Radiation-Induced High-Temperature Conversion of Cellulose

Abstract
:1. Introduction
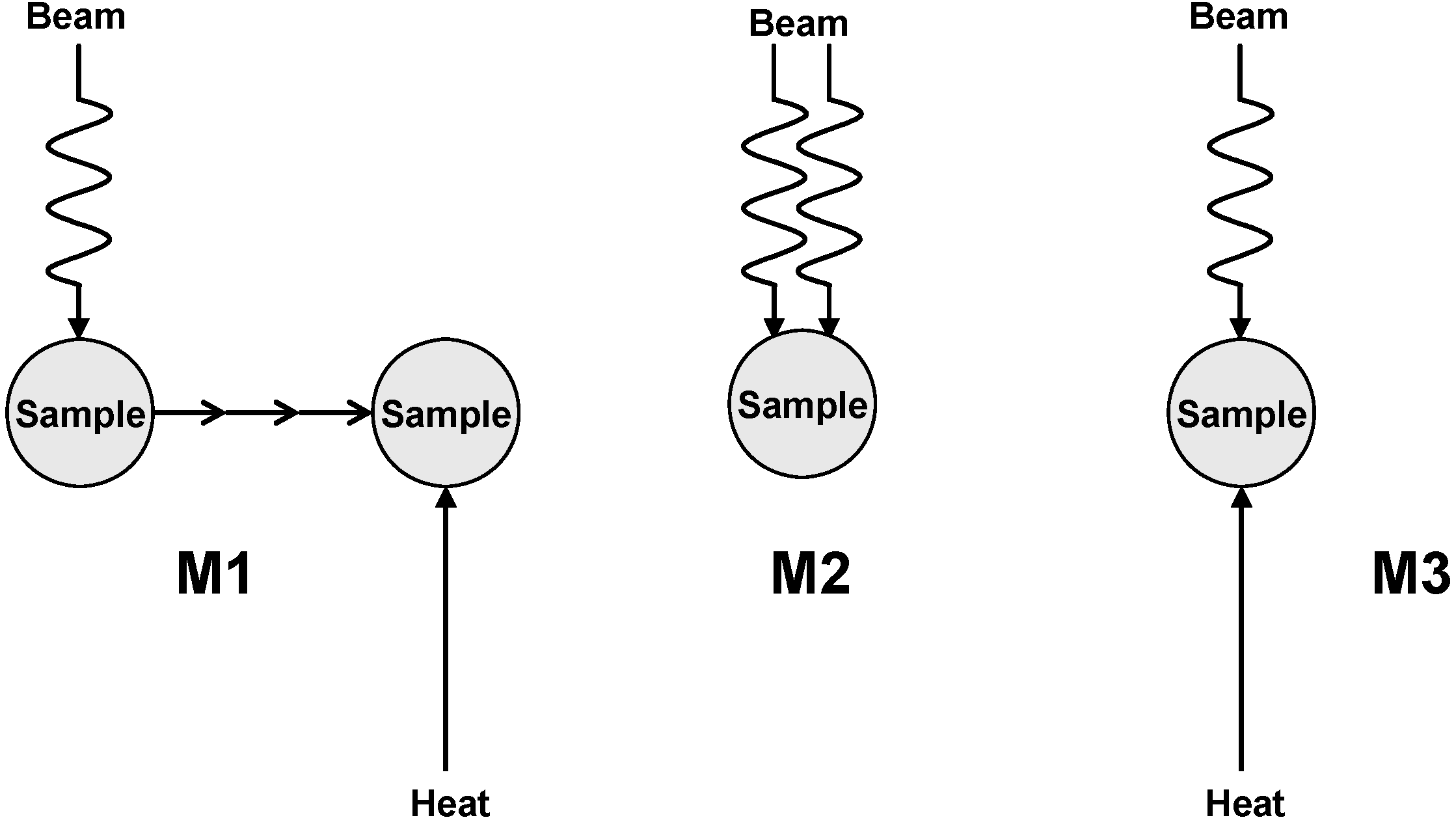
