Structural Modeling of Djenkolic Acid with Sulfur Replaced by Selenium and Tellurium

Abstract
:1. Introduction
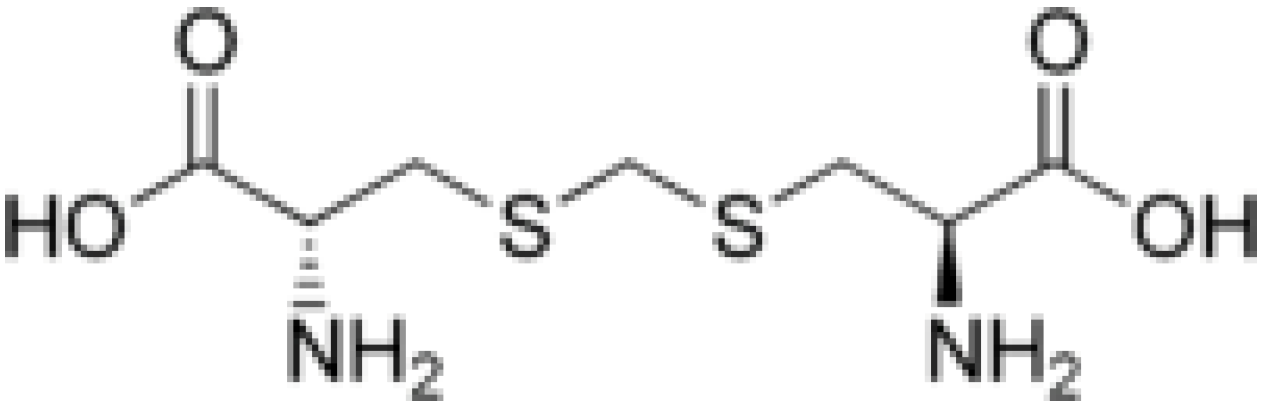
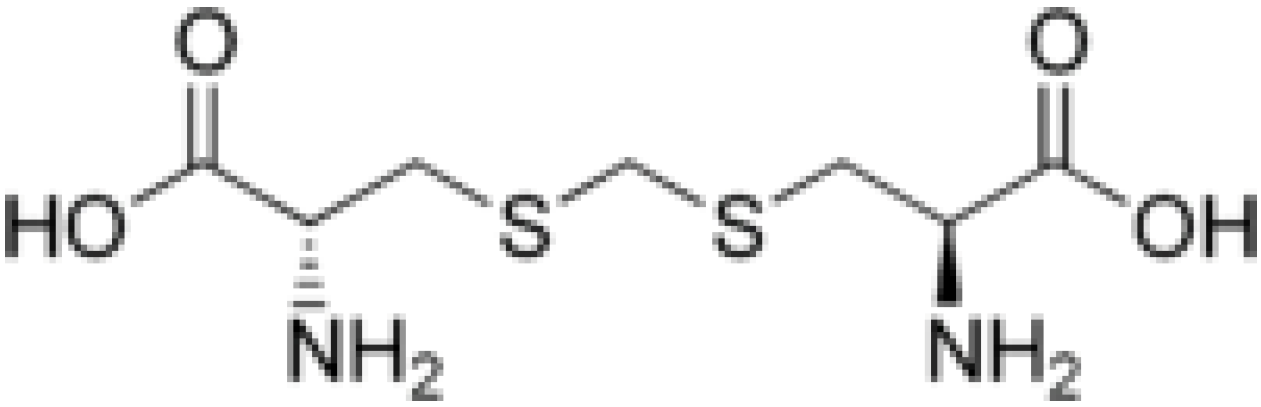
2. Results and Discussion

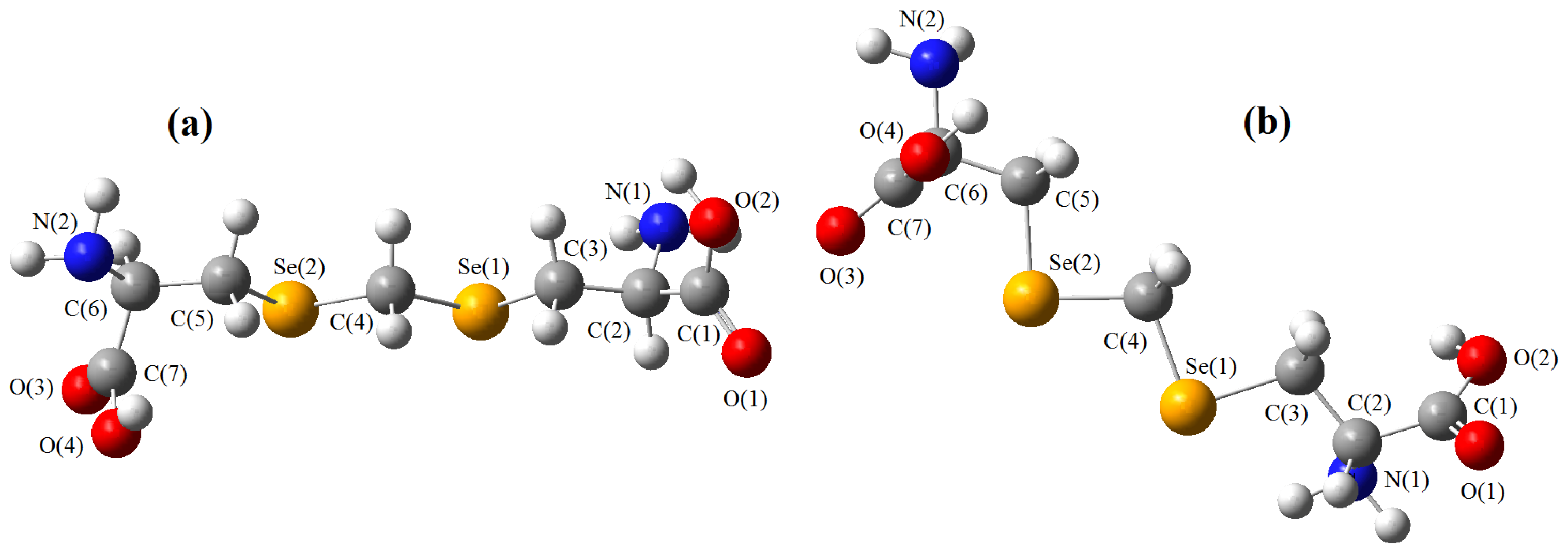
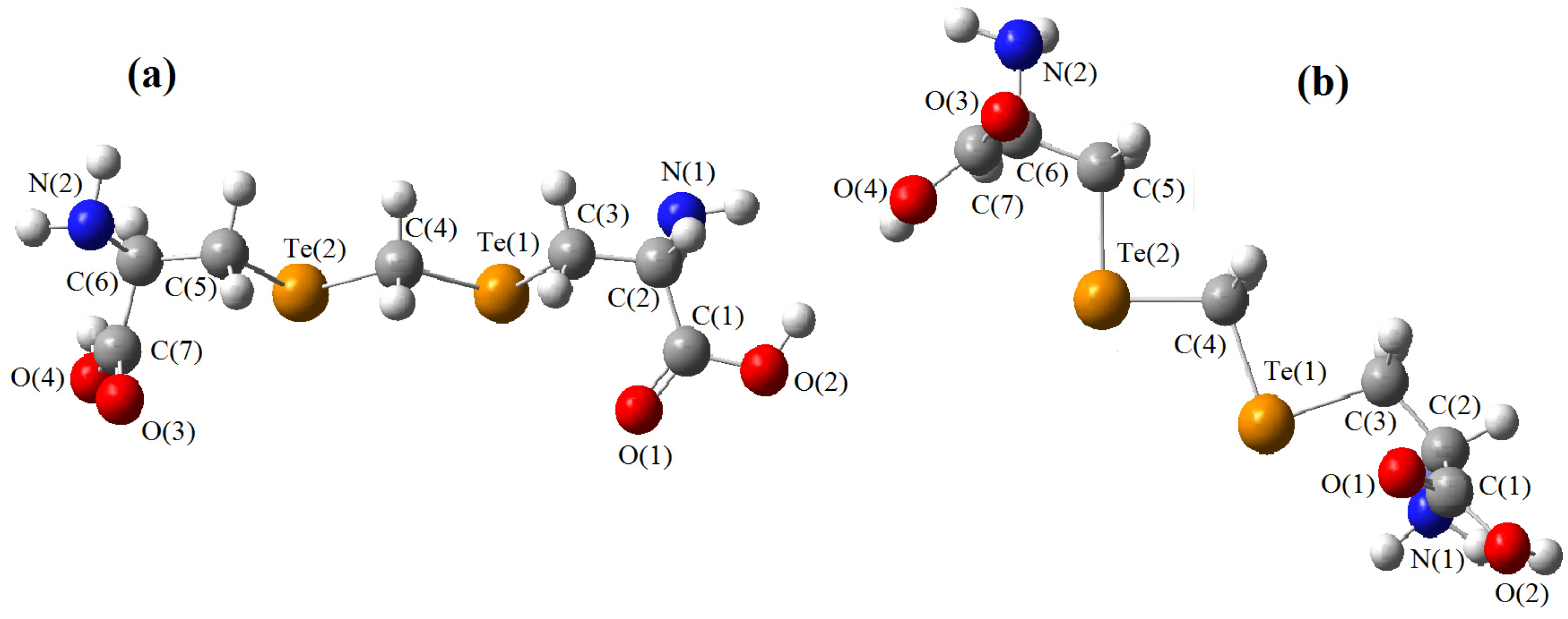

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformers | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Distances | ||||||||||
| C(1)-C(2) | 1.51 | 1.50 | 1.50 | 1.51 | 1.50 | 1.50 | 1.50 | 1.50 | 1.51 | 1.50 |
| C(2)-C(3) | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.54 | 1.53 |
| C(2)-N(1) | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 |
| C(3)-S(1) | 1.83 | 1.82 | 1.82 | 1.82 | 1.83 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 |
| C(4)-S(1) | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 |
| C(4)-S(2) | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 | 1.82 |
| C(5)-S(2) | 1.82 | 1.83 | 1.82 | 1.82 | 1.82 | 1.83 | 1.82 | 1.83 | 1.82 | 1.82 |
| C(5)-C(6) | 1.53 | 1.54 | 1.53 | 1.53 | 1.54 | 1.54 | 1.53 | 1.53 | 1.54 | 1.54 |
| C(6)-C(7) | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 | 1.51 |
| C(6)-N(2) | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 |
| C(1)-O(1) | 1.25 | 1.26 | 1.25 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 |
| C(1)-O(2) | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C(7)-O(3) | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C(7)- O(4) | 1.25 | 1.26 | 1.25 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 |
