Concise and Straightforward Asymmetric Synthesis of a Cyclic Natural Hydroxy-Amino Acid



Abstract
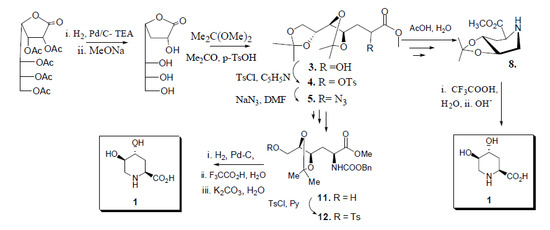
:
1. Introduction
2. Results and Discussion
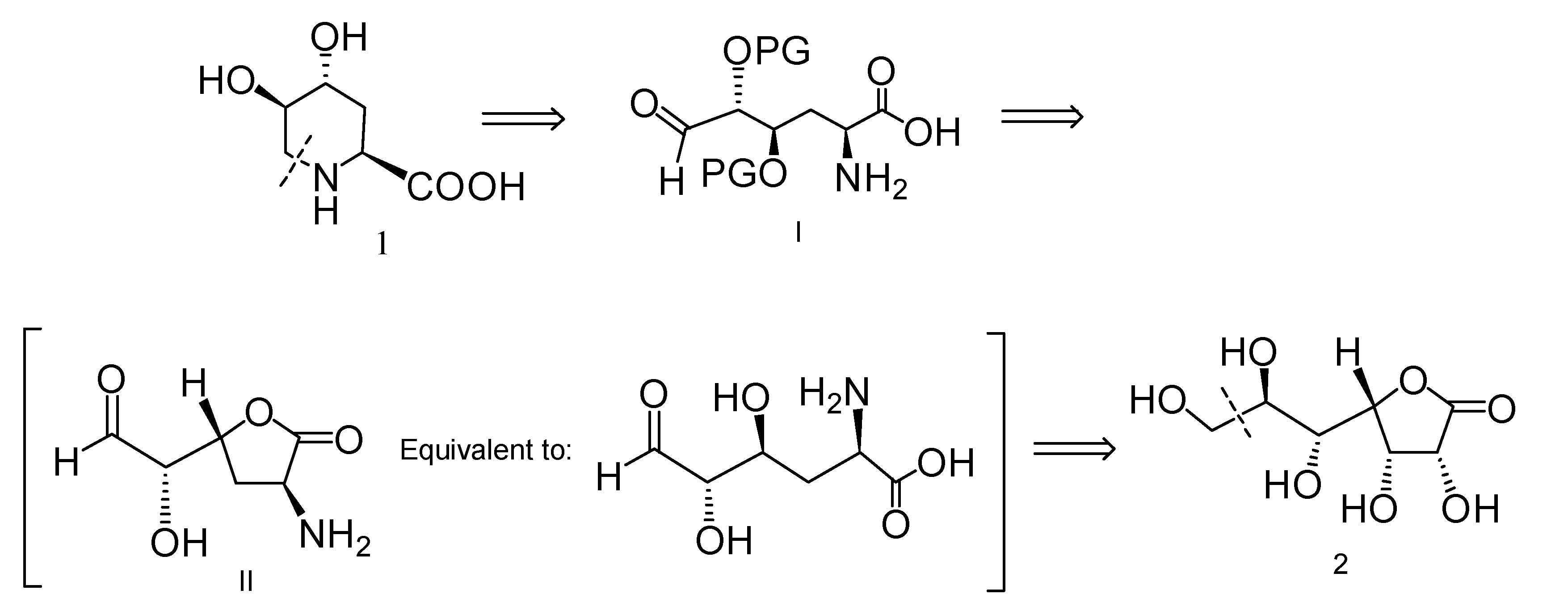
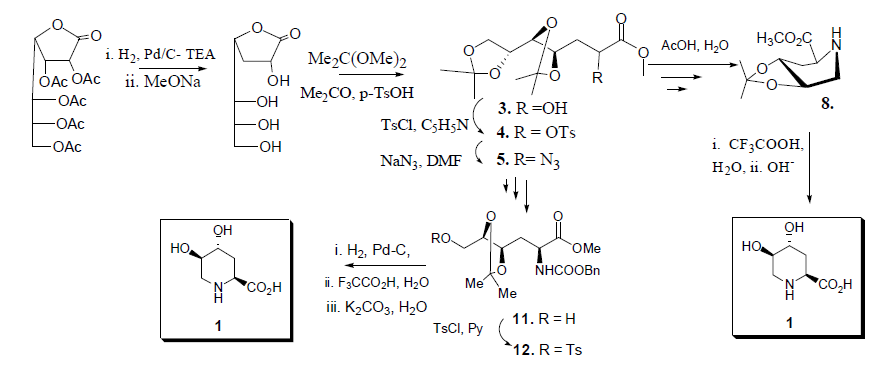
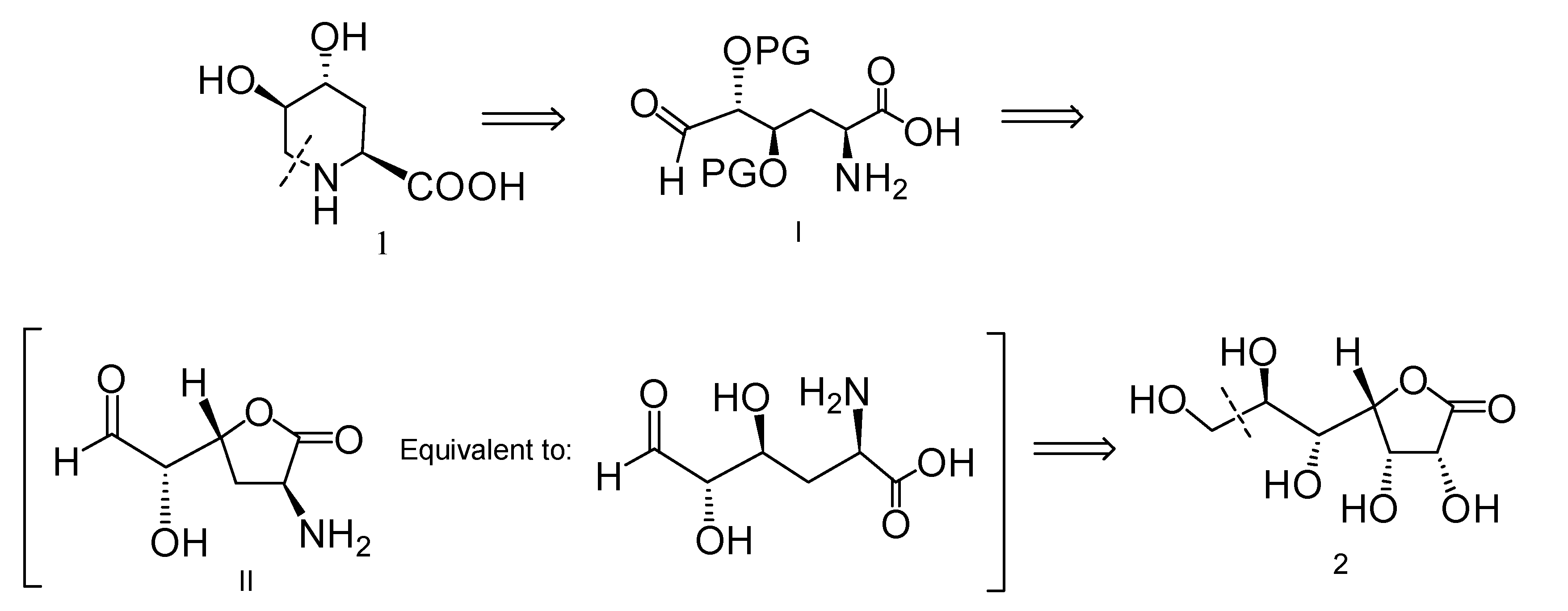
2.1. Retrosynthetic Analysis for the Synthesis of Compound 1
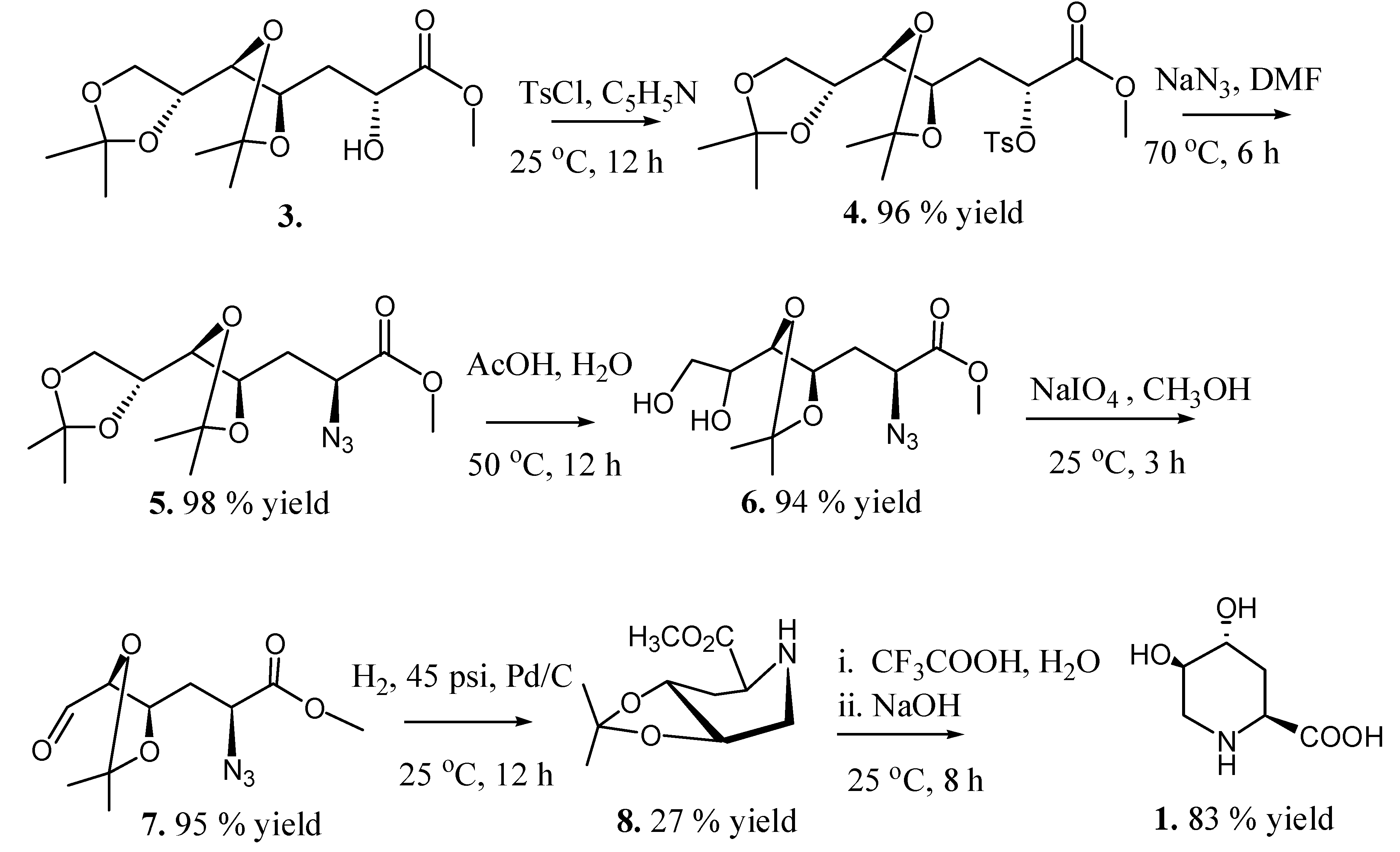
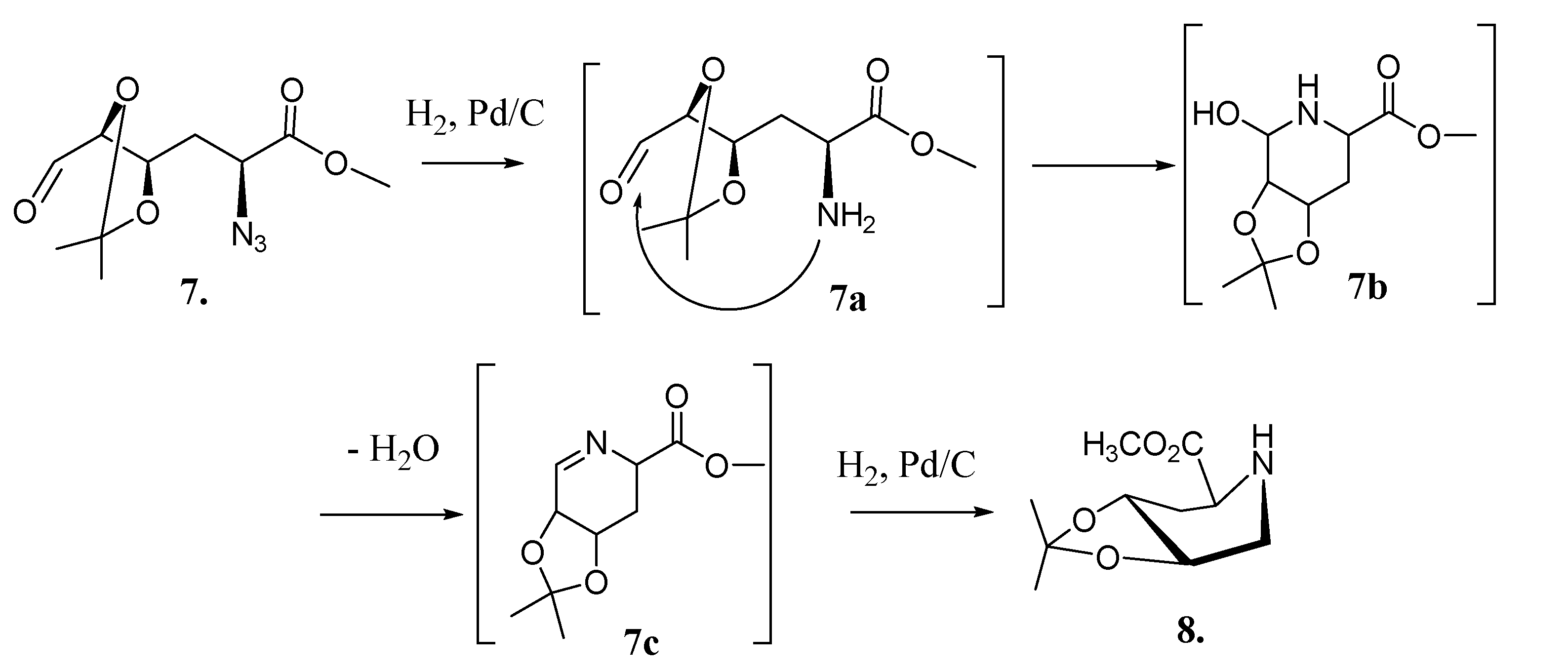
2.2. Total Synthesis of Compound 1
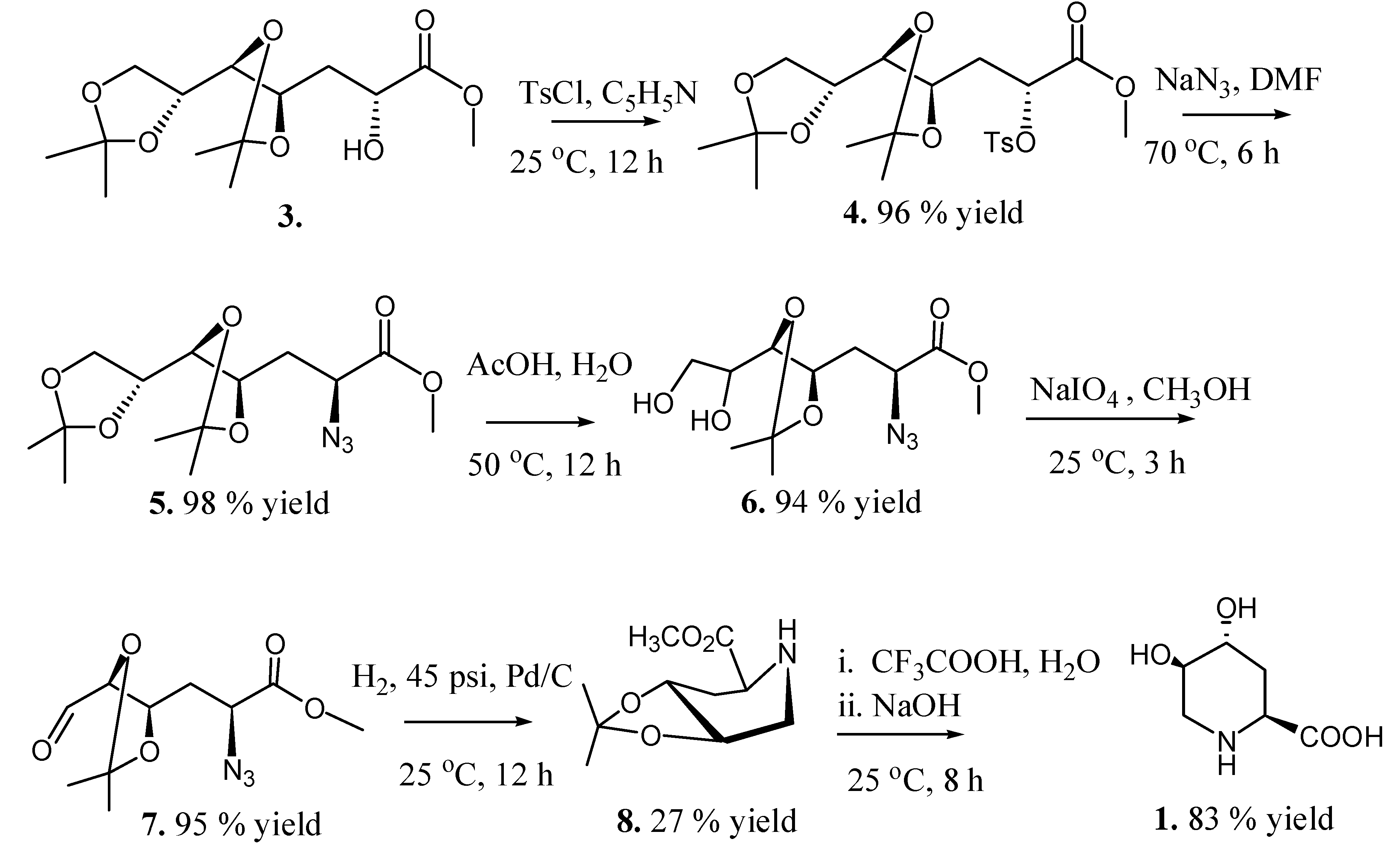
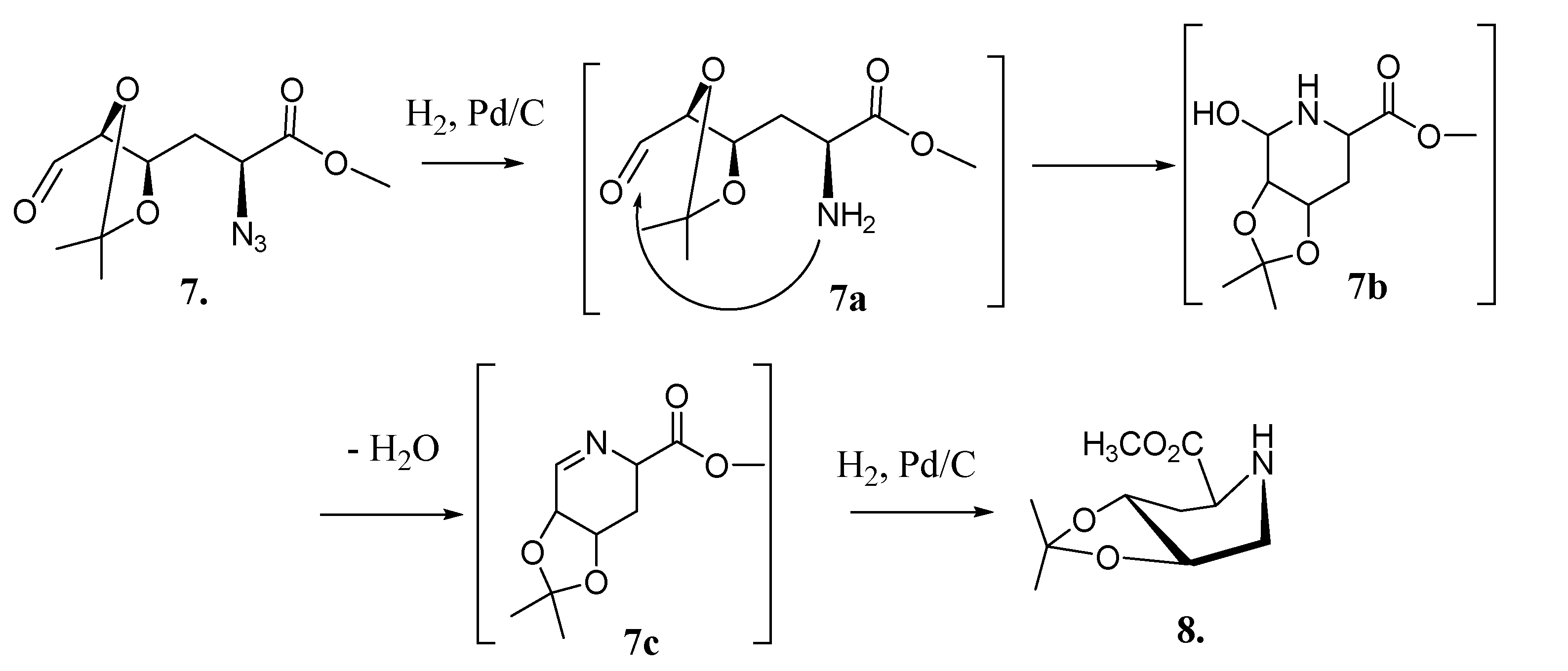
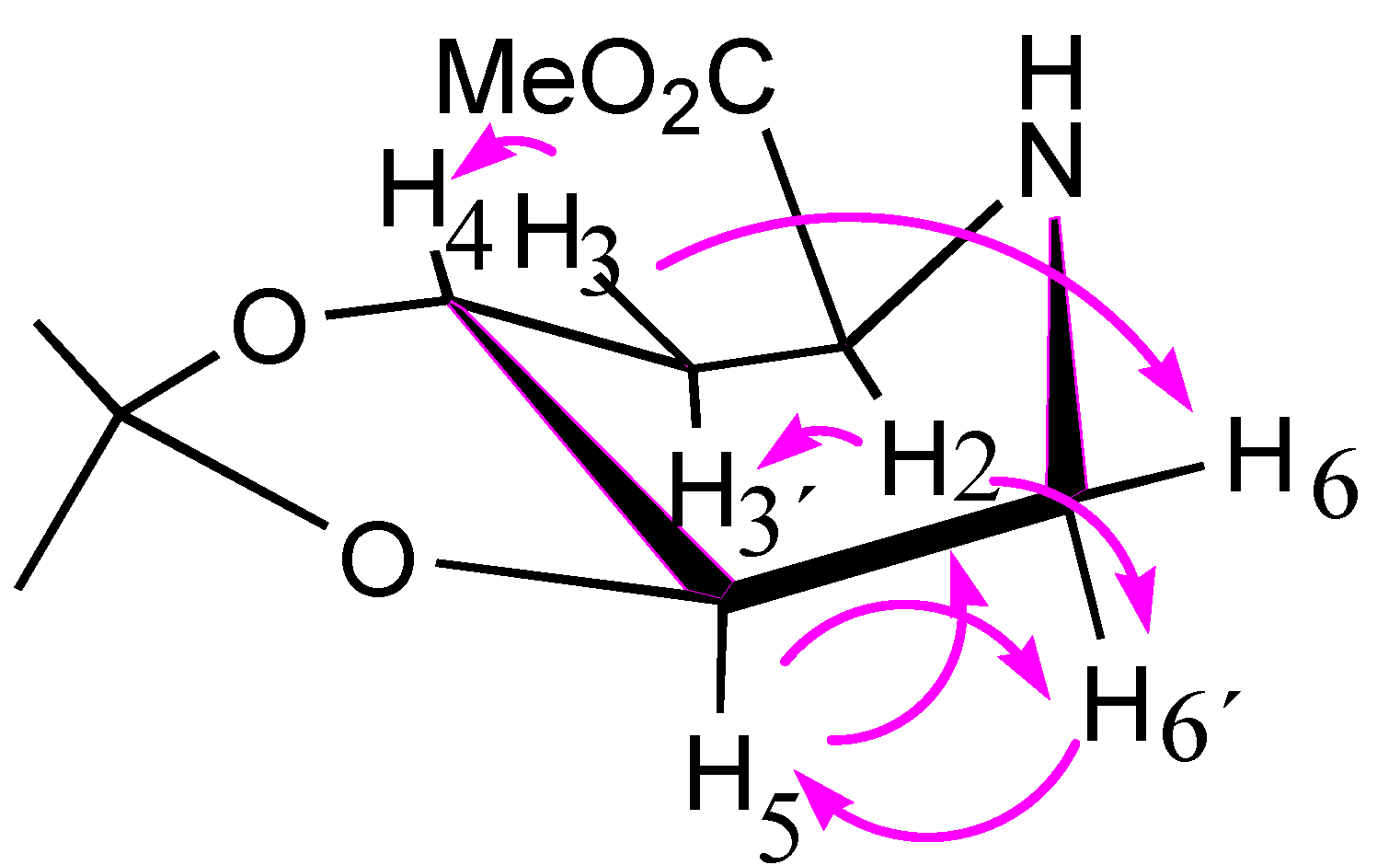


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst (Pd-C)% | Hydrogen Pressure (psi) | Time | Solvent | Yield (%) b |
|---|---|---|---|---|
| 10 | 20 | 8 h | EtOAc | 23 |
| 10 | 45 | 8 h | EtOAc | 25 |
| 10 | 45 | 24 h | EtOAc | 26 |
| 15 | 45 | 48 h | EtOAc | 27 |
| 15 | 30 | 48 h | EtOAc | 27 |
| 20 | 30 | 24 h | EtOAc | 27 |
| 25 | 45 | 24 h | EtOAc | 27 |
| 10 | 30 | 48 h | EtOAc/MeOH | 25 |
| 15 | 45 | 24 h | EtOAc/MeOH | 24 |
| 15 | 30 | 8 h | EtOAc | 23 |
| 20 | 30 | 24 h | EtOAc/MeOH | 26 |
| 20 | 45 | 24 h | EtOAc/MeOH | 25 |
| 20 | 45 | 24 h | MeOH | 25 |
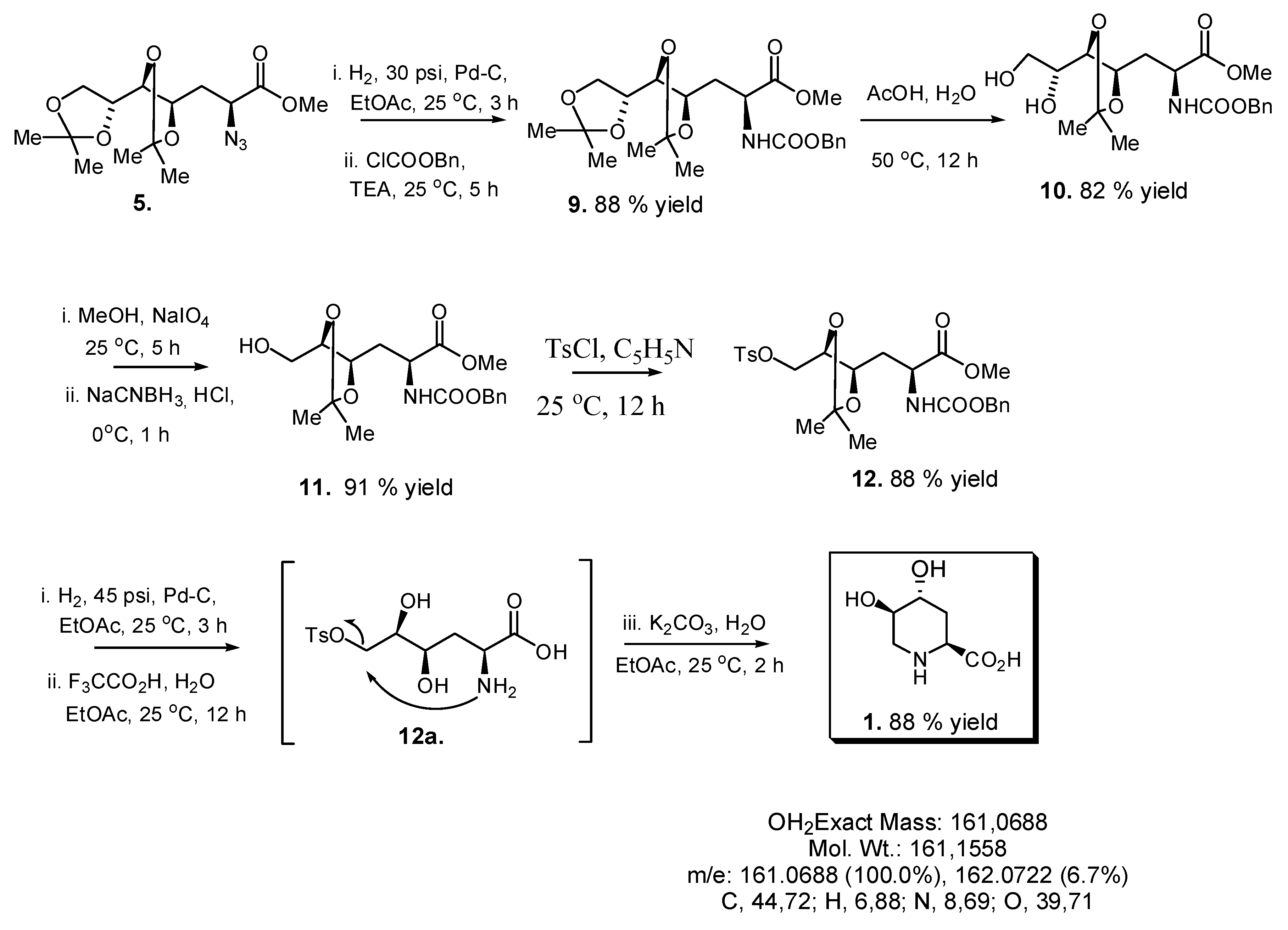
