Efficient Synthesis of Kinsenoside and Goodyeroside A by a Chemo-Enzymatic Approach

Abstract
:1. Introduction
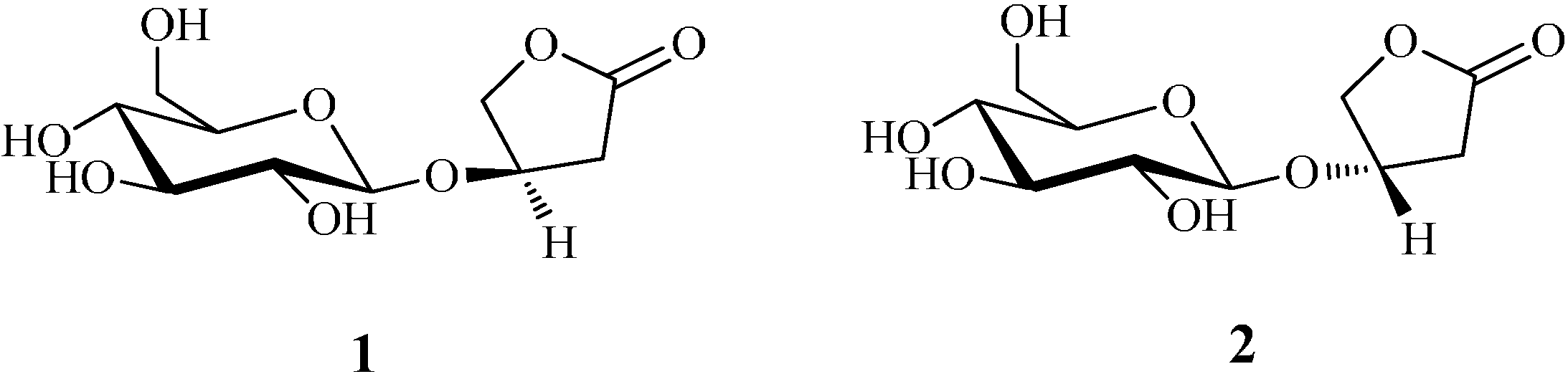
2. Results and Discussion
2.1. Improvement of the Chemical Approach
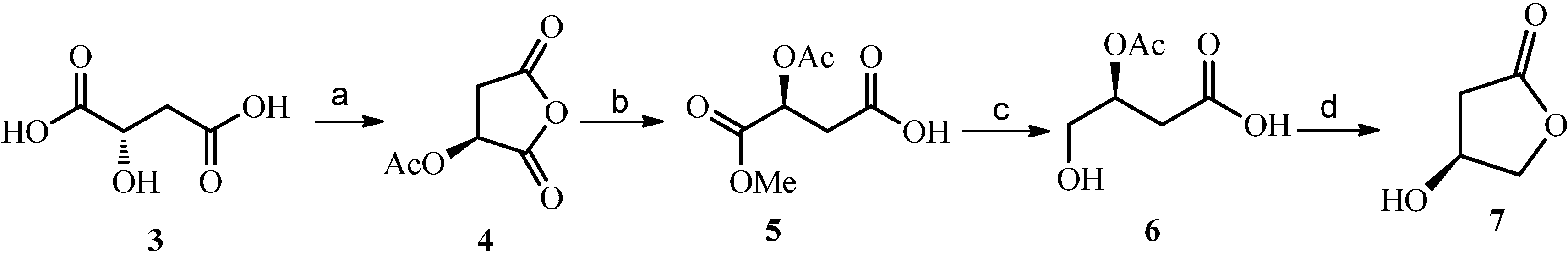
2.2. Construction and Optimization of Enzymatic Synthesis

2.2.1. Single Factor Experiments

2.2.2. Orthogonal Experiments

{kind=link}
{kind=link}
{kind=link}
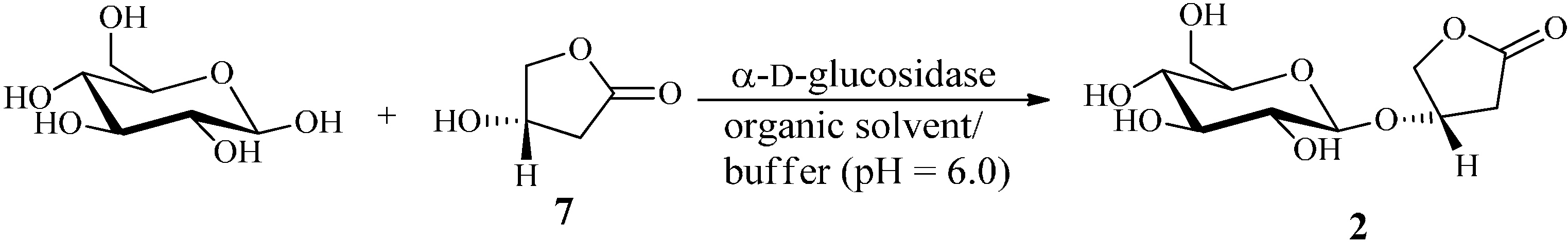{kind=link}
| Effect Factor | Enzyme Dosage (mg) | Lactone Equivalents | Reaction Time (h) | Concentration of 2 (mg/mL) | Yield (%) |
|---|---|---|---|---|---|
| 1 | 4 | 7 | 32 | 6.0 | 9.1 |
| 2 | 4 | 8 | 44 | 8.3 | 12.6 |
| 3 | 4 | 9 | 56 | 9.4 | 14.2 |
| 4 | 4 | 10 | 68 | 9.2 | 13.9 |
| 5 | 5 | 7 | 44 | 8.4 | 12.7 |
| 6 | 5 | 8 | 32 | 7. 8 | 11.8 |
| 7 | 5 | 9 | 68 | 8.8 | 13.3 |
| 8 | 5 | 10 | 56 | 11.1 | 16.8 |
| 9 | 6 | 7 | 56 | 7.5 | 11.4 |
| 10 | 6 | 8 | 68 | 9.3 | 14.1 |
| 11 | 6 | 9 | 32 | 8.7 | 13.2 |
| 12 | 6 | 10 | 44 | 9.1 | 13.8 |
| 13 | 7 | 7 | 68 | 8.1 | 12.3 |
| 14 | 7 | 8 | 56 | 8.5 | 12.9 |
| 15 | 7 | 9 | 44 | 9.6 | 14.5 |
| 16 | 7 | 10 | 32 | 9.1 | 13.8 |
| k1 | 8.217 | 7.507 | 7.876 | ||
| k2 | 8.990 | 8.478 | 8.822 | ||
| k3 | 8.641 | 9.097 | 9.115 | ||