| S-S | 2.98 | 2.96 | 2.96 | 2.96 | 2.96 | 3.00 | 2.98 | 2.96 | 2.96 | 2.96 |
| Angles | ||||||||||
| C(1)-C(2)-C(3) | 111.12 | 110.55 | 109.92 | 111.91 | 111.38 | 109.59 | 109.61 | 111.07 | 112.94 | 110.54 |
| C(2)-C(3)-S(1) | 110.69 | 110.50 | 110.97 | 110.57 | 112.35 | 110.89 | 111.04 | 110.45 | 112.10 | 110.89 |
| C(3)-S(1)-C(4) | 95.69 | 94.66 | 94.72 | 94.68 | 94.76 | 95.91 | 94.74 | 94.76 | 94.68 | 96.19 |
| S(1)-C4)-S(2) | 109.67 | 108.96 | 108.93 | 108.93 | 108.85 | 109.59 | 109.68 | 108.89 | 108.93 | 108.93 |
| C(4)-S(2)-C(5) | 94.71 | 94.71 | 94.74 | 94.73 | 94.77 | 96.91 | 95.66 | 94.78 | 94.68 | 96.17 |
| S(2)-C(5)-C(6) | 110.55 | 112.83 | 110.91 | 110.75 | 112.37 | 111.77 | 110.77 | 110.82 | 110.95 | 111.27 |
| C(5)-C(6)-C(7) | 110.37 | 113.71 | 111.00 | 111.17 | 111.71 | 112.23 | 111.20 | 111.82 | 113.08 | 110.19 |
| O(1)-C(1)-O(2) | 118.84 | 120.18 | 118.96 | 119.87 | 120.11 | 120.24 | 119.96 | 118.72 | 118.84 | 120.19 |
| O(3)-C(7)-O(4) | 119.88 | 118.64 | 118.95 | 118.83 | 118.76 | 118.72 | 118.03 | 118.76 | 118.72 | 118.82 |
| C(1)-C(2)-N(1) | 109.37 | 110.79 | 109.62 | 110.49 | 110.26 | 110.39 | 110.29 | 111.07 | 110.07 | 110.73 |
| C(5)-C(6)-N(2) | 110.10 | 111.74 | 110.07 | 110.91 | 111.55 | 111.49 | 110.89 | 111.20 | 109.30 | 111.92 |
| C(2)-C(1)-O(1) | 119.65 | 120.24 | 119.46 | 119.33 | 120.38 | 119.90 | 119.55 | 119.19 | 120.78 | 120.23 |