2. Decomposition of Cellulose at Moderate Absorbed Dose Rates
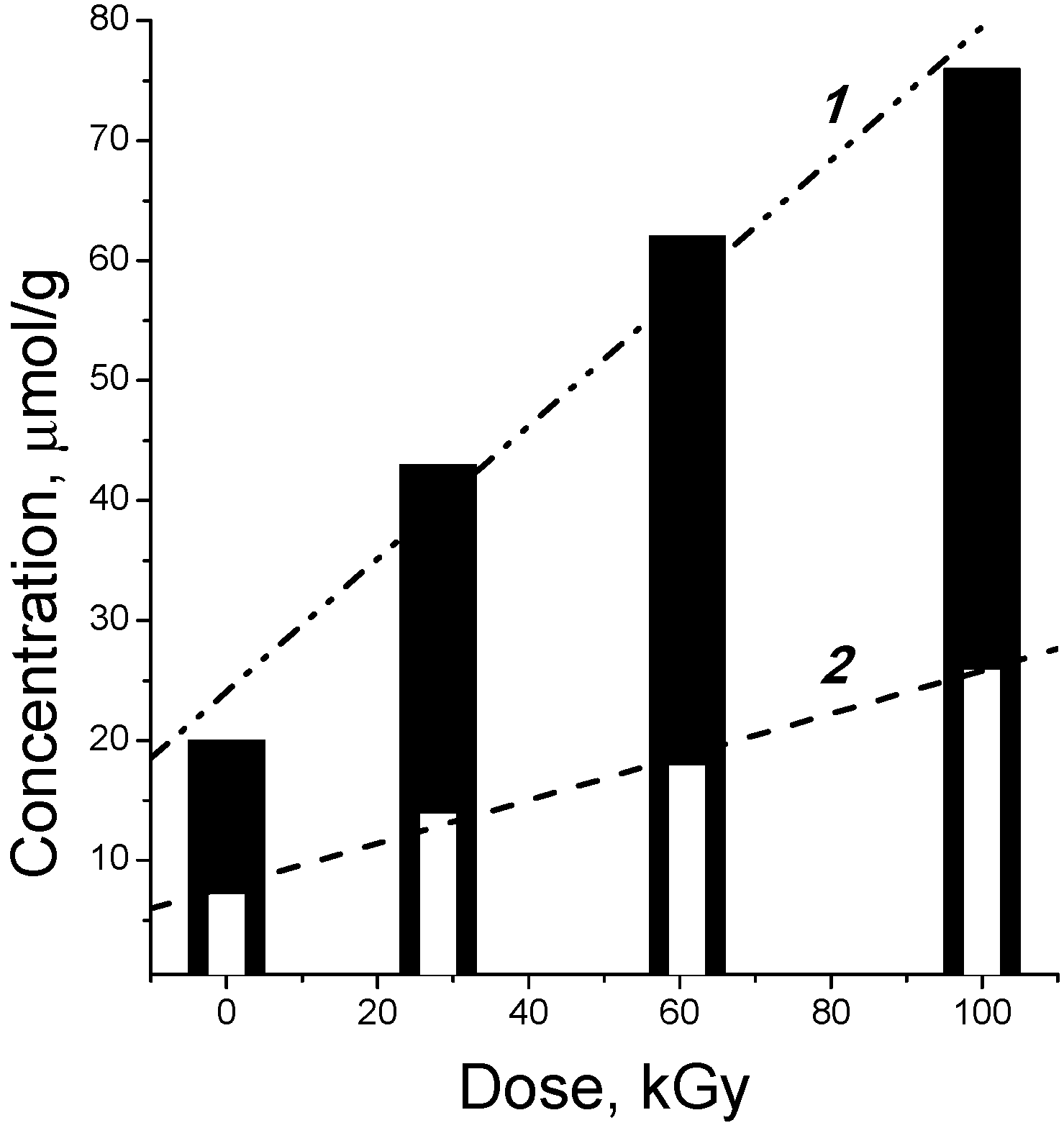
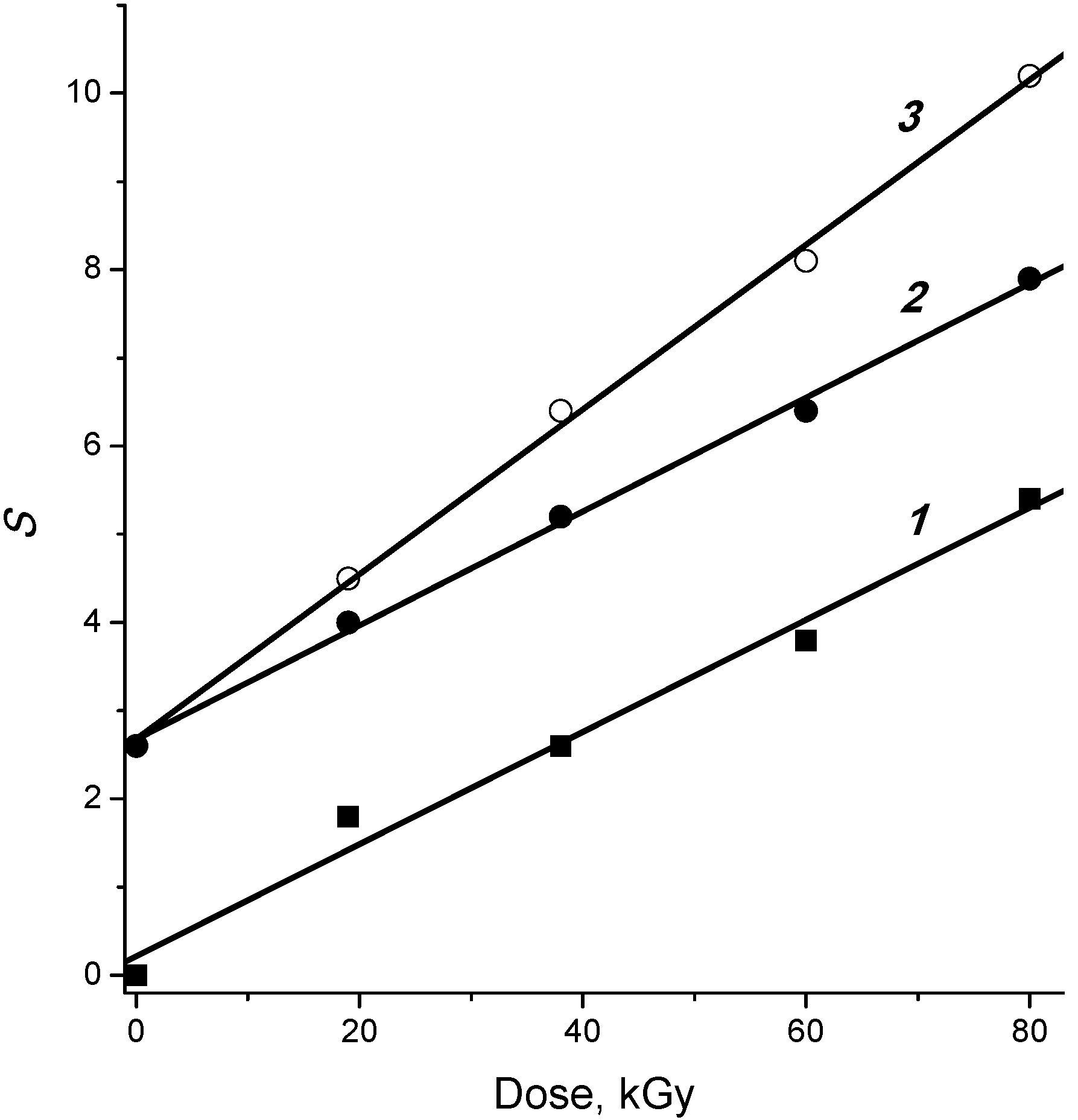
