Synthetic Fosmidomycin Analogues with Altered Chelating Moieties Do Not Inhibit 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase or Plasmodium falciparum Growth In Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
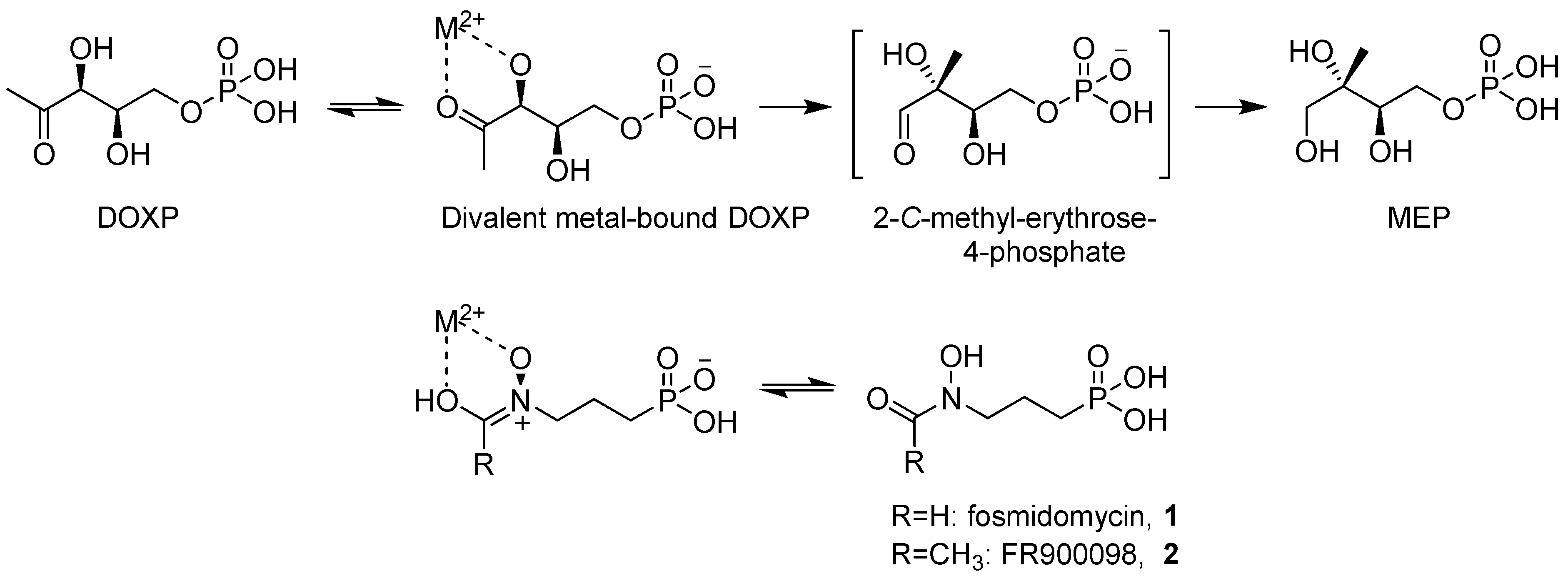
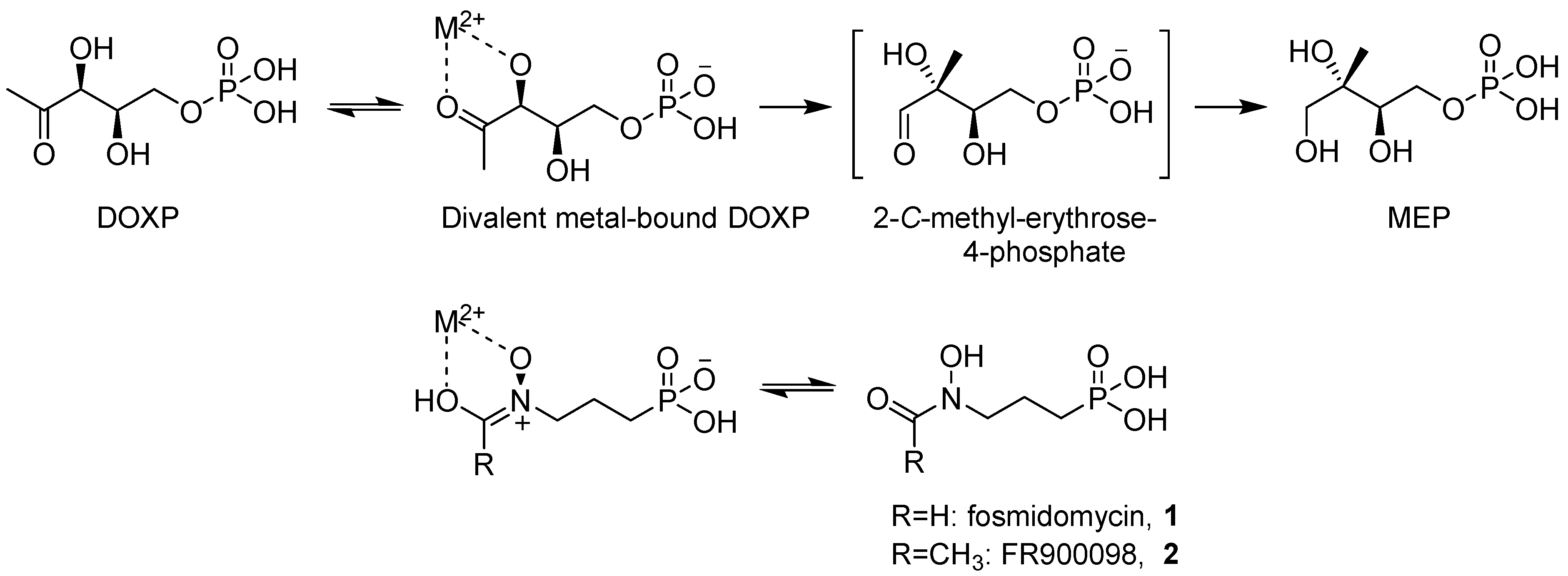
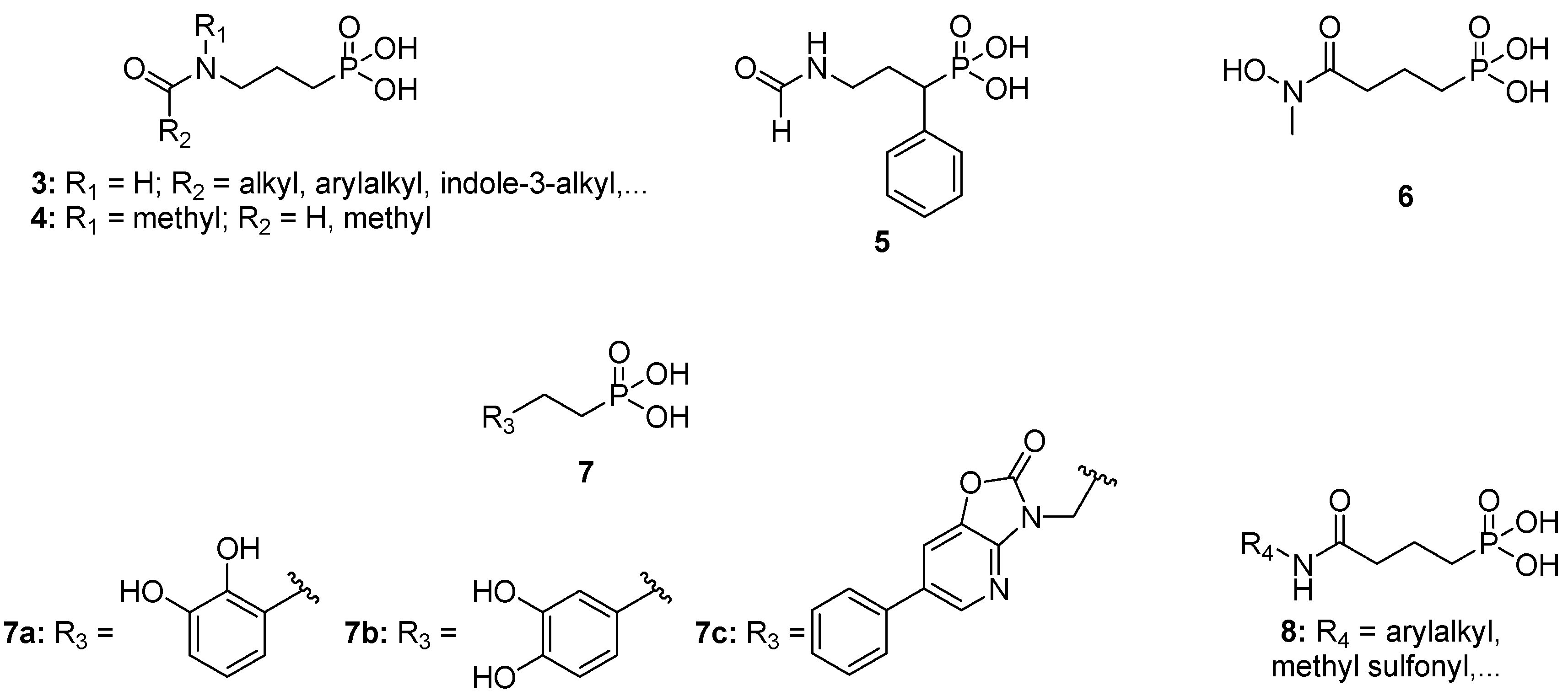
:1. Introduction
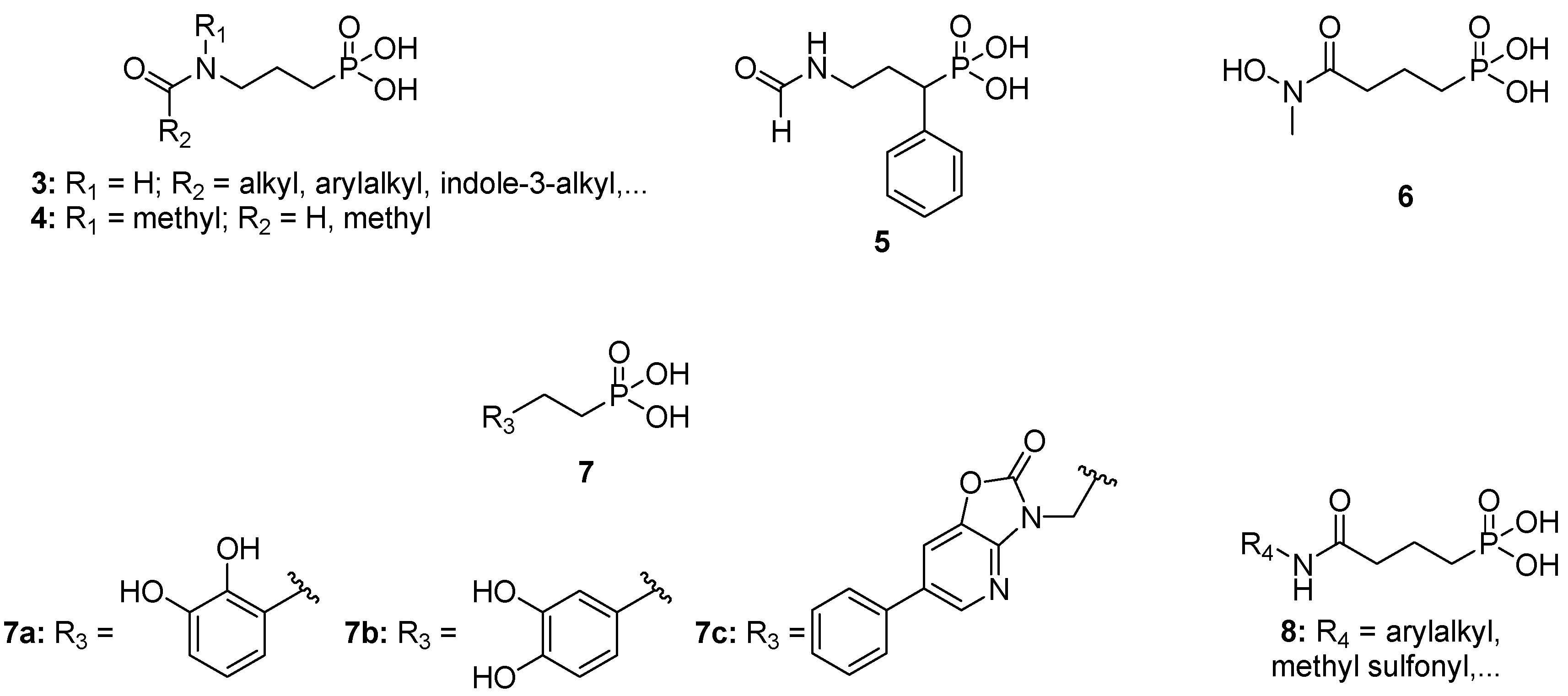
2. Results and Discussion
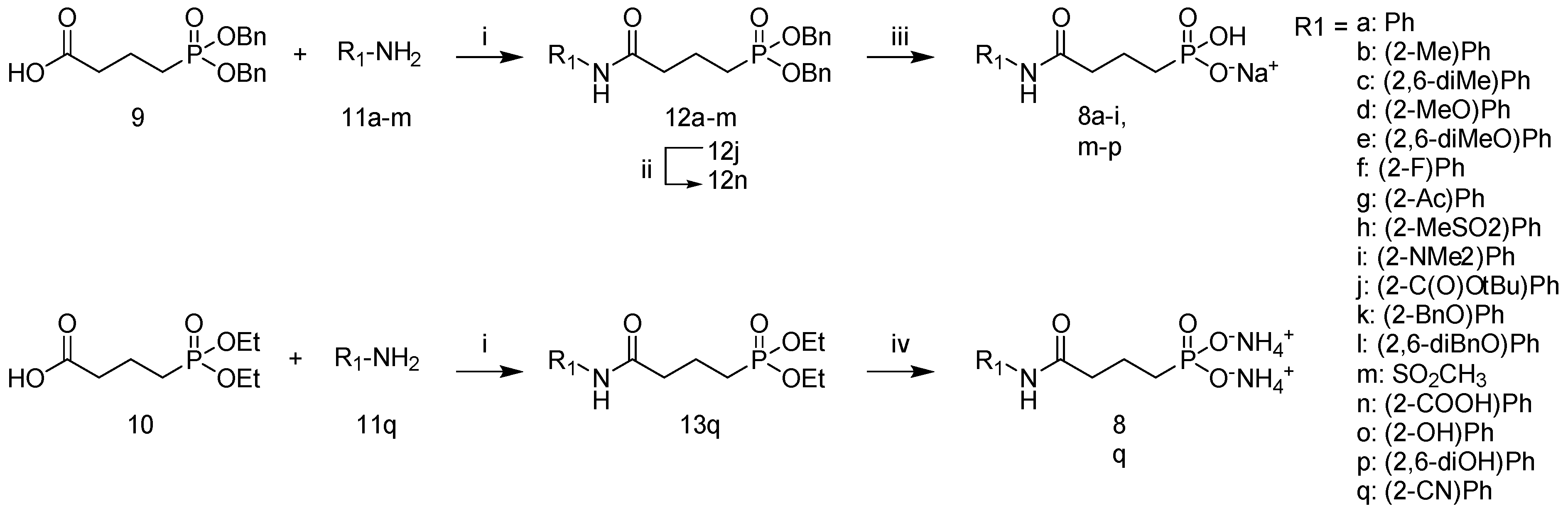
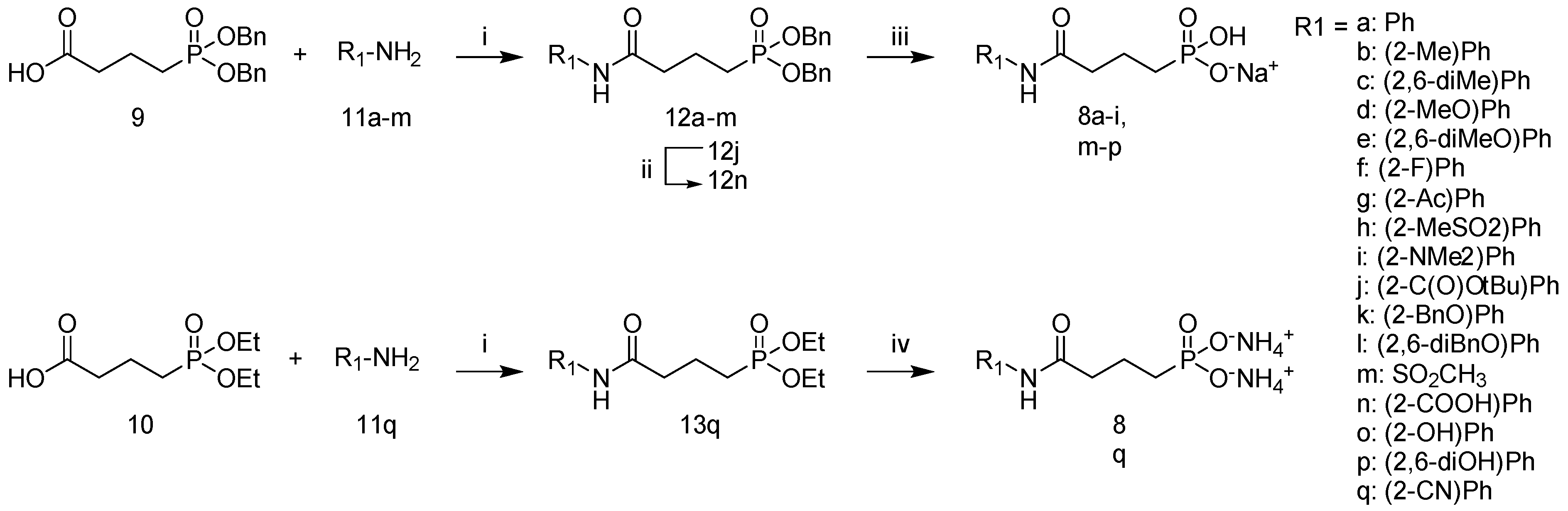
2.1. Synthesis
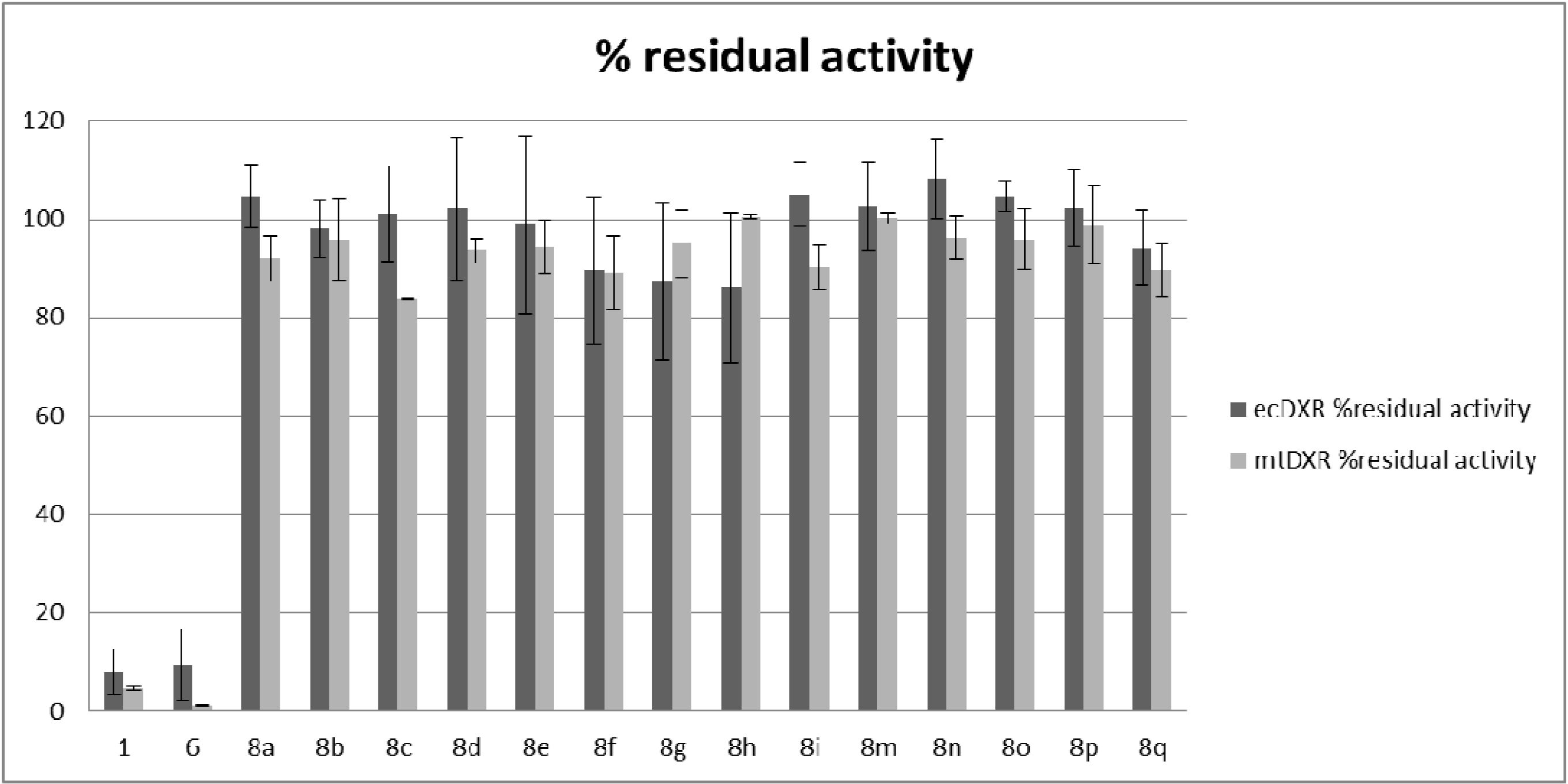
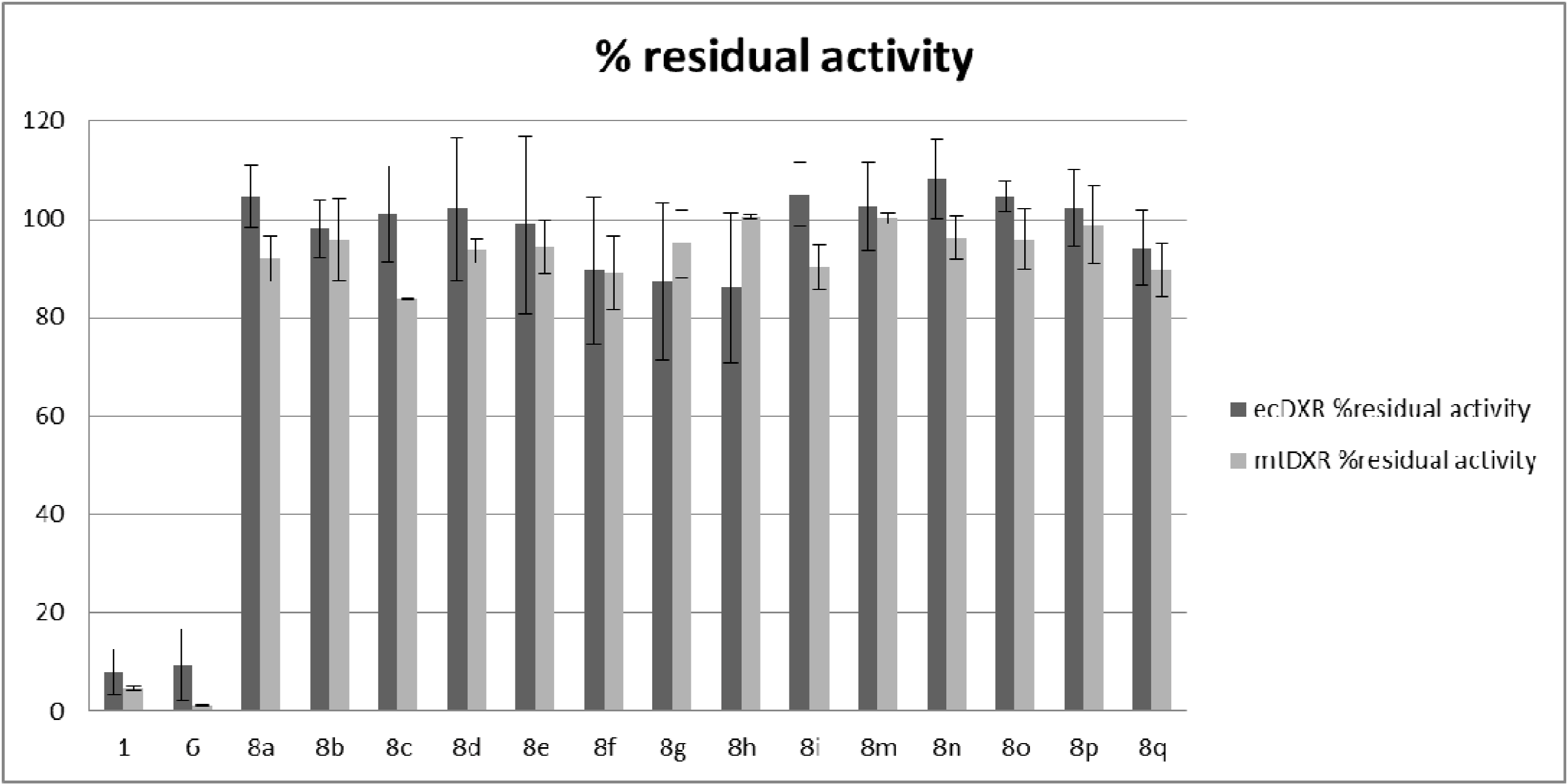
2.2. Antiplasmodial and Antitubercular Evaluation
3. Experimental
3.1. General Methods and Materials
3.2. General Procedure for the Synthesis of Protected Amides
3.3. General Procedure for Amide Deprotection Yielding Targets 8a–i, m–p
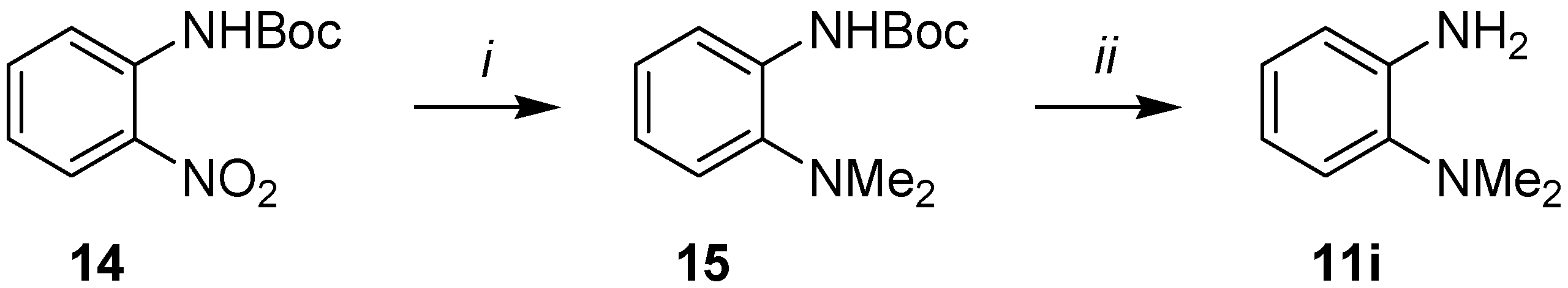
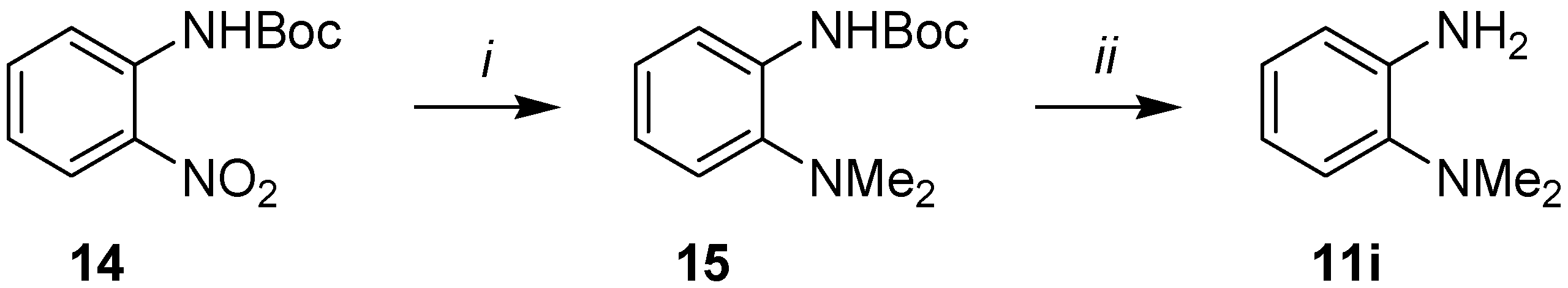
3.4. Synthesis of o-(Dimethylamino)aniline (11i)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Snow, R.W.; Guerra, C.A.; Noor, A.M.; Myint, H.Y.; Hay, S.I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 2005, 434, 214–217. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2011. WHO Press: Geneva, Switzerland, 2011. [Google Scholar]
- World Health Organization. Global Tuberculosis Control 2009: Epidemiology, Strategy, Financing; WHO Press: Geneva, Switzerland, 2009. [Google Scholar]