| k4 | 8.841 | 9.608 | 8.877 | ||
| R | 0.773 | 2.101 | 1.239 |
3. Experimental Section
3.1. Materials
3.2. NMR, MS and HPLC-ELSD Analysis
3.3. Chemical Synthesis of 3-Hydroxy-γ-butyrolactone
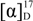 = +86.9 (c = 0.24, EtOH);
= +86.9 (c = 0.24, EtOH); 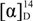 = +72.2 (c = 1.25, EtOH) in the reference [11]; 1H-NMR (CDCl3): δH 4.64–4.68 (m, 1H, H-3), 4.39 (dd, J = 10.3, 4.5 Hz, 1H, H-4b), 4.27 (d, J = 10.3 Hz, 1H, H-4a), 2.72 (dd, J = 18.0, 6.1 Hz, 1H, H-2b), 2.45–2.56 (m, 1H, H-2a). 13C-NMR (CDCl3): δC 37.8, 67.5, 76.0, 176.1. = −83.7 (c = 0.24, EtOH);
= +72.2 (c = 1.25, EtOH) in the reference [11]; 1H-NMR (CDCl3): δH 4.64–4.68 (m, 1H, H-3), 4.39 (dd, J = 10.3, 4.5 Hz, 1H, H-4b), 4.27 (d, J = 10.3 Hz, 1H, H-4a), 2.72 (dd, J = 18.0, 6.1 Hz, 1H, H-2b), 2.45–2.56 (m, 1H, H-2a). 13C-NMR (CDCl3): δC 37.8, 67.5, 76.0, 176.1. = −83.7 (c = 0.24, EtOH); 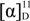 = −72.2 (c = 1.25, EtOH) in the reference [11]. 1H-NMR (CDCl3): δH 4.64–4.68 (m, 1H, H-3), 4.39 (dd, J = 10.3, 4.5 Hz, 1H), 4.24–4.29 (m, 1H, H-4a), 2.73 (dd, J = 18.0, 6.1 Hz, 1H), 2.50 (ddd, J = 18.0, 1.7, 1.1 Hz, 1H). 13C-NMR (CDCl3): δC37.8, 67.5, 75.9, 176.1.
