
Recent Advances in the Development and Application of Radiolabeled Kinase Inhibitors for PET Imaging


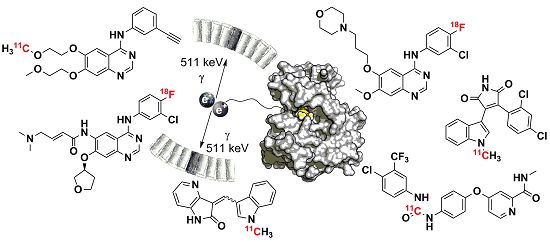
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Synthesis and Evaluation of Radiolabeled Small Molecule Kinase Inhibitors for PET Imaging
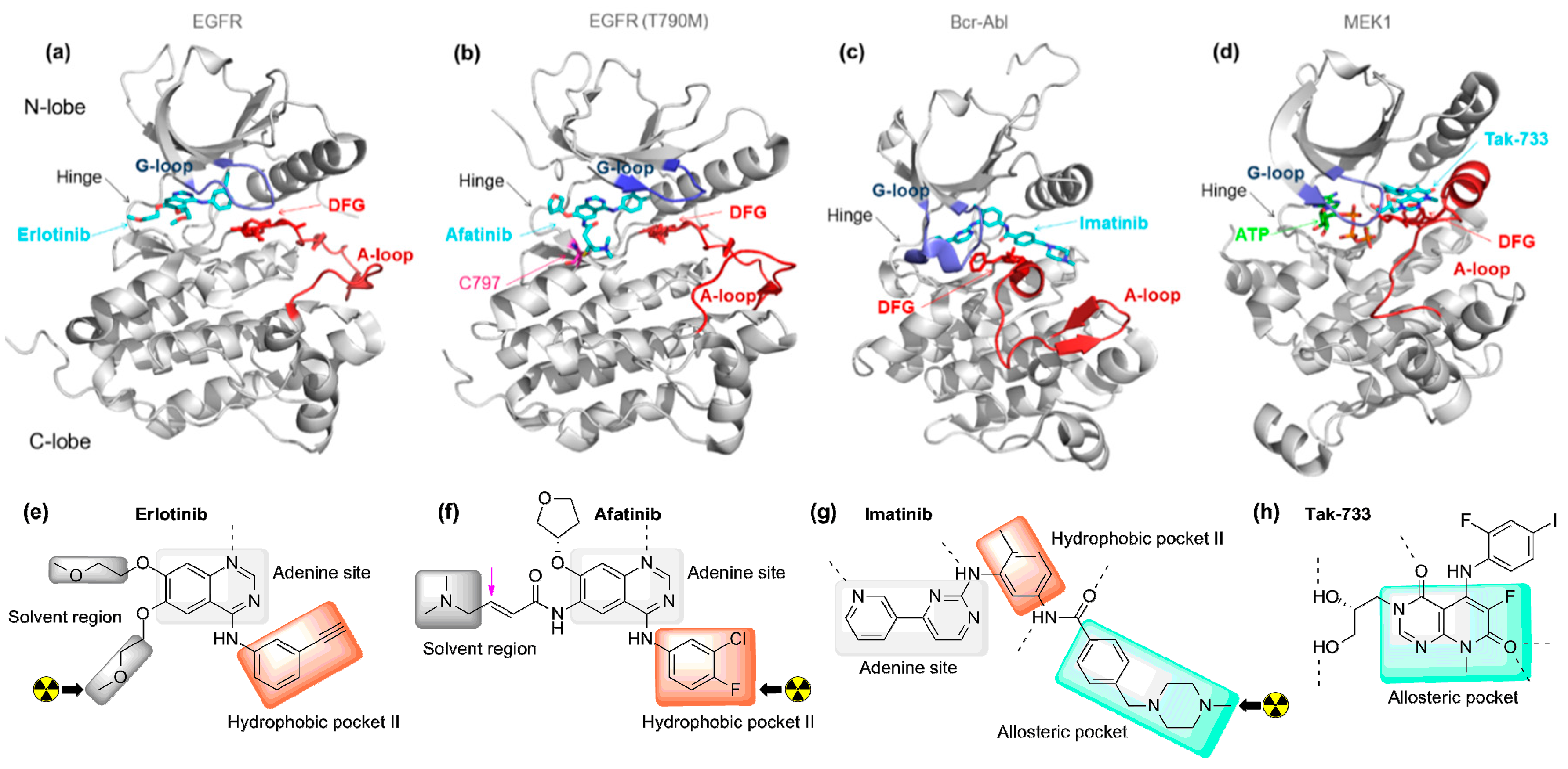
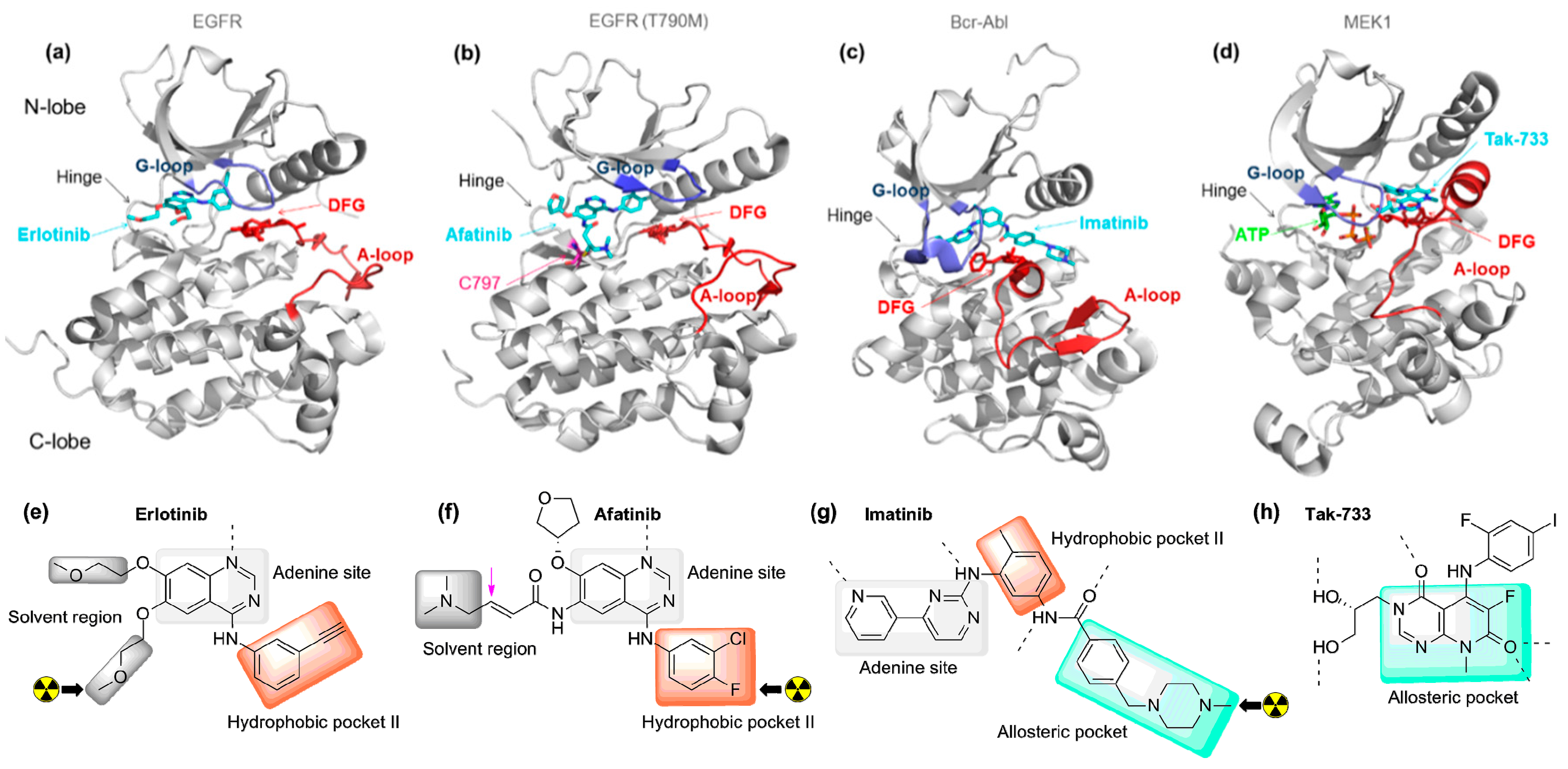
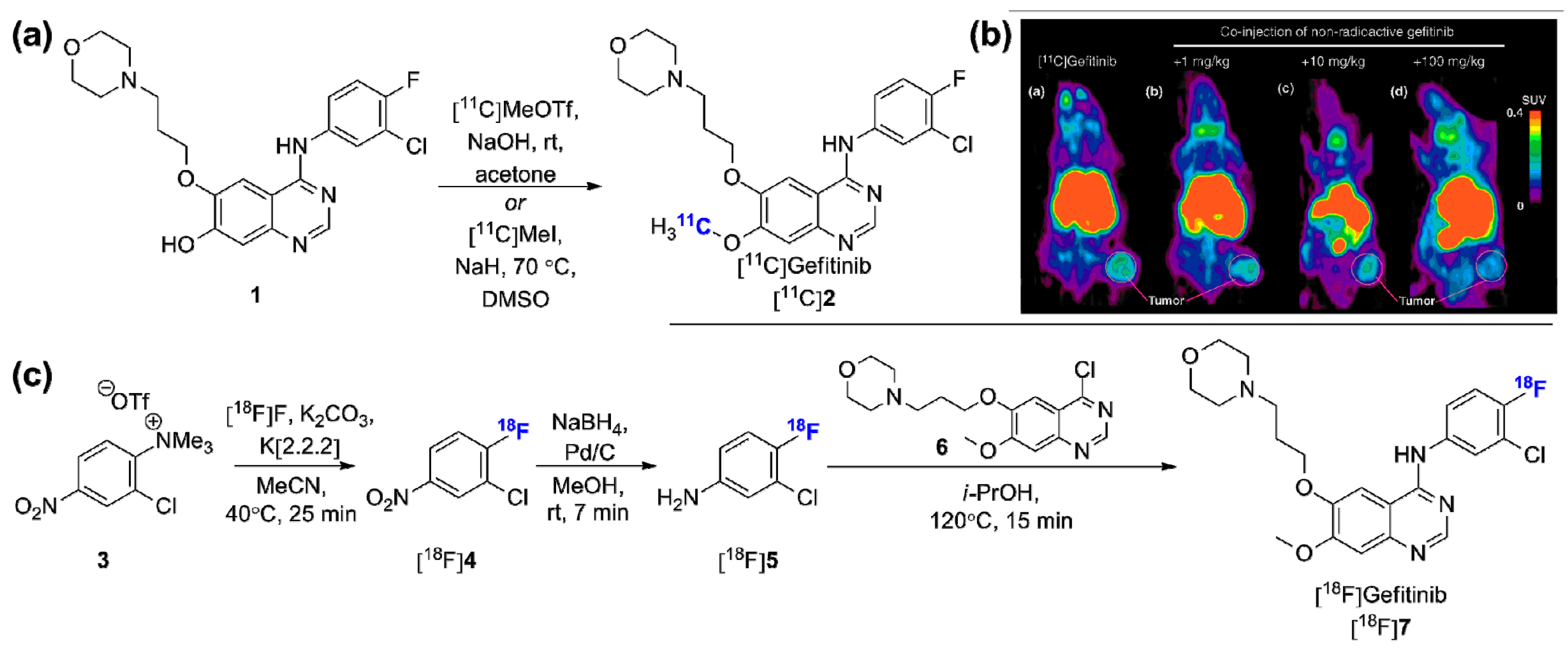
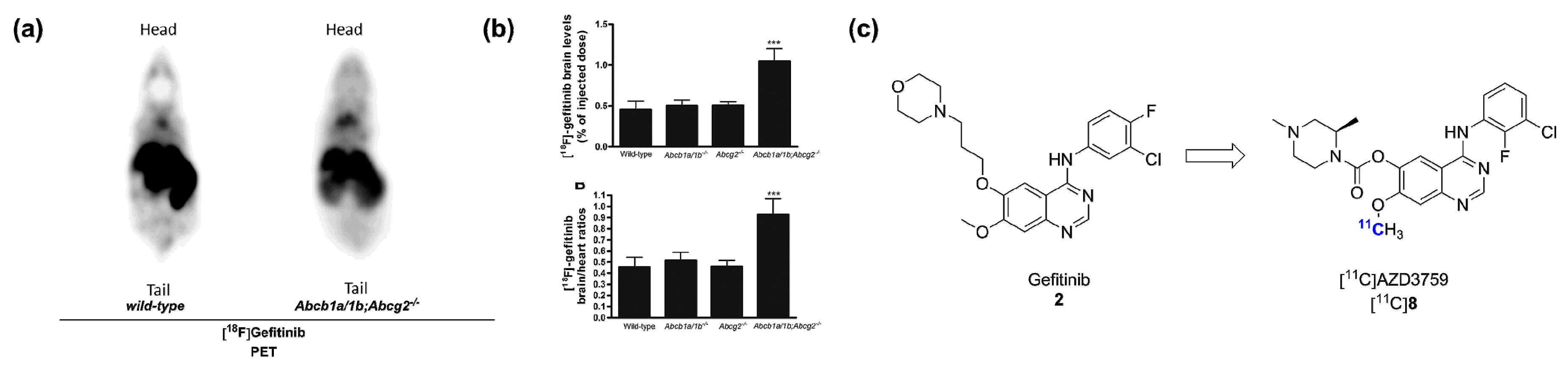
2.1. Radiolabeled Tyrosine Kinase Inhibitors
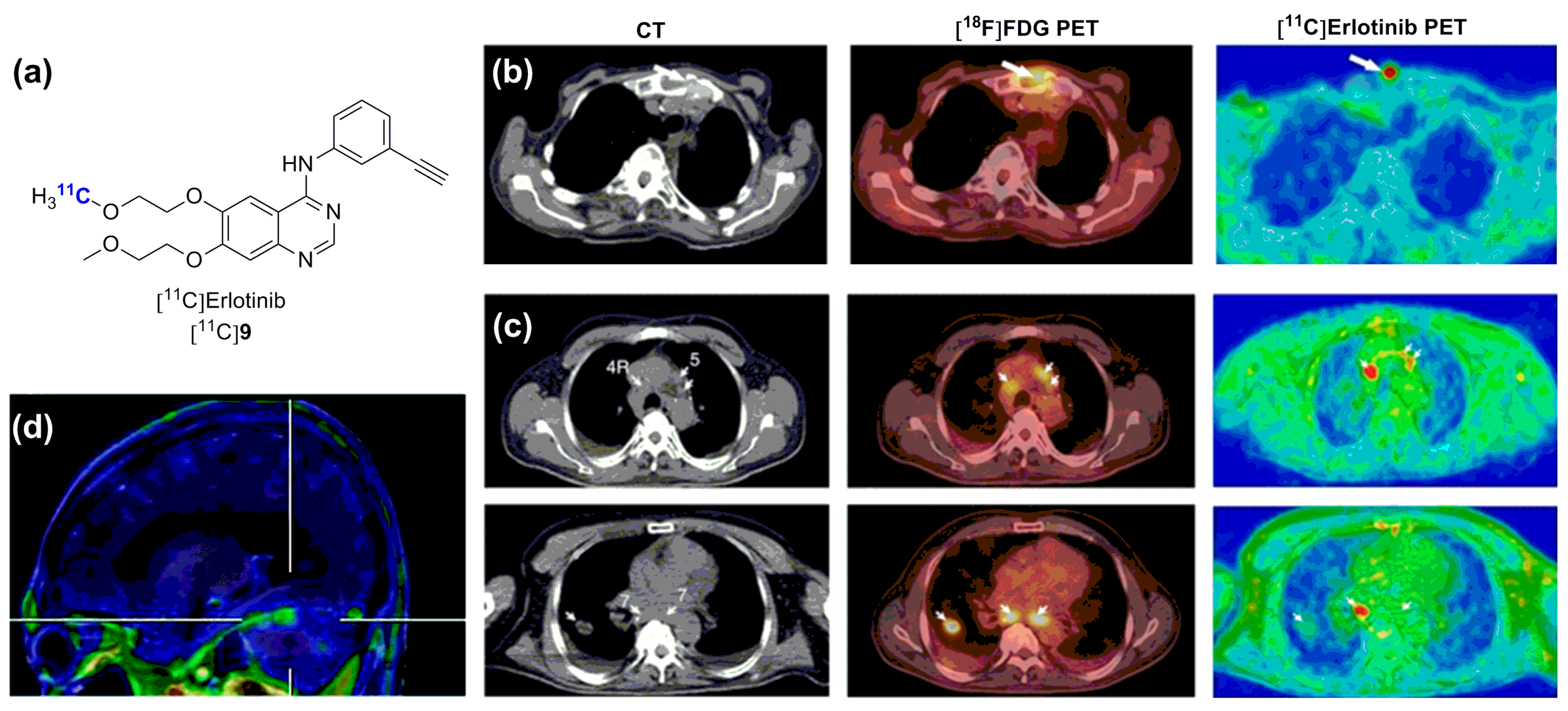
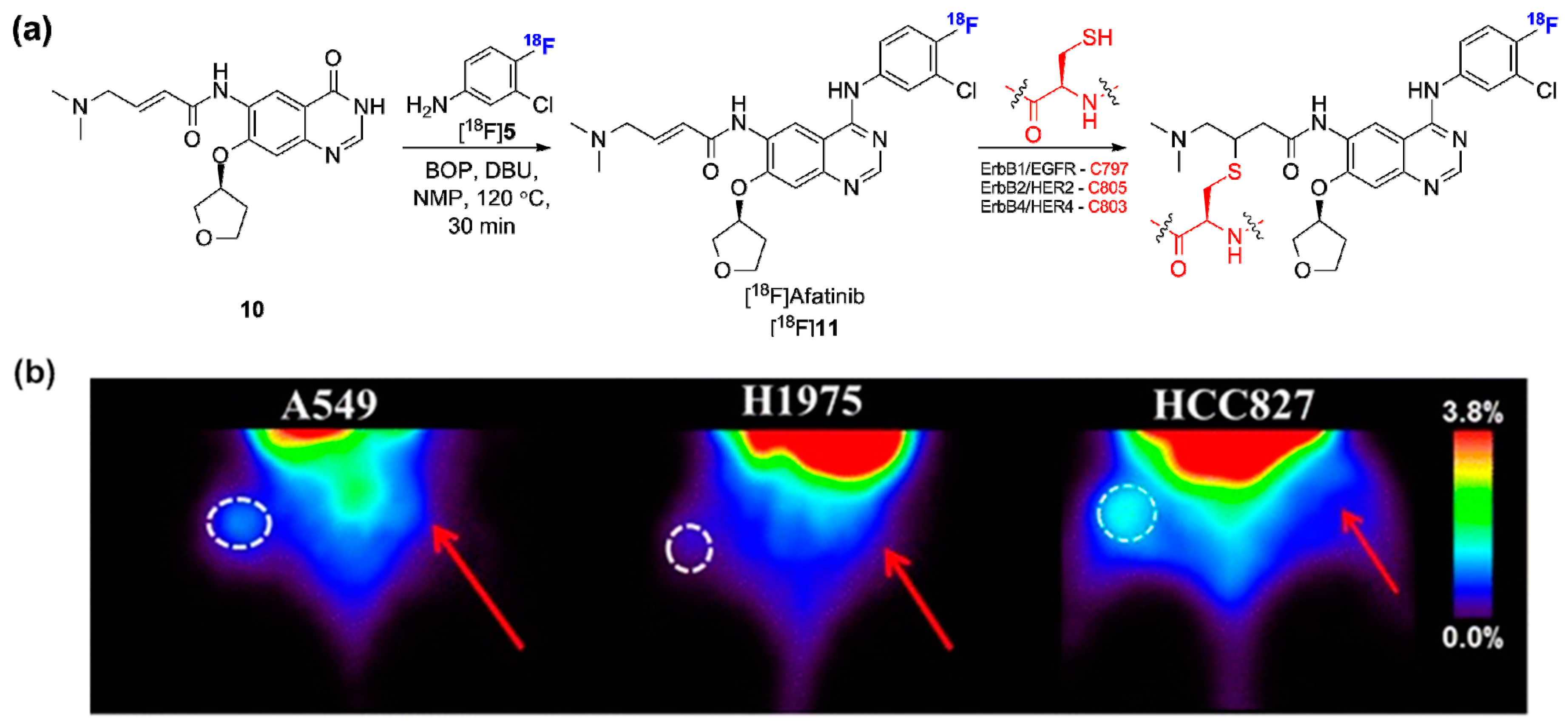

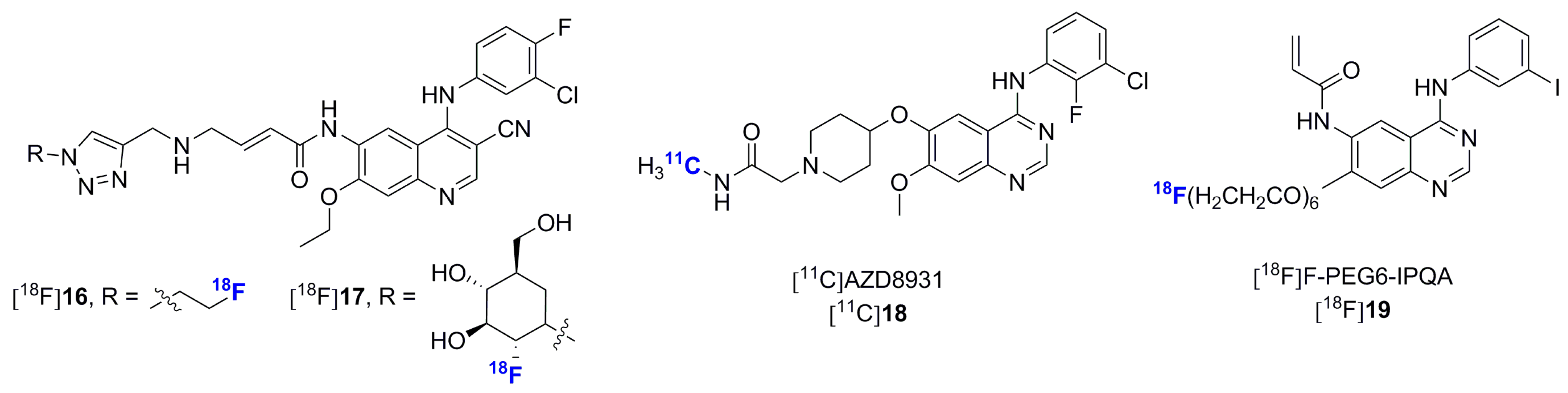
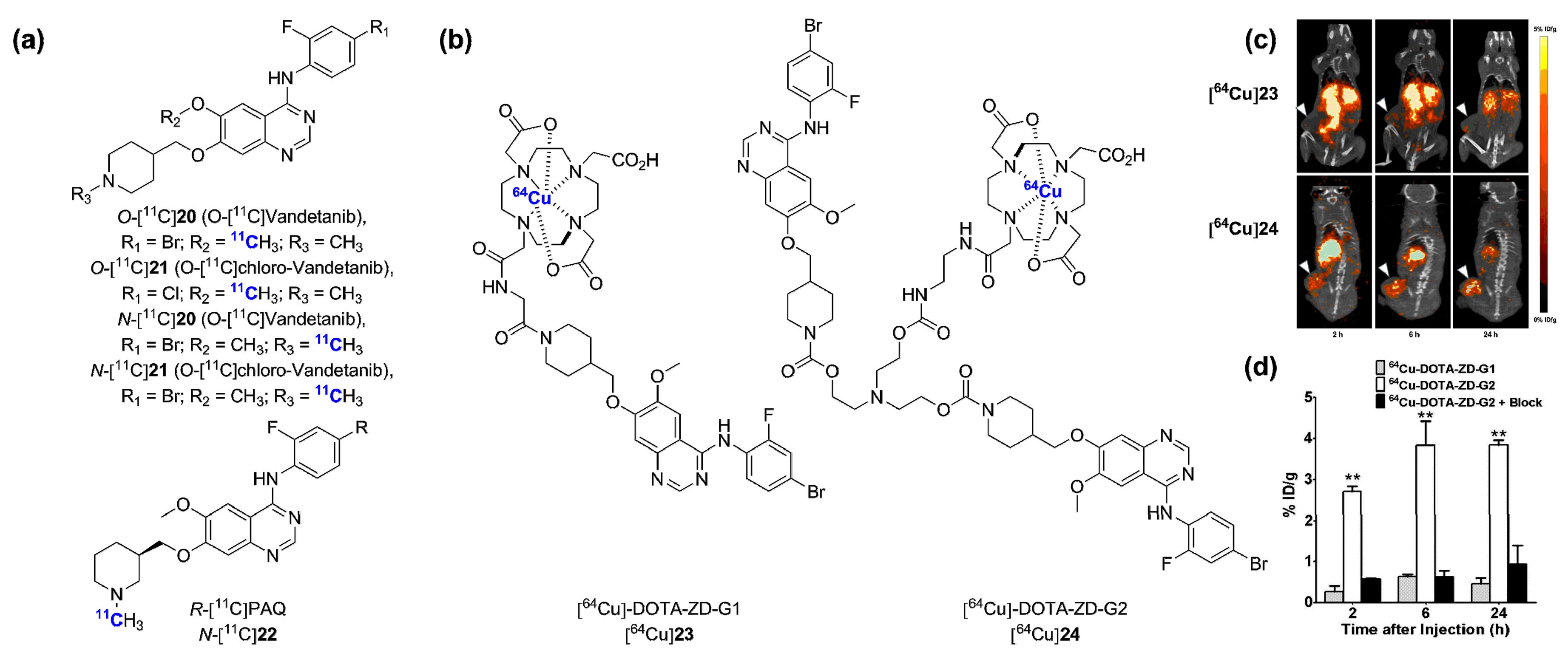

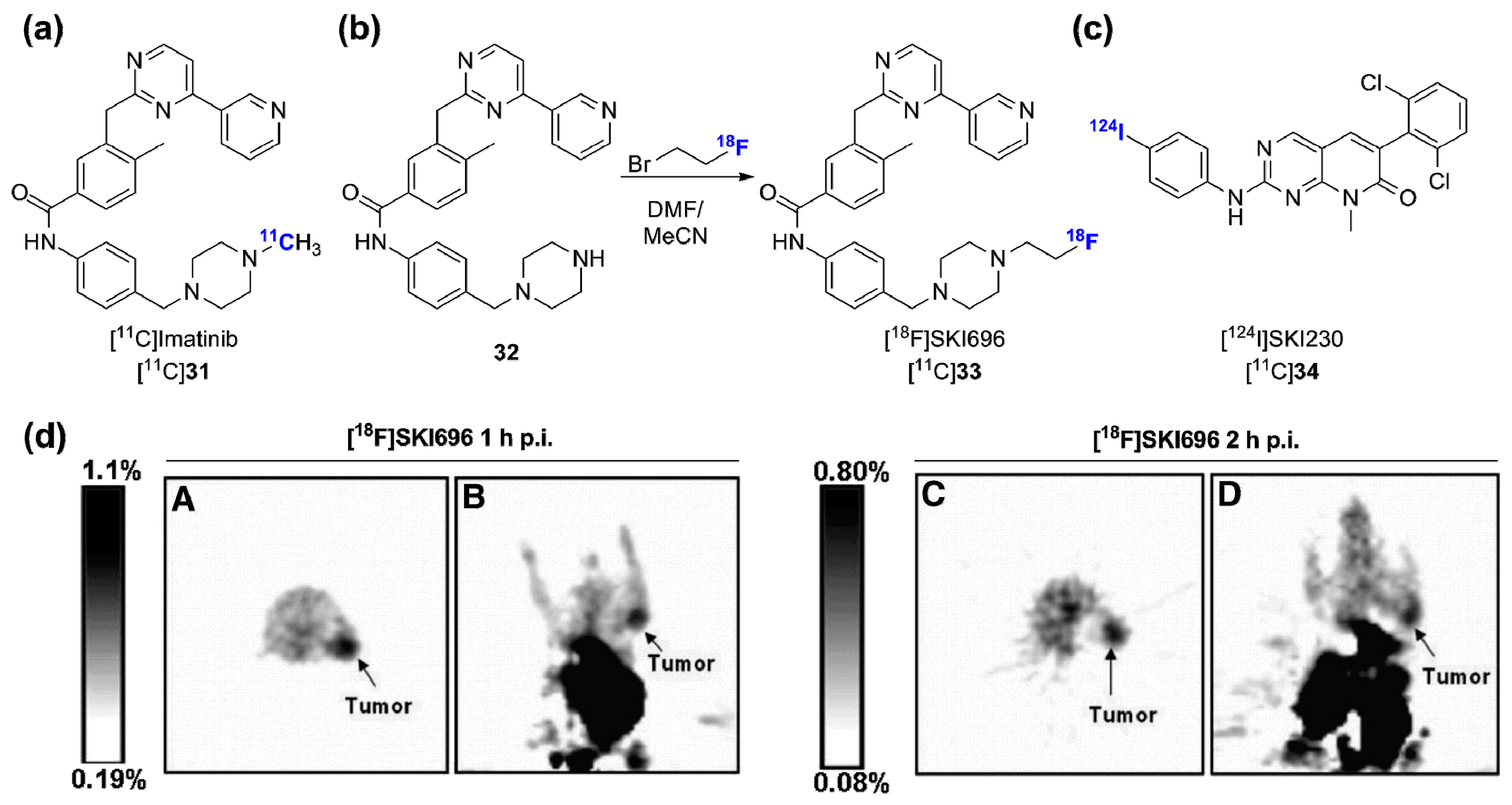
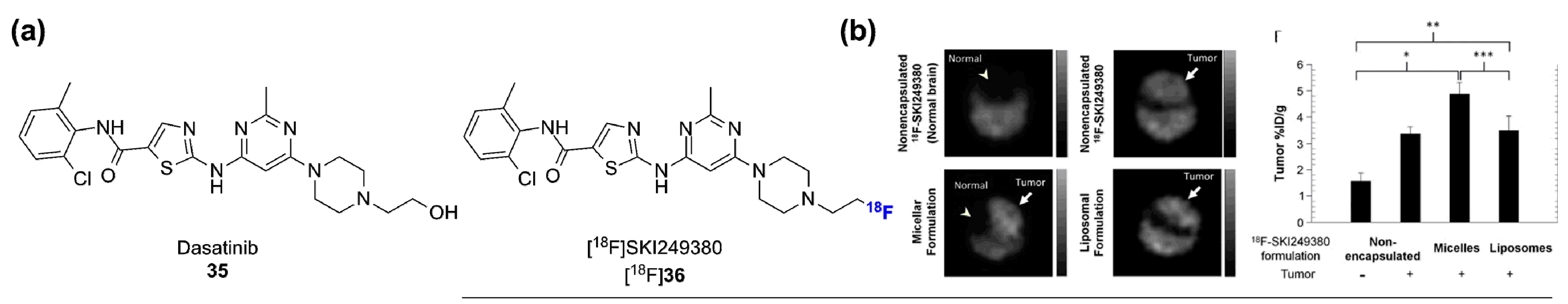
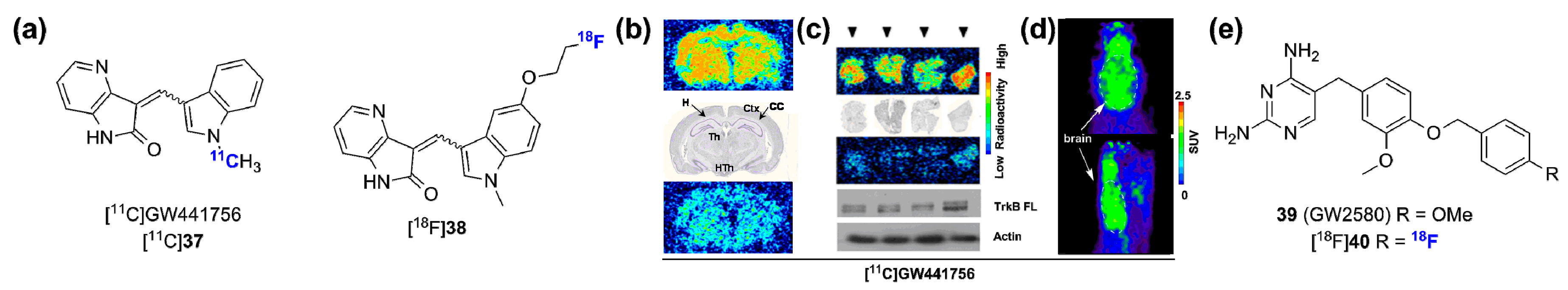
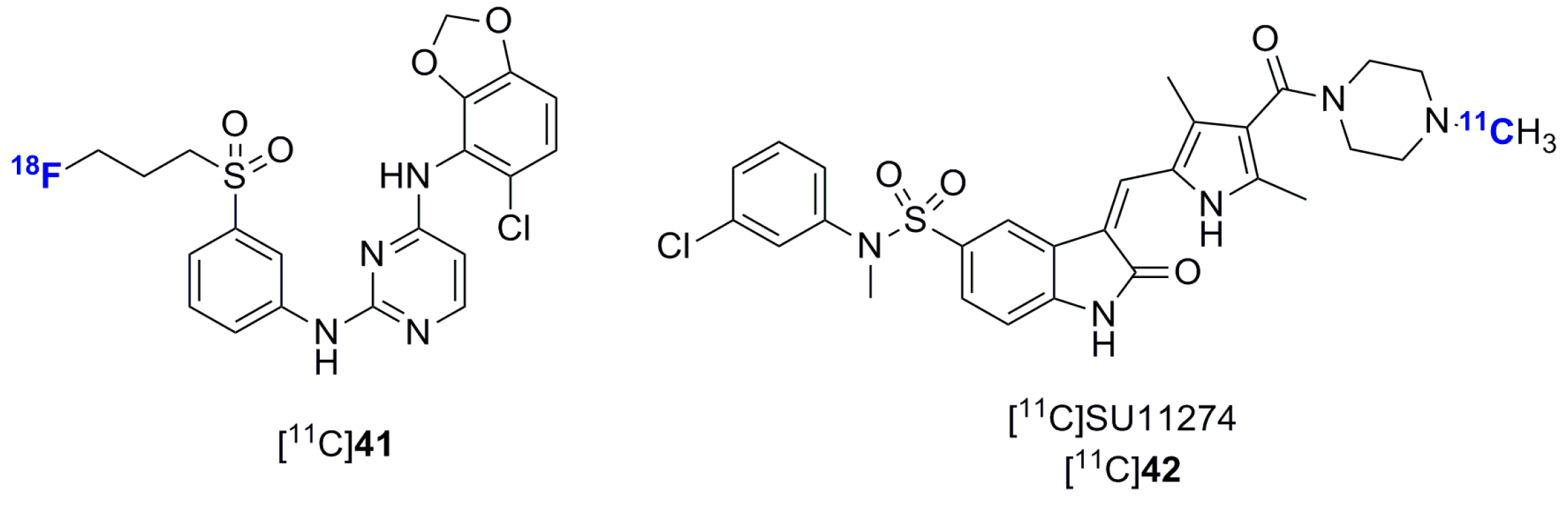
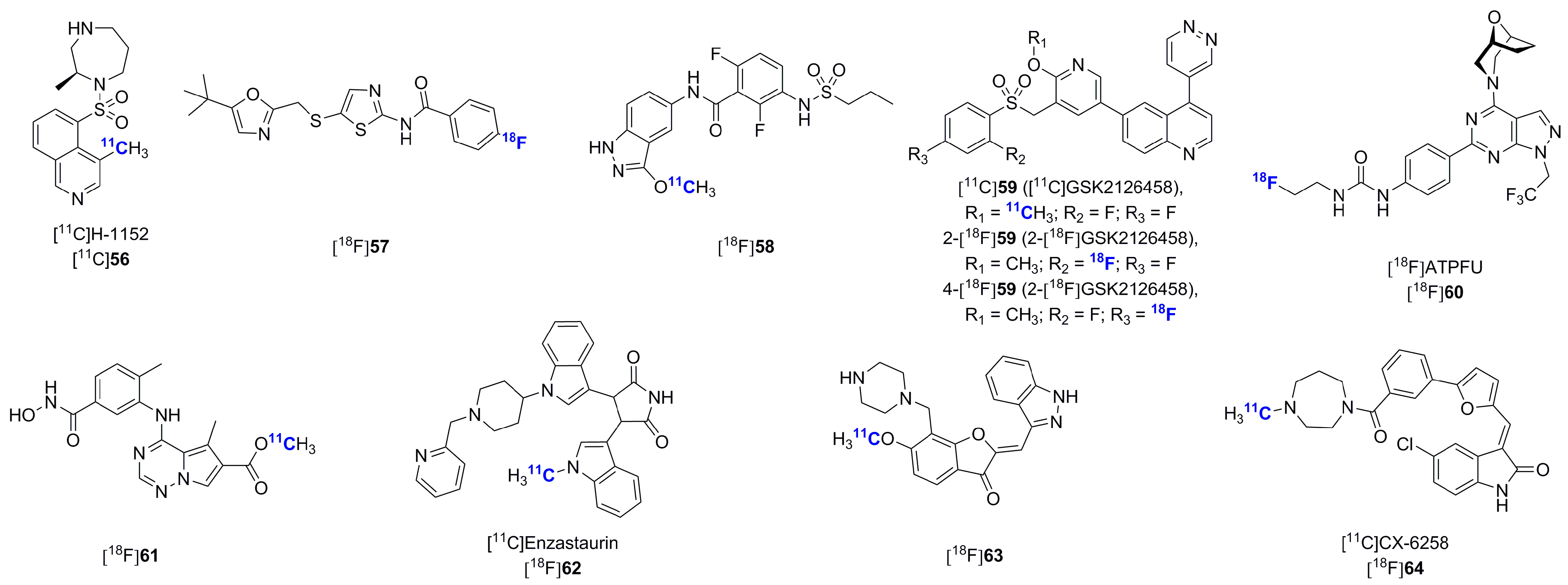
2.2. Radiolabeled Serine/Threonine Kinase Inhibitors
3. Concluding Remarks
Conflicts of Interest
References
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecules kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielson, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Available online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm455862.htm (accessed on 1 October 2015).
- Van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef] [PubMed]
- Bamborough, P.; Drewry, D.; Harper, G.; Smith, G.K.; Schneider, K. Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J. Med. Chem. 2008, 51, 7898–7914. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Furtmann, N.; Bajorath, J. Current compound coverage of the kinome. J. Med. Chem. 2015, 58, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J. A new challenging and promising era of tyrosine kinase inhibitors. ACS Med. Chem. Lett. 2014, 5, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L., Jr.; Levy, R.M. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validates type II inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitor of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.J.; Shomin, C.D.; Ghosh, I. Tinkering outside the kinase ATP box: Allosteric (type IV) and bivalent (type V) inhibitors of protein kinases. Future Med. Chem. 2010, 3, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Grutter, C.; Rauh, D. Strategies for the selective regulation of kinases with allosteric modulators: Exploiting structural features. ACS Chem. Biol. 2013, 8, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Josephs, D.H.; Fisher, D.S.; Spicer, J.; Flanagan, R.J. Clinical pharmacokinetics of tyrosine kinase inhibitors: Implication for therapeutic drug monitoring. Ther. Drug Monit. 2013, 35, 562–587. [Google Scholar] [CrossRef] [PubMed]
- Barouch-Bentov, R.; Sauer, K. Mechanism of drug resistance in kinases. Expert Opin. Investig. Drugs 2011, 20, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Lee, D.; Shim, H. Metabolic PET imaging in cancer detection and therapy response. Semin. Oncol. 2011, 38, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.M.; Rabiner, E.A.; Passchier, J.; Gunn, R.N. Positron emission tomography molecular imaging for drug development. Br. Clin. Pharmacol. 2012, 73, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.; Szigeti, K.; Mathe, D.; Metello, L.F. The role of molecular imaging in modern drug development. Drug Discov. Today 2014, 19, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Benz, M.R.; Herrmann, K.; Walter, F.; Garon, E.B.; Reckamp, K.L.; Figlin, R.; Phelps, M.E.; Weber, W.A.; Czernin, J.; Allen-Auerbach, M.S. (18F)-FDG PET/CT for monitoring treatment responses to the epidermal growth factor receptor inhibitor erlotinib. J. Nucl. Med. 2011, 52, 1689–1689. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, M.H.; Aukema, T.S.; Hartemink, K.J.; Valdes Olmos, R.A.; van Tinteren, H.; Klomp, H.M. FDG-PET/CT response evaluation during EGFR-TKI treatment in patients with NSCLC. World J. Radiol. 2014, 6, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Caldarella, C.; Muoio, B.; Isgro, M.A.; Porfiri, E.; Treglia, G.; Giovanella, L. The role of fluorine-18-fluorodeoxyglucose positron emission tomography in evaluating the response to tyrosine-kinase inhibitors in patients with metastatic primary renal cell carcinoma. Radiol. Oncol. 