), S (6.50%); Found: C (58.3%), H (3.3%), Cl (7.1%), N (11.4%), S (6.4%).
Ethyl 2-cyano-2-(4-(4-methoxyphenyl)-5,7-dioxo-6-phenyl-2-thioxo-1,2,3,4,6,7-hexahydropyrido-[4,3-d]pyrimidin-8(5H)-ylidene)acetate (13d): white crystals after crystallization from absolute ethanol; 42% yield; m.p. 188–190 °C; 1H-NMR: 1.20 (t, 3h, CH3), 3.00 (s, 1H, NH, D2O exchangeable), 3.70 (s, 3H, CH3), 4.30 (q, 2H, CH2), 5.10 (s, 1H, CH), 6.90–7.60 (m, 9H, Ar-H), 12.30 (s, 1H, NH, D2O exchangeable); 13C-NMR: 16.7 (CH3), 53.1 (OCH3) 55.5 (pyrimidine C-4), 59.0 (CH2), 91.7 (methylidine C), 108.1 (CN), 117.7, 125.1, 126.9, 132.1, 132.9, 133.8, 133.6, 135.1, 137.3, 148.8, 156.6 (sp2 + aromatic C), 156.9, 160.7, 165.0 (3CO), 178.0 (CS); IR ν: 3220, 3180 cm−1 (NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2230 (CN), 1710, 1670, 1640 (3C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 488 (13.9%); Anal. Calcd. for C25H20N4O5S (488.52): C (61.47%), H (4.13%), N (11.47%), S (6.56%); Found: C (61.5%), H (4.0%), N (11.4%), S (6.4%).
3.1.8. Reaction of 13a–d with Hydrazine Hydrate and Thiourea: Formation of 14a–d and 15a–d
Method A: To a solution of each of compounds 13a–d (0.01 mol) in absolute ethanol (30 mL) containing a catalytic amount of piperidine was added hydrazine (0.32 mL, 0.01 mol) or thiourea (0.76 g, 0.01 mol). The reaction mixture was heated under reflux for 3 h, under TLC monitoring, then cooled and poured onto ice-cold water. The solid product that separated was filtered off, dried and crystallized from ethanol.
Method B: The same reactants of method A were heated in microwave oven at 500 W and 140 °C for 7 min. The reaction mixture was treated similar to method A to obtain compounds 14a–d and 15a–d.
5-Amino-4'-(4-chlorophenyl)-5',7'-dioxo-6'-phenyl-2'-thioxo-2,2',3',4,4',5',6',7'-octahydro-1'H-spiro[pyrazole-3,8'-pyrido[4,3-d]pyrimidine]-4-carbonitrile (14a): white crystals after crystallization from absolute dioxane; 37% yield (Method A) and 81% (Method B); m.p. 235–237 °C; 1H-NMR: 3.20 (s, 1H, NH, D2O exchangeable), 4.10 (s, 1H, pyrazole H-4), 5.10 (s, 1H, pyrimidine H-4), 6.2 (s, 1H, NH, D2O exchangeable), 7.20–7.50 (m, 9H, Ar-H), 9.1 (s, 2H, NH2, D2O exchangeable), 12.60 (s, 1H, NH, D2O exchangeable); 13C-NMR: 39.5 (pyrazole C-4), 55.4 (pyrimidine C-4), 58.9 (spiro-C), 107.1 (CN), 110.3, 116.1, 126.9, 131.8, 132.7, 133.8, 134.6, 135.1, 137.3, 143.1, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0 (CS); IR ν: 3350, 3220, 3180 cm−1 (broad, NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C);MS (70 eV): M+ m/z 477 (9.2%) and 447 (3.5%); Anal. Calcd. for C22H16ClN7O2S (477.93): C (55.29%), H (3.37%), Cl (7.42%), N (20.52%), S (6.71%); Found: C (55.1%), H (3.1%), Cl (7.3%), N (20.4%), S (6.6%).