- Kuzuyama, T.; Shimizu, T.; Takahashi, S.; Seto, H. Fosmidomycin, a specific inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett. 1998, 39, 7913–7916. [Google Scholar] [CrossRef]
- Zeidler, J.; Schwender, J.; Muller, C.; Wiesner, J.; Weidemeyer, C.; Beck, E.; Jomaa, H.Z.; Lichtenthaler, H.K. Inhibition of the non-mevalonate 1-deoxy-d-xylulose-5-phosphate pathway of plant isoprenoid biosynthesis by fosmidomycin. Z. Naturforsch. 1998, 53c, 980–986. [Google Scholar]
- Rohmer, M.; Knani, M.; Simonin, P.; Sutter, B.; Sahm, H. Isoprenoid biosynthesis in bacteria: A novel pathway for the early steps leading to isopentenyl diphosphate. Biochem. J. 1993, 295 (Pt 2), 517–524. [Google Scholar]
- Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M.; Turbachova, I.; Eberl, M.; Zeidler, J.; Lichtenthaler, H.K.; et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1575. [Google Scholar] [CrossRef]
- Umeda, T.; Tanaka, N.; Kusakabe, Y.; Nakanishi, M.; Kitade, Y.; Nakamura, K.T. Molecular basis of fosmidomycin’s action on the human malaria parasite Plasmodium falciparum. Sci. Rep. 2011, 1, 1–8. [Google Scholar]
- Steinbacher, S.; Kaiser, J.; Eisenreich, W.; Huber, R.; Bacher, A.; Rohdich, F. Structural basis of fosmidomycin action revealed by the complex with 2-C-methyl-D-erythritol 4-phosphate synthase (IspC). Implications for the catalytic mechanism and anti-malaria drug development. J. Biol. Chem. 2003, 278, 18401–18407. [Google Scholar]
