Integrated Computational Tools for Identification of CCR5 Antagonists as Potential HIV-1 Entry Inhibitors: Homology Modeling, Virtual Screening, Molecular Dynamics Simulations and 3D QSAR Analysis


Abstract
:1. Introduction
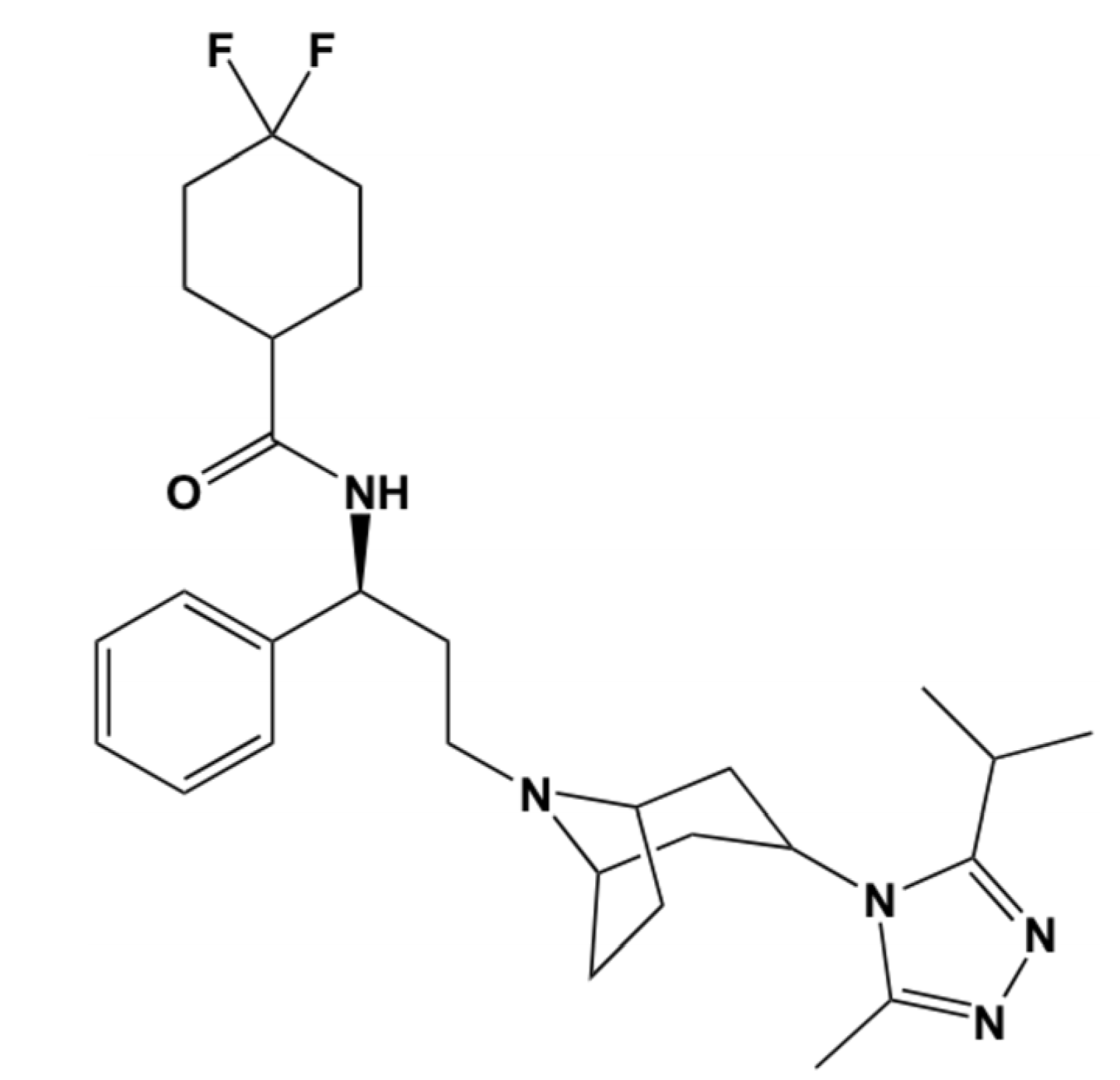
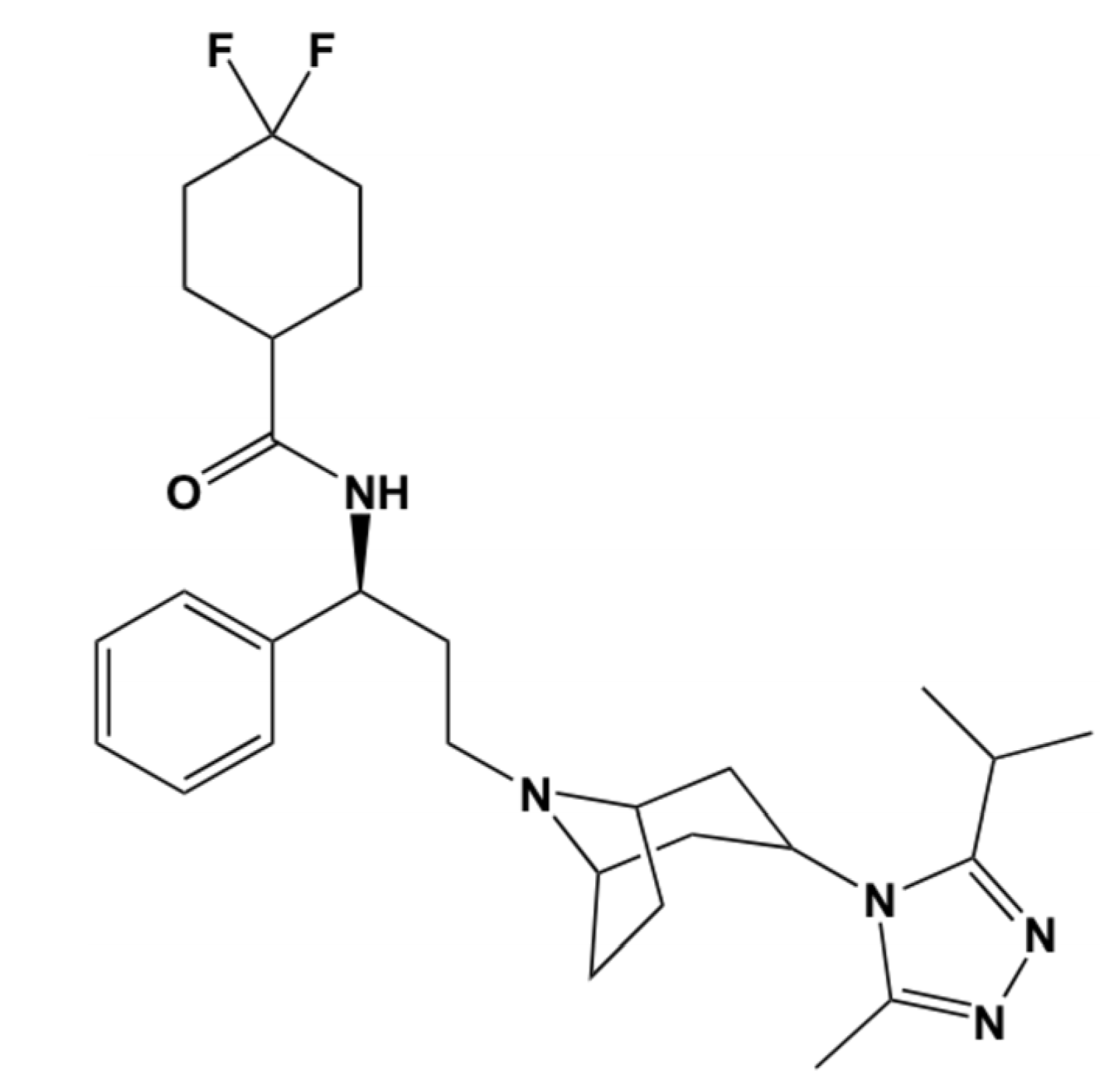
2. Computational Methods
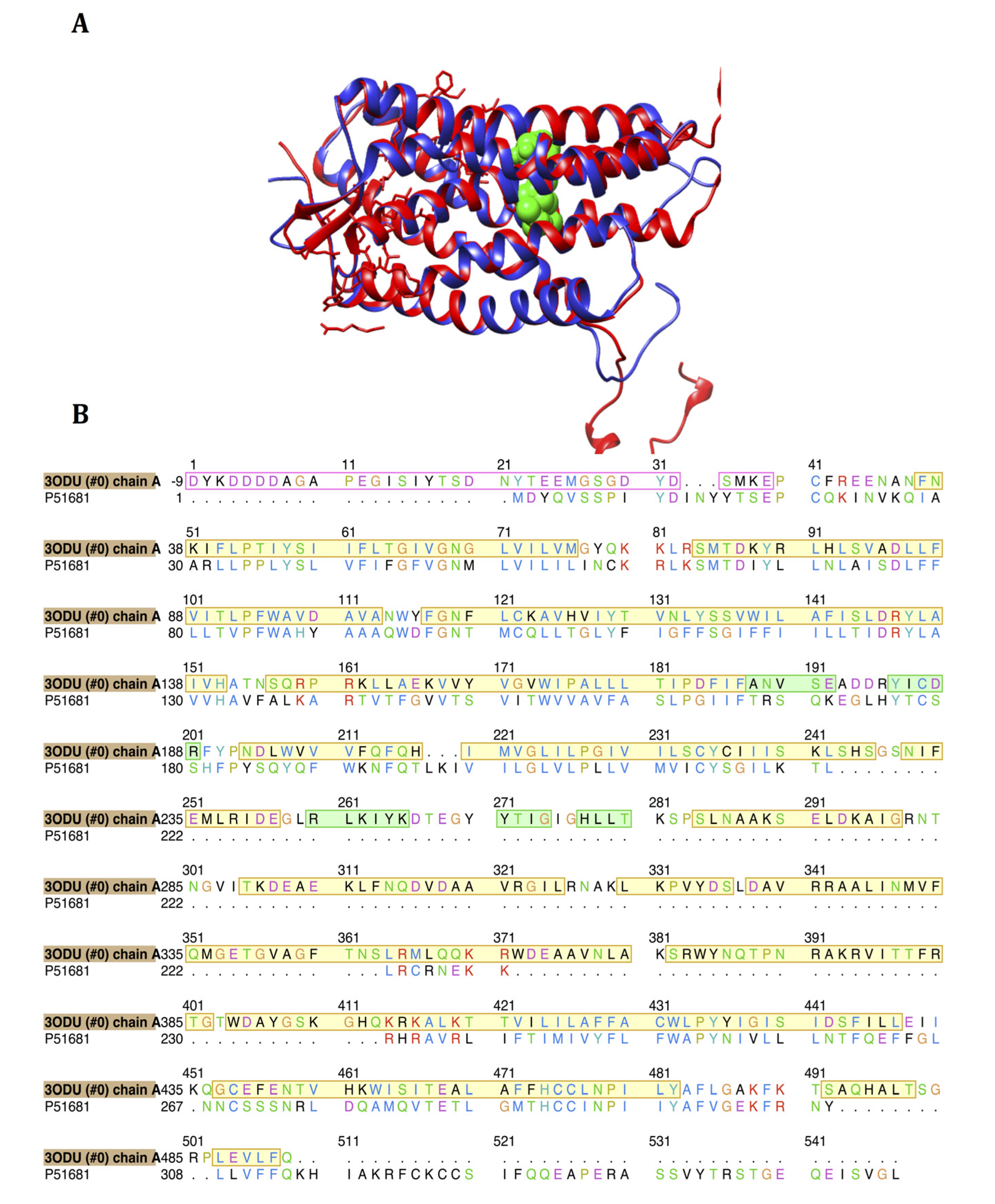
2.1. Homology Modeling of CCR5
2.2. Maraviroc Structure Acquisition and Preparation
2.3. Ligand Library Generation
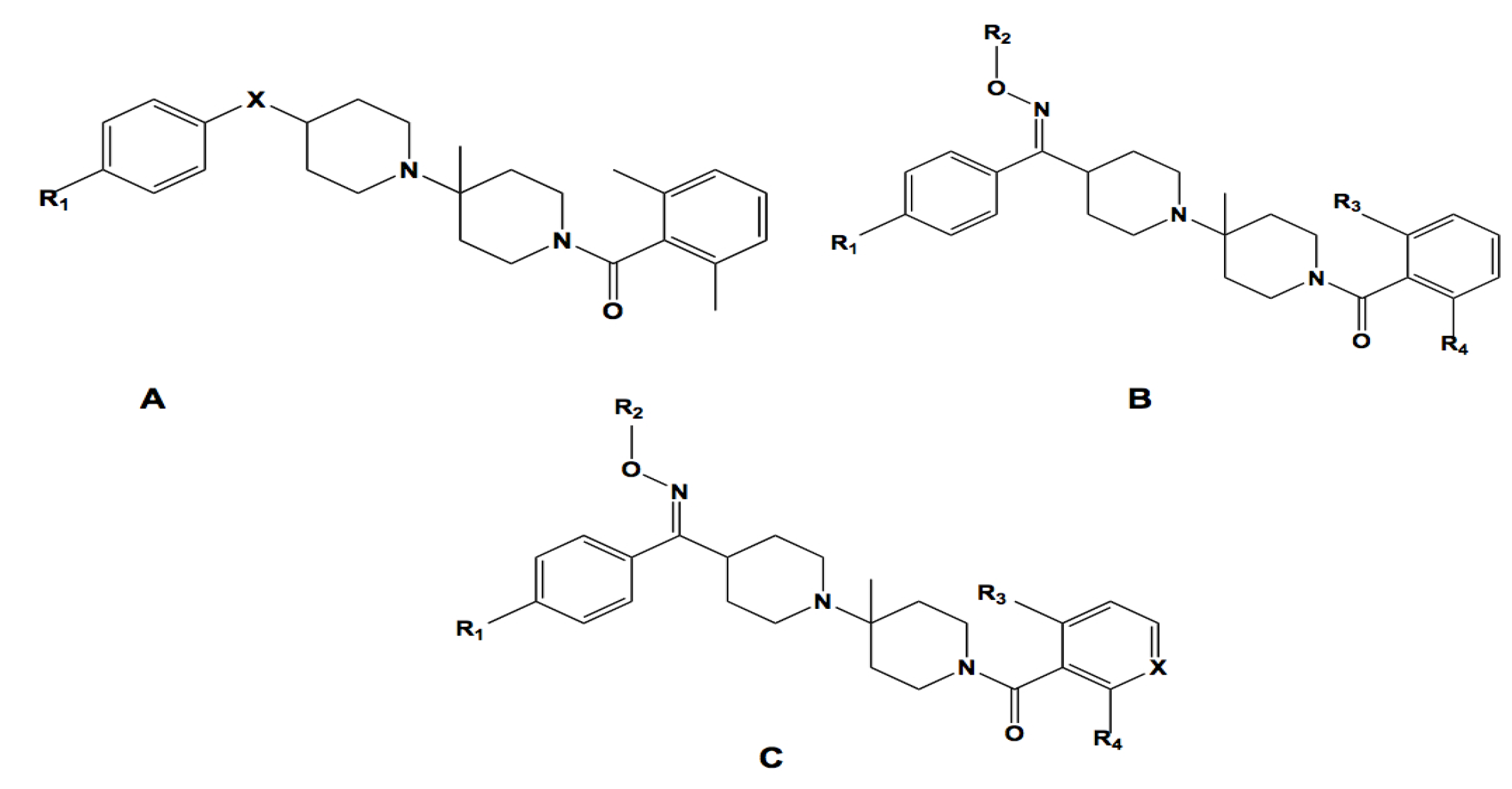
2.3.1. Structural Similarity-Based Compound Library Generation
2.3.2. Pharmacophore-Based Library Generation
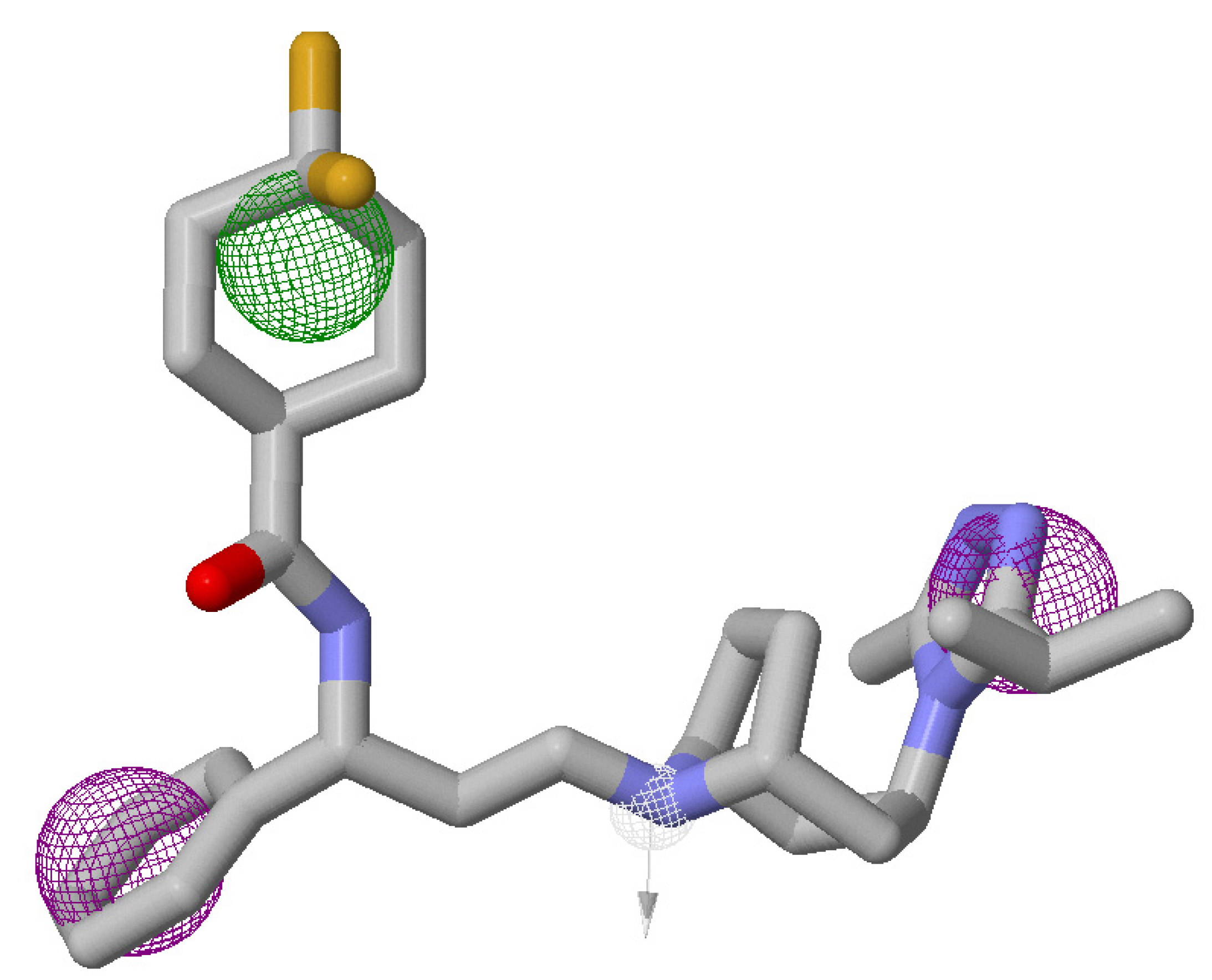
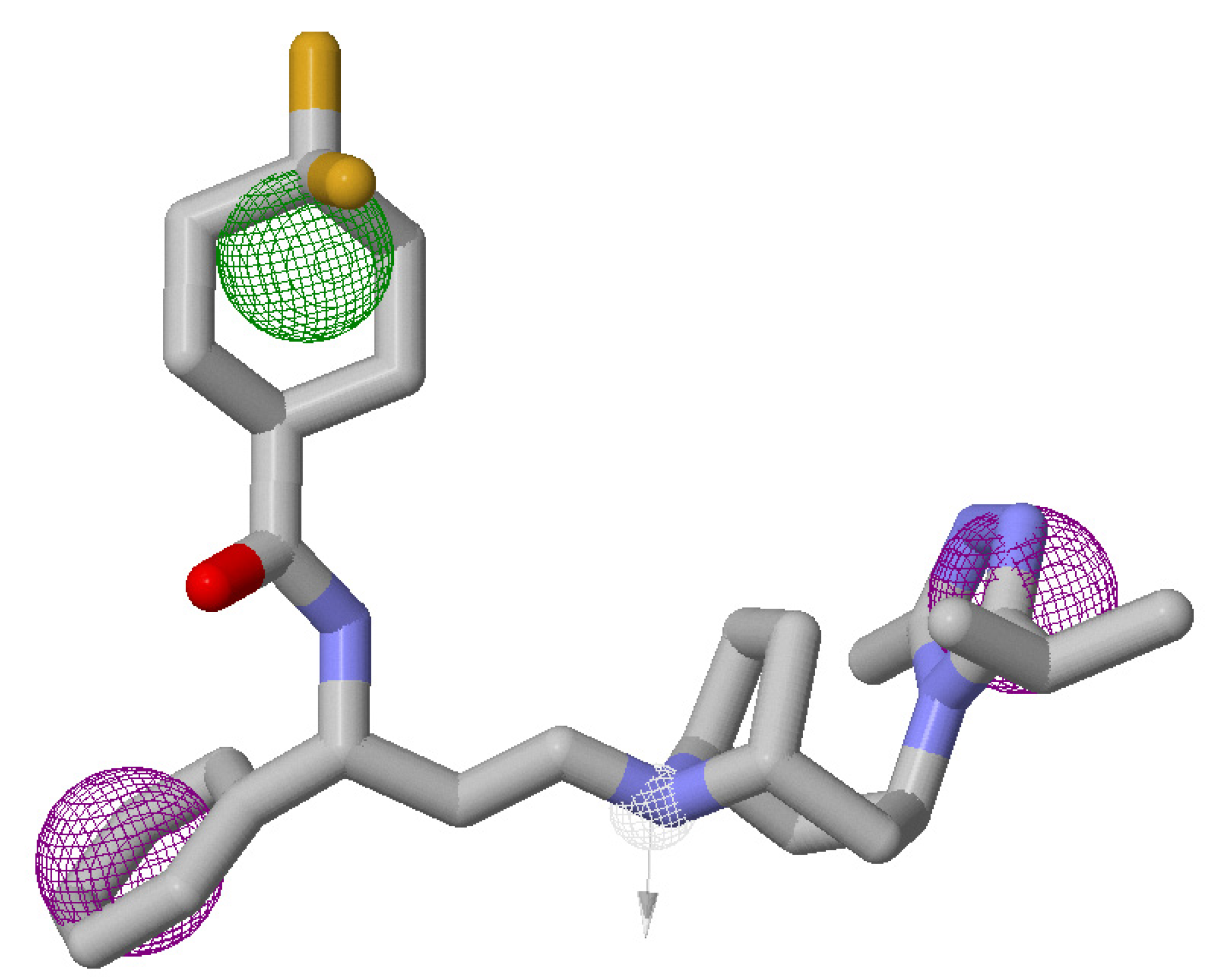
2.4. Virtual Screening and Validation of Docking Protocol
2.5. Molecular Dynamics Simulations and Post-Dynamic Analysis
2.6. Three-Dimensional (3D) QSAR Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Core | X | R1 | R2 | R3 | R4 | Expt. 1/logIC50 (mM) | Prdt. 1/logIC50 (mM) | Residual |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A | CH2 | Br | - | - | - | 0.250 | 0.259 | 0.009 |
| 2 | A | NH2 | Br | - | - | - | 0.267 | 0.265 | 0.002 |
| 3 | A(E) | =N–OCH3 | Br | - | - | - | 0.290 | 0.270 | 0.020 |
| 4 | A(E) | =N–OCH3 | Br | - | - | - | 0.252 | 0.271 | −0.019 |
| 5 | B | - | Br | CH3 | Cl | NH2 | 0.360 | 0.359 | 0.001 |
| 6 | B | - | Br | CH3 | CH3 | OH | 0.278 | 0.276 | 0.001 |
| 7 | C | N+–O | CH3 | CH3 | CH3 | CH3 | 0.267 | 0.286 | −0.018 |