2.3. Synthesis of Compound 1 Starting from Azide 5
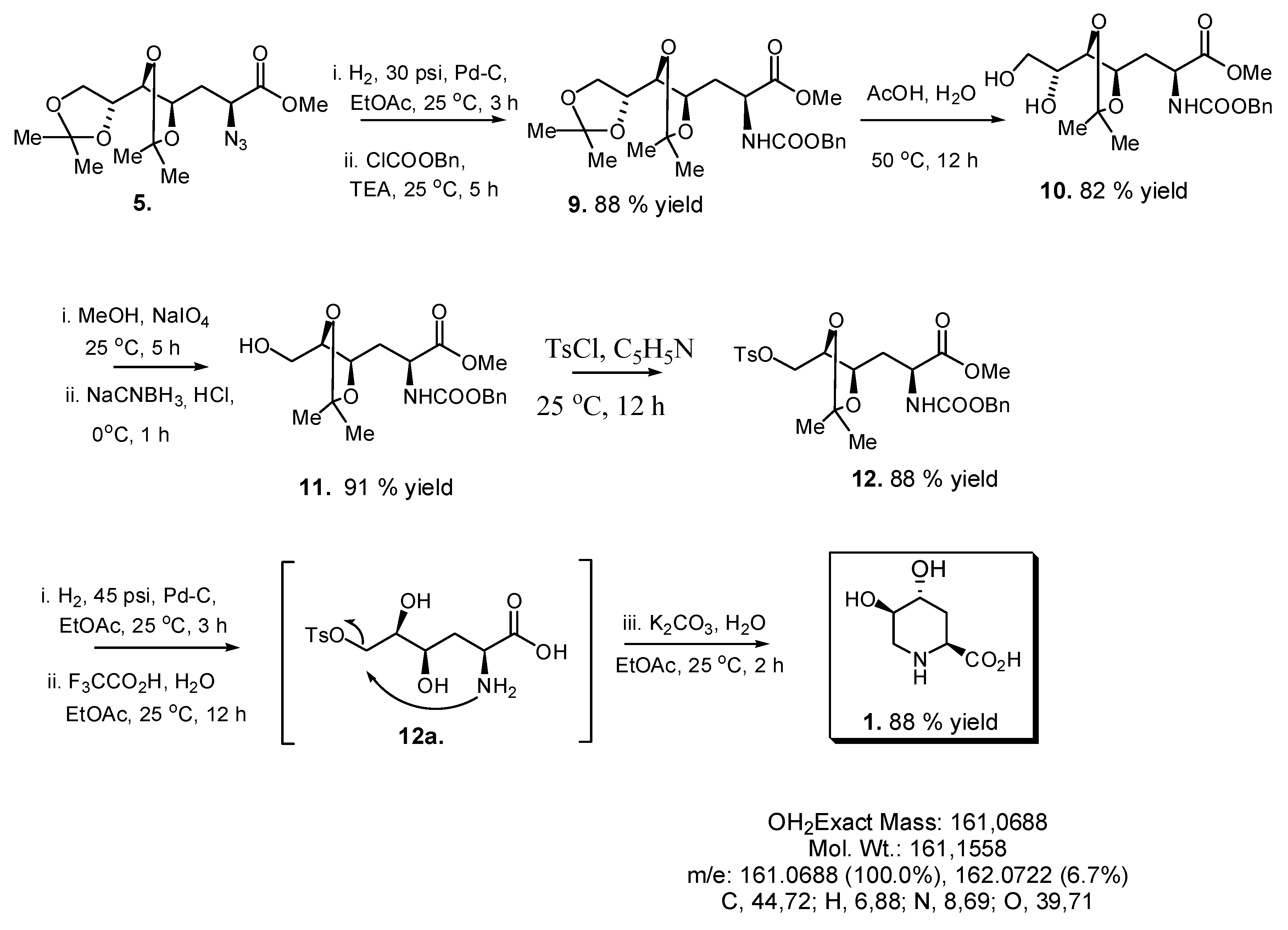
3. Experimental
3.1. General Experimental Procedures
3.2. Synthesis of Methyl 3-deoxy-4,5:6,7-di-O-isopropylidene-2-O p-toluen-sulfonyl-d-gluco-heptonate (4)
3.3. Synthesis of Methyl 2-Azido-2,3-dideoxy-4,5:6,7-di-O-isopropylidene-d-manno-heptonate (5)
3.4. Synthesis of Methyl 2-Azido-2,3-dideoxy-4,5-O-isopropylidene-d-manno-heptonate (6)
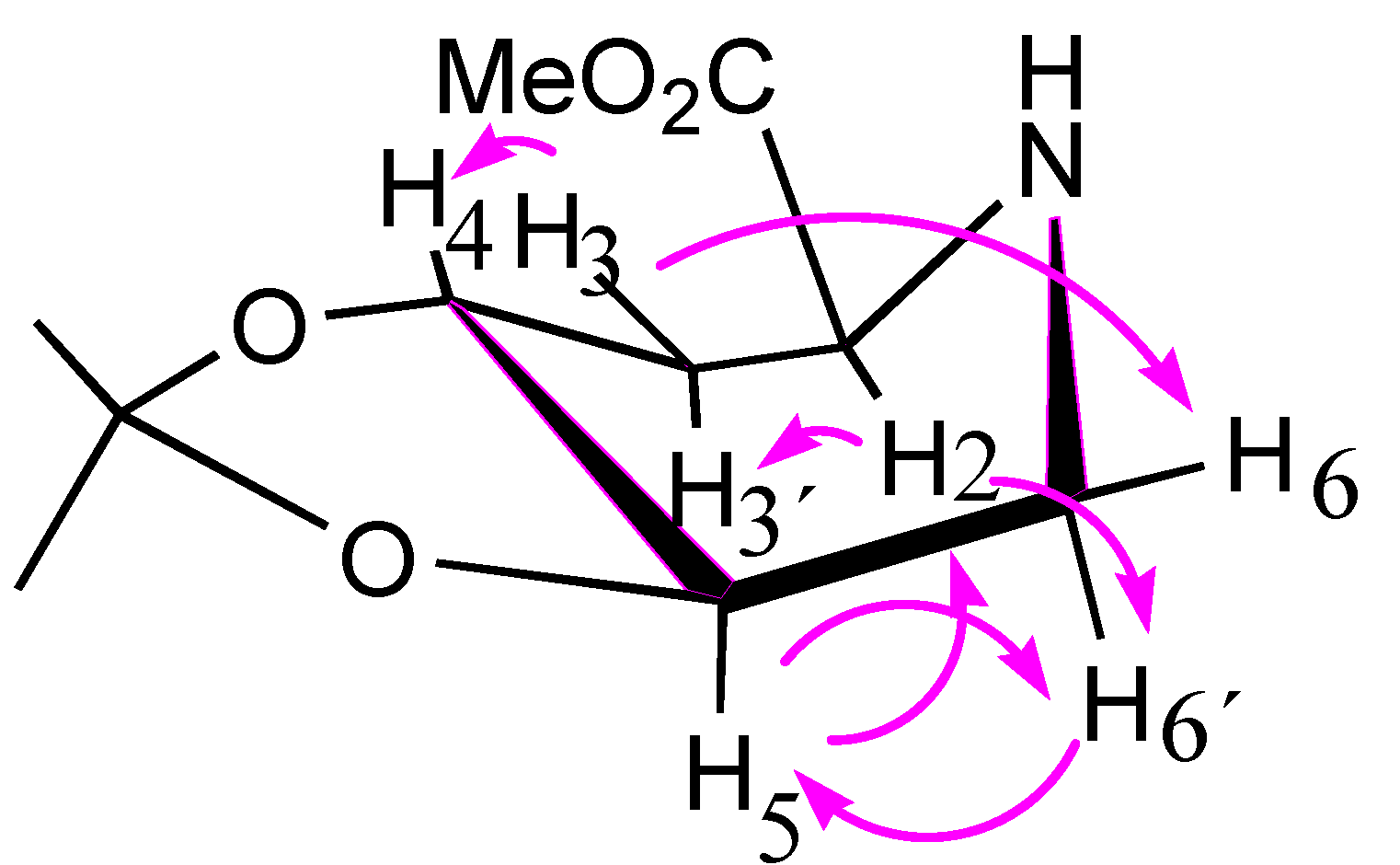
3.5. Synthesis of (2S,4R,5R) 4,5-O-Isopropylidene-methyl-pipecolate (8)
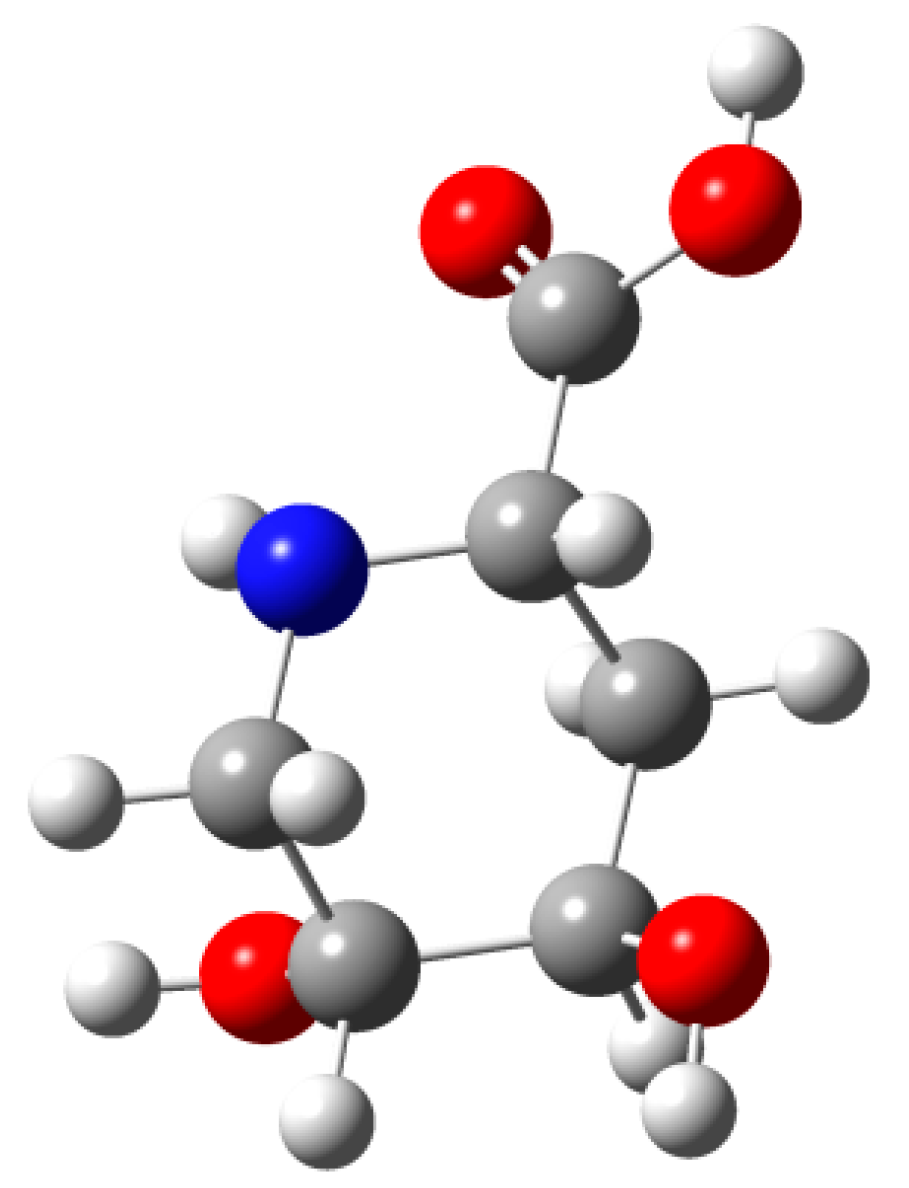
3.6. Synthesis of (2S,4R,5R) 4,5 Dihydroxy-pipecolic Acid (1) from 8
3.7. Synthesis of Methyl 2-Benzyloxycarbonylamino-2,3-dideoxy-4,5:6,7-di-O-isopropylidene-d-manno-heptonate (9)
3.8. Synthesis of Methyl 2-Benzyloxycarbonylamino-2,3-dideoxy-4,5-O-isopropylidene-d-manno-heptonate (10)
3.9. Synthesis of Methyl 2-Benzyloxycarbonyl-amino-2,3-dideoxy-4,5-O-isopropylidene-d-arabino-hexonate (11)
3.10. Synthesis of Methyl 2-Benzyloxycarbonyl-amino-2,3-dideoxy-4,5-O-isopropylidene-6-O-p-toluen-sulfonyl-d-arabino-hexonate (12)
3.11. Synthesis of Compound 1 ((2S,4R,5R) 4,5 Dihydroxy-pipecolic acid) from 12
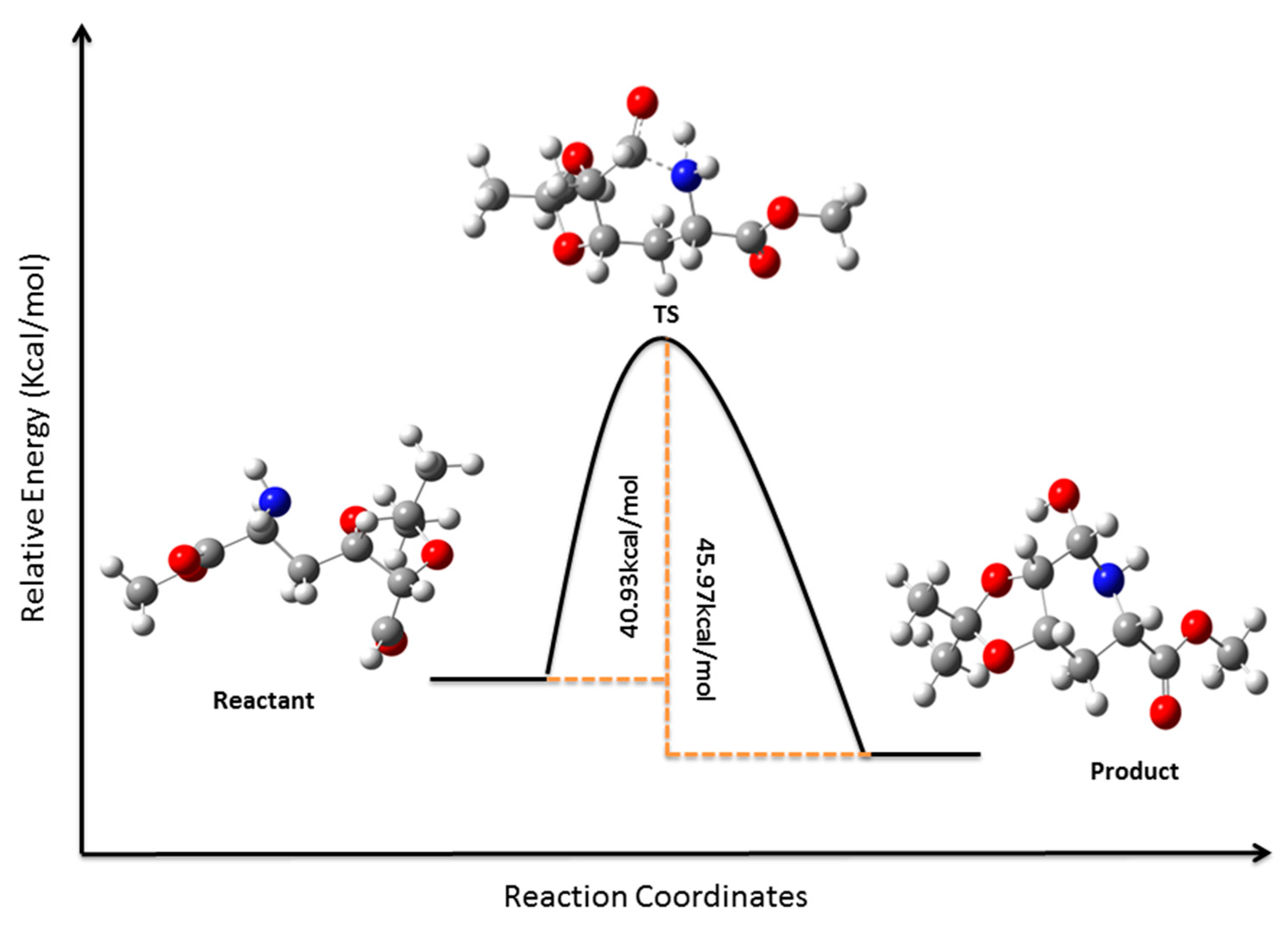