- Borrmann, S.; Issifou, S.; Esser, G.; Adegnika, A.A.; Ramharter, M.; Matsiegui, P.B.; Oyakhirome, S.; Mawili-Mboumba, D.P.; Missinou, M.A.; Kun, J.F.; et al. Fosmidomycin-clindamycin for the treatment of Plasmodium falciparum malaria. J. Infect. Dis. 2004, 190, 1534–1540. [Google Scholar] [CrossRef]
- Borrmann, S.; Adegnika, A.A.; Matsiegui, P.B.; Issifou, S.; Schindler, A.; Mawili-Mboumba, D.P.; Baranek, T.; Wiesner, J.; Jomaa, H.; Kremsner, P.G. Fosmidomycin-clindamycin for Plasmodium falciparum Infections in African Children. J. Infect. Dis. 2004, 189, 901–908. [Google Scholar] [CrossRef]
- Dhiman, R.K.; Schaeffer, M.L.; Bailey, A.M.; Testa, C.A.; Scherman, H.; Crick, D.C. 1-Deoxy-d-xylulose 5-phosphate reductoisomerase (IspC) from Mycobacterium tuberculosis: Towards understanding mycobacterial resistance to fosmidomycin. J. Bacteriol. 2005, 187, 8395–8402. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Furukawa, S.; Ogihara, H.; Yamasaki, M. Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli. Biosci. Biotechnol. Biochem. 2003, 67, 2030–2033. [Google Scholar] [CrossRef]
- Lou, B.; Yang, K. Molecular diversity of hydroxamic acids. Part II. Potential therapeutic applications. Mini Rev. Med. Chem. 2003, 3, 609–620. [Google Scholar] [CrossRef]
- O’Brien, E.C.; Farkas, E.; Gil, M.J.; Fitzgerald, D.; Castineras, A.; Nolan, K.B. Metal complexes of salicylhydroxamic acid (H2Sha), anthranilic hydroxamic acid and benzohydroxamic acid. Crystal and molecular structure of [Cu(phen)2(Cl)]ClPH2Sha, a model for a peroxidaseinhibitor complex. J. Inorg. Biochem. 2000, 79, 47–51. [Google Scholar] [CrossRef]