2014, 48, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Aboagye, E. Development of radiotracers for oncology—The interface with pharmacology. Br. J. Pharmacol. 2011, 163, 1565–1585. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Poot, A.J.; Windhorst, A.D.; van Dongen, G.A.M.S. PET imaging with small-molecule tyrosine kinase inhibitors: TKI-PET. Drug Discov. Today 2012, 17, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.D.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Van de Bittner, G.C.; Ricq, E.L.; Hooker, J.M. A philosophy for CNS radiotracer design. Acc. Chem. Res. 2014, 47, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.W.; VanBrocklin, H.F.; Wilson, A.A.; Houle, S.; Vasdev, N. Radiolabeled small molecule protein kinase inhibitors for imaging with PET or SPECT. Molecules 2010, 15, 8260–8278. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, G.K.; Zheng, Q.H.; Winkle, W.L.; Carlson, K.A. Synthesis and biodistribution of new C-11 and F-18 labeled epidermal growth factor receptor ligands. J. Nucl. Med. 1997, 38, 529. [Google Scholar]
- DeJesus, O.T.; Murali, D.; Flores, L.G.; Converse, A.K.; Dick, D.W.; Oakes, T.R.; Roberts, A.D.; Nickles, R.J. Synthesis of [18F]-ZD1839 as a PET imaging agent for epidermal growth factor receptors. J. Label. Compd. Radiopharm. 2003, 46, S1–S9. [Google Scholar]
- Johnstrom, P.; Fredriksson, A.; Thorell, J.O.; Stone-Elander, S. Synthesis of [methoxy-11C]PD153035, a selective EGF receptor tyrosine kinase inhibitor. J. Label. Compd. Radiopharm. 1998, 41, 623–629. [Google Scholar] [CrossRef]
- Fredriksson, A.; Johnstrom, P.; Thorell, J.O.; von Heijne, G.; Hassan, M.; Eksborg, S.; Kogner, P.; Borgstrom, P.; Ingvar, M.; Stone-Elander, S. In vivo evaluation of the biodistribution of 11C-labeled PD153035 in rats without and with neuroblastoma implants. Life Sci. 1999, 65, 165–174. [Google Scholar] [CrossRef]
- Samén, E.; Thorell, J.-O.; Fredriksson, A.; Stone-Elander, S. The tyrosine kinase inhibitor PD153035: Implication of labeling position on radiometabolites formed in vitro. Nucl. Med. Biol. 2006, 33, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, U.; Tochon-Danguy, H.J.; Scott, A.M. [11C]AG1478—A potential PET tracer for the molecular imaging of epidermal growth factor receptor (EGFR). J. Nucl. Med. 2004, 45, 112P. [Google Scholar]
- Owino, N.O.; Shao, X.; Snyder, S.E. Preparation of a reversible EGFR inhibitor, [11C]CIRC, as a PET radiotracer for tumor imaging. J. Label. Compd. Radiopharm. 2005, 48, S183. [Google Scholar] [CrossRef]
- Snyder, S.E.; Sherman, P.S.; Blair, J.B. 4-(3-chloro-4-[18F]fluorophenylamino)-6,7-dimethoxyquinazoline: A radiolabeled EGF receptor inhibitor for imaging tumor biochemistry with PET. J. Nucl. Med. 2000, 41, 233P. [Google Scholar]
- Bonasera, T.A.; Ortu, G.; Rozen, Y.; Krais, R.; Freedman, N.M.T.; Chisin, R.; Gazit, A.; Levitzki, A.; Mishani, E. Potential 18F-labeled biomarkers for epidermal growth factor receptor tyrosine kinase. Nucl. Med. Biol. 2001, 28, 359–374. [Google Scholar] [CrossRef]
- Dorff, P.N.; Vasdev, N.; O’Neil, J.P.; Nandanan, E.; Gibbs, A.R.; VanBrocklin, H.F. Synthesis of 2′-, 3′-, and 4′-[18F]fluoroanilinoquinazoline. J. Label. Compd. Radiopharm. 2003, 46, S117. [Google Scholar] [CrossRef]
- Ben-David, I.; Rozen, Y.; Ortu, G.; Mishani, E. Radiosynthesis of ML03, a novel positron emission tomography biomarker for targeting epidermal growth factor receptor via the labeling synthon: [11C]acryloyl chloride. Appl. Radiat. Isot. 2003, 58, 209–217. [Google Scholar] [CrossRef]
- Vasdev, N.; Dorff, P.N.; Gibbs, A.R.; Nandanan, E.; Reid, L.M.; O’Neill, J.P.; VanBrocklin, H.F. Synthesis of 6-acrylamido-4-(2-[18F]fluoroanilino)quinazoline: A prospective irreversible EGFR binding probe. J. Label. Compd. Radiopharm. 2005, 48, 109–115. [Google Scholar] [CrossRef]
- Vasdev, N.; Dorff, P.N.; Gibbs, A.R.; Nandanan, E.; Reid, L.M.; O’Neil, J.P.; VanBrocklin, H.F. Synthesis of 6-acrylamido-4-(2-[18F]fluoro-anilino)quinazoline. J. Nucl. Med. 2004, 45, 168P. [Google Scholar]