| C(2)-C(1)-O(2) | 121.50 | 119.56 | 121.56 | 120.79 | 119.47 | 119.85 | 120.49 | 122.07 | 120.67 | 119.57 |
| C(6)-C(7)-O(3) | 120.63 | 120.49 | 121.62 | 121.51 | 120.30 | 120.48 | 121.51 | 119.98 | 120.65 | 121.57 |
| C(6)-C(7)-O(4) | 119.42 | 120.81 | 119.41 | 119.65 | 120.92 | 120.77 | 119.65 | 119.25 | 120.61 | 119.59 |
| Energy (×10−8) | −3.01 | −6.19 | −4.55 | −2.72 | −2.06 | −3.27 | −3.86 | −4.18 | −2.47 | −4.75 |
| Conformers | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Distances | ||||||||||
| C(1)-C(2) | 1.50 | 1.50 | 1.50 | 1.51 | 1.50 | 1.50 | 1.51 | 1.51 | 1.51 | 1.50 |
| C(2)-C(3) | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.54 | 1.54 |
| C(2)-N(1) | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 |
| C(3)-Se(1) | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.96 | 1.95 |
| C(4)-Se(1) | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 |
| C(4)-Se(2) | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 | 1.94 |
| C(5)-Se(2) | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 | 1.95 |
| C(5)-C(6) | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 |
| C(6)-C(7) | 1.51 | 1.50 | 1.51 | 1.51 | 1.51 | 1.50 | 1.50 | 1.51 | 1.51 | 1.50 |
| C(6)-N(2) | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 |
| C(1)-O(1) | 1.26 | 1.26 | 1.25 | 1.26 | 1.26 | 1.25 | 1.25 | 1.25 | 1.26 | 1.25 |