| 8 | C | N | Cl | C2H5 | CH3 | CH3 | 0.435 | 0.392 | 0.043 |
| 9 | A | CH2 | Cl | - | - | - | 0.229 | −0.248 | −0.018 |
| 10 | A | CH2 | I | - | - | - | 0.253 | 0.230 | 0.023 |
| 11 | A | CH2 | CF3 | - | - | - | 0.333 | 0.269 | 0.064 |
| 12 | A | CH2 | CH3 | - | - | - | 0.218 | 0.233 | −0.015 |
| 13 | A | CH2 | OCH3 | - | - | - | 0.227 | 0.233 | −0.006 |
| 14 | A | CH2 | SO2CH3 | - | - | - | 0.260 | 0.273 | −0.013 |
| 15 | A | C=CH2 | Br | - | - | - | 0.333 | 0.313 | 0.020 |
| 16 | B(Z) | - | Br | H | CH3 | CH3 | 0.250 | 0.245 | 0.005 |
| 17 | B(Z) | - | Br | C4H9 | CH3 | CH3 | 0.266 | 0.275 | −0.009 |
| 18 | B | N | Br | CH2-CO-NHCH3 | CH3 | CH3 | 0.274 | 0.292 | −0.016 |
| 19 | B | N | Br | C2H5 | F | CF3 | 0.301 | 0.305 | −0.004 |
| 20 | C | N+–O | Br | C2H5 | H | CH3 | 0.318 | 0.289 | 0.290 |
| 21 | C | N+–O | Br | C2H5 | CH3 | CH3 | 0.338 | 0.382 | −0.044 |
| 22 | C | N+–O | Br | C2H5 | H | CH3 | 0.310 | 0.307 | 0.003 |
| 23 | C | N+–O | CF3 | CH3 | CH3 | CH3 | 0.329 | 0.324 | 0.005 |
| 24 | C | N+–O | OCF3 | CH3 | CH3 | CH3 | 0.270 | 0.298 | −0.028 |
| 25 | C | C=O | OCF3 | C2H5 | CH3 | CH3 | 0.307 | 0.314 | −0.007 |
| 26 | C | - | Cl | C2H5 | CH3 | CH3 | 0.324 | 0.324 | −0.006 |
| 27 | A | N | Br | - | - | - | 0.243 | 0.261 | −0.018 |
| 28 t | B | N+–O | Br | CH3 | CH3 | NH2 | 0.371 | 0.385 | 0.086 |
| 29 t | C | CH2 | Br | CH3 | CH3 | CH3 | 0.371 | 0.329 | 0.076 |
| 30 t | C | CH–OH | Br | C2H5 | CH3 | CH3 | 0.301 | 0.302 | 0.001 |
| 31 t | A | - | - | - | - | - | 0.270 | 0.277 | −0.007 |