- Waldherr, C.; Satyamurthy, N.; Toyokuni, T.; Wang, S.; Mellinghoff, I.; Tran, C.; Stout, D.; Halpern, B.; Silverman, D.H.; Barrio, J.R.; Phelps, M.E.; Sawyers, C.L.; Czernin, J. Evaluation of N-{4-[(3′-[18F]fluoroethylphenyl)amino]-6-quinazolinyl} acrylamide ([18F]FEQA), a labeled tyrosine kinase inhibitor, for imaging epidermal growth factor receptor density. J. Nucl. Med. 2003, 44, 372P. [Google Scholar]
- Abourbeh, G.; Dissoki, S.; Jacobson, O.; Litchi, A.; Ben Daniel, R.; Laki, D.; Levitzki, A.; Mishani, E. Evaluation of radiolabeled ML04, a putative irreversible inhibitor of epidermal growth factor receptor, as a bioprobe for PET imaging of EGFR-overexpressing tumors. Nucl. Med. Biol. 2007, 34, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Mishani, E.; Abourbeh, G.; Rozen, Y. Novel carbon-11 labeled 4-dimethylamino-but-2-enoic acid [4-(phenylamino)-quinazoline-6-yl]-amides: Potential PET bioprobes for molecular imaging of EGFR-positive tumors. Nucl. Med. Biol. 2004, 31, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Dissoki, S.; Eshet, R.; Billauer, H.; Mishani, E. Modified PEG-anilinoquinazoline derivatives as potential EGFR PET agents. J. Label. Compd. Radiopharm. 2009, 52, 41–52. [Google Scholar] [CrossRef]
- Pantaleo, M.; Mishani, E.; Nanni, C.; Landuzzi, L.; Boschi, S.; Nicoletti, G.; Dissoki, S.; Paterini, P.; Piccaluga, P.; Lodi, F.; et al. Evaluation of modified Evaluation of modified PEG-anilinoquinazoline derivatives as potential agents for EGFR imaging in cancer by small animal PET. Mol. Imaging Biol. 2010, 12, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Dissoki, S.; Aviv, Y.; Laky, D.; Abourbeh, G.; Levitzki, A.; Mishani, E. The effect of the [18F]- PEG group on tracer qualification of [4-(phenylamino)-quinazoline-6-yl]-amide moiety—An EGFR putative irreversible inhibitor. Appl. Radiat. Isot. 2007, 65, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Kobus, D.; Giesen, Y.; Ullrich, R.; Backes, H.; Neumaier, B. A fully automated two-step synthesis of an 18F-labelled tyrosine kinase inhibitor for EGFR kinase activity imaging in tumors. Appl. Radiat. Isot. 2009, 67, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- VanBrocklin, H.F.; Vasdev, N.; Dorff, P.N.; O’Neil, J.P.; Taylor, S.E. Metabolism of [18F]fluoroanilinoquinazolines by human hepatocytes. J. Label. Compd. Radiopharm. 2003, 46, S390. [Google Scholar] [CrossRef]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Seimbille, Y.; Phelps, M.E.; Czernin, J.; Silverman, D.H.S. Fluoriue-18 labeling of 6,7-disubstituted anilinoquinazoline derivatives for positron emission tomography (PET) imaging of tyrosine kinase receptors: Synthesis of F-18-Iressa and related molecular probes. J. Label. Comp. Radiopharm. 2005, 48, 829–843. [Google Scholar] [CrossRef]
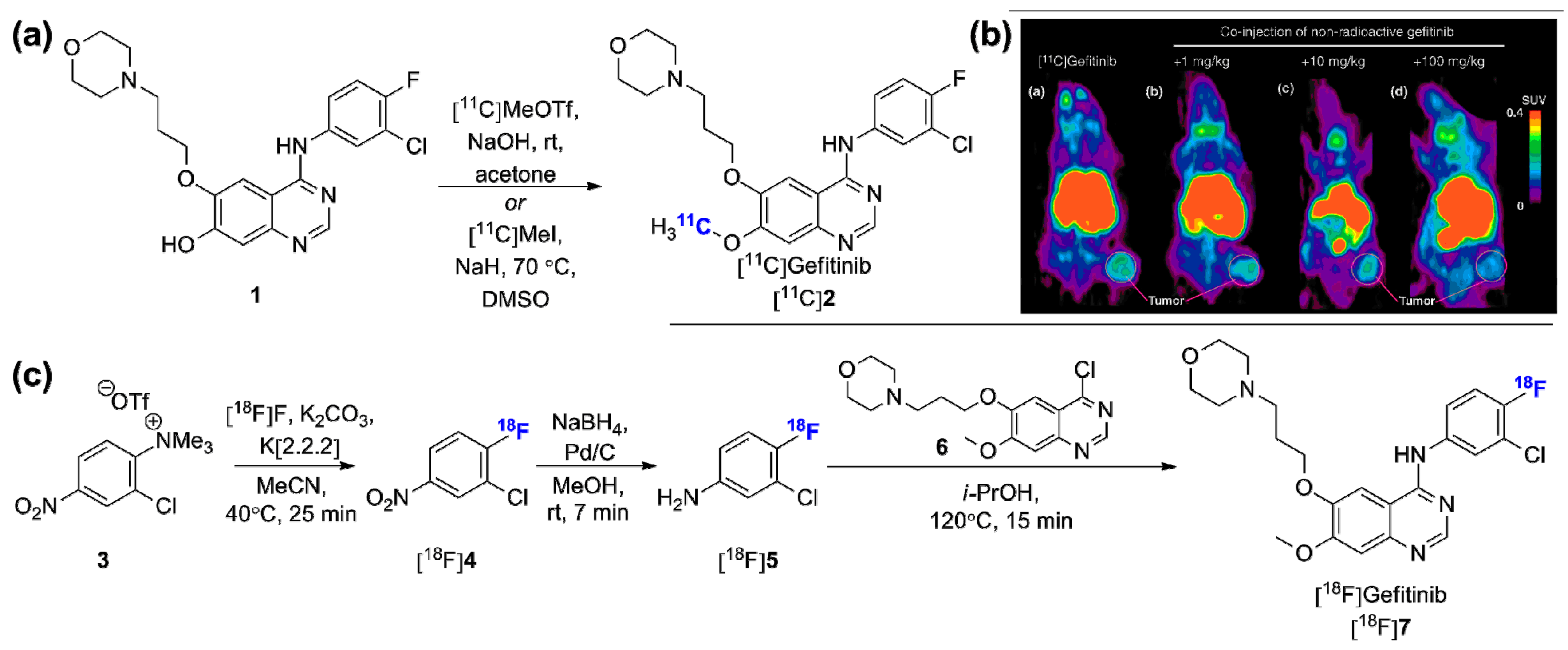
- Su, H.; Seimbille, Y.; Ferl, G.Z.; Bodenstein, C.; Fueger, B.; Kim, K.J.; Hsu, Y.T.; Dubinett, S.M.; Phelps, M.E.; Czernin, J.; et al. Evaluation of F-18 Gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur. J. Nucl. Mol. Imaging 2008, 35, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Gao, M.Z.; Miller, K.D.; Sledge, G.W.; Zheng, Q.H. Synthesis of [C-11]Iressa as a new potential PET cancer imaging agent for epidermal growth factor receptor tyrosine kinase. Bioorg. Med. Chem. 2006, 16, 4102–4106. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.P.; Ravert, H.T.; Dannals, R.F.; Pomper, M.G. Synthesis of [C-11]Gefitinib for imaging epidermal growth factor receptor tyrosine kinase with positron emission tomography. J. Label. Compd. Radiopharm. 2006, 49, 883–888. [Google Scholar] [CrossRef]
- Zhang, M.R.; Kumata, K.; Hatori, A.; Takai, N.; Toyohara, J.; Yamasaki, T.; Yanamoto, K.; Yui, J.; Kawamura, K.; Koike, S.; Ando, K.; Suzuki, K. [11C]Gefitinib ([11C]Iressa): Radiosynthesis, in vitro uptake, and in vivo imaging of intact murine fibrosarcoma. Mol. Imaging Biol. 2010, 12, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Lappchen, T.; Vlaming, M.L.; Custers, E.; Lub, J.; Sio, C.F.; DeGroot, J.; Steinbach, O.C. Automated synthesis of [18F]Gefitinib on a modular system. Appl. Radiat. Isot. 2012, 70, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Elkind, N.B.; Szentpetery, Z.; Apari, A.; Ozvegy-Laczka, C.; Varady, G.; Ujhelly, O.; Szabó, K.; Homolya, L.; Váradi, A.; Buday, L.; et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, gefitinib). Cancer Res. 2005, 65, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