- Sanderson, L.; Taylor, G.W.; Aboagye, E.O.; Alao, J.P.; Latigo, J.R.; Coombes, R.C.; Vigushin, D.M. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab. Dispos. 2004, 32, 1132–1138. [Google Scholar] [CrossRef]
- Jackson, E.R.; Dowd, C.S. Inhibition of 1-deoxy-d-xylulose-5-phosphate reductoisomerase (Dxr): A review of the synthesis and biological evaluation of recent inhibitors. Curr. Top. Med. Chem. 2012, 12, 706–728. [Google Scholar] [CrossRef]
- Giessmann, D.; Heidler, P.; Haemers, T.; van Calenbergh, S.; Reichenberg, A.; Jomaa, H.; Weidemeyerd, C.; Sanderbrand, S.; Wiesner, J.; Link, A. Towards new antimalarial drugs: Synthesis of non-hydrolyzable phosphate mimics as feed for a predictive QSAR study on 1-deoxy-d-xylulose-5-phosphate reductoisomerase inhibitors. Chem. Biodivers. 2008, 5, 643–656. [Google Scholar] [CrossRef]
- Woo, Y.; Fernandes, R.; Proteau, P. Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp PCC6803 1-deoxy-d-xylulose 5-phosphate reductoisomerase. Bioorg. Med. Chem. 2006, 14, 2375–2385. [Google Scholar]
- Haemers, T.; Wiesner, J.; Busson, R.; Jomaa, H.; van Calenbergh, S. Synthesis of α-aryl-substituted and conformationally restricted fosmidomycin analogues as promising antimalarials. Eur. J. Org. Chem. 2006, 17, 3856–3863. [Google Scholar]
- Kuntz, L.; Tritsch, D.; Grosdemange-Billiard, C.; Hemmerlin, A.; Willem, A.; Bacht, T.; Rohmer, M. Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: Phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reducto-isomerase. Biochem. J. 2005, 386, 127–135. [Google Scholar] [CrossRef]