| 32 t | A | - | Br | - | - | - | 0.205 | 0.254 | −0.54 |
| 33 t | B(Z) | N+ = O | Br | C3H7 | CH3 | CH3 | 0.310 | 0.271 | 0.38 |
| 34 t | C | CH3 | CH3 | - | - | 0.263 | 0.254 | −0.091 | |
| 35 t | C | CF3 | C2H5 | CH3 | CH3 | 0.321 | 0.306 | 0.015 |

3. Results and Discussion
3.1. Homology Modeling of CCR5
| Active site residues (3ODU) | Corresponding modeled active site residues |
|---|---|
| Glu283 | Glu283 |
| Ile198 | Ile198 |
| Leu204 | Leu104 # |
| Leu213 | * |
| Phe85 | Phe85 |
| Phe109 | Phe109 |
| Phe113 | * |
| Thr195 | Thr195 |
| Trp85 | Trp86 # |
| Trp94 | Trp94 |
| Trp248 | Trp248 |
| Tyr89 | Tyr89 |
| Tyr108 | Tyr108 |
| Tyr251 | Tyr251 |
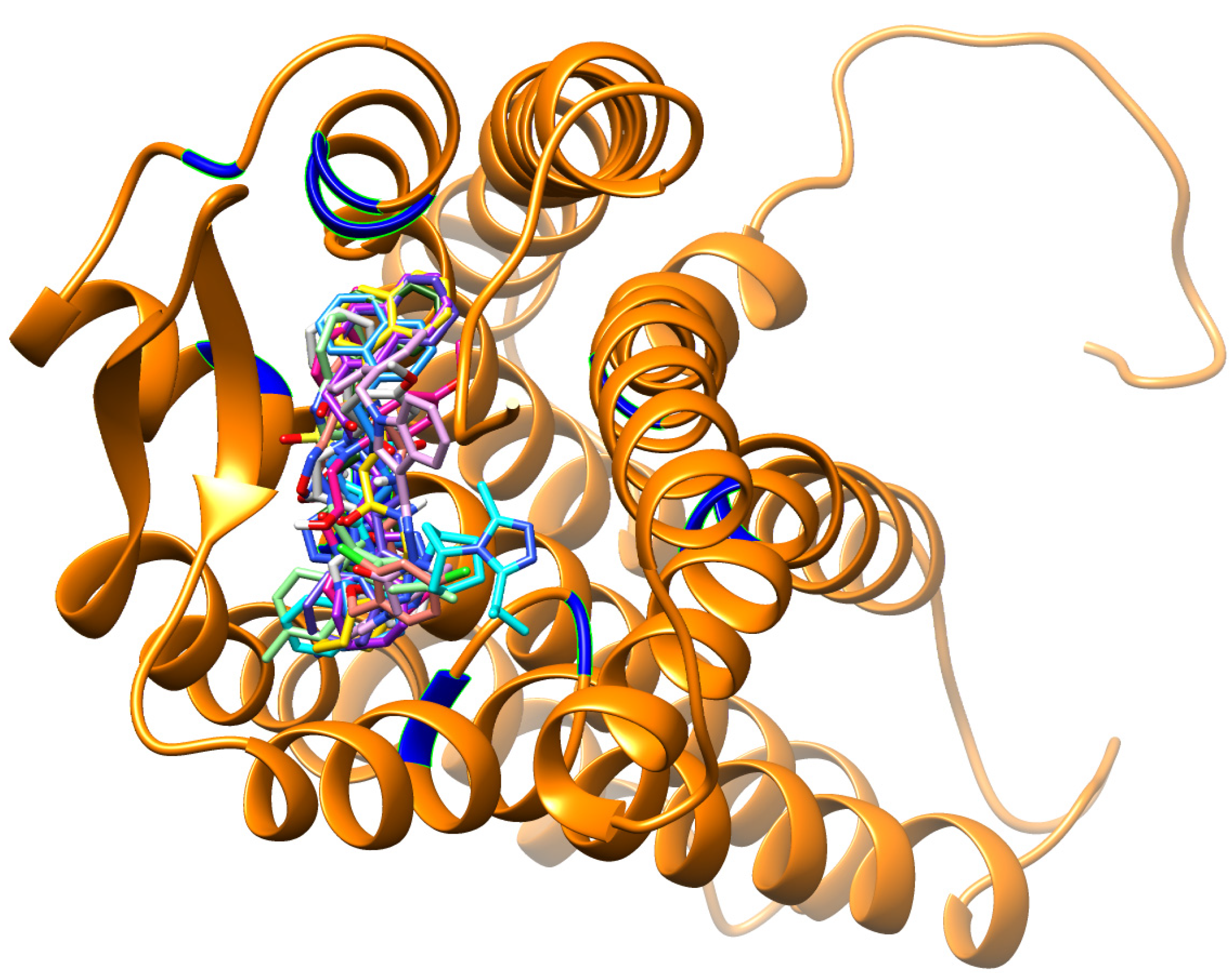
3.2. Virtual Screening
| Library | Rank | ZINC ID | Structure | Binding Energy (kcal/mol) | xlogP | H-bond Donors | H-bond Acceptors | Molecular Weight (g/mol) |
|---|---|---|---|---|---|---|---|---|
| Ref | R | ZINC03817234 |  | −10.2 | −3.50 | 2 | 6 | 514.69 |
| S * | 1 | ZINC71849549 |  | −12.2 | 2.27 | 2 | 6 | 318.89 |
| P ** | 2 | ZINC00825224 |  | −12.0 | 4.11 | 3 | 5 | 397.488 |
| P | 3 | ZINC00634884 |  | −12.0 | 5.96 | 1 | 6 | 481.60 |
| S | 4 | ZINC32760563 |  | −11.9 | 3.47 | 0 | 5 | 388.51 |
| S | 5 | ZINC32760533 |  | −11.8 | 3.44 | 0 | 5 | 388.52 |
| S | 6 | ZINC25010434 |  | −11.8 | 2.16 | 1 | 7 | 431.54 |
| P | 7 | ZINC00851466 |  | −11.8 | 5.52 | 3 | 10 | 536.38 |
| S | 8 | ZINC71818945 |  | −11.7 | 3.35 | 0 | 6 | 434.58 |
| P | 9 | ZINC00895646 |  | −11.7 | 3.99 | 2 | 7 | 451.55 |
