Two Polycyclic Geranylhydroquinone-Derived Metabolites from Roots of Arnebia hispidissima (Lehm.) DC.


Abstract
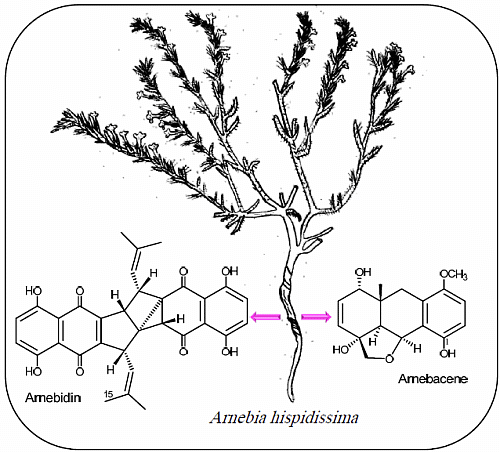
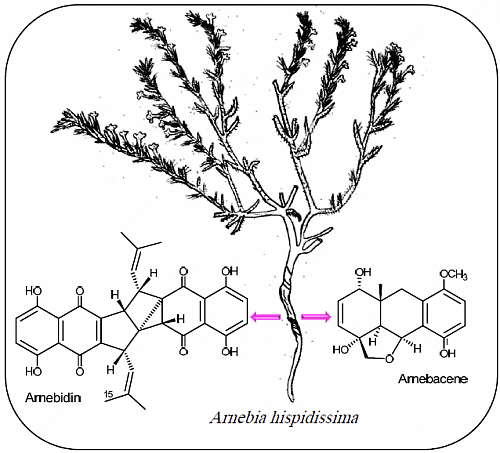
:
1. Introduction
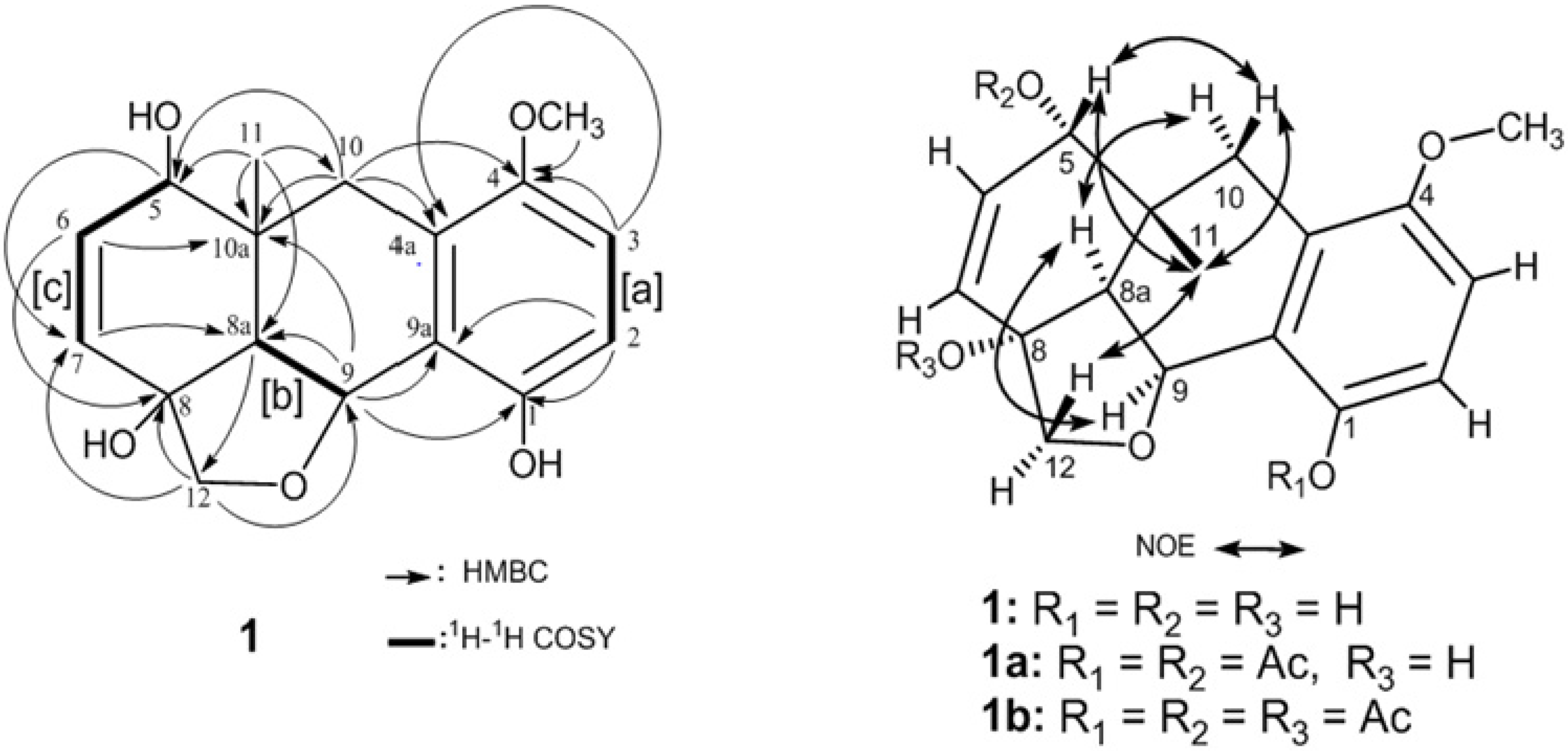
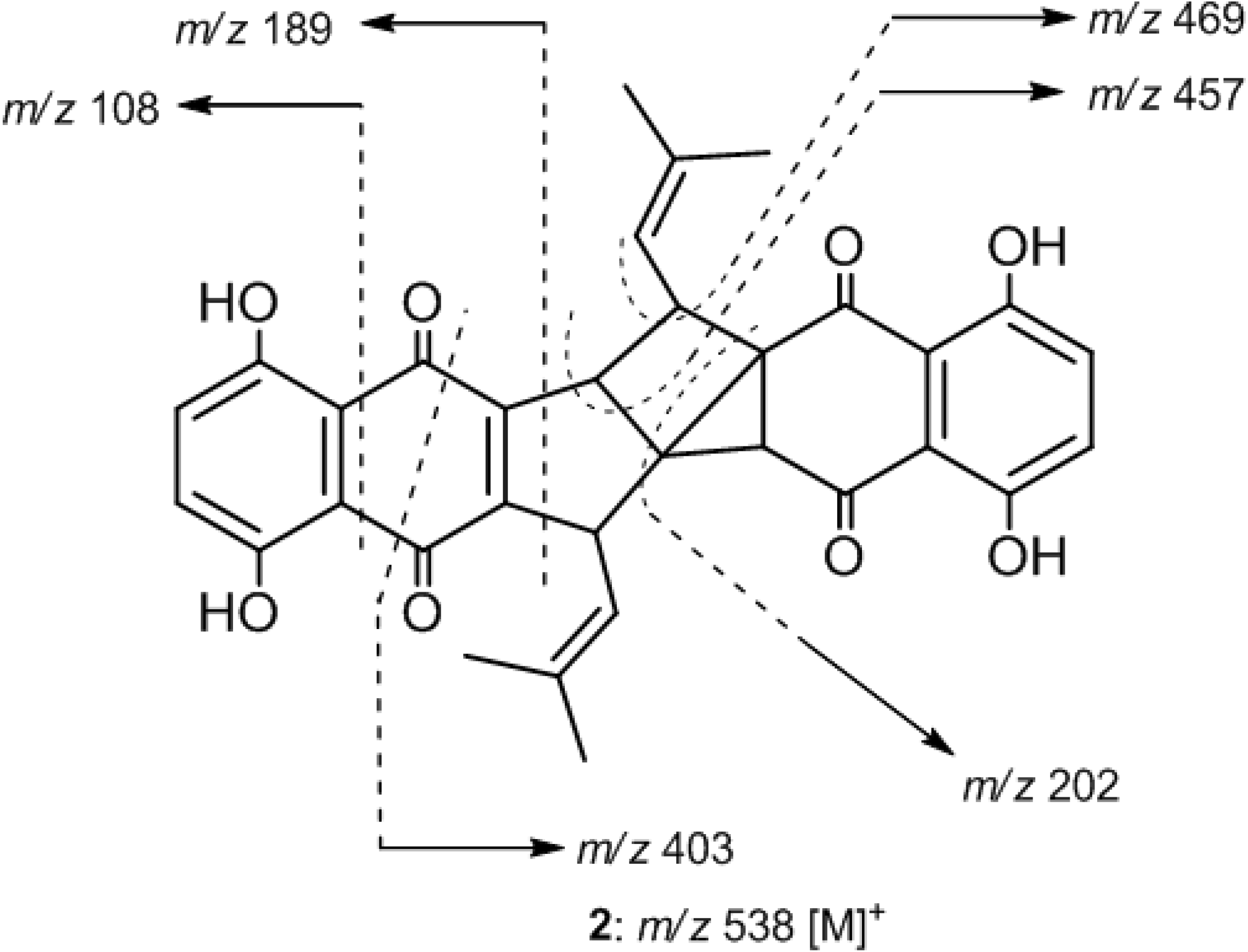
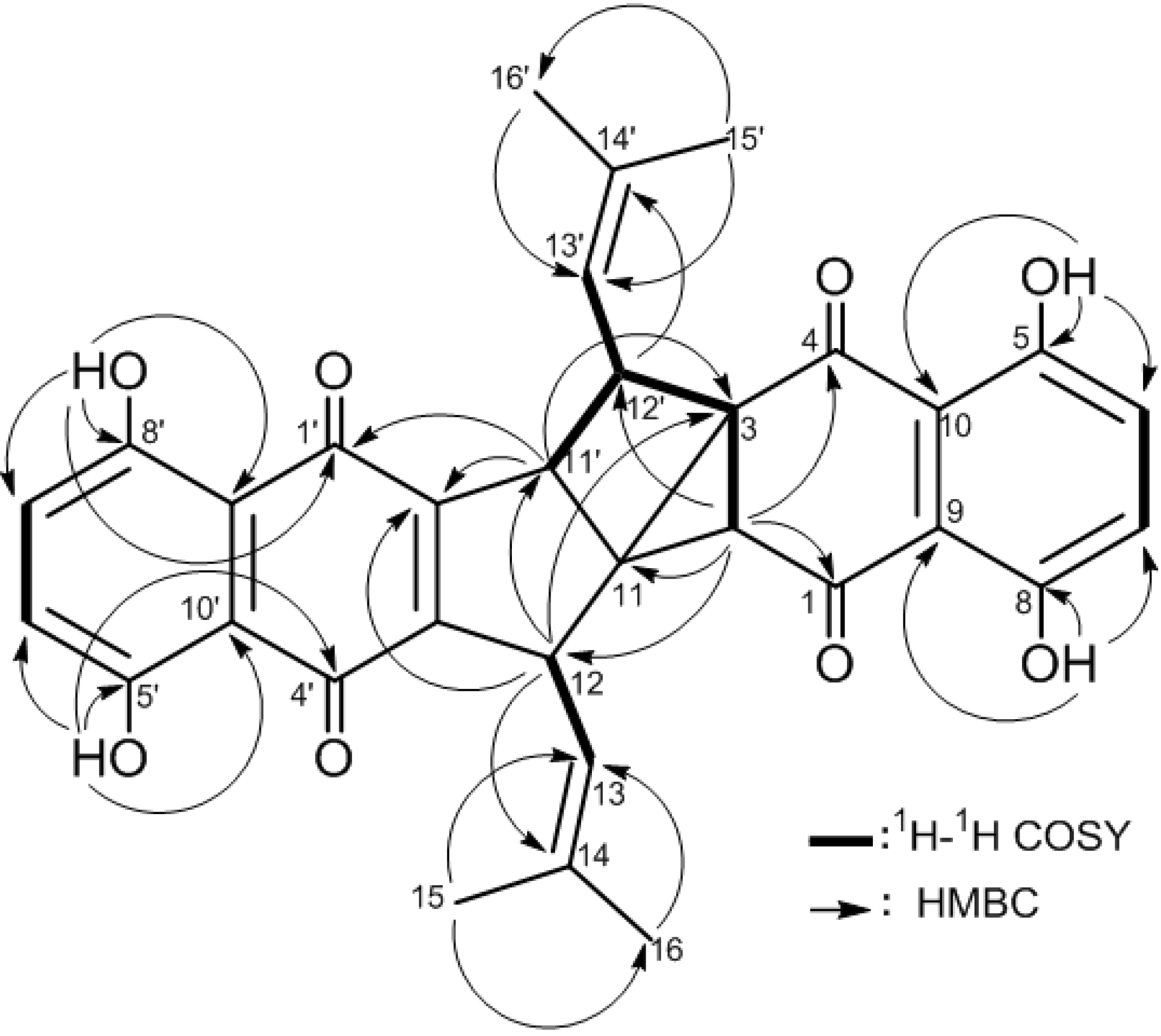
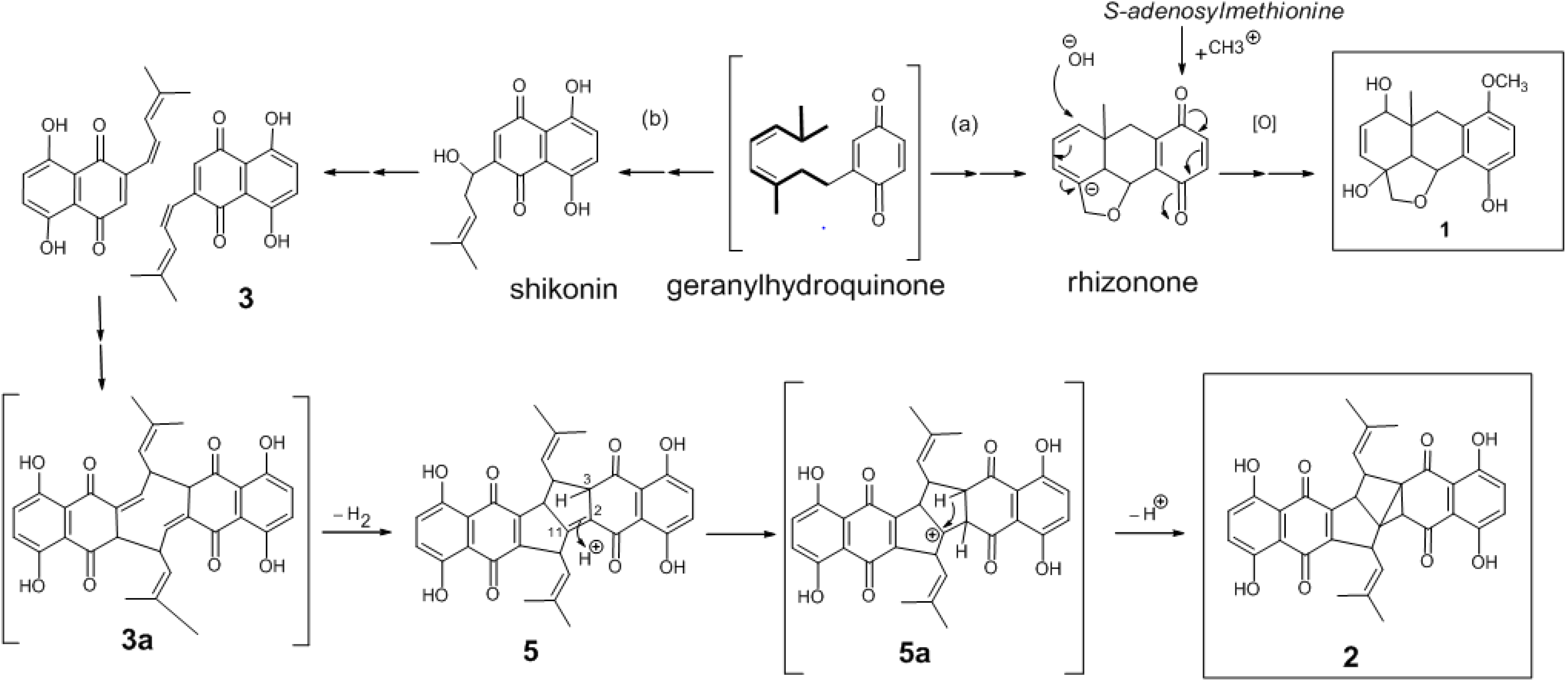
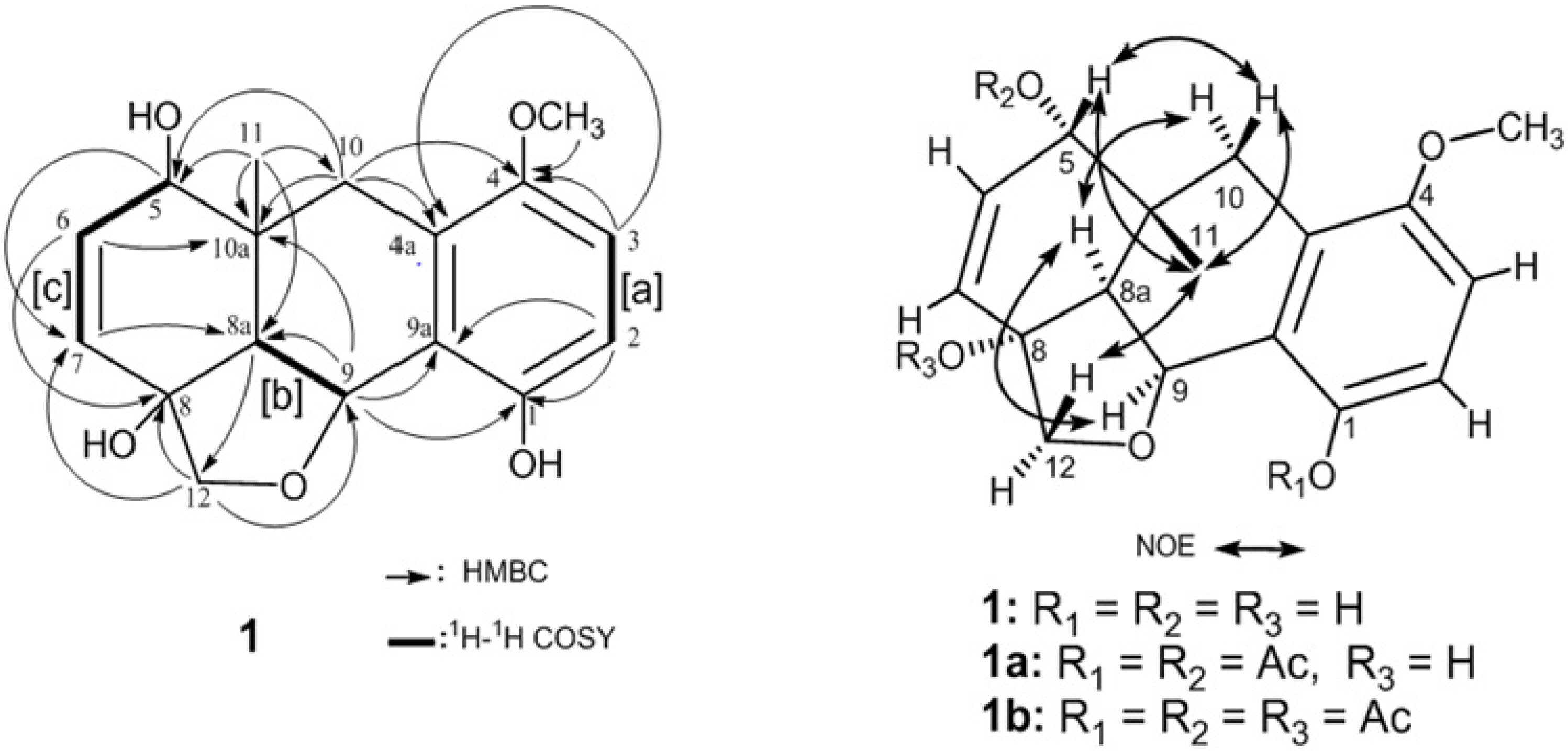
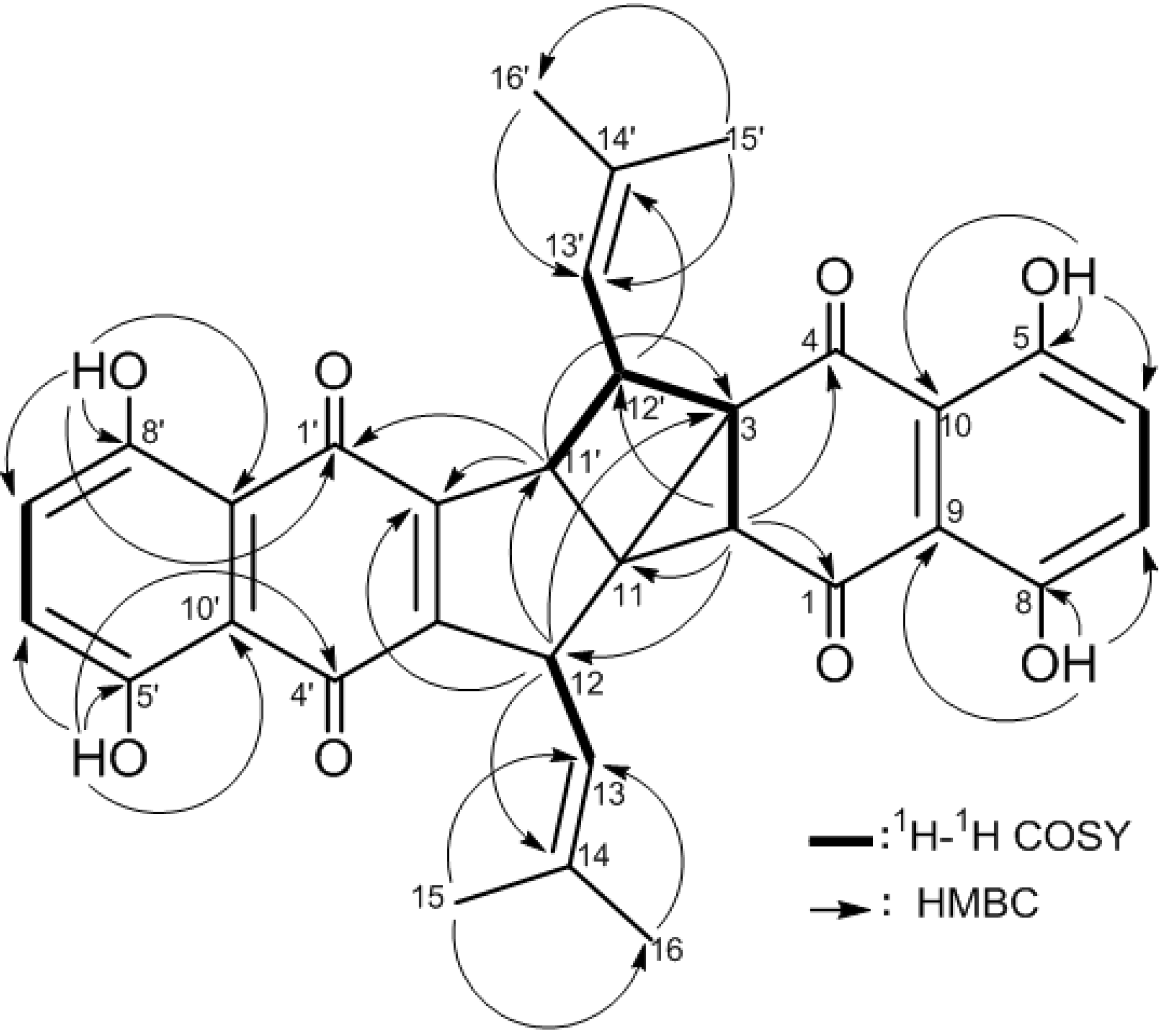
2. Results and Discussion
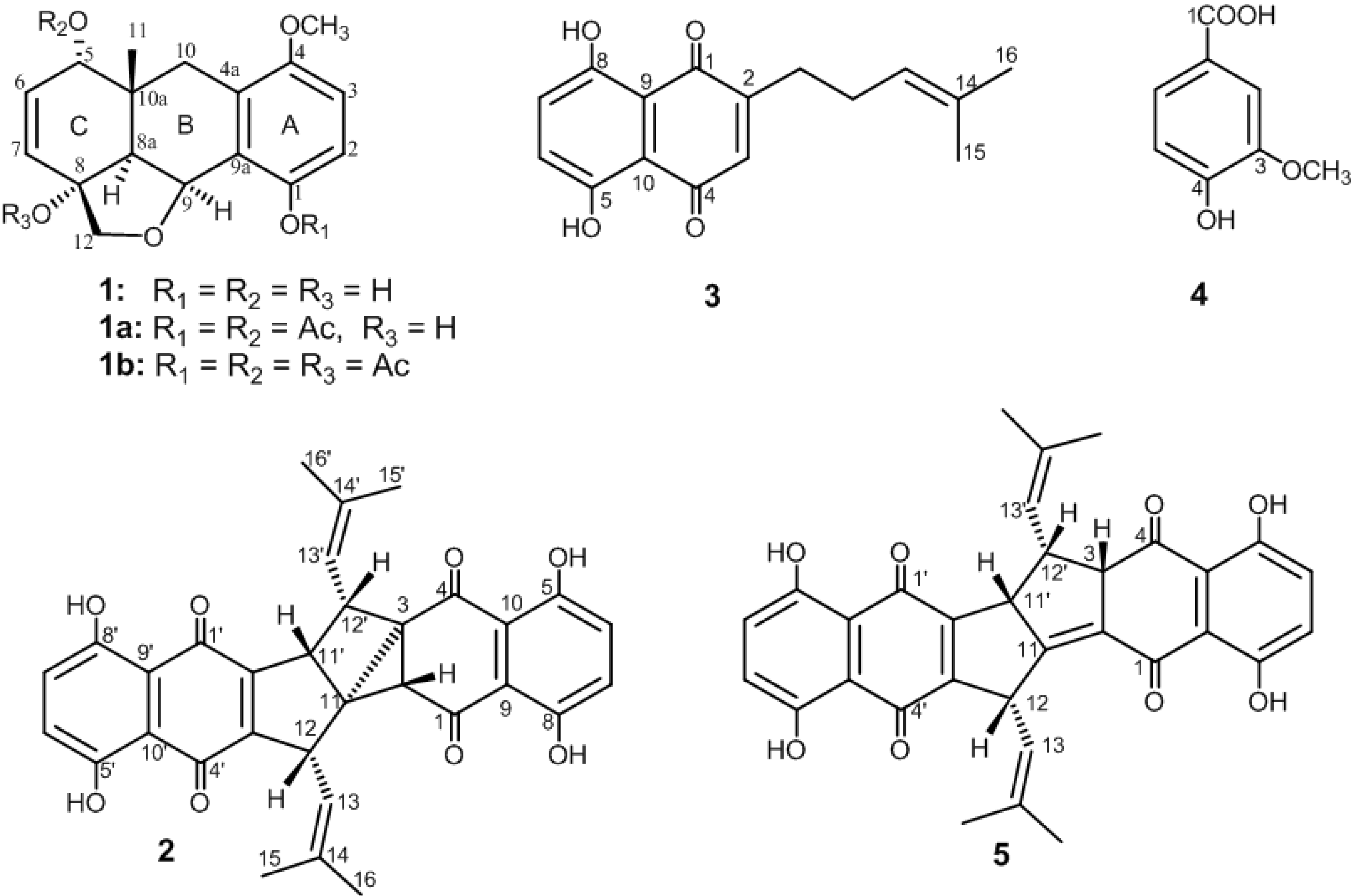

{kind=link}
{kind=link}
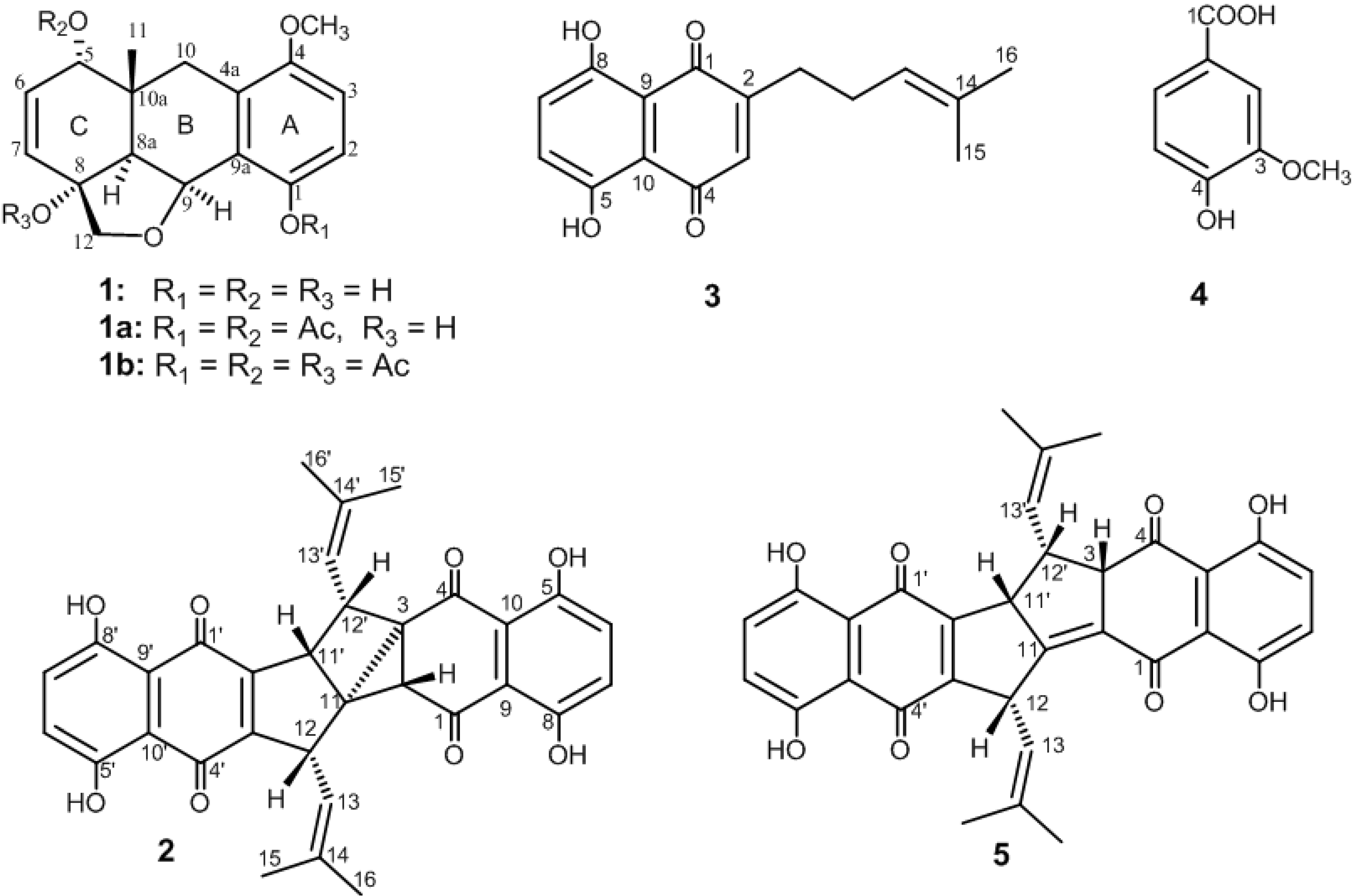{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | 1 a | |
|---|---|---|
| δC | δH | |
| 1 | 149.9 (qC) | |
| 2 | 113.3 (CH) b | 6.80, d (8.8) c |
| 3 | 111.4 (CH) | 6.77, d (8.8) |
| 4 | 151.3 (qC) | |
| 4a | 124.7 (qC) | |
| 5 | 70.3 (CH) | 3.84, d (6.0) |
| 6 | 129.7 (CH) | 6.11, dd (10.0, 6.0) |
| 7 | 131.2 (CH) | 6.01, d (10.0) |
| 8 | 78.8 (qC) | |
| 8a | 49.8 (CH) | 2.59, d (6.0) |