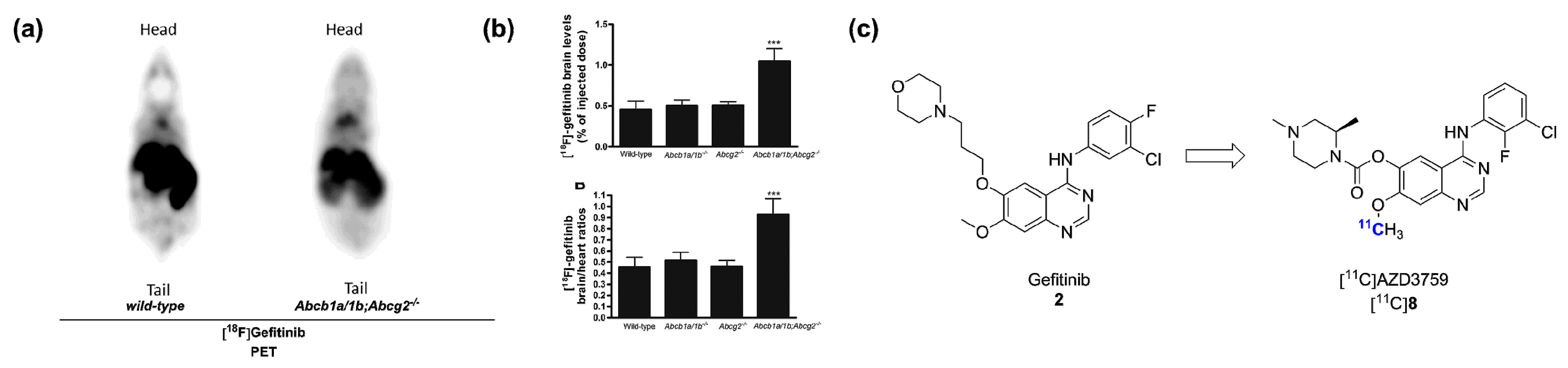
- Zeng, Q.; Wang, J.; Cheng, Z.; Chen, K.; Johnstrom, P.; Varnas, K.; Li, D.Y.; Yang, Z.F.; Zhang, X. Discovery and evaluation of clinical candidate AZD3759, a potent, oral active, central nervous system-penetrant, epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI). J. Med. Chem. 2015. [Google Scholar] [CrossRef]
- Kawamura, K.; Yamasaki, T.; Yui, J.; Hatori, A.; Konno, F.; Kumata, K.; Irie, T.; Fukumura, T.; Suzuki, K.; Kanno, I.; et al. In vivo evaluation of p-glycoprotein and breast cancer resistance protein modulation in the brain using [11C]Gefitinib. Nucl. Med. Biol. 2009, 36, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, M.L.H.; Lappchen, T.; Jansen, H.T.; Kivits, S.; van Driel, A.; van de Steeg, E.; van der Hoorn, J.W.; Sio, C.F.; Steinbach, O.C.; DeGroot, J. PET-ET imaging with [18F]-gefitinib to measure Abcb1a/1b (P-gp) and Abcg2 (Bcrp1) mediated drug-drug interaction at the murine blood-brain barrier. Nucl. Med. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02228369 (accessed on 3 October 2015).
- Dowell, J.; Minna, J.D.; Kirkpatrick, P. Erlotinib hydrochloride. Nat. Rev. Drug Discov. 2005, 4, 13–14. [Google Scholar] [CrossRef] [PubMed]
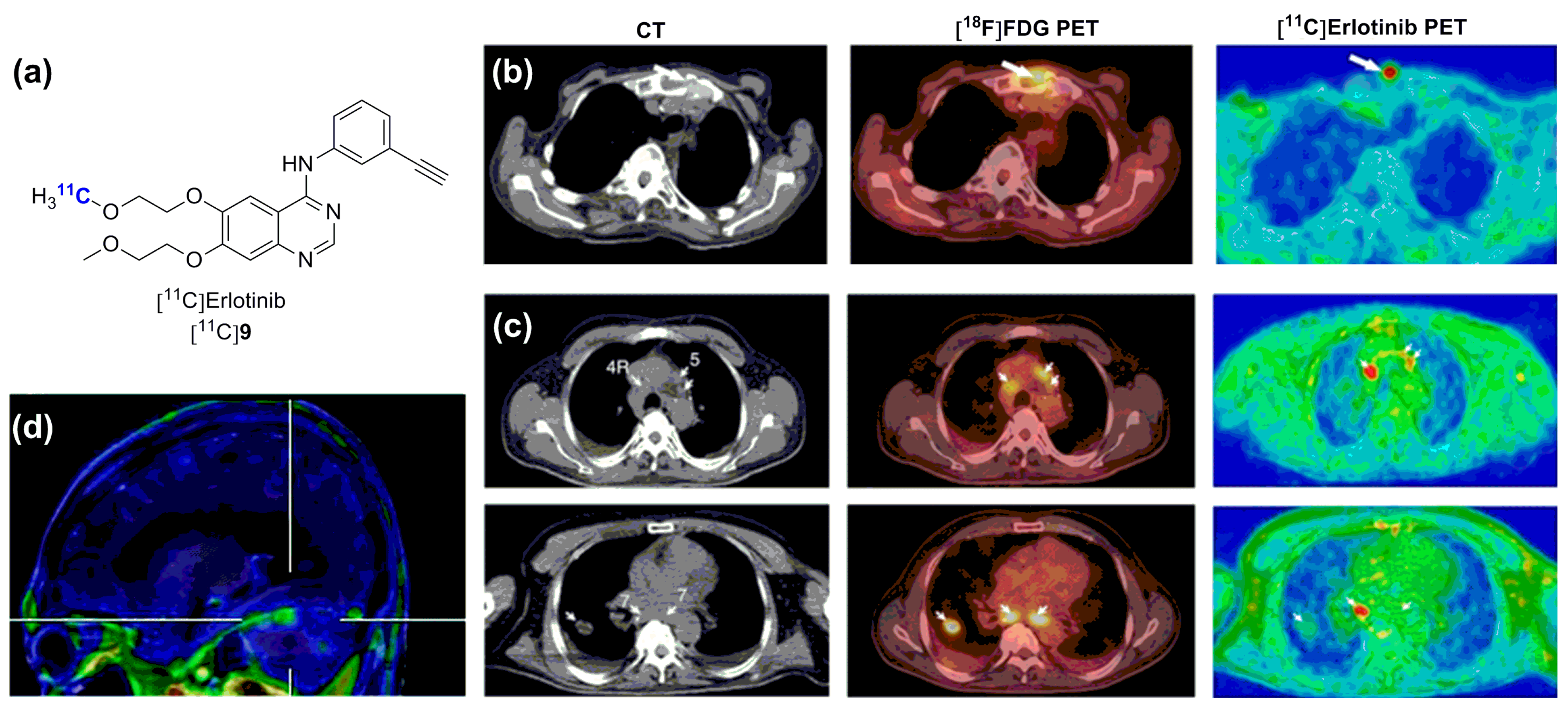
- Memon, A.A.; Jakobsen, S.; Dagnaes-Hansen, F.; Sorensen, B.S.; Keiding, S.; Nexo, E. Positron emission tomography (PET) imaging with C-11 -labeled Erlotinib: A micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009, 69, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Petrulli, J.R.; Sullivan, J.M.; Zheng, M.Q.; Bennett, D.C.; Charest, J.; Huang, Y.; Morris, E.D.; Contessa, J.N. Quantitative analysis of [11C]-Erlotinib PET demonstrates specific binding for activating mutations of the EGFR kinase domain. Neoplasia 2013, 15, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Abourbeh, G.; Itamar, B.; Salnikov, O.; Beltsov, S.; Mishani, E. Identifying Erlotinib-sensitive non-small cell lung carcinoma tumors in mice using [11C]Erlotinib PET. EJNMMI Res. 2015, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Memon, A.A.; Weber, B.; Winterdahl, M.; Jakobsen, S.; Meldgaard, P.; Madsen, H.H.T.; Keiding, S.; Nexo, E.; Sorensen, B.S. PET imaging of patients with non-small cell lung cancer employing an egf receptor targeting drug as tracer. Br. J. Cancer 2011, 105, 1850–1855. [Google Scholar] [CrossRef] [PubMed]
- Bahce, I.; Smit, E.F.; Lubberink, M.; van der Veldt, A.A.M.; Yaqub, M.; Windhorst, A.D.; Schuit, R.C.; Thunnissen, E.; Heideman, D.A.M.; Postmus, P.E.; et al. Development of [11C]erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutation status. Clin. Cancr Res. 2013, 19, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Winterdahl, M.; Memon, A.; Sorensen, B.S.; Keiding, S.; Sorensen, L.; Nexo, E.; Meldgaard, P. Erlotinib accumulation in brain metastases from non-small cell lung cancer: Visualization by positron emission tomography in a patient harboring a mutation in the epidermal growth factor receptor. J. Thorac. Oncol. 2011, 6, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Traxl, A.; Wanek, T.; Mairinger, S.; Stanek, J.; Filip, T.; Sauberer, M.; Muller, M.; Kuntner, C.; Langer, O. Breast cancer resistance protein and p-glycoprotein influence in vivo disposition of 11C-erlotinib. J. Nucl. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
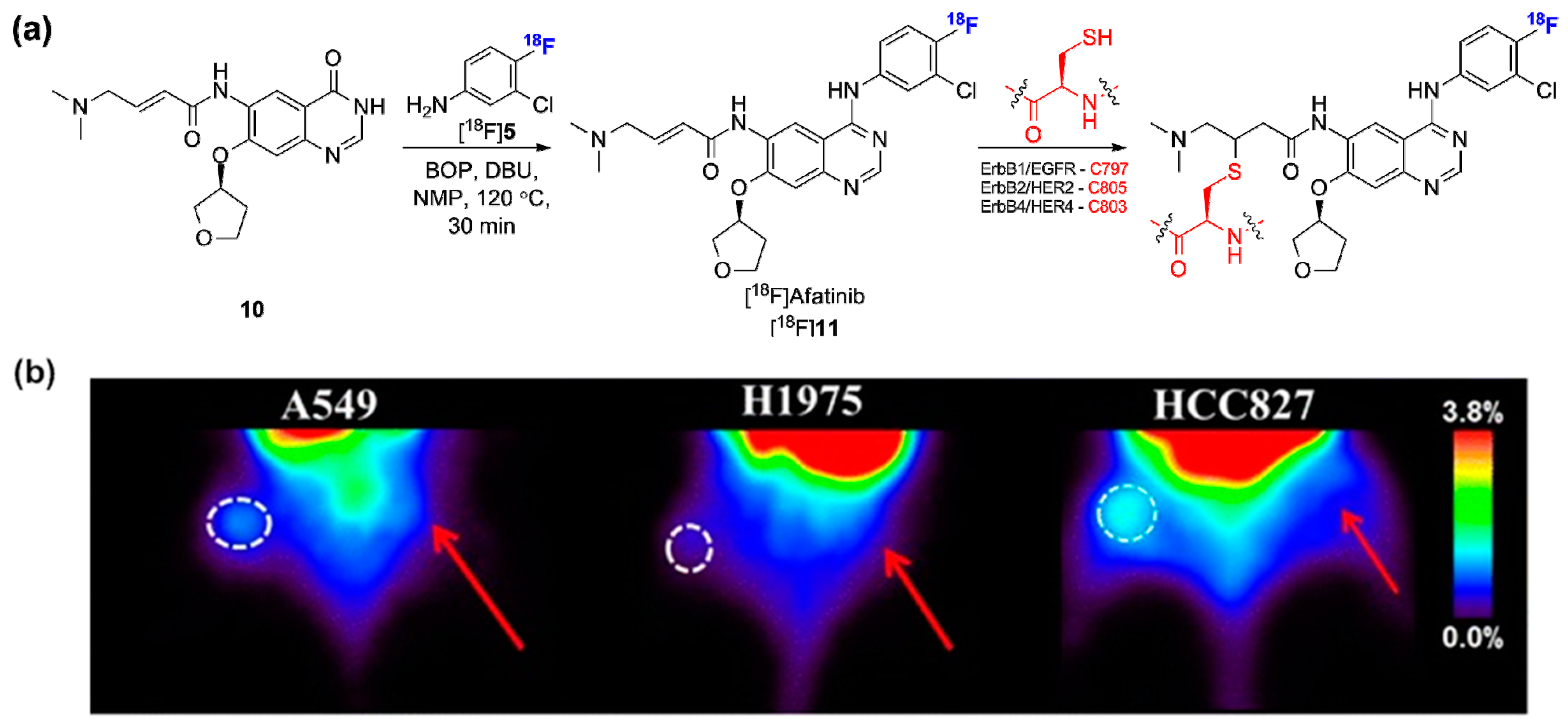
- Slobbe, P.; Windhorst, A.D.; Stigter-van Walsum, M.; Smit, E.F.; Niessen, H.G.; Solca, F.; Stehle, G.; van Dongen, G.A.; Poot, A.J. A comparative PET imaging study with the reversible and irreversible EGFR tyrosine kinase inhibitors [11C]Erlotinib and [18F]Afatinib in lung cancer-bearing mice. EJNMMI Res. 2015, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Windhorst, A.D.; Stigter-van Walsum, M.; Schuit, R.C.; Smit, E.F.; Niessen, H.G.; Solca, F.; Stehle, G.; van Dongen, G.A.; Poot, A.J. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl. Med. Biol. 2014, 41, 749–757. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) Assessment Report for Giotrif (afatinib). 16 October 2013. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002280/WC500152392.pdf (accessed on 20 November 2013).