| P | 10 | ZINC00895634 |  | −11.7 | 3.54 | 2 | 7 | 438.53 |


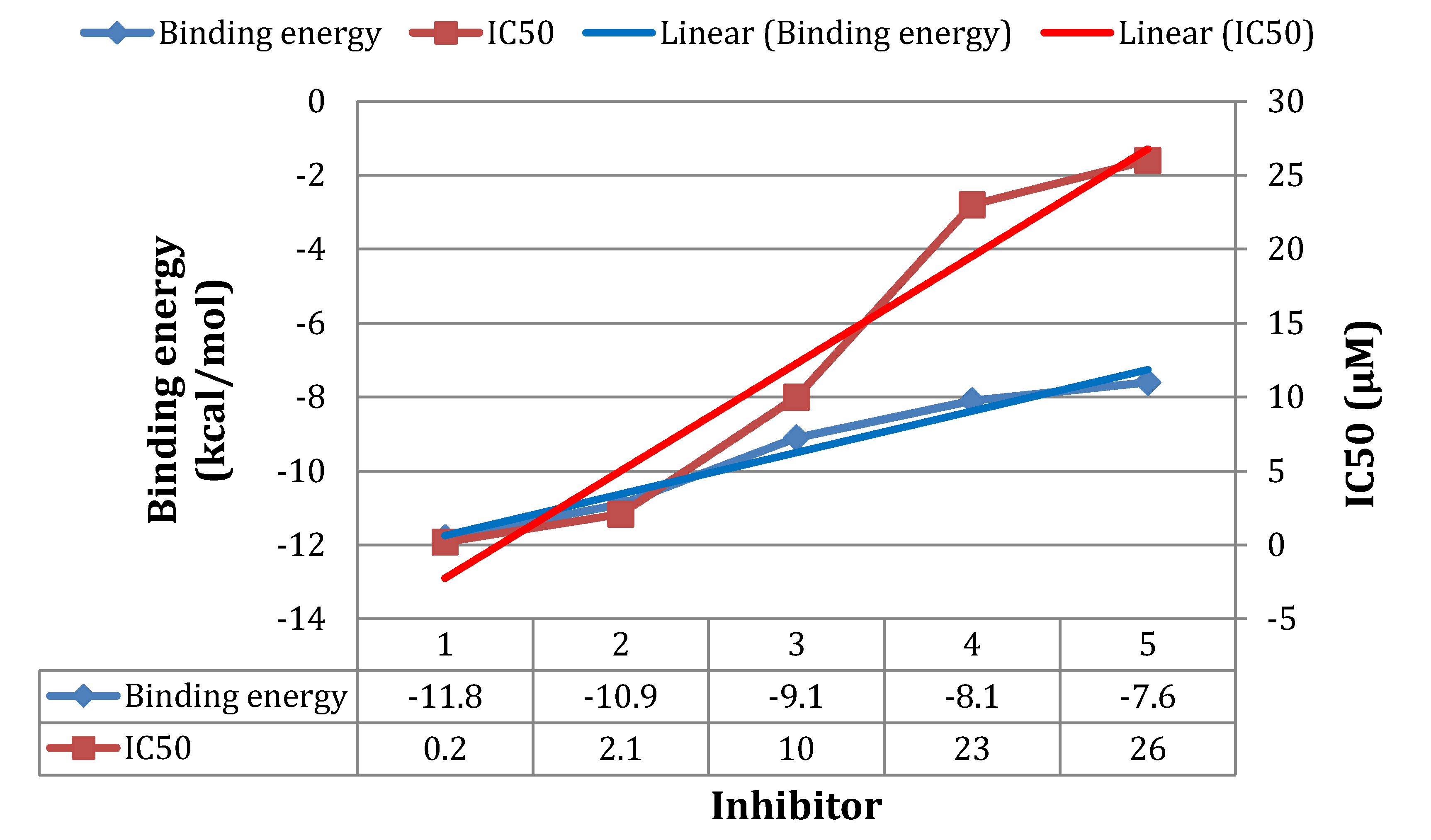
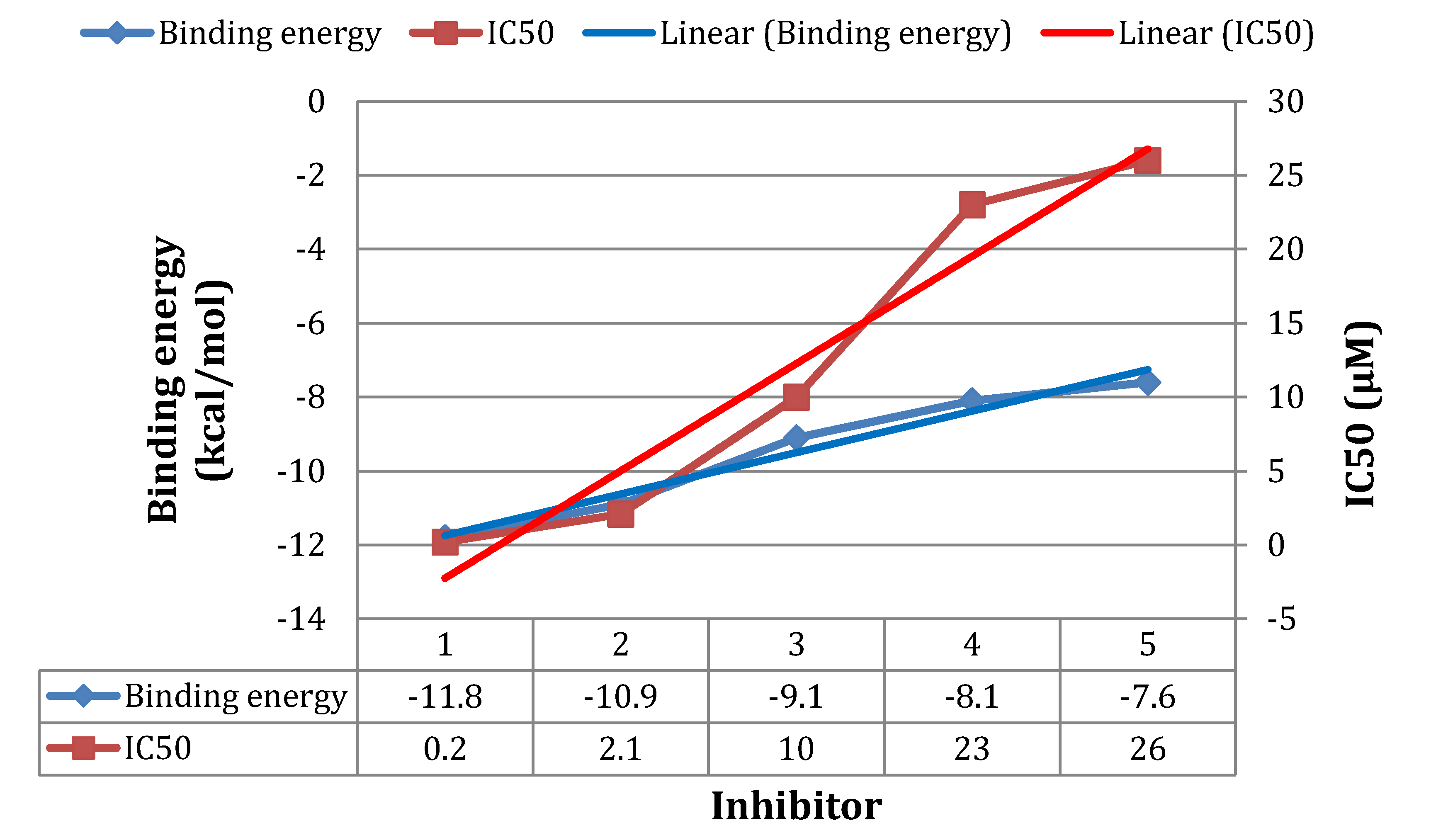
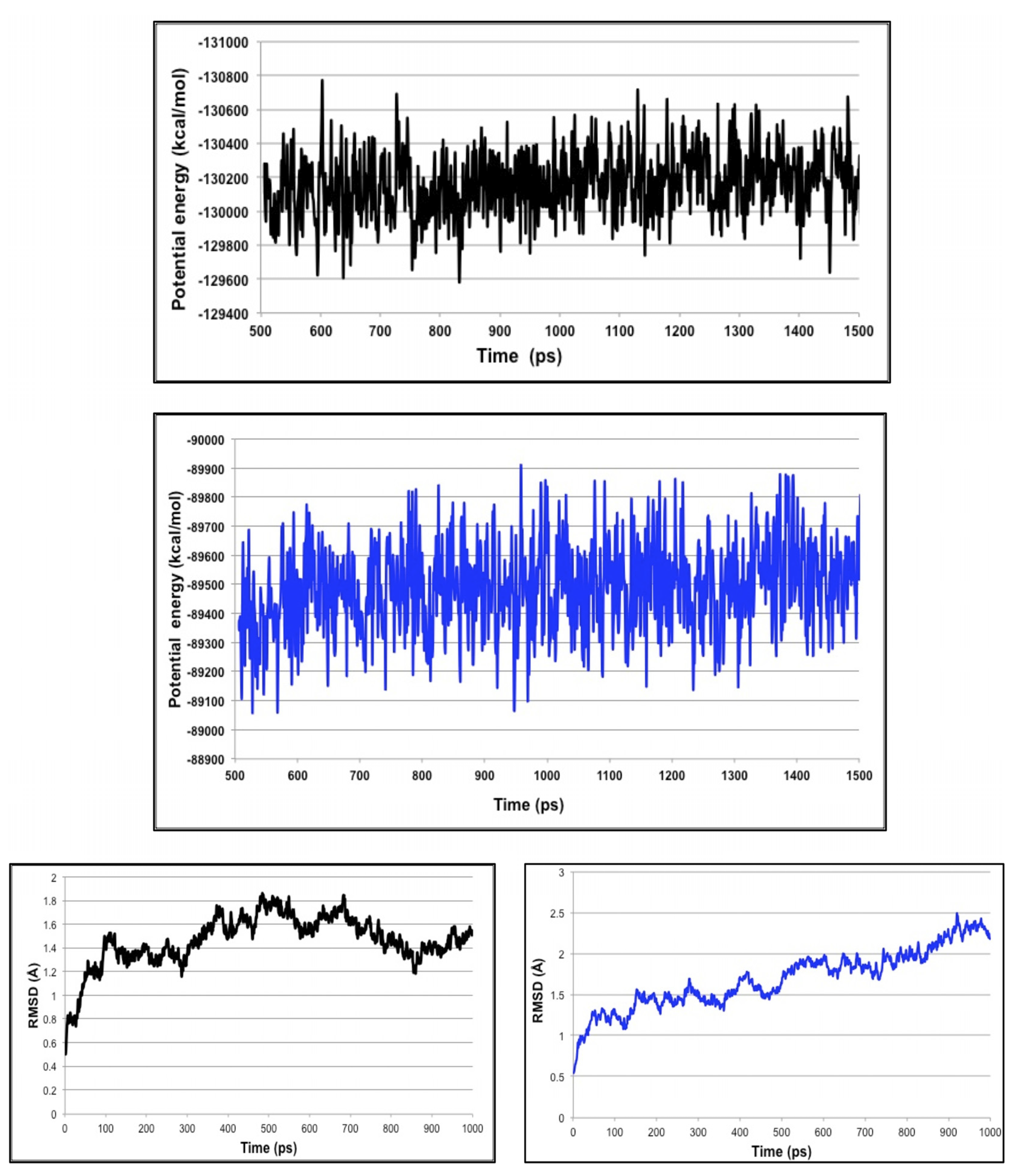
3.3. Molecular Dynamics Simulations
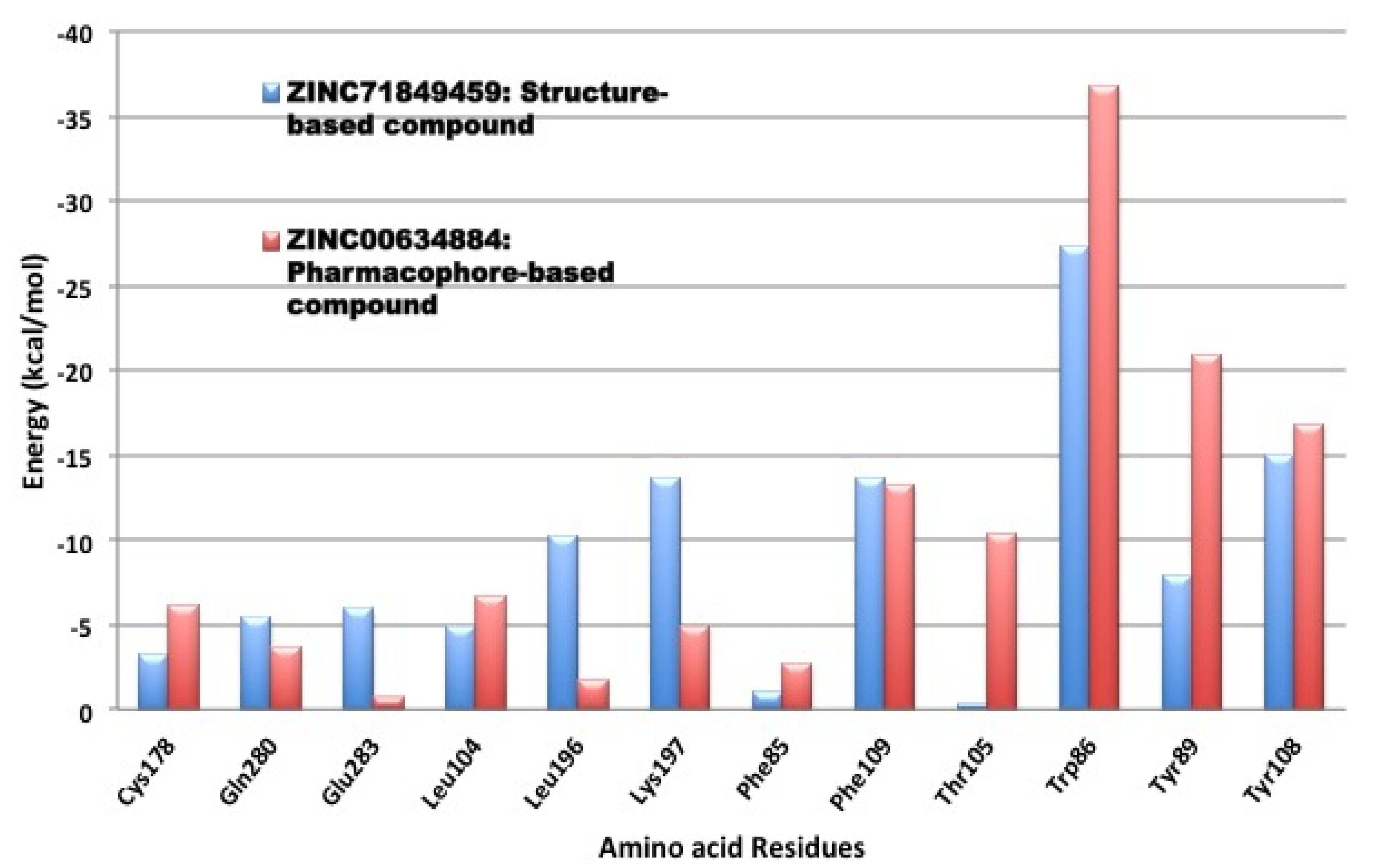
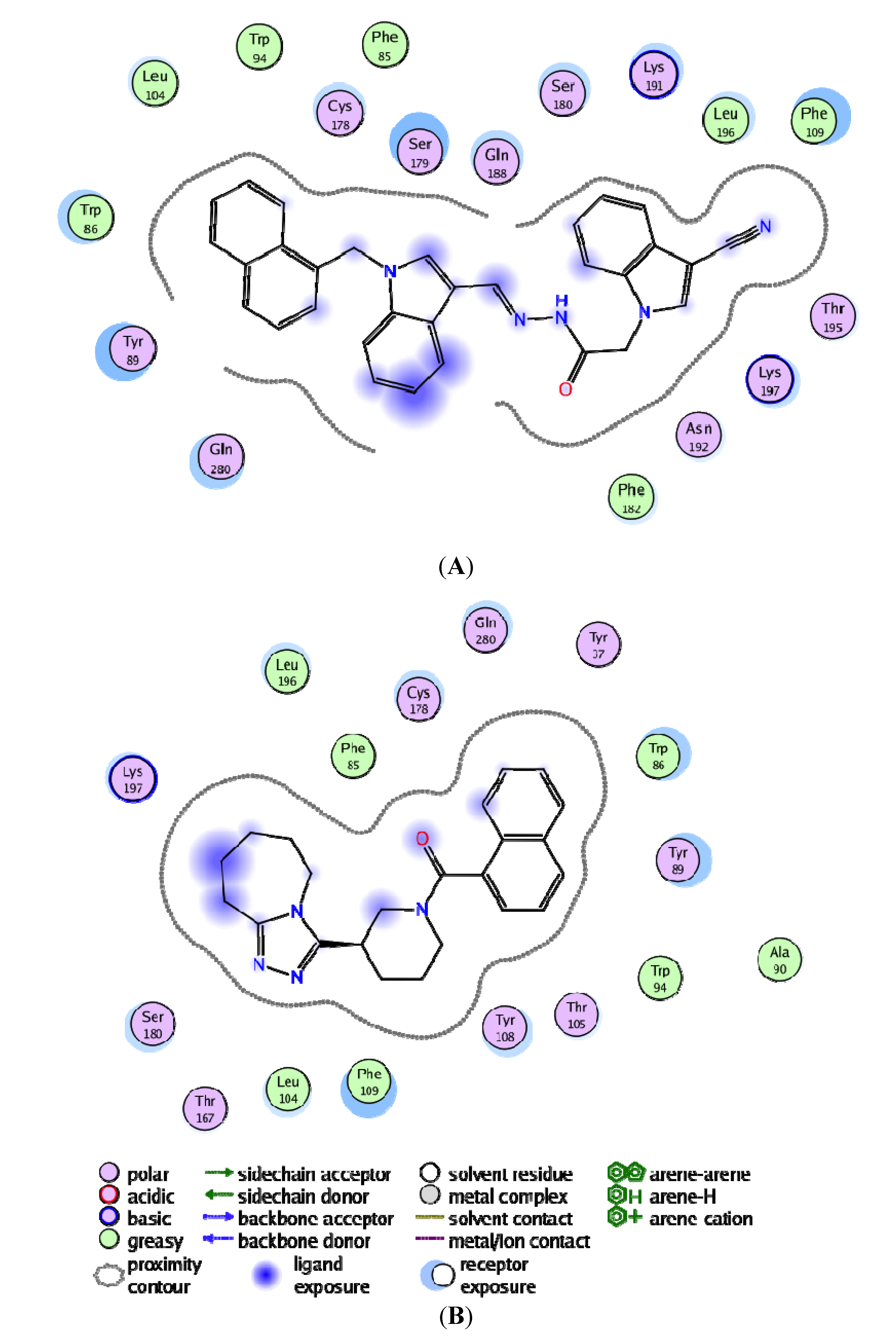
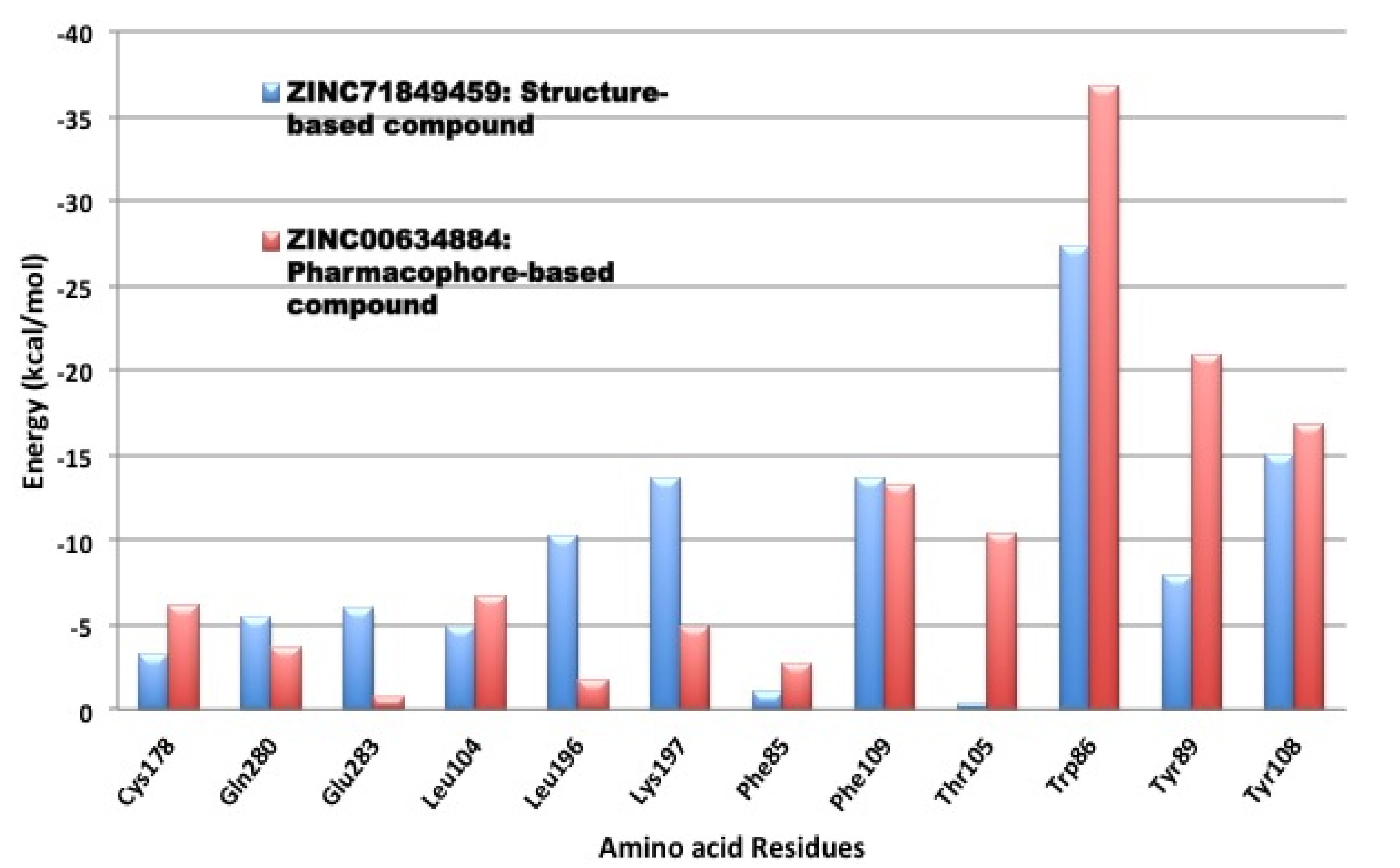
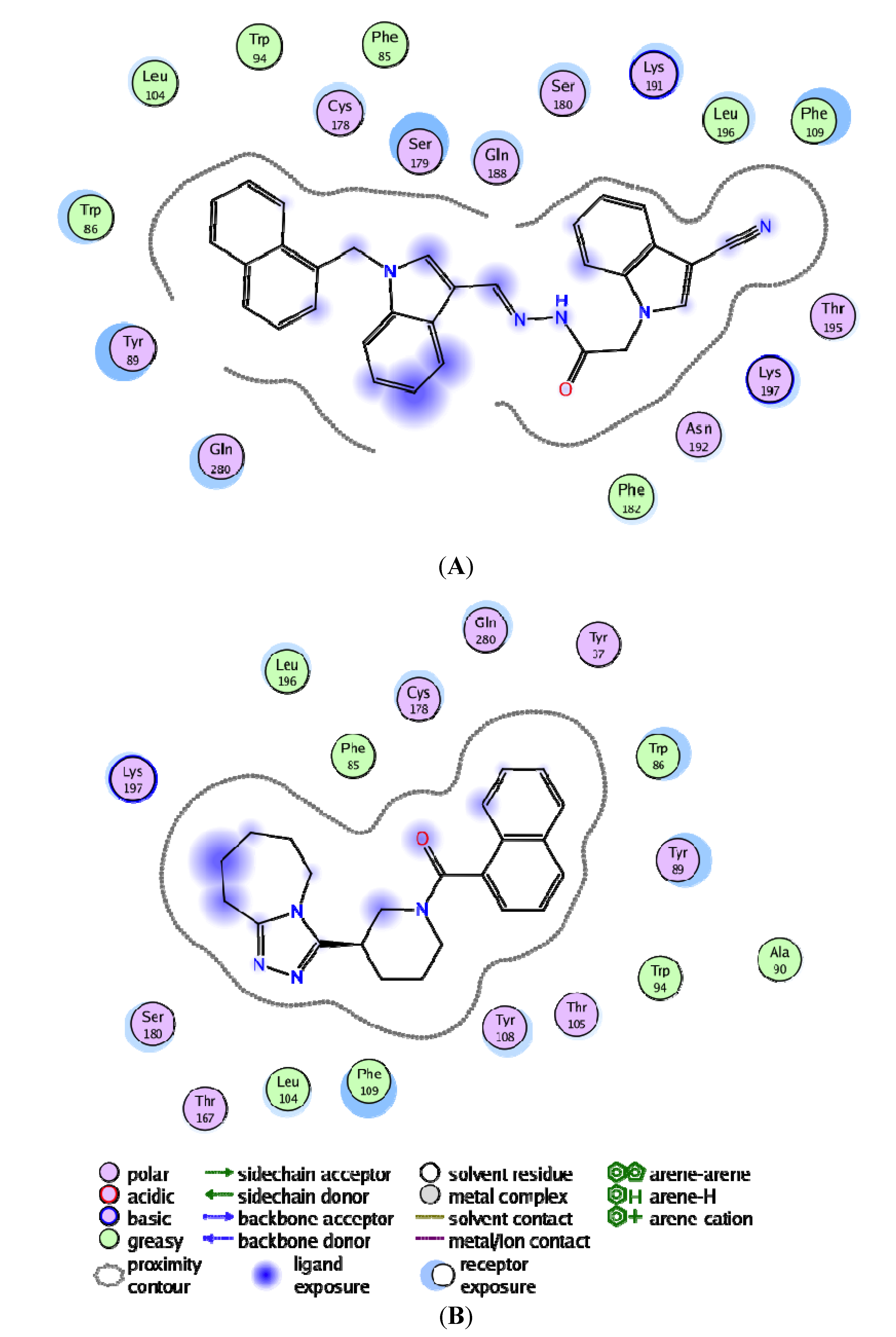
3.4. Per-Residue Interactions
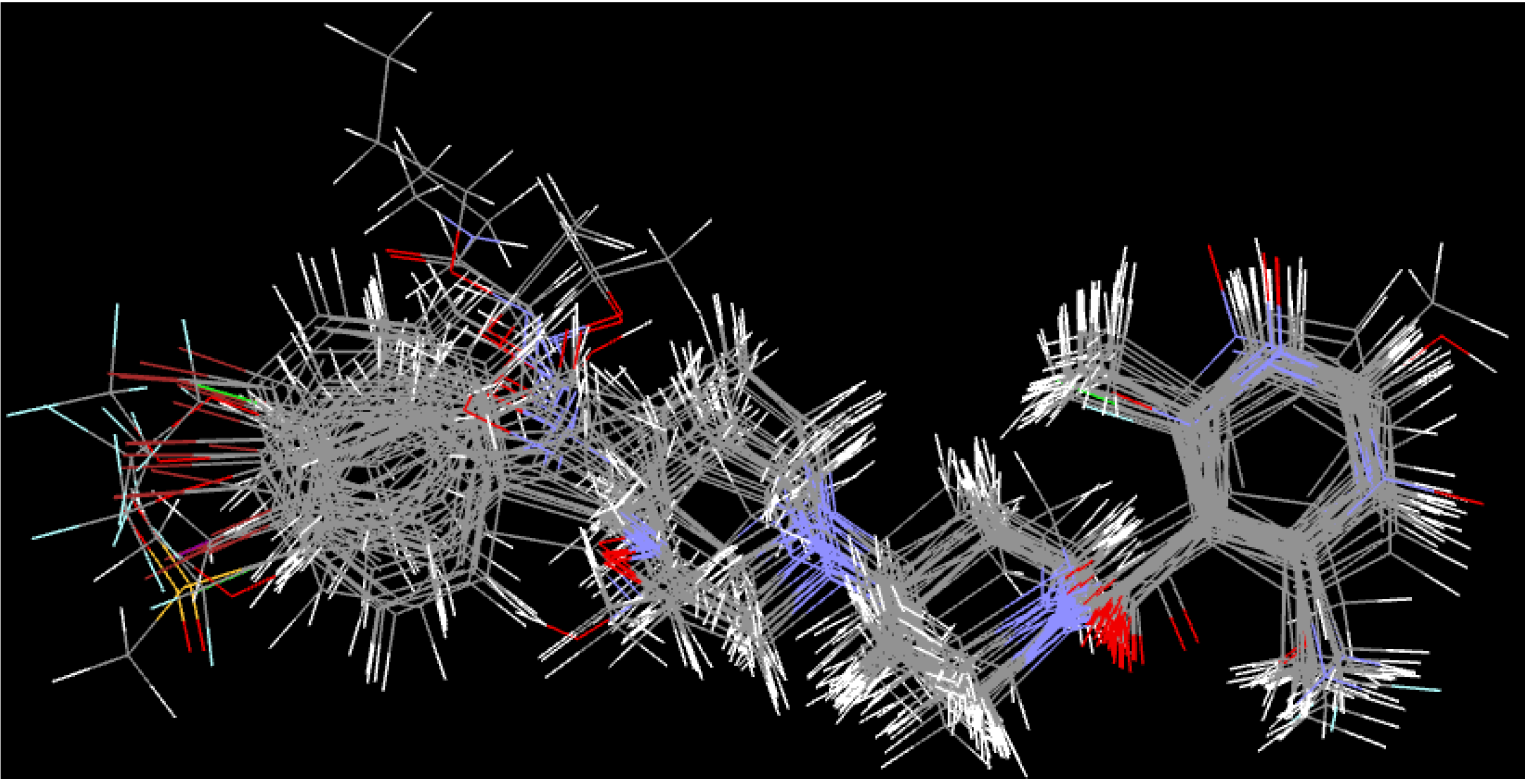
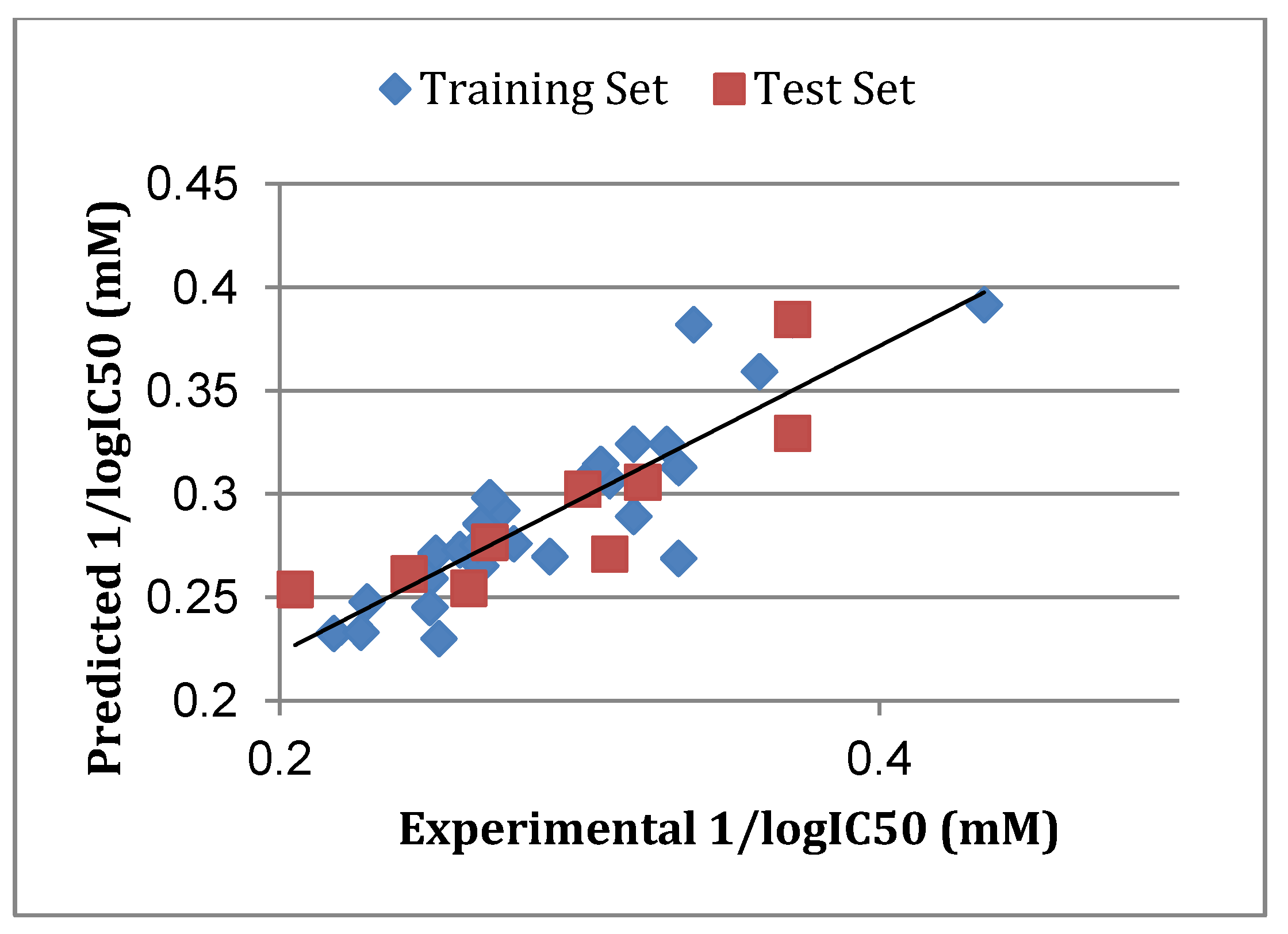
3.5. Atom-Based 3D-QSAR

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Gadhe, C.G.; Kothandan, G.; Cho, S.J. Computational modeling of human coreceptor CCR5 antagonist as a HIV-1 entry inhibitor: Using an integrated homology modeling, docking, and membrane molecular dynamics simulation analysis approach. J. Biomol. Struct. Dyn. 2013, 31, 1251–1279. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, H.; Niu, C.Y.; Luo, C.; Luo, X.M.; Shen, J.H.; Chen, K.X.; Jiang, H.L. Molecular docking and 3D QSAR studies on 1-amino-2-phenyl-4-(piperidin-1-yl)-butanes based on the structural modeling of human CCR5 receptor. Bioorgan. Med. Chem. 2004, 12, 6193–6208. [Google Scholar] [CrossRef]
- Soliman, M.E.S. A Hybrid Structure/Pharmacophore-Based Virtual Screening Approach to Design Potential Leads: A Computer-Aided Design of South African HIV-1 Subtype C Protease Inhibitors. Drug Dev. Res. 2013, 74, 283–295. [Google Scholar] [CrossRef]