| C(1)-O(2) | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C(7)-O(3) | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C(7)-O(4) | 1.26 | 1.26 | 1.25 | 1.26 | 1.26 | 1.26 | 1.26 | 1.25 | 1.25 | 1.26 |
| Se-Se | 3.18 | 3.16 | 3.17 | 3.17 | 3.17 | 3.17 | 3.18 | 3.17 | 3.17 | 3.17 |
| Angles | ||||||||||
| C(1)-C(2)-C(3) | 110.40 | 109.54 | 109.98 | 111.95 | 111.36 | 110.17 | 111.15 | 110.08 | 112.87 | 110.18 |
| C(2)-C(3)-Se(1) | 110.69 | 111.23 | 111.13 | 110.68 | 112.85 | 112.24 | 111.02 | 110.63 | 112.40 | 111.44 |
| C(3)-Se(1)-C(4) | 92.32 | 92.44 | 92.25 | 92.27 | 91.21 | 92.30 | 93.12 | 92.25 | 92.21 | 93.63 |
| Se(1)-C4)-Se(2) | 109.61 | 108.94 | 109.14 | 109.06 | 109.15 | 109.06 | 109.59 | 109.14 | 109.10 | 109.11 |
| C(4)-Se(2)-C(5) | 93.09 | 93.63 | 92.17 | 92.31 | 92.20 | 92.30 | 92.97 | 92.27 | 92.18 | 93.63 |
| Se(2)-C(5)-C(6) | 110.99 | 111.64 | 111.22 | 110.94 | 110.98 | 111.93 | 111.24 | 111.04 | 111.07 | 110.93 |
| C(5)-C(6)-C(7) | 111.09 | 111.07 | 110.98 | 111.12 | 111.10 | 110.69 | 109.64 | 111.76 | 110.59 | 110.59 |
| O(1)-C(1)-O(2) | 119.88 | 119.97 | 118.97 | 119.87 | 120.11 | 118.83 | 118.84 | 118.72 | 118.48 | 118.83 |
| O(3)-C(7)-O(4) | 118.83 | 120.20 | 118.94 | 118.84 | 118.82 | 119.99 | 119.96 | 118.75 | 118.88 | 120.19 |
| C(1)-C(2)-N(1) | 111.60 | 110.59 | 108.62 | 110.48 | 110.29 | 109.26 | 109.40 | 111.09 | 110.09 | 109.21 |
| C(5)-C(6)-N(2) | 110.90 | 110.89 | 109.62 | 110.94 | 110.91 | 110.38 | 110.37 | 111.21 | 109.91 | 110.63 |