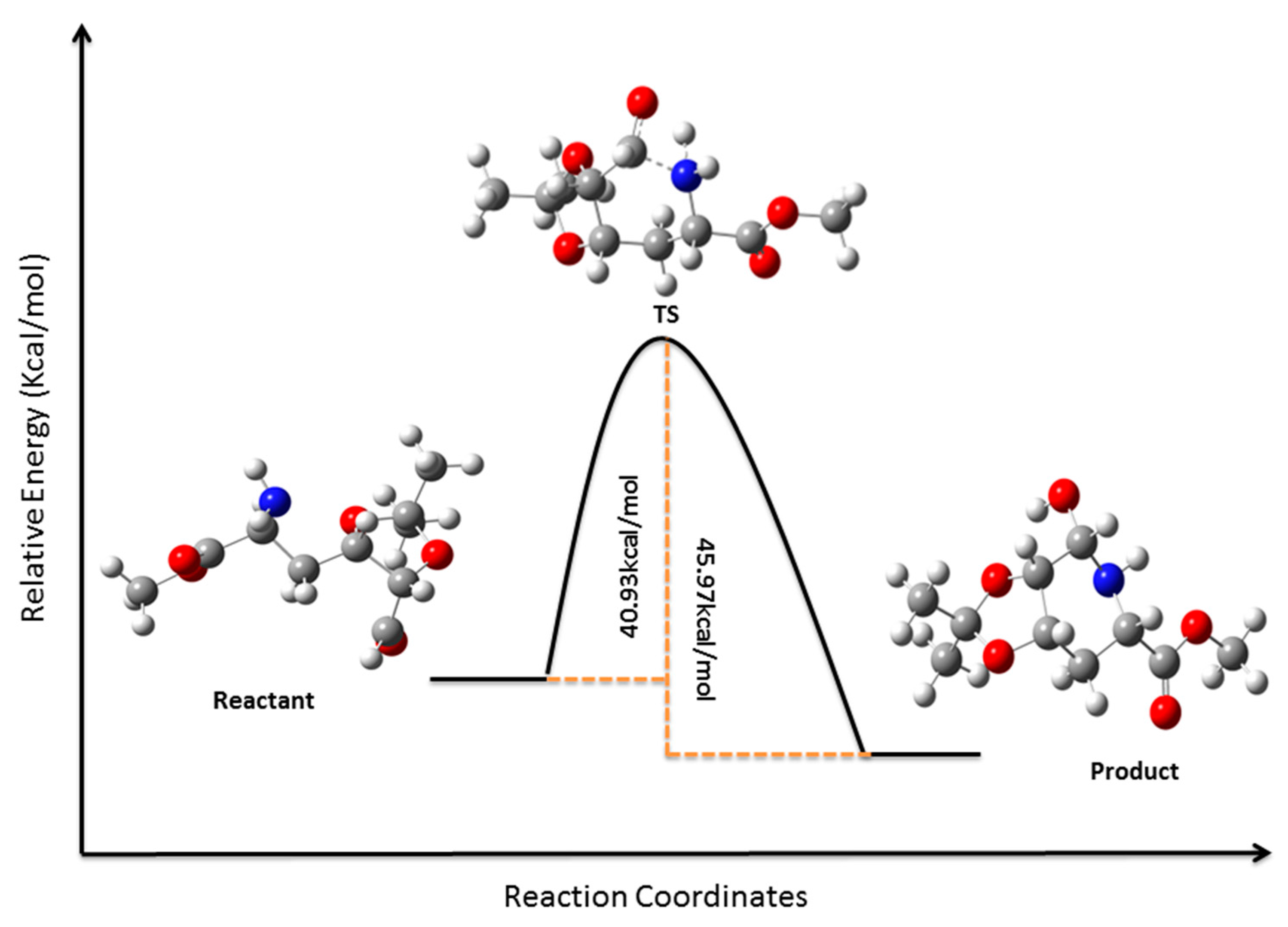
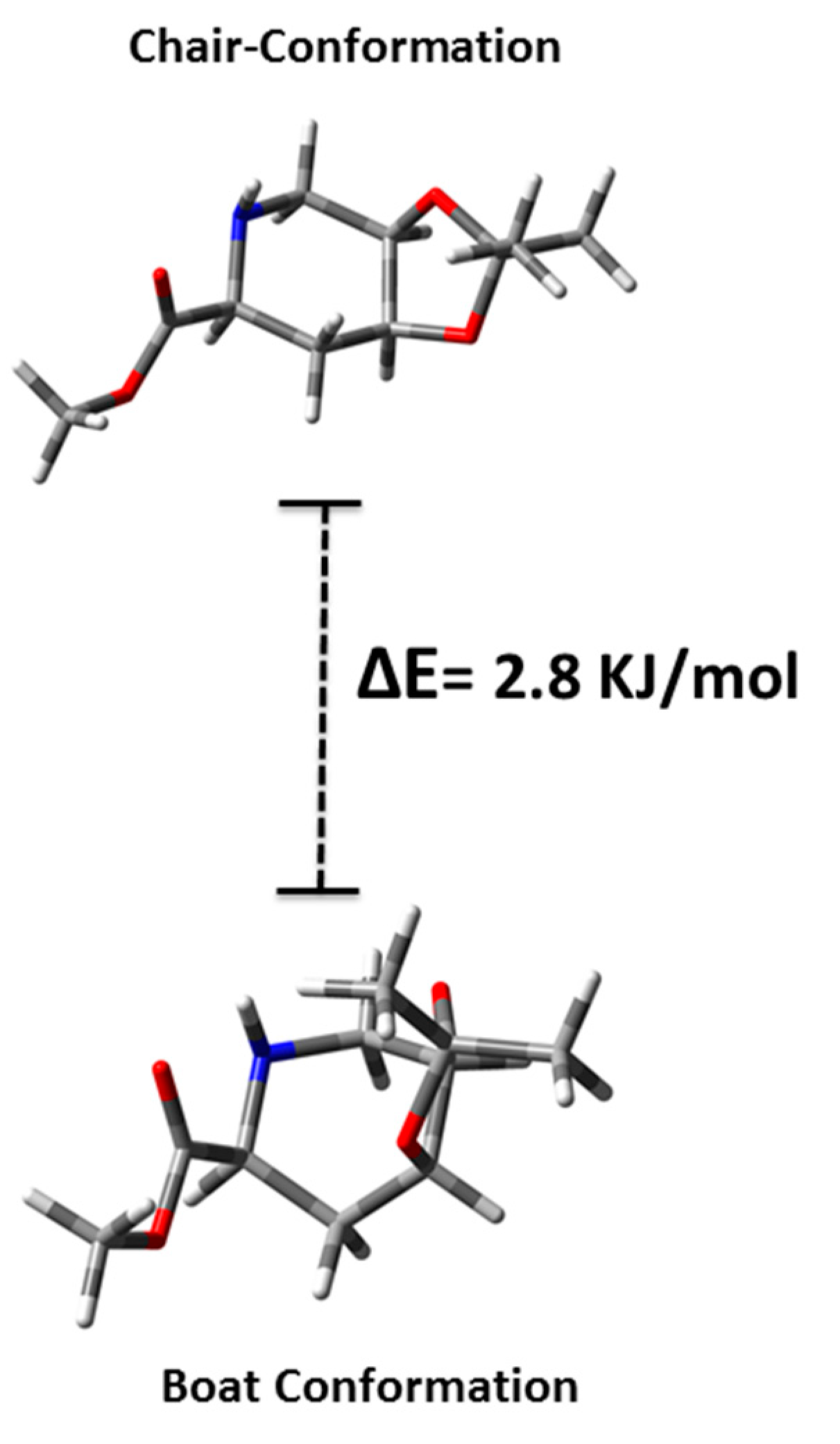
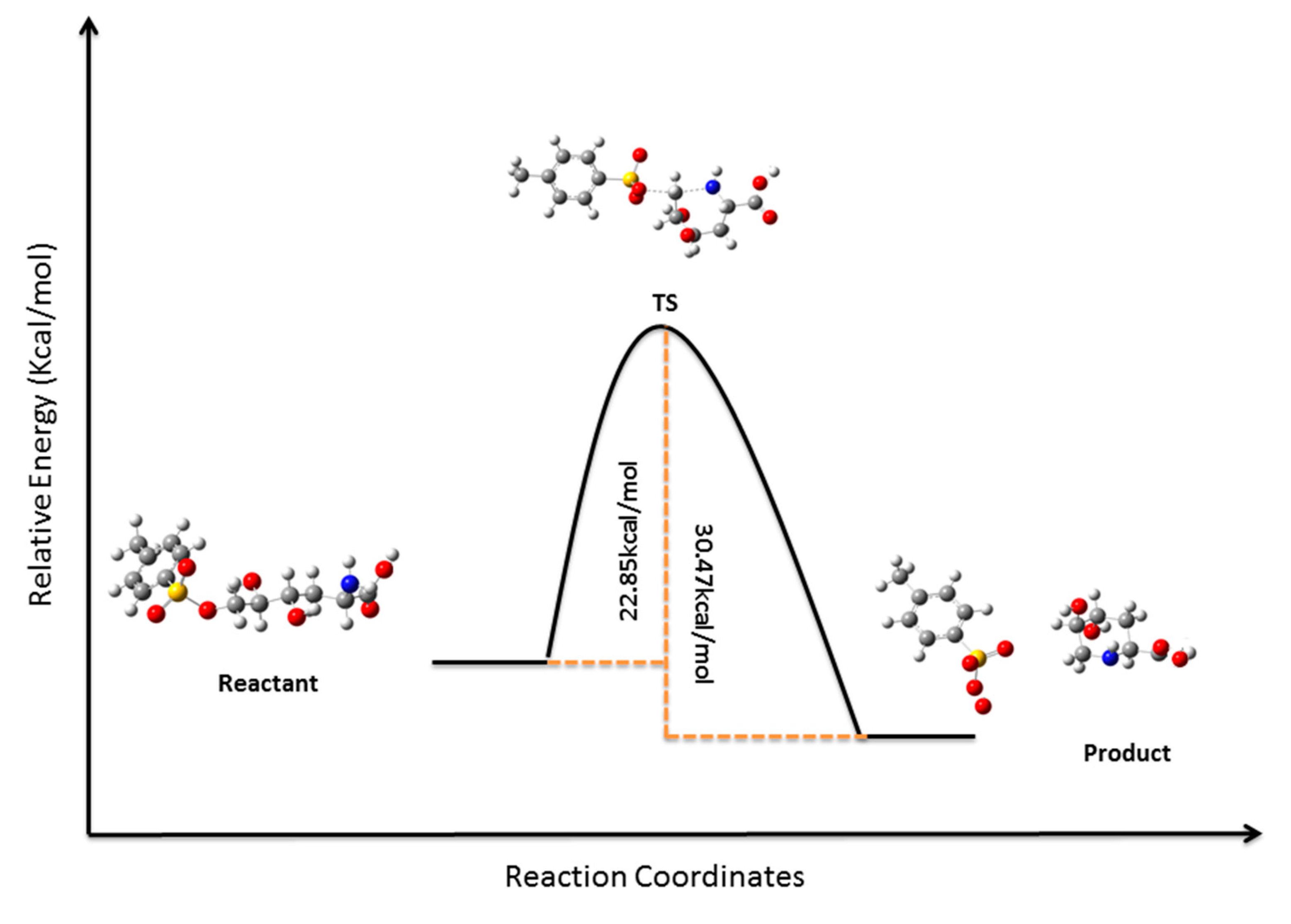
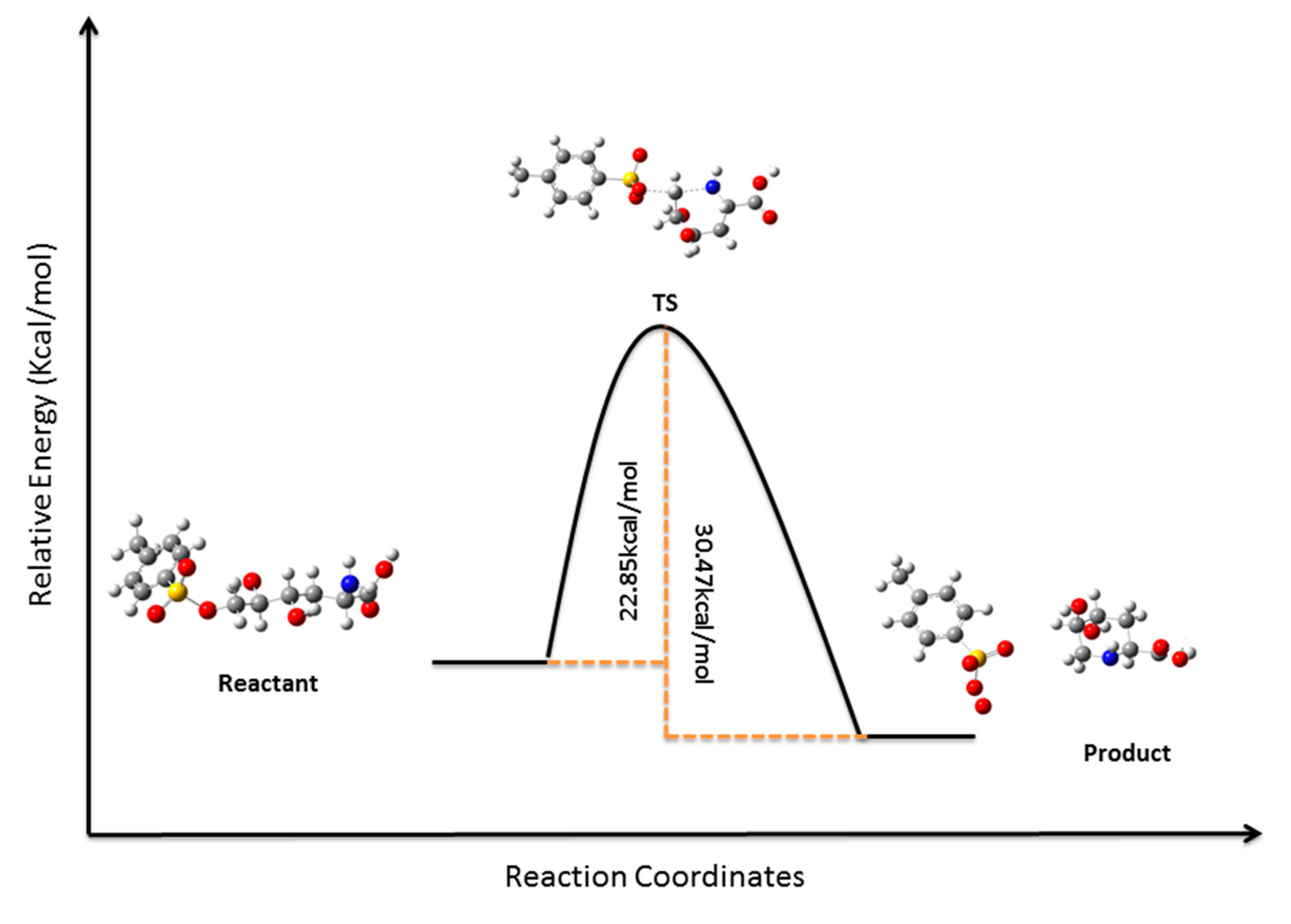
3.12. Computational Details
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fadel, A.; Lahrache, N. An efficient synthesis of enantiomerically pure (R)-pipecolic acid, (S)-proline, and their N-alkylated derivatives. J. Org. Chem. 2007, 72, 1780–1784. [Google Scholar]
- Hanson, G.J.; Vuletich, J.L.; Bedell, L.J.; Bono, C.P.; Howard, S.C.; Welphy, J.K.; Woulfe, S.L.; Zacheis, M.L. Design of MHC Class II (DR4) ligands using conformationally restricted imino acids at p3 and p5. Bioorg. Med. Chem. Lett. 1996, 6, 1931–1936. [Google Scholar]
- Matsoukas, J.M.; Agelis, G.; Hendrelis, J.; Yamdagni, R.; Wu, Q.; Ganter, R.; Smith, J.R.; Moore, D.; Morre, G.J. Synthesis and biological activities of angiotensin H, sarilesin and sarmesin analogues containing Aze or Pip at position 7. J. Med. Chem. 1993, 36, 904–911. [Google Scholar]
- Galeazzi, R.; Mobbili, G.; Orena, M. Modelling and synthesis of conformationally resticted amino acids. Curr. Org. Chem. 2004, 8, 1799–1829. [Google Scholar]