- Johnson, B.C.; Pauly, G.T.; Rai, G.; Patel, D.; Bauman, J.D.; Baker, H.L.; Das, K.; Schneider, J.P.; Maloney, D.J.; Arnold, E.; et al. A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology 2012, 9, 1–23. [Google Scholar] [CrossRef]
- Patel, J.R.; Prajapati, L.M. Predictive QSAR modeling on tetrahydropyrimidine-2-one derivatives as HIV-1 protease enzyme inhibitors. Med. Chem. Res. 2013, 22, 2795–2801. [Google Scholar] [CrossRef]
- Zhan, P.; Chen, X.; Li, D.; Fang, Z.; de Clercq, E.; Liu, X. HIV-1 NNRTIs: Structural diversity, pharmacophore similarity, and impliations for drug design. Med. Res. Rev. 2013, 33, E1–E72. [Google Scholar] [CrossRef]
- Johnson, B.C.; Metifiot, M.; Ferris, A.; Pommier, Y.; Hughes, S.H. A Homology Model of HIV-1 Integrase and Analysis of Mutations Designed to Test the Model. J. Mol. Biol. 2013, 425, 2133–2146. [Google Scholar] [CrossRef]
- Pani, A.; Loi, A.G.; Mura, M.; Marceddu, T.; la Colla, P.; Marongiu, M.E. Targeting HIV: Old and new players. Curr. Drug Targets 2002, 2, 17–32. [Google Scholar] [CrossRef]
- Fano, A.; Ritchie, D.W.; Carrieri, A. Modeling the structural basis of human CCR5 chemokine receptor function: From homology model building and molecular dynamics validation to agonist and antagonist docking. J. Chem. Inf. Model. 2006, 46, 1223–1235. [Google Scholar] [CrossRef]
- Manikandan, S.; Malik, B.K. Modeling of human CCR5 as target for HIV-I and virtual screening with marine therapeutic compounds. Bioinformation 2008, 3, 89–94. [Google Scholar] [CrossRef]