| 9 | 73.9 (CH) | 5.39, d (6.0) |
| 9a | 122.0 (qC) | |
| 10 | 33.2 (CH2) | 2.67, 2 H, s |
| 10a | 35.9 (qC) | |
| 11 | 19.5 (CH3) | 0.85, 3H, s |
| 12 | 77.9 (CH2) | 4.08, d (10.0), α |
| 3.92, d (10.0), β | ||
| 4-OMe | 56.1 (CH3) | 3.79, 3H, s |
| Atom | 2 a | 3 a | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 1 | 193.1 (qC) | |||
| 2 | 45.3 (CH) b | 2.68, d (2.5) c | ||
| 3 | 49.2 (qC) | |||
| 4 | 195.5 (qC) | |||
| 5 | 156.3 (qC) | |||
| 6 | 129.1 (CH) | 7.29, d (8.5) | ||
| 7 | 128.9 (CH) | 7.30, d (8.5) | ||
| 8 | 157.4 (qC) | |||
| 9 | 111.7 (qC) | |||
| 10 | 112.7 (qC) | |||
| 11 | 38.3 (qC) | |||
| 12 | 40.7 (CH) | 3.82, dd (8.5, 4.5) | ||
| 13 | 116.9 (CH) | 4.39, ddd (8.5, 1.5, 1.5) | ||
| 14 | 138.6 (qC) | |||
| 1' | 182.2 (qC) | 183.0 (qC) | ||
| 2' | 144.3 (qC) | 151.5 (qC) | ||
| 3' | 140.0 (qC) | 134.5 (CH) | 6.84, s | |
| 4' | 182.6 (qC) | 183.0 (qC) | ||
| 5' | 159.1 (qC) | 162.3 (qC) | ||
| 6' | 129.3 (CH) | 7.25, d (8.5) | 131.2 (CH) | 7.19, s |