- Copeland, R.A. The dynamics of drug-target interactions: Drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Discov. 2010, 5, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [PubMed]
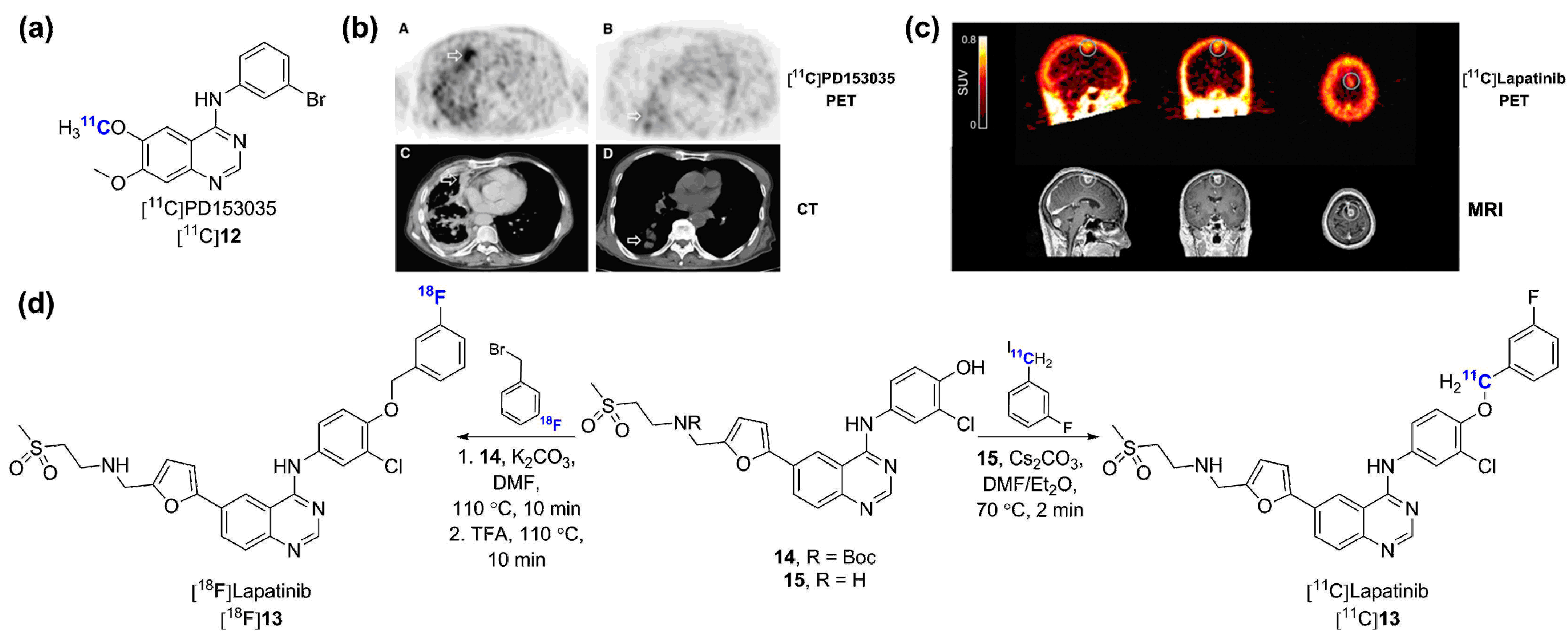
- Bos, M.; Mendelsohn, J.; Kim, Y.M.; Albanell, J.; Fry, D.W.; Baselga, J. PD153035, a tyrosine kinase inhibitor, prevents epidermal growth factor receptor activation and inhibits growth of cancer cells in a receptor number-dependent manner. Clin. Cancer Res. 1997, 3, 2099–2106. [Google Scholar] [PubMed]
- Sasaki, T.; Ishii, S.I.; Senda, M.; Akinaga, S.; Murakata, C. Synthesis of [7β−methoxy 11C]methoxy staurosporine for imaging protein kinase C localization in the brain. Appl. Radiat. Isot. 1996, 47, 67–69. [Google Scholar] [CrossRef]
- Wang, H.; Yu, J.M.; Yang, G.; Song, X.; Sun, X.; Zhao, S.; Mu, D. Assessment of 11C-labeled-4-N-(3-bromoanilino)-6,7-dimethoxyquinazoline as a positron emission tomography agent to monitor epidermal growth factor receptor expression. Cancer Sci. 2007, 9, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, J.M.; Yang, G.R.; Song, X.R.; Sun, X.R.; Zhao, S.Q.; Wang, X.W.; Zhao, W. Further characterization of the epidermal growth factor receptor ligand 11C-PD153035. Chin. Med. J. 2007, 120, 960–964. [Google Scholar] [PubMed]
- Meng, X.; Yu, J.M.; Yang, G.R.; Zhao, S.Q.; Sun, X.D.l. 11C-PD153035 PET/CT for molecular imaging of EGFR in patients with non-small cell lung cancer (NSCLC) [abstract]. J. Clin. Oncol. 2008, 26. [Google Scholar]
- Liu, N.; Li, M.; Li, X.; Meng, X.; Yang, G.; Zhao, S.; Yang, Y.; Ma, L.; Fu, Z.; Yu, J. PET-based biodistribution and radiation dosimetry of epidermal growth factor receptor-selective tracer 11C-PD153035 in humans. J. Nucl. Med. 2009, 50, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Loo, B.W., Jr.; Ma, L.; Murphy, J.D.; Sun, X.; Yu, J. Molecular imaging with 11C-PD153035 PET/CT predicts survival in non-small cell lung cancer treated with EGFR-TKI: A pilot study. J. Nucl. Med. 2011, 52, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Samen, E.; Arnberg, F.; Lu, L.; Olofsson, M.H.; Tegnebratt, T.; Thorell, J.O.; Holmin, S.; Stone-Elander, S. Metabolism of epidermal growth factor receptor targeting probe [11C]PD153035: Impact on biodistribution and tumor uptake in rats. J. Nucl. Med. 2013, 54, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cai, L.; Zhang, K.; Zhang, A.; Pu, P.; Yang, W.; Gao, S. A pilot study on EGFR-targeted molecular imaging of PET/CT with tracer 11C-PD153035 in human gliomas. Clin. Nucl. Med. 2014, 39, e20–e26. [Google Scholar] [CrossRef] [PubMed]
- Basuli, F.; Wu, H.; Li, C.; Shi, Z.-D.; Sulima, A.; Griffiths, G.L. A first synthesis of 18F-radiolabeled Lapatinib: A potential tracer for positron emission tomographic imaging of erbb1/erbb2 tyrosine kinase activity. J. Label. Compd. Radiopharm. 2011, 54, 633–636. [Google Scholar] [CrossRef]
- Saleem, A.; Searle, G.E.; Kenny, L.M.; Huiban, M.; Kozlowski, K.; Waldman, A.D.; Woodley, L.; Palmieri, C.; Lowdell, C.; Kaneko, T.; et al. Lapatinib access into normal brain and brain metastases in patients with HER-2 overexpressing breast cancer. EJNMMI Res. 2015, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.W.; Humphreys, J.E.; Harmon, K.A.; Castellino, S.; O’Mara, M.J.; Olson, K.L.; John-Williams, L.S.; Koch, K.M.; Serabjit-Singh, C.J. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab. Dispos. 2008, 36, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Pivot, X.; Manikhas, A.; Z’urawski, B.; Chmielowska, E.; Karaszewska, B.; Allerton, R.; Chan, S.; Fabi, A.; Bidoli, P.; Gori, S.; et al. CEREBEL (EGF111438): A Phase III, Randomized, Open-Label Study of Lapatinib Plus Capecitabine Versus Trastuzumab Plus Capecitabine in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2015, 33, 1–9. [Google Scholar] [CrossRef] [PubMed]
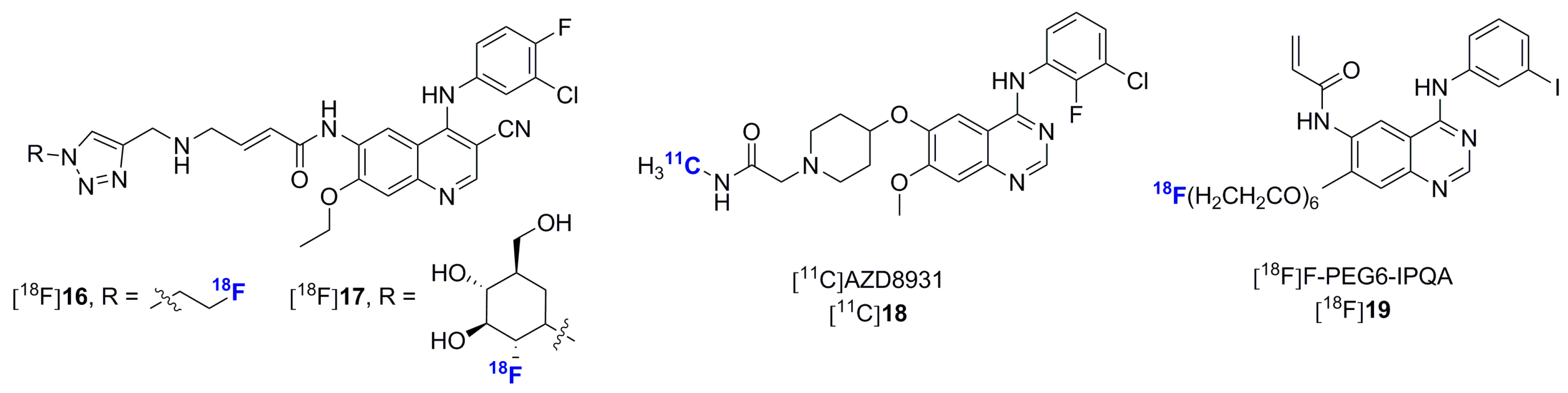
- Pisaneschi, F.; Nguyen, Q.-D.; Shamsaei, E.; Glaser, M.; Robins, E.; Kaliszczak, M.; Smith, G.; Spivey, A.C.; Aboagye, E.O. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010, 18, 6634–6645. [Google Scholar] [CrossRef] [PubMed]
- Pisaneschi, F.; Slade, R.L.; Iddon, L.; George, G.P.; Nguyen, Q.D.; Spivey, A.C.; Aboagye, E.O. Synthesis of a new fluorine-18 glycosylated “click” cyanoquinoline for the imaging of epidermal growth factor receptor. J. Label. Comp. Radiopharm. 2014, 57, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Balatoni, J.; Mukhopadhyay, U.; Ogawa, K.; Gonzalez-Lepera, C.; Shavrin, A.; Volgin, A.; Tong, W.; Alauddin, M.; Gelovani, J. Radiosynthesis and initial in vitro evaluation of [18F]FPEG6-IPQA—A novel PET radiotracer for imaging EGFR expression-activity in lung carcinomas. Mol. Imaging Biol. 2011, 13, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Ogawa, K.; Balatoni, J.; Mukhapadhyay, U.; Pal, A.; Gonzalez-Lepera, C.; Shavrin, A.; Soghomonyan, S.; Flores, L., 2nd; Young, D.; et al. Molecular imaging of active mutant l858R EGF receptor (EGFR) kinase-expressing nonsmall cell lung carcinomas using PET/ct. Proc. Natl. Acad. Sci. USA 2011, 108, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.H.; Lin, C.F.; Kong, F.L.; Wang, H.E.; Hsieh, Y.J.; Gelovani, J.G.; Liu, R.S. Molecular imaging of nonsmall cell lung carcinomas expressing active mutant EGFR kinase using PET with [124I]-morpholino-ipqa. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Zheng, Q.H. The first radiosynthesis of [11C]AZD8931 as a new potential PET agent for imaging of EGFR, HER2 and HER3 signaling. Bioorg. Med. Chem. Lett. 2014, 24, 4455–4459. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, N.; Dorff, P.N.; O’Neil, J.P.; Chin, F.T.; Hanrahan, S.; VanBrocklin, H.F. Metabolic stability of 6,7-dialkoxy-4-(2-, 3- and 4-[18F]fluoroanilino)quinazolines, potential EGFR imaging probes. Bioorg. Med. Chem. 2011, 19, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Medina, O.P.; Pillarsetty, N.; Glekas, A.; Punzalan, B.; Longo, V.; Gonen, M.; Zanzonico, P.; Smith-Jones, P.; Larson, S.M. Optimizing tumor targeting of the lipophilic EGFR-binding radiotracer SKI 243 using a liposomal nanoparticle delivery system. J. Control. Release 2011, 149, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Neto, C.; Fernandes, C.; Oliveira, M.C.; Gano, L.; Mendes, F.; Kniess, T.; Santos, I. Radiohalogenated 4-anilinoquinazoline-based EGFR-TK inhibitors as potential cancer imaging agents. Nucl. Med. Biol. 2012, 39, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Zhang, L.; Xu, S.; Luo, J.; Lu, X.; Zhang, Z.; Xu, T.; Liu, Y.; Tu, Z.; Xu, Y.; et al. Design, synthesis, and biological evaluation of novel conformationally constrained inhibitors targeting epidermal growth factor receptor threonine⁷⁹⁰→methionine⁷⁹⁰ mutant. J. Med. Chem. 2012, 55, 2711–2723. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Huang, Z.; Zheng, C.; Wan, L.; Zhang, L.; Peng, S.; Ding, K.; Ji, H.; Tian, J.; Zhang, Y. Novel hybrids of (phenylsulfonyl)furoxan and anilinopyrimidine as potent and selective epidermal growth factor receptor inhibitors for intervention of non-small-cell lung cancer. J. Med. Chem. 2013, 56, 4738–4748. [Google Scholar] [CrossRef] [PubMed]