- Cormier, E.G.; Persuh, M.; Thompson, D.A.D.; Lin, S.W.; Sakmar, T.P.; Olson, W.C.; Dragic, T. Specific interaction of CCR5 amino-terminal domain peptides containing sulfotyrosines with HIV-1 envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA 2000, 97, 5762–5767. [Google Scholar]
- Farzan, M.; Vasilieva, N.; Schnitzler, C.E.; Chung, S.; Robinson, J.; Gerard, N.P.; Gerard, C.; Choe, H.; Sodroski, J. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J. Biol. Chem. 2000, 275, 33516–33521. [Google Scholar] [CrossRef]
- Cormier, E.G.; Tran, D.N.; Yukhayeva, L.; Olson, W.C.; Dragic, T. Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J. Virol. 2001, 75, 5441–5449. [Google Scholar]
- Cocchi, F.; DEVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar]
- Pease, J.; Horuk, R. Chemokine receptor antagonists. J. Med. Chem. 2012, 55, 9363–9392. [Google Scholar] [CrossRef]
- Ditzel, H.J.; Rosenkilde, M.M.; Garred, P.; Wang, M.; Koefoed, K.; Pedersen, C.; Burton, D.R.; Schwartz, T.W. The CCR5 receptor acts as an alloantigen in CCR5Delta32 homozygous individuals: Identification of chemokineand HIV-1-blocking human antibodies. Proc. Natl. Acad. Sci. USA 1998, 95, 5241–5245. [Google Scholar] [CrossRef]
- Kothandan, G.; Gadhe, C.G.; Cho, S.J. Structural Insights from Binding Poses of CCR2 and CCR5 with Clinically Important Antagonists: A Combined In Silico Study. PLoS One 2012, 7, e32864. [Google Scholar] [CrossRef]
- Perez-Nueno, V.I.; Ritchie, D.W.; Rabal, O.; Pascual, R.; Borrell, J.I.; Teixido, J. Comparison of ligand-based and receptor-based virtual screening of HIV entry inhibitors for the CXCR4 and CCR5 receptors using 3D ligand shape matching and ligand-receptor docking. J. Chem. Inf. Model. 2008, 48, 509–533. [Google Scholar] [CrossRef]