= −72.2 (c = 1.25, EtOH) in the reference [11]. 1H-NMR (CDCl3): δH 4.64–4.68 (m, 1H, H-3), 4.39 (dd, J = 10.3, 4.5 Hz, 1H), 4.24–4.29 (m, 1H, H-4a), 2.73 (dd, J = 18.0, 6.1 Hz, 1H), 2.50 (ddd, J = 18.0, 1.7, 1.1 Hz, 1H). 13C-NMR (CDCl3): δC37.8, 67.5, 75.9, 176.1.3.4. Enzymatic Synthesis of Compounds 1 and 2
= +21.0 (c = 0.10, EtOH ); = +17.9 (c = 1.24, EtOH) in the reference [11]. 1H-NMR (pyridine-d5): δH 4.91 (d, J = 7.8 Hz, 1H, H-1'), 4.84–4.87 (m, 1H, H-3), 4.70 (d, J = 10.1 Hz, 1H, H-4a), 4.55 (d, J = 11.8 Hz, 1H, H-6'a), 4.34–4.45 (m, 2H, H-4b and H-6'b), 4.20–4.24 (m, 2H, H-4' and H-5'), 3.93–4.01 (m, 2H, H-3' and H-2'), 2.83–2.92 (m, 2H, H-2a and H-2b). 13C-NMR (pyridine-d5): δC 36.0, 63.0, 71.8, 75.1, 75.2, 75.6, 78.7, 79.1, 104.4, 176.4. ESI-MS: m/z 309.10 [M+HCOO]− (Calcd for C11H17O10: 309.08). = −68.7 (c = 0.10, H2O); = −69.9 (c = 0.55, H2O) in the reference [11]. 1H-NMR (pyridine-d5) δH 4.94 (d, J = 7.7 Hz, 1H, H-1'), 4.87–4.91 (m, 1H, H-3), 4.66 (d, J = 10.1 Hz, 1H, H-4a), 4.55 (d, J = 11.8 Hz, 1H, H-6'a), 4.33–4.41 (m, 2H, H-4b and H-6'b), 4.19–4.26 (m, 2H, H-4' and H-5'), 3.92–4.01 (m, 2H, H-3' and H-2'), 2.86–2.95 (m, 2H, H-2a and H-2b). 13C-NMR (pyridine-d5): δC36.8, 63.0, 71.8, 74.4, 75.0, 75.1, 78.7, 79.1, 104.0, 176.7. ESI-MS: m/z 309.10 [M+HCOO]− (Calcd for C11H17O10: 309.08).4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ito, A.; Kasa, R. Aliphatic and aromatic glucosides from Anoectochilus koshunensis. Phytochemistry 1993, 33, 1133–1137. [Google Scholar] [CrossRef]
- Du, X.M.; Yoshizawa, T.; Shoyama, Y. Butanoic acid glucoside composition of whole body and in vitro plantlets of Anoectochilus formosanus. Phytochemistry 1998, 49, 1925–1928. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Cai, J.Y.; Ruan, H.L. Antihyperglycemic activity of kinsenoside, a high yielding constituent from Anoectochilus roxburghii in streptozotocin diabetic rats. J. Ethnopharmacol. 2007, 114, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.Y.; Li, Y.M.; Zhu, D.Y. Phenolic glucosides and a γ-lactone glucoside from the sprouts of Crocus sativus. Planta Med. 1999, 65, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Du, X.M.; Sun, N.Y.; Chen, Y. Hepatoprotective aliphatic glycosides from three Goodyera species. Biol. Pharm. Bull. 2000, 23, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.B.; Lin, W.L.; Hsieh, C.C. The hepatoprotective activity of kinsenoside from Anoectochilus formosanus. Phytother. Res. 2007, 21, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Du, X.M.; Sun, N.; Tamura, T. Higher yielding isolation of kinsenoside in Anoectochilus and its antihyperliposis effect. Biol. Pharm. Bull. 2001, 24, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Wu, J.B.; Lin, H.; Lin, W.C. Kinsenoside isolation from anoectochilus formosanus suppresses LPS-stimulated inflammatory reactions inmacrophages and endotoxin shock