- Behrendt, C.T.; Kunfermann, A.; Illarionova, V.; Matheeussen, A.; Pein, M.K.; Gräwert, T.; Kaiser, J.; Bacher, A.; Eisenreich, W.; Illarionov, B.; et al. Reverse fosmidomycin derivatives against the antimalarial drug target IspC (Dxr). J. Med. Chem. 2011, 54, 6796–6802. [Google Scholar] [CrossRef]
- Brücher, K.; Illarionov, B.; Held, J.; Tschan, S.; Kunfermann, A.; Pein, M.K.; Bachar, A.; Gräwert, T.; Maes, L.; Mordmüller, B.; et al. α-ubstituted β-Oxa isosteres of fosmidomycin: Synthesis and biological evaluation. J. Med. Chem. 2012, 55, 6566–6575. [Google Scholar] [CrossRef]
- Kunfermann, A.; Lienau, C.; Illarionov, B.; Held, J.; Gräwert, T.; Behrendt, C.T.; Werner, P.; Hähn, S.; Eisenreich, W.; Riederer, U.; et al. IspC as target for antiinfective drug discovery: Synthesis, enantiomeric separation and structural biology of fosmidomycin thia isosters. J. Med. Chem. 2013, 56, 8151–8162. [Google Scholar] [CrossRef]
- Deng, L.; Sundriyal, S.; Rubio, V.; Shi, Z.Z.; Song, Y. Coordination chemistry based approach to lipophilic inhibitors of 1-deoxy-d-xylulose-5-phosphate reductoisomerase. J. Med. Chem. 2009, 52, 6539–6542. [Google Scholar] [CrossRef]
- Andaloussi, M.; Lindh, M.; Bjorkelid, C.; Suresh, S.; Wieckowska, A.; Iyer, H.; Karlen, A.; Larhed, M. Substitution of the phosphonic acid and hydroxamic acid functionalities of the Dxr inhibitor FR900098: An attempt to improve the activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2011, 21, 5403–5407. [Google Scholar] [CrossRef]