- Afantitis, A.; Melagraki, G.; Sarimveis, H.; Koutentis, P.A.; Markopoulos, J.; Igglessi-Markopoulou, O. Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques. J. Comput. Aided Mol. Des. 2006, 20, 83–95. [Google Scholar] [CrossRef]
- Aher, Y.D.; Agrawal, A.; Bharatam, P.V.; Garg, P. 3D-QSAR studies of substituted 1-(3,3-diphenylpropyl)-piperidinyl amides and ureas as CCR5 receptor antagonists. J. Mol. Model. 2007, 13, 519–529. [Google Scholar] [CrossRef]
- Kellenberger, E.; Springael, J.-Y.; Parmentier, M.; Hachet-Haas, M.; Galzi, J.-L.; Rognan, D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J. Med. Chem. 2007, 50, 1294–1303. [Google Scholar]
- Consortium, T. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for “Omics” research on drugs. Nucleic Acids Res. 2011, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4. [Google Scholar] [CrossRef]
- Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 5.12.1 (version 5). 2013. ChemAxon http://www.chemaxon.com.
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Forli, S. AutoDock|Raccoon: An automated tool for preparing AutoDock virtual screenings. 2013.
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Kruger, H.G.; Govender, T.; Maguire, G.E.M.; Sayed, Y.; Ibrahim, M.A.A.; Naicker, P.; Soliman, M.E.S. Comparison of the Molecular dynamics and calculated binding free energies for nine FDA-approved HIV-1 PR drugs against subtype B and C-SA HIV PR. Chem. Biol. Drug Des. 2013, 81, 208–218. [Google Scholar] [CrossRef]