- Ohara, C.; Takahashi, R.; Miyagawa, T.; Yoshimura, Y.; Kato, A.; Adachi, I.; Takahata, H. Synthesis of all stereoisomers of 3-hydroxypipecolic acid and 3-hydroxy-4,5-dehydropipecolic acid and their evaluation as glycosidase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1810–1813. [Google Scholar]
- Fantur, K.; Hofer, D.; Schitter, G.; Steiner, A.J.; Pabst, B.M.; Wrodnigg, T.M.; Stütz, A.E.; Paschke, E. DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human GM1-gangliosidosis fibroblasts. Mol. Gen. Met. 2010, 100, 262–268. [Google Scholar]
- Boucheron, C.; Compain, P.; Martin, O.R. A stereodivergent approach to 1-deoxynojirimycin, 1-deoxygalactonojirimycin and 1-deoxymannojirimycin derivatives. Tetrahedron Lett. 2006, 47, 3081–3084. [Google Scholar]
- Steiner, A.J.; Schitter, G.; Stütz, A.E.; Wrodnigg, T.M.; Tarling, C.A.; Withers, S.G.; Fantur, K.; Mahuran, D.; Paschke, E.; Tropak, M. 1-Deoxygalactonojirimycin-lysine hybrids as potent d-galactosidase inhibitors. Bioorg. Med. Chem. 2008, 16, 10216–10220. [Google Scholar]
- Thorstensson, F.; Wångsell, F.; Kvarnström, I.; Vrang, L.; Hamelink, E.; Jansson, K.; Hallberg, A.; Rosenquist, A.; Samuelsson, B. Synthesis of novel potent hepatitis C virus NS3 protease inhibitors: Discovery of 4-hydroxy-cyclopent-2-ene-1,2-dicarboxylic acid as a N-acyl-l-hydroxyproline bioisostere. Bioorg. Med. Chem. 2007, 15, 827–838. [Google Scholar]