| C(2)-C(1)-O(1) | 119.45 | 120.73 | 119.46 | 119.32 | 120.39 | 119.59 | 119.64 | 119.18 | 120.77 | 119.59 |
| C(2)-C(1)-O(2) | 120.63 | 119.29 | 119.46 | 120.79 | 119.46 | 121.57 | 121.51 | 122.08 | 120.68 | 121.57 |
| C(6)-C(7)-O(3) | 121.51 | 119.92 | 121.55 | 121.50 | 121.52 | 120.42 | 120.46 | 121.98 | 121.49 | 119.57 |
| C(6)-C(7)-O(4) | 119.65 | 119.82 | 119.42 | 119.65 | 119.64 | 119.57 | 119.56 | 119.25 | 119.61 | 120.23 |
| Energy (×10−8) | −3.45 | −4.32 | −2.73 | −3.01 | −2.83 | −3.36 | −6.96 | −4.67 | −4.13 | −4.31 |
| Conformers | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Distances | ||||||||||
| C(1)-C(2) | 1.50 | 1.50 | 1.50 | 1.51 | 1.50 | 1.50 | 1.50 | 1.51 | 1.51 | 1.50 |
| C(2)-C(3) | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 |
| C(2)-N(1) | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 |
| C(3)-Te(1) | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 |
| C(4)-Te(1) | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 |
| C(4)-Te(2) | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 | 2.14 |
| C(5)-Te(2) | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 | 2.15 |
| C(5)-C(6) | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 |
| C(6)-C(7) | 1.53 | 1.50 | 1.50 | 1.51 | 1.51 | 1.50 | 1.50 | 1.50 | 1.50 | 1.50 |
| C(6)-N(2) | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 |
| C(1)-O(1) | 1.26 | 1.26 | 1.25 | 1.26 | 1.26 | 1.26 | 1.25 | 1.25 | 1.25 | 1.26 |