5-Amino-4'-(4-methoxyphenyl)-5',7'-dioxo-6'-phenyl-2'-thioxo-2,2',3',4,4',5',6',7'-octahydro-1'H-spiro[pyrazole-3,8'-pyrido[4,3-d]pyrimidine]-4-carbonitrile (14b): white crystals after crystallization from dil. dioxane; 42% yield (Method A) and 84% (Method B); m.p. 229–231 °C; 1H-NMR: 3.00 (s, 1H, NH, D2O exchangeable), 3.70 (s, 3H, OCH3), 4.10 (s, 1H, pyrazole H-4), 5.10 (s, 1H, pyrimidine H-4), 6.2 (s, 1H, NH, D2O exchangeable), 7.20–7.50 (m, 9H, Ar-H), 9.4 (s, 2H, NH2, D2O exchangeable), 12.50 (s, 1H, NH, D2O exchangeable); 13C-NMR: 39.5 (pyrazole C-4), 53.7 (OCH3), 55.4 (pyrimidine C-4), 58.9 (spiro-C), 107.1 (CN), 112.7, 114.3, 126.9, 131.8, 132.7, 133.8, 134.6, 135.1, 137.3, 143.1, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0 (CS); IR ν: 3350, 3220, 3180 cm−1 (broad, NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 473 (10.8%); Anal. Calcd. for C23H19N7O3S (477.93): C (58.43%), H, 4.04; N, 20.71; S, 6.77; Found: C (58.3%), H (4.1%), N (20.5%), S (6.6%).
4'-(4-Chlorophenyl)-5-hydroxy-5',7'-dioxo-6'-phenyl-2'-thioxo-2,2',3',4,4',5',6',7'-octahydro-1'H-spiro-[pyrazole-3,8'-pyrido[4,3-d]pyrimidine]-4-carbonitrile (14c): white crystals after crystallization from absolute ethanol; 45% yield (Method A) and 89% (Method B); m.p. 258–260 °C; 1H-NMR: 3.40 (s, 1H, NH, D2O exchangeable), 4.50 (s, 1H, pyrazole H-4), 5.40 (s, 1H, pyrimidine H-4), 6.2 (s, 1H, NH, D2O exchangeable), 7.20–7.50 (m, 9H, Ar-H), 11.1 (s, 1H, OH, D2O exchangeable), 12.60 (s, 1H, NH, D2O exchangeable); 13C-NMR: 41.1 (pyrazole C-4), 55.4 (pyrimidine C-4), 58.9 (spiro-C), 108.9 (CN), 110.3, 115.1, 126.9, 128.8, 130.1, 132.5, 134.6, 135.1, 137.3, 143.1, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0 (CS); IR ν: 3400, 3270, 3180 cm−1 (broad, NH + OH), 3060 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 478 (10.7%) and 480 (3.8%); Anal. Calcd. for C22H15ClN6O3S (478.91): C (55.17%), H (3.16%), Cl (7.40%), N (17.55%), S (6.70%); Found: C (55.0%), H (3.2%), Cl (7.3%), N (17.3%), S (6.6%).
5-Hydroxy-4'-(4-methoxyphenyl)-5',7'-dioxo-6'-phenyl-2'-thioxo-2,2',3',4,4',5',6',7'-octahydro-1'H-spiro[pyrazole-3,8'-pyrido[4,3-d]pyrimidine]-4-carbonitrile (14d): white crystals after crystallization from absolute ethanol; 44% yield (Method A) and 82% (Method B); m.p. 237–239 °C; 1H-NMR: 3.30 (s, 1H, NH, D2O exchangeable), 3.80 (s, 3H, OCH3), 4.10 (s, 1H, pyrazole H-4), 5.10 (s, 1H, pyrimidine H-4), 6.2 (s, 1H, NH, D2O exchangeable), 7.20–7.50 (m, 9H, Ar-H), 10.8 (s, 1H, OH, D2O exchangeable, 12.50 (s, 1H, NH, D2O exchangeable); 13C-NMR: 39.5 (pyrazole C-4), 53.8 (OCH3), 55.4 (pyrimidine C-4), 58.9 (spiro-C), 108.9 (CN), 112.7, 124.8, 126.9, 128.8, 132.1, 133.3, 134.6, 135.1, 137.3, 144.6, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0 (CS); IR ν: 3350, 3200, 3150 cm−1 (broad, NH + OH), 3060 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 474 (12.0%); Anal. Calcd. for C23H18N6O4S (474.49): C (58.22%), H (3.82%), N (17.71%), S (6.76%); Found: C (58.1%), H (3.6%), N (17.5%), S (6.7%).