- Stöckel-Maschek, A.; Stiebitz, B.; Koelsch, R.; Neubert, K. Novel 3-amino-2-hydroxy acids containing protease inhibitors. Part 1: Synthesis and kinetic characterization as aminopeptidase P inhibitors. Bioorg. Med. Chem. 2005, 13, 4806–4820. [Google Scholar]
- Manning, K.S.; Lynn, D.G.; Shabanowitz, J.; Fellows, L.E.; Singh, M.; Schrire, B.D. A glucuronidase inhibitor from the seeds of Baphia racemosa: Application of fast atom bombardment coupled with collision activated dissociation in natural product structure assignment. J. Chem. Soc. Chem. Commun. 1985, 127–129. [Google Scholar]
- Mallick, A.; Kumari, N.; Roy, R.; Palanivel, A.; Vankar, Y.D. A concise synthesis of (2R,3R)- and (2R,3S)-3-hydroxypipecolic acids, and total synthesis of (–)-deoxoprosopinine and (+)-2-epi-deoxoprosopinine from d-glycals. Eur. J. Org. Chem. 2014, 5557–5563. [Google Scholar]
- Fleet, G.W.J.; Witty, D.R. Synthesis of homochiral β-hydroxy-α-aminoacids [(2S,3R,4R)-3,4-dihydroxyproline and (2S,3R,4R)-3,4-dihydroxypipecolic aicd] and of 1,4-dideoxy-1,4-imino-d-arabinitol [DAB1] and fagomine [1,5-imino-1,2,5-trideoxy-d-arabino-hexitol]. Tetrahedron Asymmetry 1990, 1, 119–136. [Google Scholar]