- Heald, R.A.; Chan, B.K.; Bryan, C.; Eigenbrot, C.; Yu, C.; Burdick, D.; Hanan, E.J.; Chan, E.; Schaefer, G.; La, H.; et al. Noncovalent Mutant Selective Epidermal Growth Factor Receptor Inhibitors: A Lead Optimization Case History. J. Med. Chem. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Crouthamel, M.-C.; Rominger, D.H.; Gontarek, R.R.; Tummino1, P.J.; Levin, R.A.; King, A.G. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br. J. Cancer 2009, 101, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical Antiangiogenesis and Antitumor Activities of Axitinib (AG-013736), an Oral, Potent, and Selective Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [PubMed]
- Boss, D.S.; Glen, H.; Beijnen, J.H.; Keesen, M.; Morrison, R.; Tait, B.; Copalu, W.; Mazur, A.; Wanders, J.; O’Brien, J.P.; Schellens, J.H.M.; Evans, T.R.J. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; O’Reilly, M.S.; Onn, A.; Ryan, A.J. Vandetanib (ZD6474): An orally available receptor tyrosine kinase inhibitor that selectively targets pathways critical for tumor growth and angiogenesis. Expert Opin. Investig. Drugs 2007, 16, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-U. Polypharmacology—Foe or friend. J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, S.; Zu, Y.; Lee, D.Y.; Li, Z. A tyrosine kinase inhibitor-based high-affinity PET radiopharmaceutical targets vascular endothelial growth factor receptor. J. Nucl. Med. 2014, 55, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Samen, E.; Lu, L.; Mulder, J.; Thorell, J.O.; Damberg, P.; Tegnebratt, T.; Holmgren, L.; Rundqvist, H.; Stone-Elander, S. Visualization of angiogenesis during cancer development in the polyoma middle t breast cancer model: Molecular imaging with (R)-[11C]PAQ. EJNMMI Res. 2014, 4, 17. [Google Scholar] [CrossRef] [PubMed]
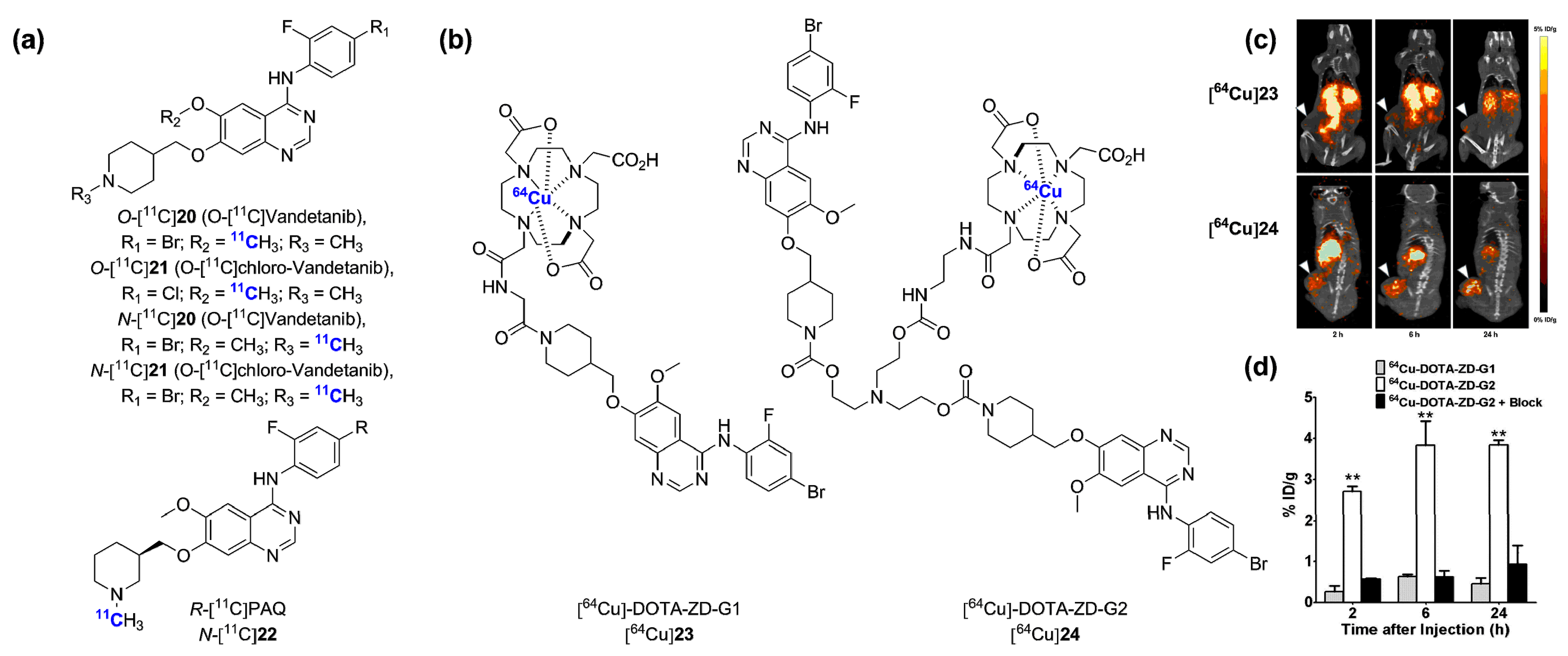
- Gao, M.Z.; Lola, C.M.; Wang, M.; Miller, K.D.; Sledge, G.W.; Zheng, Q.H. Radiosynthesis of C-11 Vandetanib and C-11 chloro-Vandetanib as new potential PET agents for imaging of VEGFR in cancer. Bioorg. Med. Chem. Lett. 2011, 21, 3222–3226. [Google Scholar] [CrossRef] [PubMed]
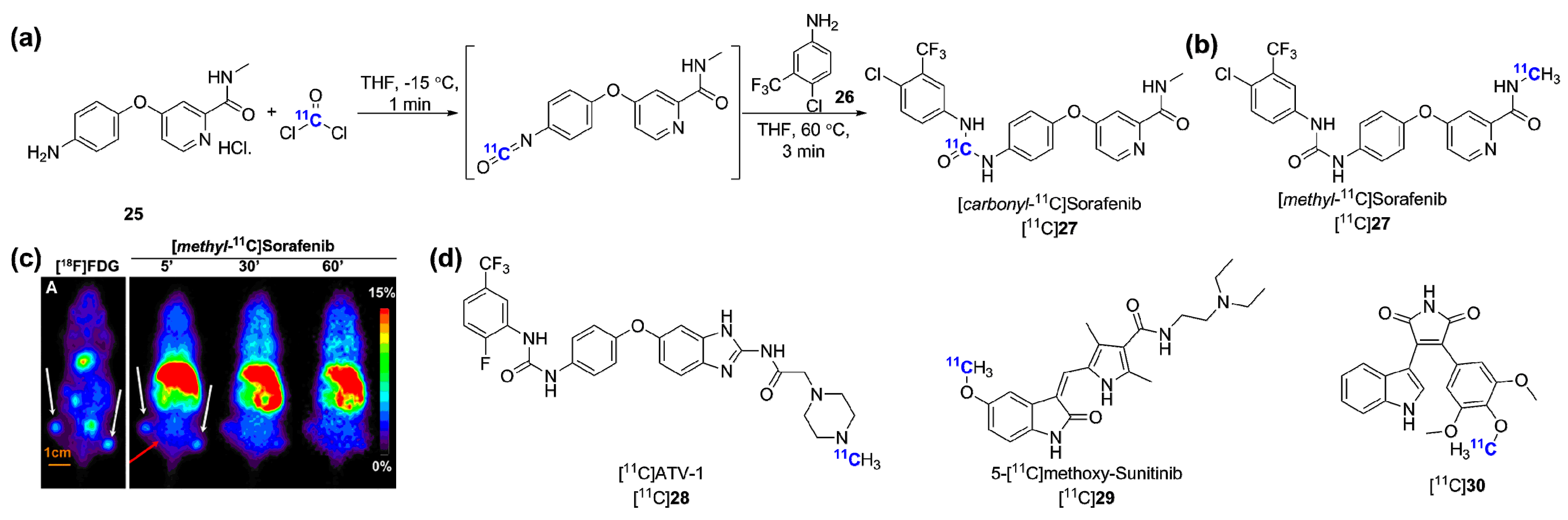
- Asakawa, C.; Ogawa, M.; Kumata, K.; Fujinaga, M.; Kato, K.; Yamasaki, T.; Yui, J.; Kawamura, K.; Hatori, A.; Fukumura, T.; et al. [11C]Sorafenib: Radiosynthesis and preliminary PET study of brain uptake in p-gp/bcrp knockout mice. Bioorg. Med. Chem. Lett. 2011, 21, 2220–2223. [Google Scholar] [CrossRef] [PubMed]
- Poot, A.J.; van der Wildt, B.; Stigter-van Walsum, M.; Rongen, M.; Schuit, R.C.; Hendrikse, N.H.; Eriksson, J.; van Dongen, G.A.; Windhorst, A.D. [11C]Sorafenib: Radiosynthesis and preclinical evaluation in tumor-bearing mice of a new tki-PET tracer. Nucl. Med. Biol. 2013, 40, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Muñoz, C.; Alzate-Morales, J.H.; Cunha, S.; Gano, L.; Bergmann, R.; Steinbach, J.; Kniess, T. Synthesis, in silico, in vitro, and in vivo investigation of 5-[¹¹C]methoxy-substituted sunitinib, a tyrosine kinase inhibitor of VEGFR-2. Eur. J. Med. Chem. 2012, 58, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Windhorst, A.D.; Haumann, R.; Schuit, R.; van Dongen, G.A.; Poot, A.J. Development of an LC-MS method to analyze the primary metabolite of [11C]nintedanib in vivo. J. Label. Compd. Radiopharm. 2015, S30. [Google Scholar] [CrossRef]
- Ilovich, O.; Billauer, H.; Dotan, S.; Mishani, E. Labeled 3-aryl-4-indolylmaleimide derivatives and their potential as angiogenic PET biomarkers. Bioorg. Med. Chem. 2010, 18, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Ilovich, O.; Åberg, O.; Långström, B.; Mishani, E. Rhodium-mediated [11C]carbonylation: A library of n-phenyl-n′-{4-(4-quinolyloxy)-phenyl}-[11c]-urea derivatives as potential pet angiogenic probes. J. Label. Compd. Radiopharm. 2009, 52, 151–157. [Google Scholar] [CrossRef]
- Dissoki, S.; Abourbeh, G.; Salnikov, O.; Mishani, E.; Jacobson, O. PET molecular imaging of angiogenesis with a multiple tyrosine kinase receptor-targeted agent in a rat model of myocardial infarction. Mol. Imaging Biol. 2015, 17, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Santos, I.C.; Santos, I.; Pietzsch, H.-J.; Kunstler, J.-U.; Kraus, W.; Rey, A.; Margaritis, N.; Bourkoula, A.; Chiotellis, A.; et al. Rhenium and technetium complexes bearing quinazoline derivatives: Progress towards a 99mTc biomarker for EGFR-TK imaging. Dalton Trans. 2008, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.T.; Persson, M.; Madsen, J.; Kjær, A. High tumor uptake of 64Cu: implications for molecular imaging of tumor characteristics with copper-based PET tracers. Nucl. Med. Biol. 2013, 40, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; van Waterschoot, R.A.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol. Cancer Ther. 2010, 9, 319–326. [Google Scholar] [CrossRef] [PubMed]
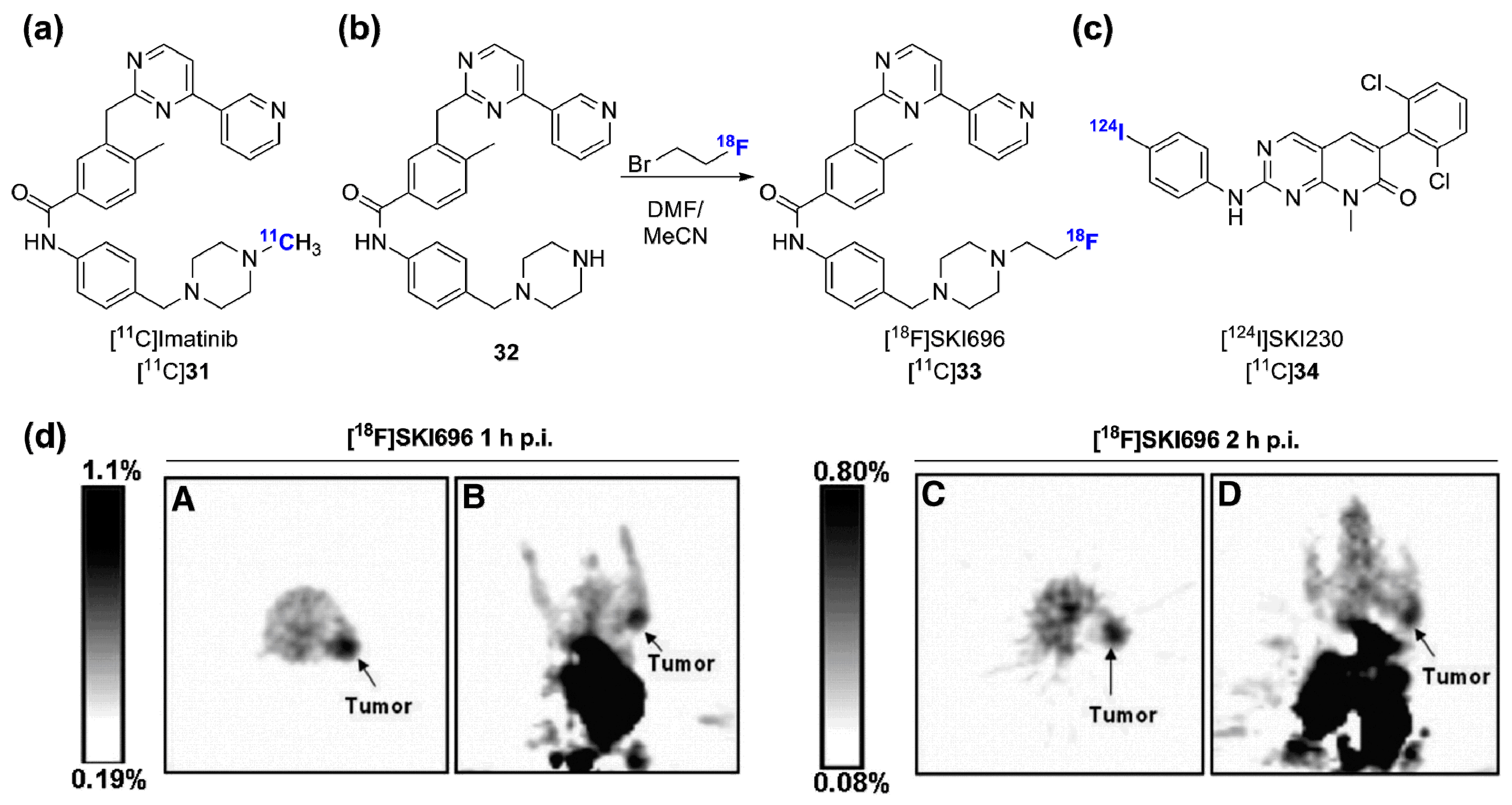
- Kil, K.-E.; Ding, Y.-S.; Lin, K.-S.; Alexoff, D.; Kim, S.W.; Shea, C.; Xu, Y.; Muench, L.; Fowler, J.S. Synthesis and positron emission tomography studies of carbon-11-labeled Imatinib (Gleevec). Nucl. Med. Biol. 2007, 34, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Bornman, W.; Pal, A.; Ghosh, P.; Lim, S.T.; Gelovani, J.; Maxwell, D.; Alauddin, M.M. STI571 analogs: 18F-STI571as potential agents for PET imaging of c-kit expression at a kinase level. In Proceedings of the 233rd ACS National Meeting, Chicago, IL, USA, 25 March 2007.