| 7' | 129.9 (CH) | 7.22, d (8.5) | 130.9 (CH) | 7.19, s |
| 8' | 158.6 (qC) | 163.0 (qC) | ||
| 9' | 112.5 (qC) | 111.7 (qC) | ||
| 10' | 111.4 (qC) | 112.0 (qC) | ||
| 11' | 38.1 (CH) | 3.89, dd (4.5, 1.5) | 26.6 (CH2) | 2.64, 2H, ddd (7.5, 1) |
| 12' | 37.6 (CH) | 3.35, ddd (8.5, 4.5, 2.5) | 29.7 (CH2) | 2.30, 2H, q, (13.5, 7.5) |
| 13' | 117.4 (CH) | 4.63, ddd (8.5, 1.5, 1.5) | 122.4 (CH) | 5.13, ddd (7.5, 1.5, 1.5) |
| 14' | 137.8 (qC) | 133.6 (qC) | ||
| Me-15 | 18.6 (CH3) | 1.83, 3H, d (1.5) | 17.8 (CH3) | 1.69, 3H, d (1.5) |
| Me-16 | 26.0 (CH3) | 1.54, 3H, d (1.5) | 25.7 (CH3) | 1.60, 3H, d (1.5) |
| Me-15' | 18.7 (CH3) | 1.61, 3H, d (1.5) | ||
| Me-16' | 25.8 (CH3) | 1.72, 3H, d (1.5) | ||
| OH-5 | 12.29, s | 12.46, s | ||
| OH-8 | 12.48, s | 12.56, s | ||
| OH-5' | 12.40, s | |||
| OH-8' | 12.07, s | |||


3. Experimental
3.1. General
3.2. Plant Materials
3.3. Extraction and Isolation of Compounds