- Romeo, J.T.; Swain, L.A.; Bleecker, A.B. Cis-4-hydroxypipecolic acid and 2,4-cis-4,5-trans-4,5-dihydroxypipecolic acid from Calliandra. Phytochemistry 1983, 22, 1615–1617. [Google Scholar]
- Despontin, J.; Marlier, M.; Dardenn, G. l-cis-5-hydroxypipecolic acid from seeds of Gymnocladus dioicus. Phytochemistry 1977, 16, 387–388. [Google Scholar]
- Hatanaka, S.-I.; Kaneko, S. Cis-5-hydroxy-L-pipecolic acid from Morus alba and Lathyrus japonicus. Phytochemistry 1977, 16, 1041–1042. [Google Scholar]
- Kumar, P.S.; Baskaran, S. A regioselective reductive cleavage of benzylidene acetal: Stereoselective synthesis of N-Boc-protected cis-(2R,3S)-3-hydroxy pipecolic acid. Tetrahedron Lett. 2009, 50, 3489–3492. [Google Scholar]
- Hoarau, S.; Fauchere, J.L.; Pappalardo, L.; Roumestant, M.L.; Viallefont, P. Synthesis of enantiomerically pure (2R, 5S)- and (2R, 5R)-5-hydroxypipecolic acid from glycinate schiff bases. Tetrahedron Asymmetry 1996, 7, 2585–2593. [Google Scholar]
- Botman, P.N.M.; Dommerholt, F.J.; de Gelder, R.; Broxterman, Q.B.; Schoemaker, H.E.; Rutjes, F.; Blaauw, R.H. Diastereoselective synthesis of (2S,5R)-5-hydroxypipecolic acid and 6-substituted derivatives. Org. Lett. 2004, 6, 4941–4944. [Google Scholar]