- Williams, S.L.; de Oliveira, C.A.F.; Vazquez, H.; McCammon, J.A. From Zn to Mn: The Study of novel manganese-binding groups in the search for new drugs against tuberculosis. Chem. Biol. Drug Des. 2011, 77, 117–123. [Google Scholar] [CrossRef]
- Bodill, T.; Conibear, A.C.; Blatch, G.L.; Lobb, K.A.; Kaye, P.T. Synthesis and evaluation of phosphonated N-heteroarylcarboxamides as DOXP-reductoisomerase (Dxr) inhibitors. Bioorg. Med. Chem. 2011, 19, 1321–1327. [Google Scholar] [CrossRef]
- San Jose, G.; Jackson, E.R.; Uh, E.; Johny, C.; Haymond, A.; Lundberg, L.; Pinkham, C.; Kehn-Hall, K.; Boshoff, H.I.; Couch, R.D.; et al. Design of potential bisubstrate inhibitors against Mycobacterium tuberculosis (Mtb) 1-deoxy-d-xylulose 5-phosphate reductoisomerase (Dxr)—evidence of a novel binding mode. Med. Chem. Commun. 2013, 4, 1099–1104. [Google Scholar] [CrossRef]
- Gavalda, S.; Braga, R.; Dax, C.; Vigroux, A. N-Sulfonyl hydroxamate derivatives as inhibitors of class II fructose-1,6-diphosphate aldolase. Bioorg. Med. Chem. Lett. 2005, 15, 5375–5377. [Google Scholar] [CrossRef]
- McNeil, D.W.; Kelly, T.A. Asimple method for the protection of aryl amines as their t-butylcarbamoyl (Boc) derivatives. Tetrahedron Lett. 1994, 35, 9003–9006. [Google Scholar] [CrossRef]