| C(1)-O(2) | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C(7)-O(3) | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 | 1.39 |
| C(7)-O(4) | 1.25 | 1.26 | 1.25 | 1.25 | 1.26 | 1.26 | 1.25 | 1.25 | 1.26 | 1.26 |
| Te-Te | 3.49 | 3.50 | 3.49 | 3.49 | 3.49 | 3.49 | 3.50 | 3.50 | 3.49 | 3.49 |
| Angles | ||||||||||
| C(1)-C(2)-C(3) | 110.40 | 109.81 | 109.96 | 111.94 | 111.34 | 110.66 | 109.64 | 110.07 | 113.51 | 110.57 |
| C(2)-C(3)-Te(1) | 111.05 | 111.67 | 111.57 | 111.08 | 113.57 | 112.52 | 111.78 | 110.81 | 113.82 | 111.18 |
| C(3)-Te(1)-C(4) | 90.47 | 90.40 | 90.41 | 90.32 | 90.55 | 90.43 | 90.59 | 90.52 | 90.49 | 91.54 |
| Te(1)-C4)-Te(2) | 109.36 | 109.79 | 109.37 | 109.41 | 109.24 | 109.36 | 109.65 | 108.50 | 109.36 | 109.35 |
| C(4)-Te(2)-C(5) | 90.47 | 92.62 | 90.39 | 90.41 | 90.53 | 90.45 | 91.33 | 91.99 | 90.54 | 91.48 |
| Te(2)-C(5)-C(6) | 113.36 | 111.85 | 111.55 | 111.34 | 111.26 | 111.73 | 111.32 | 111.33 | 111.43 | 111.89 |
| C(5)-C(6)-C(7) | 111.09 | 111.04 | 110.89 | 111.06 | 111.04 | 110.16 | 111.10 | 111.68 | 110.54 | 110.14 |
| O(1)-C(1)-O(2) | 119.88 | 120.25 | 118.98 | 119.88 | 120.09 | 119.99 | 119.96 | 118.73 | 118.54 | 120.20 |
| O(3)-C(7)-O(4) | 118.84 | 120.19 | 118.95 | 119.82 | 118.83 | 118.83 | 118.83 | 118.75 | 118.89 | 118.83 |
| C(1)-C(2)-N(1) | 111.62 | 109.56 | 109.62 | 110.51 | 110.27 | 110.78 | 110.23 | 111.15 | 111.23 | 110.78 |