- Le Corre, L.; Dhimane, H. Synthesis of 5-substituted pipecolic acid derivatives as new conformationally constrained ornithine and arginine analogues. Tetrahedron Lett. 2005, 46, 7495–7497. [Google Scholar]
- Occhiato, E.G.; Scarpi, D.; Guarna, A.; Tabasso, S.; Deagostino, A.; Prandi, C. A short and convenient synthesis of enantiopure cis- and trans-4-hydroxypipecolic acid. Synthesis 2009, 3611–3616. [Google Scholar]
- Di Nardo, C.; Varela, O. Enantioselective synthesis of (2R,4S)-4-hydroxy-pipecolic acid from d-gluconoheptono-1,4-lactone. J. Org. Chem. 1999, 64, 6119–6125. [Google Scholar]
- Scarpi, D.; Bartali, L.; Casini, A.; Occhiato, E.G. Complementary and stereodivergent approaches to the synthesis of 5-Hydroxy- and 4,5-dihydroxypipecolic acids from enantiopure hydroxylated lactams. Eur. J. Org. Chem. 2013, 1306–1317. [Google Scholar]
- Shewry, P.R.; Fowden, L. 4,5-Dihydroxypipecolic acids in the seed of Julbernardia isoberlinia and Brachystegia. Phytochemistry 1976, 15, 1981–1983. [Google Scholar]
- Marlier, M.; Dardenne, G.; Casimir, M. 2S-Carboxy-4R,5S-dihydroxypiperidine et 2S-carboxy-4S,5S-dihydroxypiperidine a partir de Derris elliptica. Phytochemistry 1976, 15, 183–185. [Google Scholar]