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boulos, L. Flora of Egypt, Geraniaceae-Boraginaceae; Al Hadara Publishing: Cairo, Egypt, 2000; Volume 2, pp. 291–352. [Google Scholar]
- Hamdard, M.E.; Badar, Y.; Khan, M.S.; Shamsi, M.A. Revised phytochemical study of Arnebia hispidissima. Pak. J. Pharm. Sci. 1988, 1, 19–20. [Google Scholar]
- Jain, S.C.; Singh, B.; Jain, R. Arnebins and antimicrobial activities of Arnebia hispidissima DC. cell cultures. Phytomedicine 2000, 6, 474–476. [Google Scholar] [CrossRef]
- Shukla, Y.N.; Srivastava, A.; Singh, S.C.; Kumar, S. New naphthoquinones from Arnebia hispidissima roots. Planta Med. 2001, 67, 575–577. [Google Scholar] [CrossRef]
- Singh, B.; Sahu, P.M.; Jain, S.C.; Singh, S. Estimation of naphthaquinones from Arnebia hispidissima (Lehm.) DC. In vivo and in vitro. I. Anti-inflammatory screening. Phytother. Res 2004, 18, 154–159. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, M.K.; Meghwal, P.R.; Sahu, P.M.; Singh, S. Anti-inflammatory activity of shikonin derivatives from Arnebia