| C(5)-C(6)-N(2) | 110.92 | 110.92 | 110.07 | 110.91 | 110.97 | 111.73 | 110.92 | 111.20 | 109.94 | 111.88 |
| C(2)-C(1)-O(1) | 119.45 | 120.11 | 119.46 | 119.33 | 120.40 | 119.58 | 119.56 | 119.18 | 120.70 | 120.23 |
| C(2)-C(1)-O(2) | 120.64 | 119.62 | 121.54 | 120.78 | 119.46 | 120.42 | 120.46 | 122.07 | 120.69 | 119.56 |
| C(6)-C(7)-O(3) | 119.65 | 119.87 | 121.60 | 121.51 | 121.51 | 121.57 | 121.50 | 119.25 | 119.61 | 119.58 |
| C(6)-C(7)-O(4) | 121.50 | 199.92 | 119.42 | 119.66 | 119.65 | 119.59 | 119.65 | 121.98 | 121.48 | 121.57 |
| Energy (×10−8) | −2.09 | −3.52 | −2.66 | −4.08 | −3.73 | −2.95 | −3.50 | −3.08 | −2.03 | −3.63 |

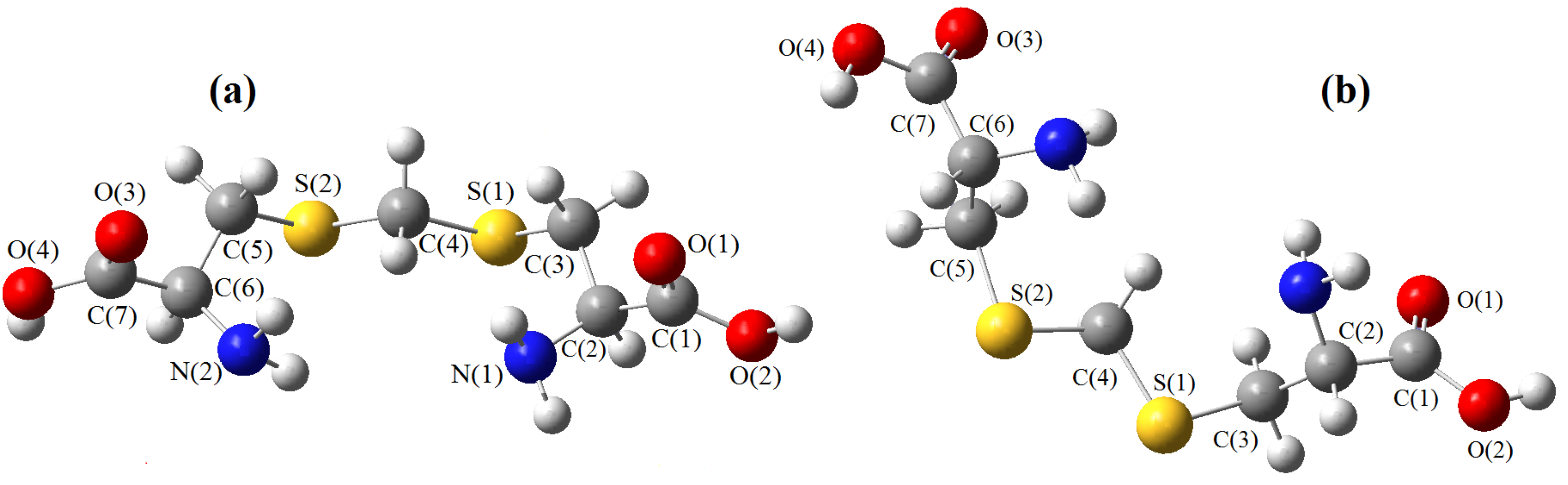
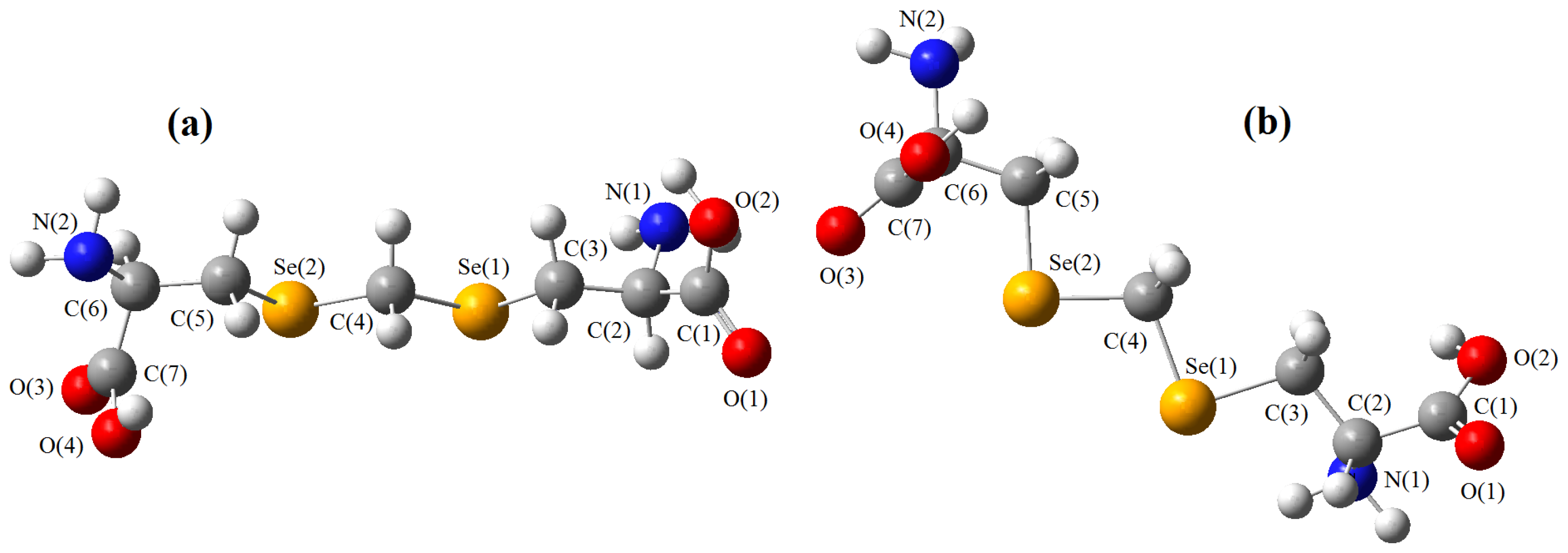
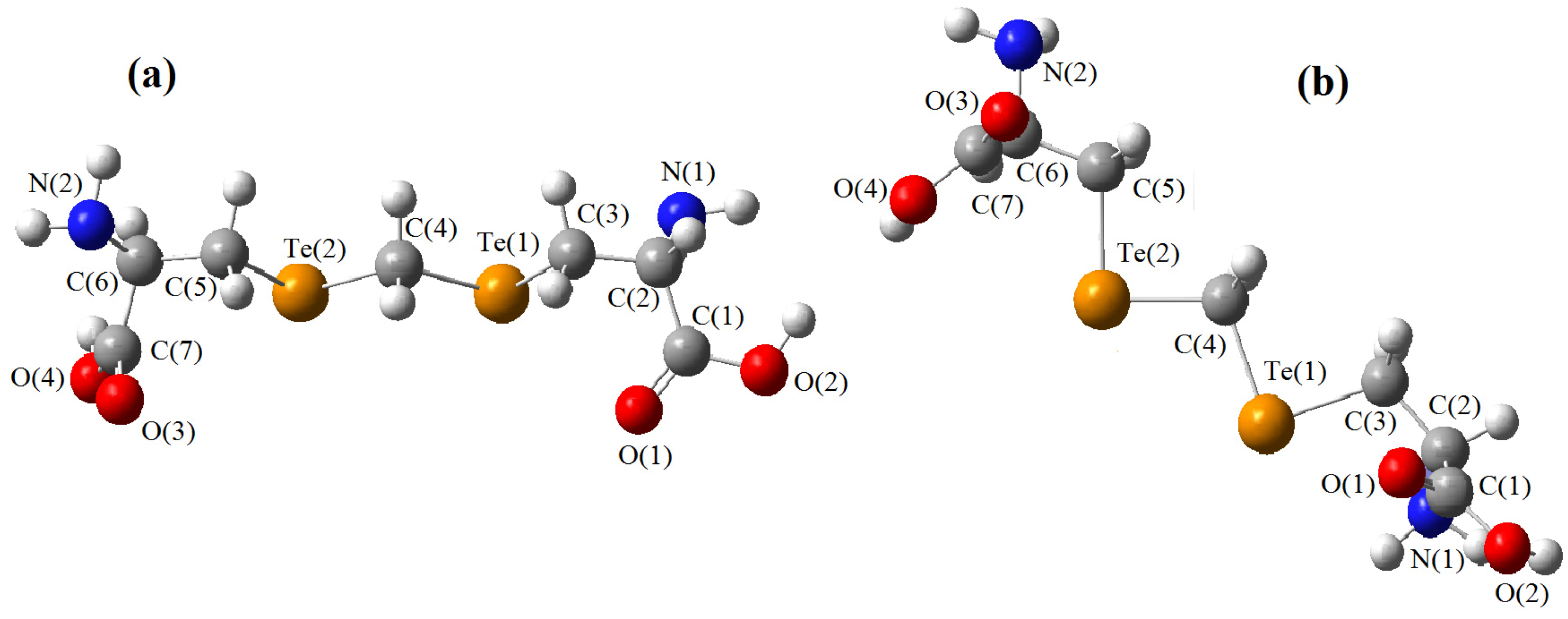
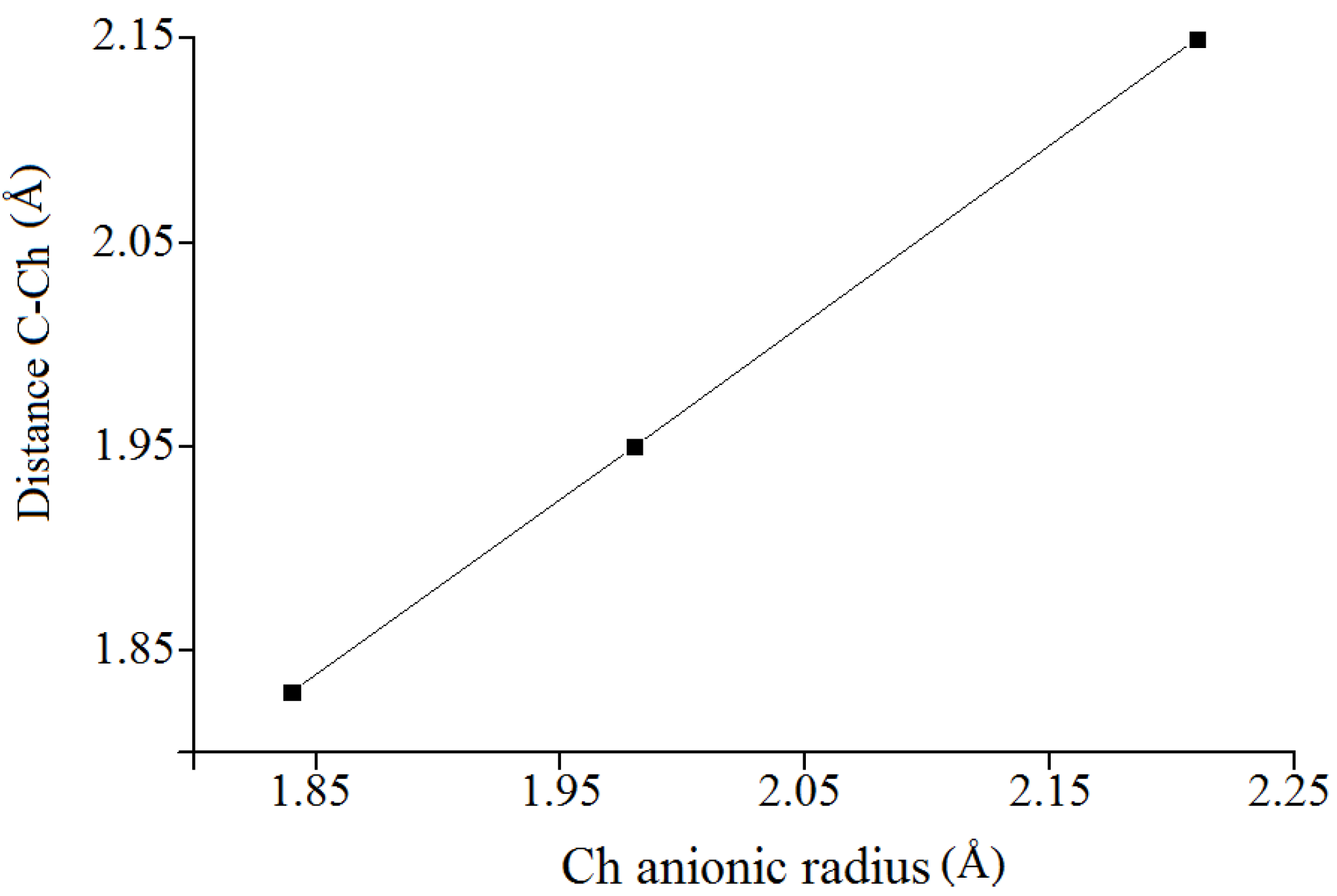
 must be located in the same plane. This condition limits the number of possible conformers because of hindered rotation about C(4), so most of the rotamers must be generated in the peripheral areas. It is interesting that the aforementioned blocks are reminiscent of the fragments S····S belonging R-type ring motifs, characteristic for the structure of l-cysteines obtained at high pressures [16]. The main effects of compression were to reduce S····S distances from 3.846 to 3.450 Å. In our case of two cysteine molecules linked through a methylene group these distances are even shorter: 2.96–3.50 Å, coinciding only at the upper limit.