- The Molecular Operating Environment (MOE) available under license from Chemical Computing Group Inc., 1010 Sherbrooke St. W. Suite 910, Montreal, Quebec, Canada H3A 2R7.
- Discovery Studio Modeling Environment; Release 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2012.
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucl. Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moonsamy, S.; Dash, R.C.; Soliman, M.E.S. Integrated Computational Tools for Identification of CCR5 Antagonists as Potential HIV-1 Entry Inhibitors: Homology Modeling, Virtual Screening, Molecular Dynamics Simulations and 3D QSAR Analysis. Molecules 2014, 19, 5243-5265. https://doi.org/10.3390/molecules19045243
Moonsamy S, Dash RC, Soliman MES. Integrated Computational Tools for Identification of CCR5 Antagonists as Potential HIV-1 Entry Inhibitors: Homology Modeling, Virtual Screening, Molecular Dynamics Simulations and 3D QSAR Analysis. Molecules. 2014; 19(4):5243-5265. https://doi.org/10.3390/molecules19045243
Chicago/Turabian StyleMoonsamy, Suri, Radha Charan Dash, and Mahmoud E. S. Soliman. 2014. "Integrated Computational Tools for Identification of CCR5 Antagonists as Potential HIV-1 Entry Inhibitors: Homology Modeling, Virtual Screening, Molecular Dynamics Simulations and 3D QSAR Analysis" Molecules 19, no. 4: 5243-5265. https://doi.org/10.3390/molecules19045243