hispidissima. Phytomedicine 2003, 10, 375–380. [Google Scholar]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Couladouros, E.A.; Hepworth, D.; Nicolaou, K.C. Chemistry and biology of alkannins, shikonins and related naphthazarin natural products. Angew. Chem. Int. Ed. 1999, 38, 270–300. [Google Scholar] [CrossRef]
- Ghazanfar, S.A. Handbook of Arabian Medicinal Plants; CRC Press: London,UK, 1994; pp. 52–58. [Google Scholar]
- Jain, S.C.; Jain, R.; Singh, B. Antimicrobial principles from Arnebia hispidissima. Pharmaceut. Biol. 2003, 41, 231–233. [Google Scholar] [CrossRef]
- Wassel, G.; el-Menshawi, B.; Saeed, A.; Mahran, G.; el-Merzabani, M. Screening of selected plants for pyrrolizidine alkaloids and antitumor activity. Pharmazie 1987, 42, 709. [Google Scholar]
- Shukla, Y.N.; Tandon, J.S.; Dhar, M.M. Arnebin-7 A new Naphthaquinone from Arnebia nobilis. Indian J. Chem. 1973, 11, 528–529. [Google Scholar]
- Damianakos, H.; Kretschmer, N.; Syklowska-Baranek, K.; Pietrosiuk, A.; Bauer, R.; Chinou, I. Antimicrobial and cytotoxic isohexenylnaphthazarins from Arnebia euchroma (Royle) Jonst. (Boraginaceae) callus and cell suspension culture. Molecules 2012, 17, 14310–14322. [Google Scholar] [CrossRef]
- Chang, S.W.; Kim, K.H.; Lee, I.K.; Choi, S.U.; Ryu, S.Y.; Lee, K.R. Phytochemical constituents of Bistorta manshuriensis. Nat. Prod. Sci. 2009, 15, 234–240. [Google Scholar]
- Naz, S.; Ahmad, S.; Ajaz, R.S.; Asad, S.S.; Siddiqi, R. Antibacterial activity directed isolation of compounds from Onosma hispidum. Microbiol. Res. 2006, 161, 43–48. [Google Scholar] [CrossRef]
- Meselhy, M.R.; Kadota, S.; Tsubono, K.; Hattori, M.; Namba, T. Biotransformation of shikonin by human intestinal bacteria. Tetrahedron Lett. 1994, 50, 3081–3098. [Google Scholar] [CrossRef]
- Meselhy, M.R.; Kadota, S.; Tsubono, K.; Kusai, A.; Hattori, M.; Namba, T. Shikometabolins A, B, C and D, novel dimeric naphthoquinone metabolites obtained from shikonin by human intestinal bacteria. Tetrahedron Lett. 1994, 3, 583–586. [Google Scholar]