- Doubrovin, M.; Kochetkova, T.; Santos, E.; Veach, D.R.; Smith-Jones, P.; Pillarsetty, N.; Balatoni, J.; Bornmann, W.; Gelovani, J.; Larson, S.M. (124)i-iodopyridopyrimidinone for PET of abl kinase-expressing tumors in vivo. J. Nucl. Med. 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Glekas, A.P.; Pillarsetty, N.K.; Punzalan, B.; Khan, N.; Smith-Jones, P.; Larson, S.M. In vivo imaging of bcr-abl overexpressing tumors with a radiolabeled Imatinib analog as an imaging surrogate for Imatinib. J. Nucl. Med. 2011, 52, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Maxwell, D.S.; Sun, D.; Bhanu Prasad, B.A.; Pal, A.; Wang, S.; Balatoni, J.; Ghosh, P.; Lim, S.T.; Volgin, A.; et al. Imatinib analogs as potential agents for PET imaging of bcr-abl and c-kit expression at a kinase level. Bioorg. Med. Chem. 2014, 22, 623–632. [Google Scholar] [CrossRef] [PubMed]
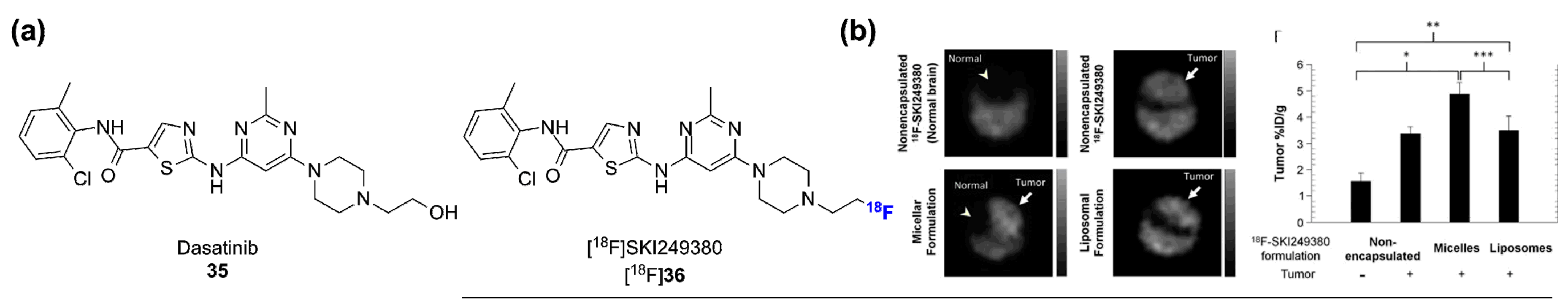
- Veach, D.R.; Namavari, M.; Pillarsetty, N.; Santos, E.B.; Beresten-Kochetkov, T.; Lambek, C.; Punzalan, B.J.; Antczak, C.; Smith-Jones, P.M.; Djaballah, H.; et al. Synthesis and biological evaluation of a fluorine-18 derivative of Dasatinib. J. Med. Chem. 2007, 50, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, M.P.; Zanzonico, P.; Veach, D.; Somwar, R.; Pillarsetty, N.; Lewis, J.; Larson, S. Dosimetry of 18F-labeled tyrosine kinase inhibitor ski-249380, a Dasatinib-tracer for PET imaging. Mol. Imaging Biol. 2012, 14, 25–31. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01916135 (accessed on 17 October 15).
- Benezra, M.; Hambardzumyan, D.; Penate-Medina, O.; Veach, D.R.; Pillarsetty, N.; Smith-Jones, P.; Phillips, E.; Ozawa, T.; Zanzonico, P.B.; Longo, V.; et al. Fluorine-labeled Dasatinib nanoformulations as targeted molecular imaging probes in a pdgfb-driven murine glioblastoma model. Neoplasia 2012, 14, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, R.; Hong, H.; Cai, W. Positron emission tomography image-guided drug delivery: Current status and future perspectives. Mol. Pharm. 2014, 11, 3777–3797. [Google Scholar] [CrossRef] [PubMed]
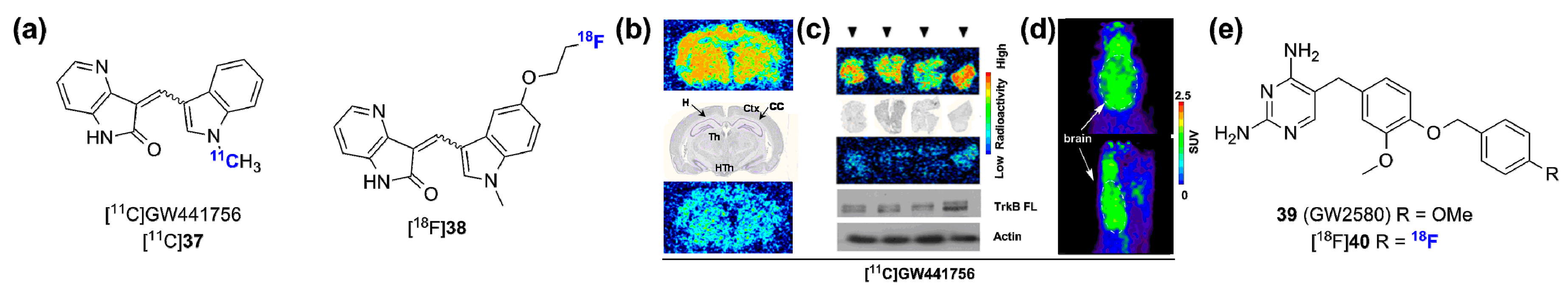
- Bernard-Gauthier, V.; Aliaga, A.; Aliaga, A.; Boudjemeline, M.; Hopewell, R.; Kostikov, A.; Rosa-Neto, P.; Thiel, A.; Schirrmacher, R. Syntheses and evaluation of carbon-11 and fluorine-18-radiolabeled pan-tropomyosin receptor kinase (Trk) inhibitors : Exploration of the 4-aza-2-oxindole scaffold as Trk PET imaging agents. ACS Chem. Neurosci. 2015, 6, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gauthier, V.; Schirrmacher, R. 5-(4-((4-[18F]fluorobenzyl)oxy)-3-methoxybenzyl)pyrimidine-2,4-diamine: A selective dual inhibitor for potential PET imaging of Trk/CSF-1R. Bioorg. Med. Chem. Lett. 2014, 24, 4784–4790. [Google Scholar] [CrossRef] [PubMed]
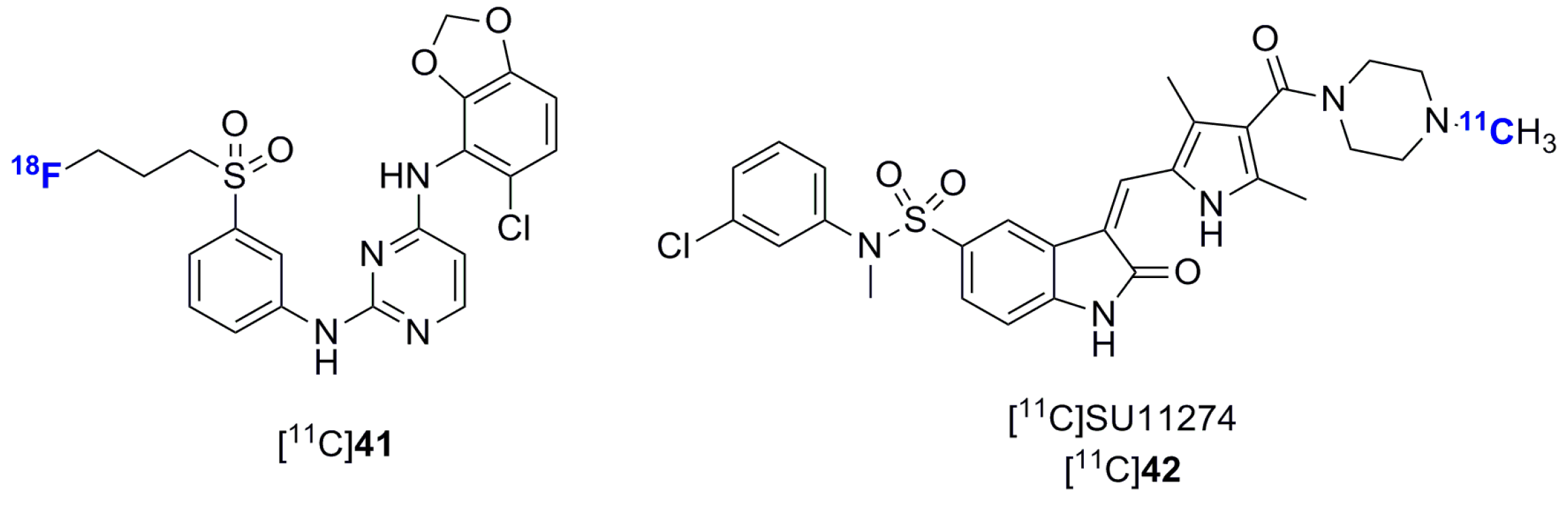
- Wu, C.; Tang, Z.; Fan, W.; Zhu, W.; Wang, C.; Somoza, E.; Owino, N.; Li, R.; Ma, P.C.; Wang, Y. In vivo positron emission tomography (PET) imaging of mesenchymal-epithelial transition (MET) receptor. J. Med. Chem. 2010, 53, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Mamat, C.; Mosch, B.; Neuber, C.; Köckerling, M.; Bergmann, R.; Pietzsch, J. Fluorine-18 radiolabeling and radiopharmacological characterization of a benzodioxolylpyrimidine-based radiotracer targeting the receptor tyrosine kinase EphB4. ChemMedChem 2012, 7, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Neurosci. 2003, 72, 609–642. [Google Scholar]
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015, 5, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Tabernero, J. BRAF as a target for cancer therapy. Anticancer Agents Med. Chem. 2011, 11, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Gavriilidis, P.; Giakoustidis, A.; Giakoustidis, D. Aurora Kinases and Potential Medical Applications of Aurora Kinase Inhibitors: A Review. J. Clin. Med. Res. 2015, 7, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; di Sanza, C.; Incani, U.C.; Milella, M. The mTOR pathway: A new target in cancer therapy. Curr. Cancer Drug Targets 2010, 10, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Arfeen, M.; Bharatam, P.V. Design of glycogen synthase kinase-3 inhibitors: An overview on recent advancements. Curr. Pharm. Des. 2013, 19, 4755–4775. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, N.; LaRonde, F.J.; Woodgett, J.R.; Garcia, A.; Rubie, E.A.; Meyer, J.H.; Houle, S.; Wilson, A.A. Rationally designed PKA inhibitors for positron emission tomography: Synthesis and cerebral biodistribution of N-(2-(4-bromocinnamylamino)ethyl)-N-[11C]methyl-isoquinoline-5-sulfonamide. Bioorg. Med. Chem. 2008, 16, 5277–5284. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D.; Zheng, Q.-H. The first design and synthesis of [11C]MKC-1 ([11C]Ro 31–7453), a new potential PET cancer imaging agent. Nucl. Med. Biol. 2010, 37, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kudo, K.; Okada, M.; Yanamoto, K.; Hatori, A.; Irie, T.; Suzuki, K.; Miura, S. Synthesis and biodistribution of [11C]methyl-bisindolylmareimide III, an inhibitor of protein kinase C. J. Label. Compd. Radiopharm. 2007, 50, S398. [Google Scholar]
- Cai, L.; Ozaki, H.; Fujita, M.; Hong, J.S.; Bukhari, M.; Innis, R.B.; Pike, V.W. [11C]GO6976 as a potential radioligand for imaging protein kinase C with PET. J. Label. Compd. Radiopharm. 2009, 52, S337. [Google Scholar]
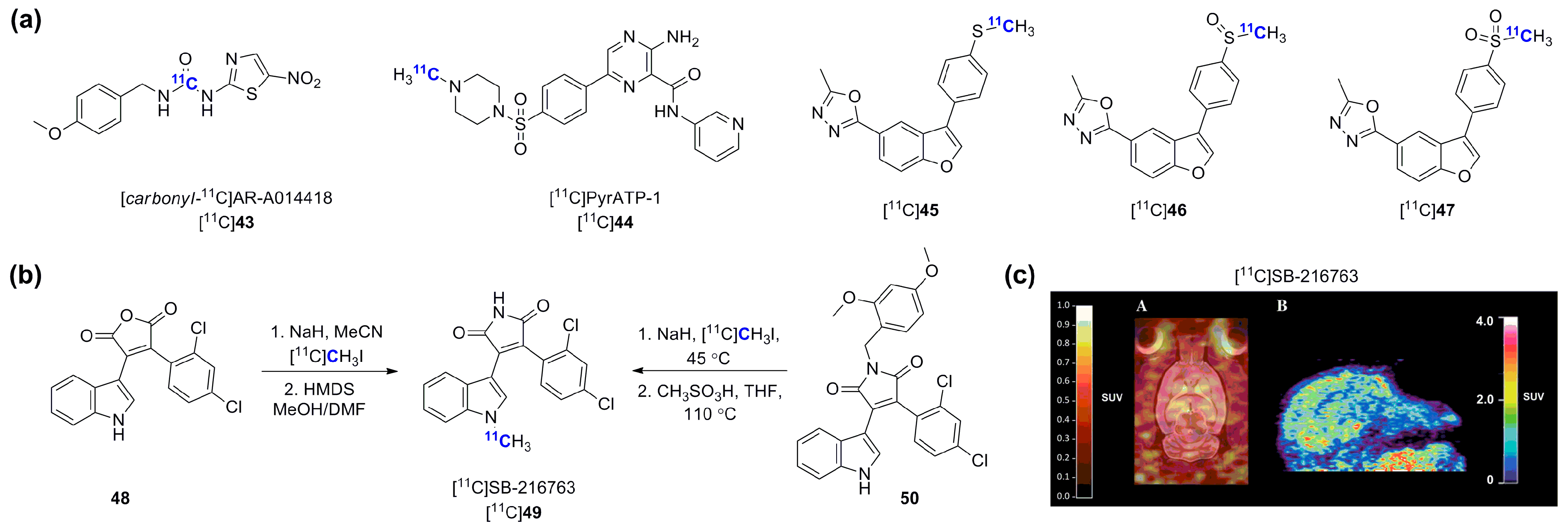
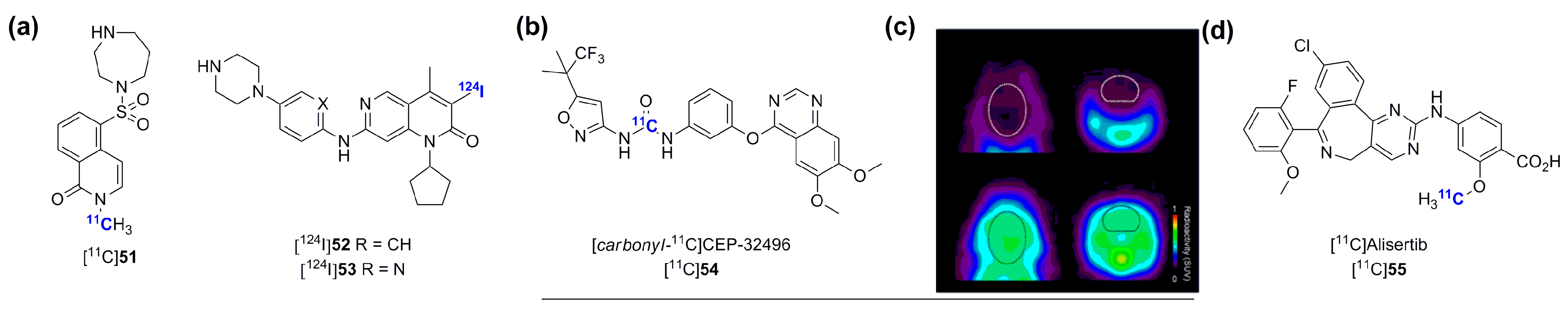
- Vasdev, N.; Garcia, A.; Stableford, W.T.; Young, A.B.; Meyer, J.H.; Houle, S.; Wilson, A.A. Synthesis and ex vivo evaluation of carbon-11 labelled N-(4-methoxybenzyl)-]N′-(5-nitro-1,3-thiazol-2-yl)urea ([C-11]AR-A014418): A radiolabelled glycogen synthase kinase-3 beta specific inhibitor for PET studies. Bioorg. Med. Chem. 2005, 15, 5270–5273. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.W.; Wilson, A.A.; Rubie, E.A.; Woodgett, J.R.; Houle, S.; Vasdev, N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: Radiosynthesis of [11C-carbonyl] AR-A014418 by [11C]CO2 fixation. Bioorg. Med. Chem. Lett. 2012, 22, 2099–2101. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, B.H.; Liang, S.H.; Holland, J.P.; Collier, T.L.; Hooker, J.M.; Wilson, A.A.; Vasdev, N. 11CO2 fixation: A renaissance in PET radiochemistry. Chem. Commun. 2013, 49, 5621–5629. [Google Scholar] [CrossRef] [PubMed]
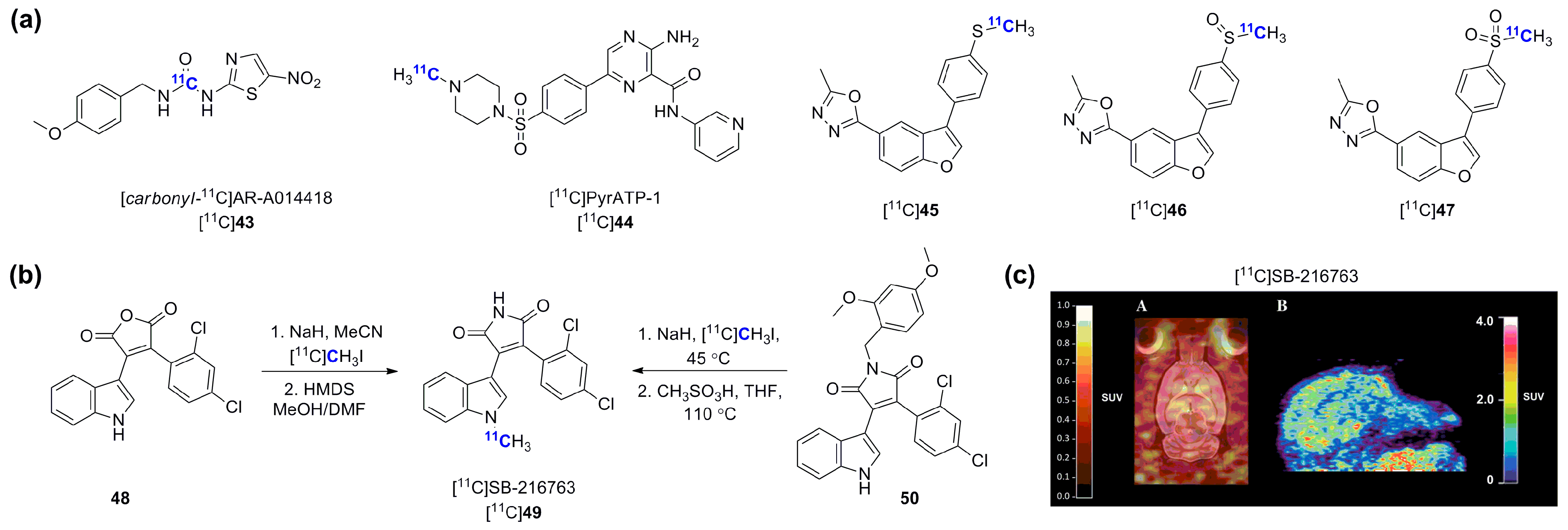