6'-Amino-4-(4-chlorophenyl)-5,7-dioxo-6-phenyl-2,2'-dithioxo-2,3,3',4,5,5',6,7-octahydro-1H,2'H-spiro[pyrido[4,3-d]pyrimidine-8,4'-pyrimidine]-5'-carbonitrile (15a): white crystals after crystallization from dil. DMF; 47% yield (Method A) and 88% (Method B); m.p. 240–242 °C; 1H-NMR: 3.00 (s, 1H, NH, D2O exchangeable), 3.40 (s, 1H, spiro-pyrimidine H-4), 5.10 (s, 1H, pyrimidine H-4), 5.50 (s, 1H, NH, D2O exchangeable), 7.20–7.50 (m, 9H, Ar-H), 8.70 (s, 2H, NH2, D2O exchangeable), 12.00 (s, 1H, NH, D2O exchangeable); 13C-NMR: 29.0 (spiro-pyrimidine C-5), 55.4 (pyrimidine C-4), 63.9 (spiro-C), 107.8 (CN), 114.3, 119.5, 126.9, 128.8, 132.7, 133.8, 134.6, 136.1, 138.1, 143.1, 154.3 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0, 181.0 (2CS); IR ν: 3350, 3220, 3180 cm−1 (broad, NH), 3060 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C);MS (70 eV): M+ m/z 522 (10.0%) and 524 (3.8%); Anal. Calcd. for C23H16ClN7O2S2 (522.00): C (52.92%), H (3.09%), Cl (6.79%), N (18.78%), S (12.29%); Found: C (52.8%), H (3.2%), Cl (6.8%), N (18.9%), S (12.1%).
6'-Amino-4-(4-methoxyphenyl)-5,7-dioxo-6-phenyl-2,2'-dithioxo-2,3,3',4,5,5',6,7-octahydro-1H,2'H-spiro[pyrido[4,3-d]pyrimidine-8,4'-pyrimidine]-5'-carbonitrile (15b): white crystals after crystallization from dil. DMF; 43% yield (Method A) and 87% (Method B); m.p. 226–228 °C; 1H-NMR: 2.80 (s, 1H, NH, D2O exchangeable), 3.50 (s, 1H, spiro-pyrimidine H-4), 3.80 (s, 1H, OCH3), 5.00 (s, 1H, pyrimidine H-4), 5.50 (s, 1H, NH, D2O exchangeable), 6.80–7.50 (m, 9H, Ar-H), 8.50 (s, 2H, NH2, D2O exchangeable), 12.20 (s, 1H, NH, D2O exchangeable); 13C-NMR: 27.8 (spiro-pyrimidine C-5), 52.6 (OCH3), 55.7 (pyrimidine C-4), 64.2 (spiro-C), 107.8 (CN), 112.5, 116.2, 125.2, 127.0, 128.4, 129.8, 131.6, 136.1, 138.1, 139.0, 154.2 (sp2 + aromatic C), 156.9, 160.7 (2CO), 178.0, 181.0 (2CS); IR ν: 3350, 3200, 3160 cm−1 (broad, NH), 3080 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1500 (aromatic C=C); MS (70 eV): M+ m/z 517 (11.3%); Anal. Calcd. for C24H19N7O3S2 (517.58): C (55.69%), H (3.70%), N (18.94%), S (12.39%); Found: C (55.6%), H (3.5%), N (18.9%), S (12.1%).