- Sumandeep, K.G.; Hao, X.; Kirchoff, P.D.; Cierpicki, T.; Turbiak, A.J.; Wan, B.; Zhang, N.; Peng, K.W.; Franzblau, S.G.; Garcia, G.A.; et al. Structure-based design of novel benzoxazinorifamycins with potent binding affinity to wild-type and rifampin-resistant mutant Mycobacterium tuberculosis RNA polymerases. J. Med. Chem. 2012, 55, 3814–3826. [Google Scholar] [CrossRef]
- Jawaid, S.; Seidle, H.; Zhou, W.; Abdirahman, H.; Abadeer, M.; Hix, J.H.; van Hoek, M.L.; Couch, R.D. Kinetic characterization and phosphoregulation of the Francisella tularensis 1-deoxy-d-xylulose 5-phosphate reductoisomerase (MEP Synthase). PLoS One 2009, 4, e8288. [Google Scholar] [CrossRef] [Green Version]
- Larsen, T.M.; Wedekind, J.E.; Rayment, I.; Reed, G.H. A Carboxylate oxygen of the substrate bridges the magnesium ions at the active Site of enolase: Structure of the yeast enzyme complexed with the equilibrium mixture of 2-phosphoglycerate and phosphoenolpyruvate at 1.8 Å Resolution. Biochemistry 1996, 35, 4351–4358. [Google Scholar]
- Bodill, T.; Conibear, A.C.; Mutorwa, M.K.M.; Goble, J.L.; Blatch, G.L.; Lobb, K.A.; Klein, R.; Kaye, P.T. Exploring DOXP-reductoisomerase binding limits using phosphonated N-aryl and N-heteroarylcarboxamides as DXR inhibitors. Bioorg. Med. Chem. 2013, 21, 4332–4341. [Google Scholar] [CrossRef]
- Zinglé, C.; Kuntz, L.; Tritsch, D.; Grosdemange, C.B.; Rohmer, M. Modifications around the hydroxamic acid chelating group of fosmidomycin, an inhibitor of the metalloenzyme 1-deoxyxylulose 5-phosphate reductoisomerase (DXR). Bioorg. Med. Chem. Lett. 2012, 22, 6563–6567. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chofor, R.; Risseeuw, M.D.P.; Pouyez, J.; Johny, C.; Wouters, J.; Dowd, C.S.; Couch, R.D.; Van Calenbergh, S. Synthetic Fosmidomycin Analogues with Altered Chelating Moieties Do Not Inhibit 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase or Plasmodium falciparum Growth In Vitro. Molecules 2014, 19, 2571-2587. https://doi.org/10.3390/molecules19022571
Chofor R, Risseeuw MDP, Pouyez J, Johny C, Wouters J, Dowd CS, Couch RD, Van Calenbergh S. Synthetic Fosmidomycin Analogues with Altered Chelating Moieties Do Not Inhibit 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase or Plasmodium falciparum Growth In Vitro. Molecules. 2014; 19(2):2571-2587. https://doi.org/10.3390/molecules19022571
Chicago/Turabian StyleChofor, René, Martijn D.P. Risseeuw, Jenny Pouyez, Chinchu Johny, Johan Wouters, Cynthia S. Dowd, Robin D. Couch, and Serge Van Calenbergh. 2014. "Synthetic Fosmidomycin Analogues with Altered Chelating Moieties Do Not Inhibit 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase or Plasmodium falciparum Growth In Vitro" Molecules 19, no. 2: 2571-2587. https://doi.org/10.3390/molecules19022571
APA StyleChofor, R., Risseeuw, M. D. P., Pouyez, J., Johny, C., Wouters, J., Dowd, C. S., Couch, R. D., & Van Calenbergh, S. (2014). Synthetic Fosmidomycin Analogues with Altered Chelating Moieties Do Not Inhibit 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase or Plasmodium falciparum Growth In Vitro. Molecules, 19(2), 2571-2587. https://doi.org/10.3390/molecules19022571