must be located in the same plane. This condition limits the number of possible conformers because of hindered rotation about C(4), so most of the rotamers must be generated in the peripheral areas. It is interesting that the aforementioned blocks are reminiscent of the fragments S····S belonging R-type ring motifs, characteristic for the structure of l-cysteines obtained at high pressures [16]. The main effects of compression were to reduce S····S distances from 3.846 to 3.450 Å. In our case of two cysteine molecules linked through a methylene group these distances are even shorter: 2.96–3.50 Å, coinciding only at the upper limit.3. Material and Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Melnikov, P.; Cuin, A.; Corbi, P.P.; Cavicchioli, M.; Massabni, A.C. Powder X-ray characterization of djenkolic acid. Powder Diffr. 2001, 16, 1–2. [Google Scholar] [CrossRef]
- Du Vigneaud, V.; Patterson, W.I. The synthesis of djenkolic acid. J. Biol. Chem. 1936, 114, 533–538. [Google Scholar]
- Tetsuji Shiseido Co Ltd. Cosmetics to promote maturation of cornified envelope and method for treatment. EP 1374832 A1, 2 January 2004. [Google Scholar]
- Bigoli, F.; Lanfranchi, M.; Leporati, E.; Nardelli, M.; Pellinghelli, M.A. The structure of S,S'-methylenebis(L-cysteine) monohydrochloride. Acta Crystallogr. 1982, B38, 498–502. [Google Scholar]
- Cavicchioli, M.; Corbi, P.P.; Melnikov, P.; Massabni, A.C. Synthesis, characterization and thermal behavior of complexes of Cu(II), Zn(II) and Cd(II) with S,S'-methylenebis (cysteine). J. Inorg. Biochem. 2002, 55, 951–959. [Google Scholar]
- Melnikov, P.; Corbi, P.P.; Diaz Aguila, C.; Zacharias, M.A.; Cavicchioli, M.; Massabni, A.C. Iron(II) djenkolate: synthesis and properties. J. Alloy. Compd. 2000, 307, 179–183. [Google Scholar] [CrossRef]
- Nascimento, V.A.; Melnikov, P.; Zanoni, L.Z. Comparative structural modeling of cysteine and selenocysteine. JP J. Solids Struct. 2011, 5, 153–161. [Google Scholar]
- Melnikov, P.; Nascimento, V.A.; Consolo, L.Z.Z.; Silva, A.F. Comparative structural modeling of telluromethionine and isosteric aminoacids. Chem. Phys. Res. J. 2013, 6, 1–12. [Google Scholar]
- Rajeswaran, M.; Parthasarathy, R. Structure of dl-selenomethionine, C5H11NO2Se. Acta Crystallogr. 1984, C40, 647–650. [Google Scholar]
- Guillot, R.; Muzet, N.; Dahaoui, S.; Lecomte, C.; Jelsch, C. Experimental and theoretical charge density of dl-alanyl-methionine. Acta Crystallogr. B 2001, 57, 567–578. [Google Scholar] [CrossRef]
- Taniguchi, T.; Takaki, Y.; Sakurai, K. The crystal structures of the α and β forms of dl-methionine. Bull. Chem. Soc. Jpn. 1980, 53, 803–804. [Google Scholar] [CrossRef]
- Mostad, A.; Natarajan, S. Crystal and molecular structure of dl-methionine nitrate. Z. Kristallogr. 2010, 172, 175–182. [Google Scholar] [CrossRef]
- Gajda, J.; Pacholczyk, J.; Bujacz, A.; Bartoszak-Adamanska, E.; Bujacz, G.; Ciesielski, W.; Potrzebowski, M. Structure and dynamics of L-selenomethionine in the solid state. J. Phys. Chem. B 2006, 110, 25692–25701. [Google Scholar] [CrossRef]
- The Cambridge Crystallography Database, version 5.31; University Press: London, UK, 2010.
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Appendix A: Typical. Interatomic Distances in Organic Compounds and Organometallic Compounds and Coordination Complexes of the d- and f-block metals. In Structure Correlations; Bürgi, H.-B., Dunitz, J.D., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; Volume 1. [Google Scholar] [CrossRef]
- Moggach, S.A.; Allan, D.R.; Clark, S.J.; Gutmann, M.J.; Parson, S.; Pulham, C.R.; Sawyer, L. High-pressure polymorphism in l-cysteine: The crystal structures of l-cysteine-III and l-cysteine-IV. Acta Crystallogr. B 2006, 62, 296–309. [Google Scholar] [CrossRef]
- Roux, M.V.; Foces-Foces, C.; Notario, R.; da Silva, M.A.V.R.; da Silva, M.D.M.C.R.; Santos, A.F.L.O.M.; Juaristi, E. Experimental and computational thermochemical study of sulfur-containing amino acids: l-cysteine, l-cystine, and l-cysteine-derived radicals. S-S, S-H, and C-S bond dissociation enthalpies. J. Phys. Chem. B 2010, 114, 10530–10540. [Google Scholar] [CrossRef]
- Frisch, A.M.J.; Trucks, G.W.; Shelelgel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C. et al. Gaussian 03; revision E.0; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Frisch, A.M.J.; Dennington, R.D., II.; Keith, T.A.; Millam, J.; Nielsen, A.B.; Holder, A.J.; Hiscocks, J. Gaussview, 4.1.2; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems; Wiley-Interscience: New York, NY, USA, 2001. [Google Scholar]
- Nascimento, V.A.; Melnikov, P.; Consolo, L.Z.Z. Computerized modeling of adenosine triphosphate, adenosine triarsenate and adenosine trivanadate. Molecules 2012, 17, 9489–9495. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Melnikov, P.; Nascimento, V.A.; Silva, A.F.; Consolo, L.Z.Z. Structural Modeling of Djenkolic Acid with Sulfur Replaced by Selenium and Tellurium. Molecules 2014, 19, 4847-4856. https://doi.org/10.3390/molecules19044847
Melnikov P, Nascimento VA, Silva AF, Consolo LZZ. Structural Modeling of Djenkolic Acid with Sulfur Replaced by Selenium and Tellurium. Molecules. 2014; 19(4):4847-4856. https://doi.org/10.3390/molecules19044847
Chicago/Turabian StyleMelnikov, Petr, Valter A. Nascimento, Anderson F. Silva, and Lourdes Z. Z. Consolo. 2014. "Structural Modeling of Djenkolic Acid with Sulfur Replaced by Selenium and Tellurium" Molecules 19, no. 4: 4847-4856. https://doi.org/10.3390/molecules19044847