- Cole, E.L.; Shao, X.; Sherman, P.; Quesada, C.; Fawaz, M.V.; Desmond, T.J.; Scott, P.J. Synthesis and evaluation of [11C]pyrATP-1, a novel radiotracer for PET imaging of glycogen synthase kinase-3beta (GSK-3β). Nucl. Med. Biol. 2014, 41, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Kumata, K.; Yui, J.; Xie, L.; Zhang, Y.; Nengaki, N.; Fujinaga, M.; Yamasaki, T.; Shimoda, Y.; Zhang, M.R. Radiosynthesis and preliminary PET evaluation of glycogen synthase kinase 3beta (GSK-3β) inhibitors containing [11C]methylsulfanyl, [11C]methylsulfinyl or [11C]methylsulfonyl groups. Bioorg. Med. Chem. Lett. 2015, 25, 3230–3233. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D.; Zheng, Q.H. The first synthesis of [11C]SB-216763, a new potential PET agent for imaging of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2011, 21, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shao, X.; Cole, E.L.; Ohnmacht, S.A.; Ferrari, V.; Hong, Y.T.; Williamson, D.J.; Fryer, T.D.; Quesada, C.A.; Sherman, P.; Riss, P.J.; et al. Synthesis and initial in vivo studies with [11C]SB-216763: The first radiolabeled brain penetrative inhibitor of GSK-3. ACS Med. Chem. Lett. 2015, 6, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, A.C.; Mason, S.; Collins, J.; Buckley, K.R.; Coletta, P.; Beanlands, R.S.; Dasilva, J.N. Radiosynthesis of N-[11C]-methyl-hydroxyfasudil as a new potential PET radiotracer for rho-kinases (ROCKs). Appl. Radiat. Isot. 2010, 68, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, J.; Seki, C.; Takuwa, H.; Kawaguchi, H.; Ikoma, Y.; Fujinaga, M.; Kanno, I.; Zhang, M.R.; Kuwabara, S.; Ito, H. Evaluation of rho-kinase activity in mice brain using N-[11C]methyl-hydroxyfasudil with positron emission tomography. Mol. Imaging Biol. 2014, 16, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Moreau, S.; DaSilva, J.N.; Valdivia, A.; Fernando, P. N-[11C]-Methyl-hydroxyfasudil is a potential biomarker of cardiac hypertrophy. Nucl. Med. Biol. 2015, 42, 192–197. [Google Scholar] [CrossRef] [PubMed]
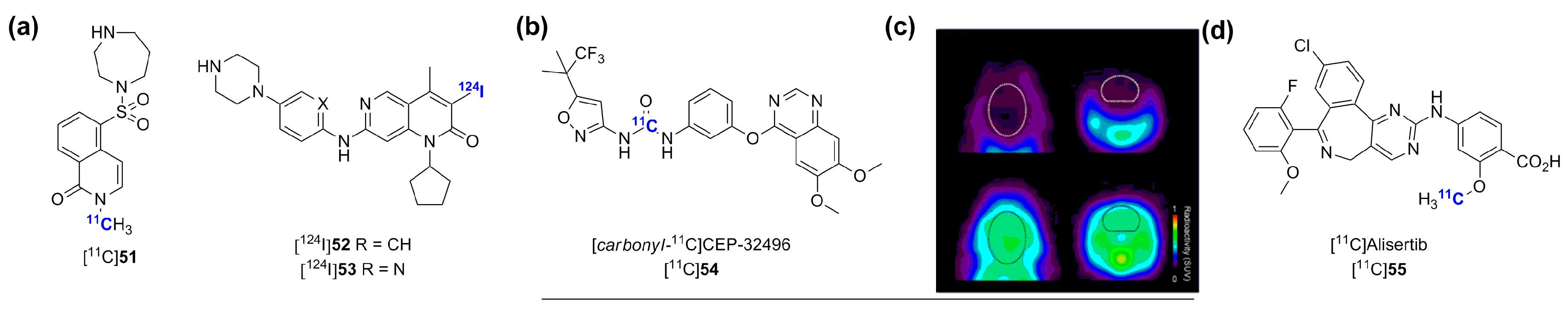
- Koehler, L.; Graf, F.; Bergmann, R.; Steinbach, J.; Pietzsch, J.; Wuest, F. Radiosynthesis and radiopharmacological evaluation of cyclin-dependent kinase 4 (CDK4) inhibitors. Eur. J. Med. Chem. 2010, 45, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, Y.; Yui, J.; Fujinaga, M.; Xie, L.; Kumata, K.; Ogawa, M.; Yamasaki, T.; Hatori, A.; Kawamura, K.; Zhang, M.R. [11C-carbonyl]CEP-32496: Radiosynthesis, biodistribution and PET study of brain uptake in p-gp/bcrp knockout mice. Bioorg. Med. Chem. Lett. 2014, 24, 3574–3577. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Windhorst, A.D.; van Dongen, G.A.; Poot, A.J. Synthesis of [11C]vemurafenib via a unique [11C]CO carbonylative Stille coupling to image V600E mutated B-Raf in cancer. J. Label. Compd. Radiopharm. 2015, 58, S281. [Google Scholar] [CrossRef] [Green Version]
- Goos, J.A.; Verbeek, J.; Geldof, A.A.; Hiemstra, A.C.; van de Wiel, M.A.; Adamzek, K.A.; Delis-Van Diemen, P.M.; Stroud, S.G.; Bradley, D.P.; Meijer, G.A.; et al. Molecular imaging of aurora kinase A (AURKA) expression: Synthesis and preclinical evaluation of radiolabeled alisertib (MLN8237). Nucl. Med. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
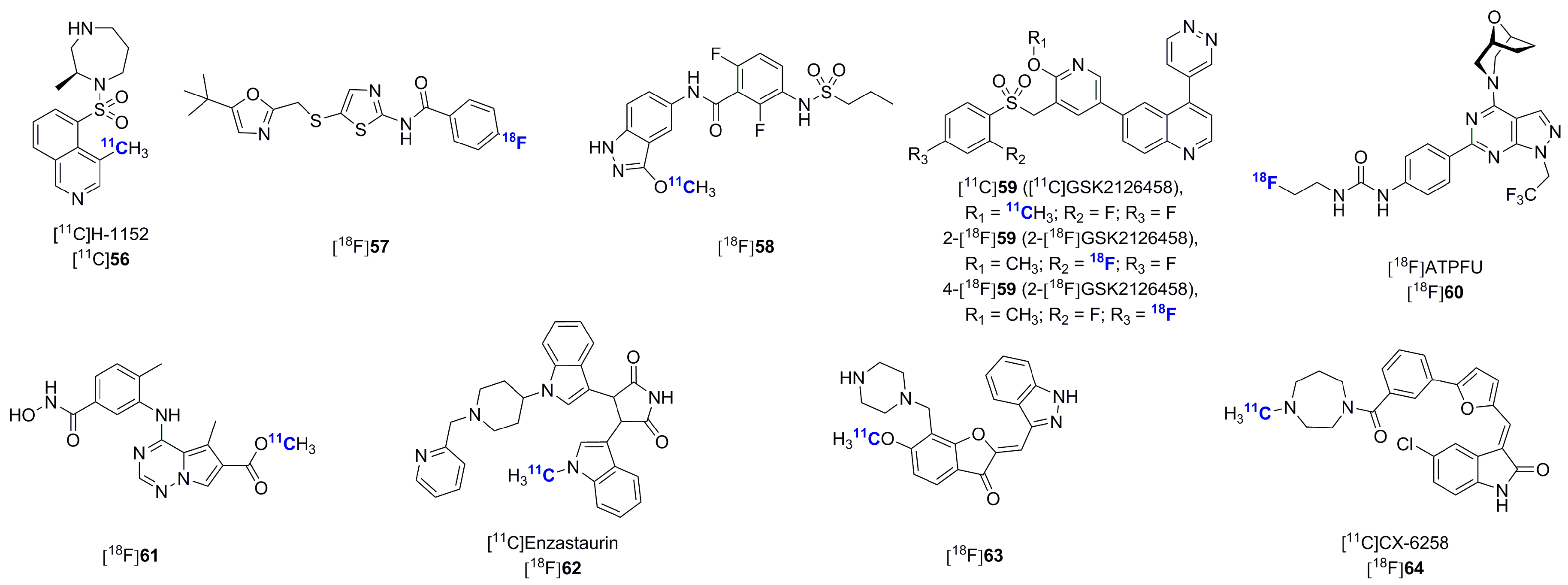
- Suzuki, M.; Takashima-Hirano, M.; Koyama, H.; Yamaoka, T.; Sumi, K.; Nagata, H.; Hidaka, H.; Doi, H. Efficient synthesis of [11C]H-1152, a PET probe specific for Rho-kinases, highly potential targets in diagnostic medicine and drug development. Tetrahedron 2012, 68, 2336–2341. [Google Scholar] [CrossRef]
- Svensson, F.; Kniess, T.; Gergmann, R.; Pietzsch, J.; Wuest, F. Synthesis of an 18F-labeled cyclin-dependant kinase-2 inhibitor. J. Label. Compd. Radiopharm. 2011, 54, 769–774. [Google Scholar] [CrossRef]
- Wang, M.; Gao, M.; Miller, K.D.; Zheng, Q.H. Synthesis of 2,6-difluoro-N-(3-[11C]methoxy-1H-pyrazolo[3,4-b]pyridine-5-yl)-3-(propylsulfonamidio)benzamide as a new potential PET agent for imaging of B-RafV6°°E in cancers. Bioorg. Med. Chem. Lett. 2013, 23, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Miller, K.D.; Sledge, G.W.; Zheng, Q.H. [11C]GSK2126458 and [18F]GSK2126458, the first radiosynthesis of new potential PET agents for imaging of PI3K and mTOR in cancers. Bioorg. Med. Chem. Lett. 2012, 22, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Majo, V.J.; Simpson, N.R.; Prabhakaran, J.; Mann, J.J.; Kumar, J.S. Radiosynthesis of [18F]ATPFU: A potential PET ligand for mTOR. J. Label. Comp. Radiopharm. 2014, 57, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Zheng, Q.H. Synthesis of carbon-11-labeled 4-(phenylamino)-pyrrolo[2,1-f][1,2,4]triazine derivatives as new potential PET tracers for imaging of p38α mitogen-activated protein kinase. Bioorg. Med. Chem. Lett. 2014, 24, 3700–3705. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, L.; Gao, M.; Miller, K.D.; Sledge, G.W.; Zheng, Q.H. [11C]enzastaurin, the first design and radiosynthesis of a new potential PET agent for imaging of protein kinase C. Bioorg. Med. Chem. Lett. 2011, 21, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, M.; Miller, K.D.; Zheng, Q.H. Synthesis of (Z)-2-((1h-indazol-3-yl)methylene)-6-[11C]methoxy-7-(piperazin-1-ylmethyl)benzofuran-3(2H)-one as a new potential PET probe for imaging of the enzyme PIM1. Bioorg. Med. Chem. Lett. 2013, 23, 4342–4346. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tzintzun, R.; Gao, M.; Xu, Z.; Zheng, Q.H. Synthesis of [11C]CX-6258 as a new PET tracer for imaging of pim kinases in cancer. Bioorg. Med. Chem. Lett. 2015, 25, 3831–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morph, R. Selectively Nonselective Kinase Inhibition: Striking the Right Balance. J. Med. Chem. 2010, 53, 1413–1437. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; Verkaar, F.; Alwan, H.; de Man, J.; Buijsman, R.C.; Zaman, G.J. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br. J. Pharmacol. 2012, 166, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.J.; Sanders, J.M.; Henze, D.A.; Rudd, M.T.; Su, H.P.; Li, Y.; Nanda, K.K.; Egbertson, M.S.; Manley, P.J.; Jones, K.L.; et al. Maximizing diversity from a kinase screen: Identification of novel and selective pan-Trk inhibitors for chronic pain. J. Med. Chem. 2014, 57, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.; Pargellis, C.A.; Cirillo, P.F.; Gilmore, T.; Hickey, E.R.; Peet, G.W.; Proto, A.; Swinamer, A.; Moss, N. The Kinetics of Binding to p38 MAP Kinase by Analogues of BIRB 796. Bioorg. Med. Chem. Lett. 2003, 13, 3101–3104. [Google Scholar] [CrossRef]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP Kinase by Utilizing a Novel Allosteric Binding Site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Gavrin, L.K.; Saiah, E. Approaches to discover non-ATP site kinase inhibitors. Med. Chem. Commun. 2013, 4, 41–51. [Google Scholar] [CrossRef]
- Breen, M.E.; Soellner, M.B. Small molecule substrate phosphorylation site inhibitors of protein kinases: Approaches and challenges. ACS Chem. Biol. 2014, 10, 175–189. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard-Gauthier, V.; Bailey, J.J.; Berke, S.; Schirrmacher, R. Recent Advances in the Development and Application of Radiolabeled Kinase Inhibitors for PET Imaging. Molecules 2015, 20, 22000-22027. https://doi.org/10.3390/molecules201219816
Bernard-Gauthier V, Bailey JJ, Berke S, Schirrmacher R. Recent Advances in the Development and Application of Radiolabeled Kinase Inhibitors for PET Imaging. Molecules. 2015; 20(12):22000-22027. https://doi.org/10.3390/molecules201219816
Chicago/Turabian StyleBernard-Gauthier, Vadim, Justin J. Bailey, Sheldon Berke, and Ralf Schirrmacher. 2015. "Recent Advances in the Development and Application of Radiolabeled Kinase Inhibitors for PET Imaging" Molecules 20, no. 12: 22000-22027. https://doi.org/10.3390/molecules201219816