4-(4-Chlorophenyl)-6'-hydroxy-5,7-dioxo-6-phenyl-2,2'-dithioxo-2,3,3',4,5,5',6,7-octahydro-1H,2'H-spiro[pyrido[4,3-d]pyrimidine-8,4'-pyrimidine]-5'-carbonitrile (15c): white crystals after crystallization from dioxane; 51% yield (Method A) and 90% (Method B); m.p. 255–257 °C; 1H-NMR: 3.20 (s, 1H, NH, D2O exchangeable), 3.50 (s, 1H, spiro-pyrimidine H-4), 5.10 (s, 1H, pyrimidine H-4), 5.80 (s, 1H, NH, D2O exchangeable), 7.20–7.50 (m, 9H, Ar-H), 11.10 (s, 1H, OH, D2O exchangeable), 12.40 (s, 1H, NH, D2O exchangeable); 13C-NMR: 29.0 (spiro-pyrimidine C-5), 55.4 (pyrimidine C-4), 63.9 (spiro-C), 107.8 (CN), 113.9, 119.8, 127.2, 128.8, 132.4, 133.8, 134.5, 136.5, 138.6, 152.1, 155.3 (sp2 + aromatic C), 158.9, 162.7 (2CO), 178.0, 180.5 (2CS); IR ν: 3350, 3220, 3180 cm−1 (broad, NH), 3050 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1490 (aromatic C=C); MS (70 eV): M+ m/z 522 (14.0%) and 524 (4.7%); Anal. Calcd. for C23H15ClN6O3S2 (522.99): C (52.82%), H (2.89%), Cl (6.78%), N (16.07%), S (12.26%); Found: C (52.6%), H (2.9%), Cl (6.6%), N (15.9%), S (12.1%).
6'-Hydroxy-4-(4-methoxyphenyl)-5,7-dioxo-6-phenyl-2,2'-dithioxo-2,3,3',4,5,5',6,7-octahydro-1H,2'H-spiro[pyrido[4,3-d]pyrimidine-8,4'-pyrimidine]-5'-carbonitrile (15d): white crystals after crystallization from dioxane; 47% yield (Method A) and 91% (Method B); m.p. 238–240 °C; 1H-NMR: 2.90 (s, 1H, NH, D2O exchangeable), 3.60 (s, 1H, spiro-pyrimidine H-4), 3.90 (s, 1H, OCH3), 5.00 (s, 1H, pyrimidine H-4), 5.50 (s, 1H, NH, D2O exchangeable), 6.80–7.50 (m, 9H, Ar-H), 10.80 (s, 1H, OH, D2O exchangeable), 12.10 (s, 1H, NH, D2O exchangeable); 13C-NMR: 27.2 (spiro-pyrimidine C-5), 54.4 (OCH3), 57.1 (pyrimidine C-4), 64.0 (spiro-C), 108.4 (CN), 116.5, 119.6, 125.0, 127.4, 128.8, 130.3, 132.6, 136.1, 138.1, 139.0, 153.1 (sp2 + aromatic C), 158.9, 160.7 (2CO), 178.0, 181.3 (2CS); IR ν: 3350, 3200, 3160 cm−1 (broad, NH), 3080 (aromatic CH), 2850 (aliphatic CH), 2200 (CN), 1670, 1640 (2C=O), 1600, 1500 (aromatic C=C);MS (70 eV): M+ m/z 518 (14.0%), 516 (5.1%); Anal. Calcd. for C24H18N6O4S2 (518.57): C (55.59%), H (3.50%), N (16.21%), S (12.37%); Found: C (55.4%), H (3.4%), N (16.3%), S (12.3%).




{kind=link}
{kind=link}
{kind=link}