2.1. Regularities and Products of Cellulose Decomposition
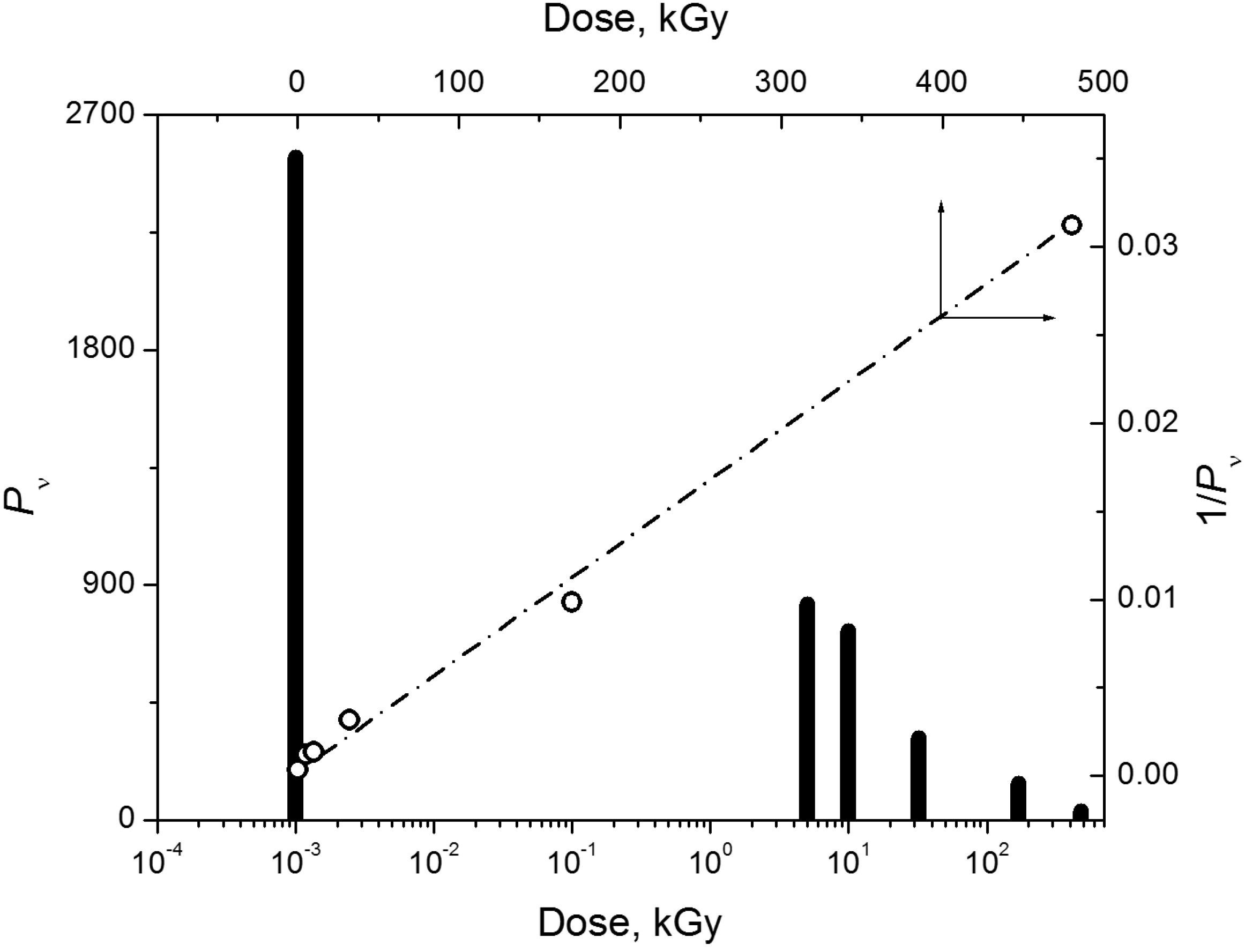

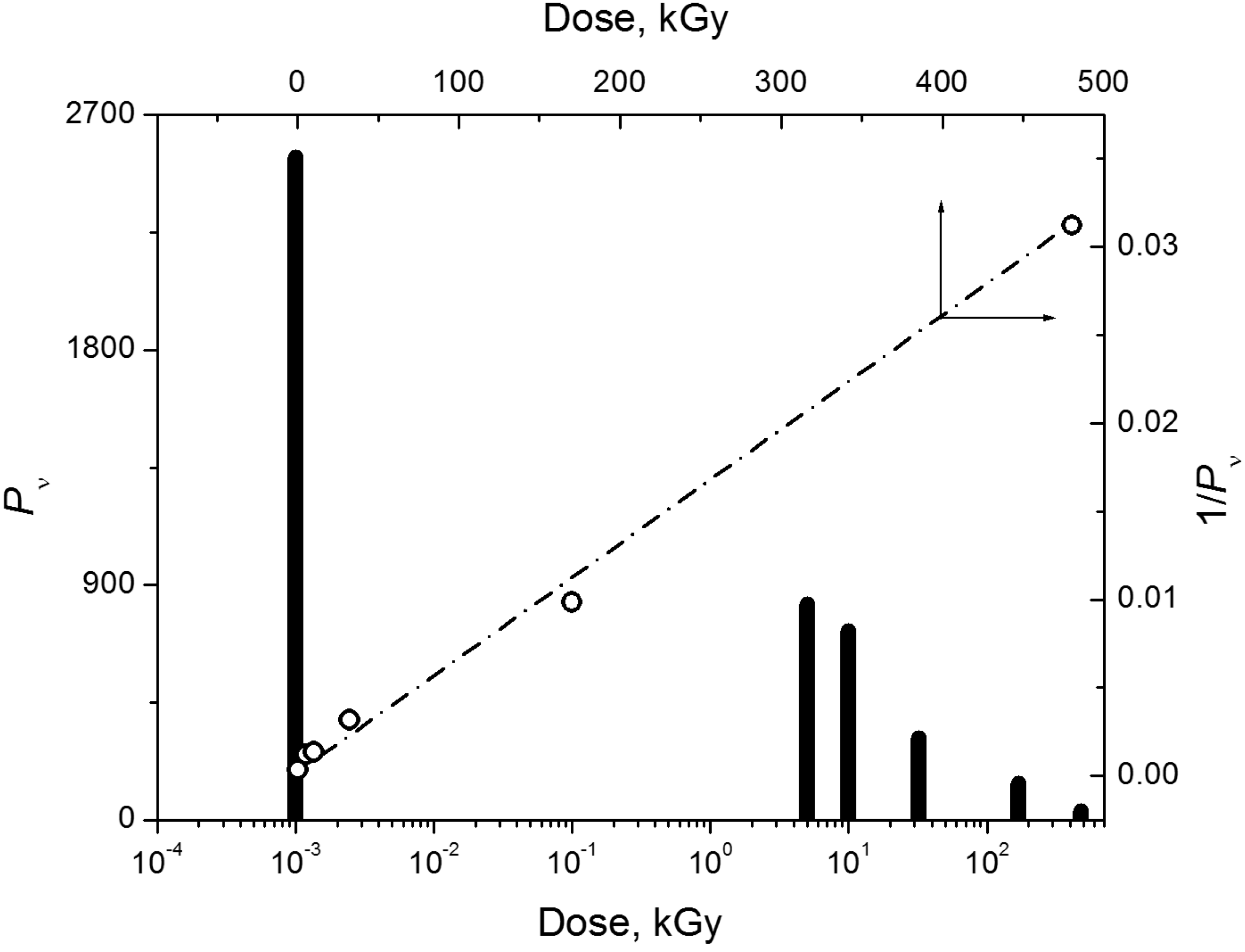