- Uhrig, M.L.; Simirgiotis, M.J.; Varela, O. Enantiospecific synthesis of the sugar amino acid (2S,5S)-5-(aminomethyl)-tetrahydrofuran-2-carboxylic acid. Tetrahedron Asymmetry 2010, 21, 2435–2440. [Google Scholar]
- Gómez, R.V.; Kolender, A.A.; Varela, O. Synthesis of polyhydroxy amino acids based on d- and l-alanine from d-glycero-d-gulo-heptono-1,4-lactone. Carbohydr. Res. 2006, 341, 1498–1504. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian Software, version 09 revision D01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simirgiotis, M.J.; Vallejos, J.; Areche, C.; Sepúlveda, B. Concise and Straightforward Asymmetric Synthesis of a Cyclic Natural Hydroxy-Amino Acid. Molecules 2014, 19, 19516-19531. https://doi.org/10.3390/molecules191219516
Simirgiotis MJ, Vallejos J, Areche C, Sepúlveda B. Concise and Straightforward Asymmetric Synthesis of a Cyclic Natural Hydroxy-Amino Acid. Molecules. 2014; 19(12):19516-19531. https://doi.org/10.3390/molecules191219516
Chicago/Turabian StyleSimirgiotis, Mario J., Javier Vallejos, Carlos Areche, and Beatriz Sepúlveda. 2014. "Concise and Straightforward Asymmetric Synthesis of a Cyclic Natural Hydroxy-Amino Acid" Molecules 19, no. 12: 19516-19531. https://doi.org/10.3390/molecules191219516