in mice. Shock 2011, 35, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Liu, Q.; Xiao, B.; Zhou, J. The vascular protective properties of kinsenoside isolated from Anoectochilus roxburghii under high glucose condition. Fitoterapia 2013, 86, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Lin, H.; Wu, J.B.; Lin, W.C. Kinsenoside prevents ovariectomy-induced bone loss and suppresses osteoclastogenesis by regulating classical NF-κB pathways. Osteoporos. Int. 2013, 24, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, H.H.; Chen, Q.H. A novel total synthesis of kinsenoside and goodyeroside A relying on the efficient reaction of the chiral 2(5H)-furanones. J. Asian Nat. Prod. Res. 2005, 7, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Suzuki, N.; Yamaura, M.; Yoshizawa, T. Synthesis of 3-O-β-d-glucopyranosyl-(3R)-hydroxybutanolide (kinsenoside) and 3-O-β-d-glucopyranosyl-(3S)-hydroxy-butanolide (goodyeroside A). J. Carbohydr. Chem. 2005, 24, 73–84. [Google Scholar] [CrossRef]
- Henrot, S.; Larcheveque, M.; Petit, Y. Aminoacids as chiral synthons: Preparation of enantiomerically pure (R) and (S) malic acids and its application to the synthesis of 3-hydroxy-4-butanolide. Synth. Commun. 1986, 16, 183–190. [Google Scholar] [CrossRef]
- Ensch, C.; Hesse, M. Enantioselective entry to the homalium alkaloid hoprominol: Synthesis of an (R, R, R)-hoprominol derivative. Helv. Chim. Acta 2003, 86, 233–246. [Google Scholar] [CrossRef]
- Tong, A.M.; Xu, J.H.; Lu, W.Y.; Lin, G.Q. Construction and optimization of a monophasic organic-water system for enzymatic synthesis of p-nitrobenzyl-β-d-glucopyranosides by reverse hydrolysis. J. Mol. Catal. B-Enzym. 2005, 32, 83–88. [Google Scholar] [CrossRef]
- Vic, G.; Thomas, D.; Crout, D.H.G. Solvent effect on enzyme-catalyzed synthesis of β-glucosides using the reverse hydrolysis method: Application to the preparative-scale synthesis of 2-hydroxybenzyl and octyl β-glucopyranosides. Enzym. Microb. Technol. 1997, 20, 597–603. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1 and 2 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Xia, Y.; Lai, Y.; Tang, F.; Luo, Z.; Xue, Y.; Yao, G.; Zhang, Y.; Zhang, J. Efficient Synthesis of Kinsenoside and Goodyeroside A by a Chemo-Enzymatic Approach. Molecules 2014, 19, 16950-16958. https://doi.org/10.3390/molecules191016950
Zhang Y, Xia Y, Lai Y, Tang F, Luo Z, Xue Y, Yao G, Zhang Y, Zhang J. Efficient Synthesis of Kinsenoside and Goodyeroside A by a Chemo-Enzymatic Approach. Molecules. 2014; 19(10):16950-16958. https://doi.org/10.3390/molecules191016950
Chicago/Turabian StyleZhang, Yang, Yihong Xia, Yongji Lai, Fang Tang, Zengwei Luo, Yongbo Xue, Guangmin Yao, Yonghui Zhang, and Jinwen Zhang. 2014. "Efficient Synthesis of Kinsenoside and Goodyeroside A by a Chemo-Enzymatic Approach" Molecules 19, no. 10: 16950-16958. https://doi.org/10.3390/molecules191016950