- Silverstein, R.M.; Webster, F.X. Spectrometric Identification of Organic Compounds, 6th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2002; pp. 185–187. [Google Scholar]
- Fukui, H.; Feroj, H.A.F.M.; Kyo, M. Formation and secretion of a uinque quinone by hairy root cultures of Lithospermum erythrorhizon. Phytochemistry 1999, 51, 511–515. [Google Scholar] [CrossRef]
- Rivera, S.B.; Swedlund, B.D.; King, G.J.; Bell, R.N.; Hussey, C.E., Jr.; Shattuck-Eidens, D.M.; Wrobel, W.M.; Peiser, G.D.; Poulter, C.D. Chrysanthemyl diphosphate synthase: Isolation of the gene and characterization of the recombinant non-head-to-tail monoterpene synthase from Chrysanthemum cinerariaefolium. Proc. Natl. Acad. Sci. USA 2001, 98, 4373–4378. [Google Scholar] [CrossRef]
- Dewick, P.M. Medicinal Natural Products. In A Biosynthetic Approach, 3th ed.; JohnWiley & Sons, Inc.: New York, NY, USA, 2009; pp. 193–198. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ahmed, A.F.; Saad, H.-E.A.; El-Karim, E.M.A. Two Polycyclic Geranylhydroquinone-Derived Metabolites from Roots of Arnebia hispidissima (Lehm.) DC. Molecules 2014, 19, 5940-5951. https://doi.org/10.3390/molecules19055940
Ahmed AF, Saad H-EA, El-Karim EMA. Two Polycyclic Geranylhydroquinone-Derived Metabolites from Roots of Arnebia hispidissima (Lehm.) DC. Molecules. 2014; 19(5):5940-5951. https://doi.org/10.3390/molecules19055940
Chicago/Turabian StyleAhmed, Atallah F., Hassan-Elrady A. Saad, and Eman M. Abd El-Karim. 2014. "Two Polycyclic Geranylhydroquinone-Derived Metabolites from Roots of Arnebia hispidissima (Lehm.) DC." Molecules 19, no. 5: 5940-5951. https://doi.org/10.3390/molecules19055940
APA StyleAhmed, A. F., Saad, H.-E. A., & El-Karim, E. M. A. (2014). Two Polycyclic Geranylhydroquinone-Derived Metabolites from Roots of Arnebia hispidissima (Lehm.) DC. Molecules, 19(5), 5940-5951. https://doi.org/10.3390/molecules19055940