{kind=link}
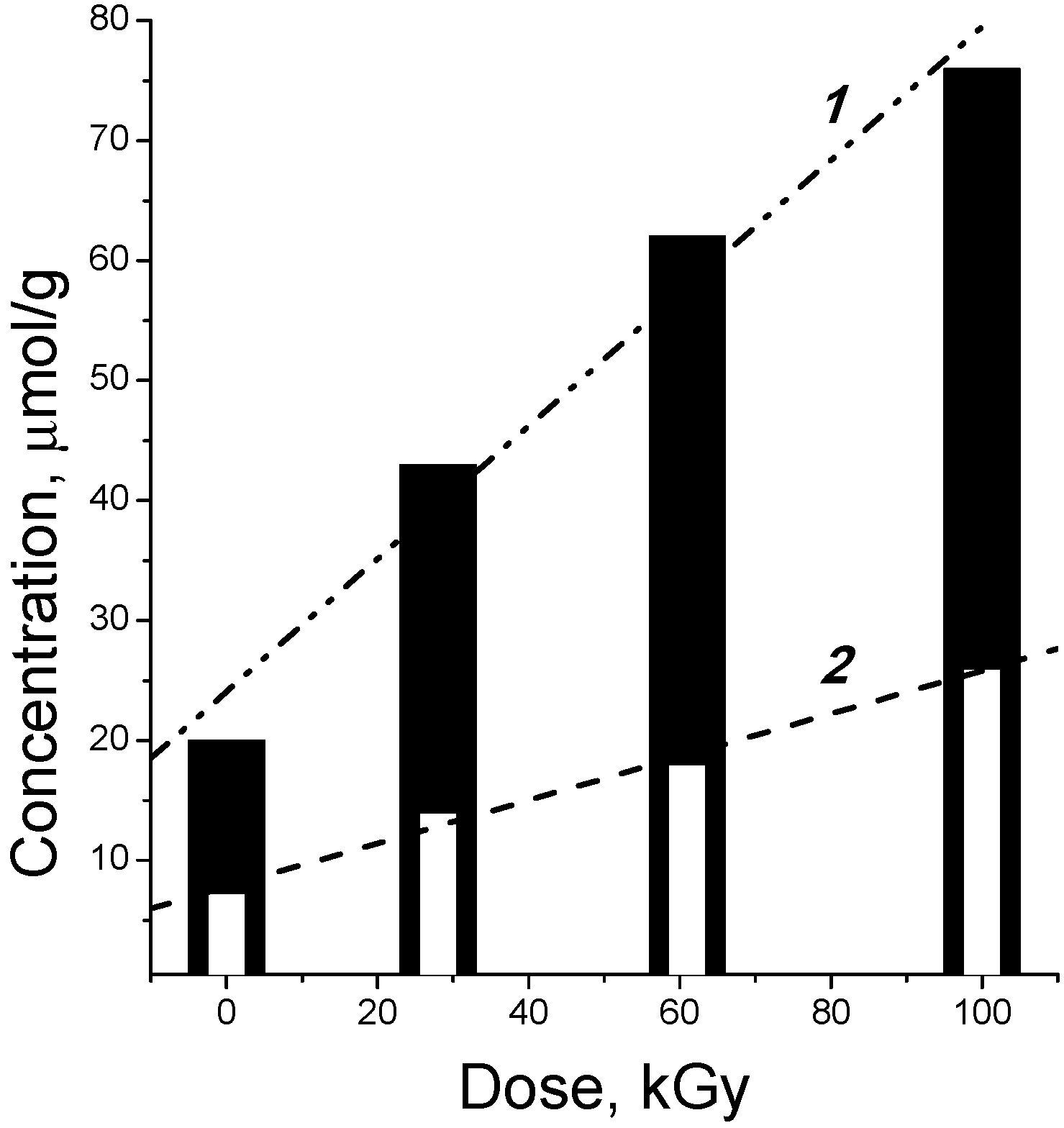
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose type | Concentration, mmol/100 g | Yield, mmol/100 eV | ||
|---|---|---|---|---|
| -CHO | -CO2H | -CHO | -CO2H | |
| Sulfite cellulose | 0.13 | 0.24 | 6.0 | 2.0 |
| High-viscosity sulfite cellulose | 0.53 | 3.16 | 5.4 | 1.8 |
| Cotton cellulose | 0.45 | 0.45 | 5.9 | 2.5 |
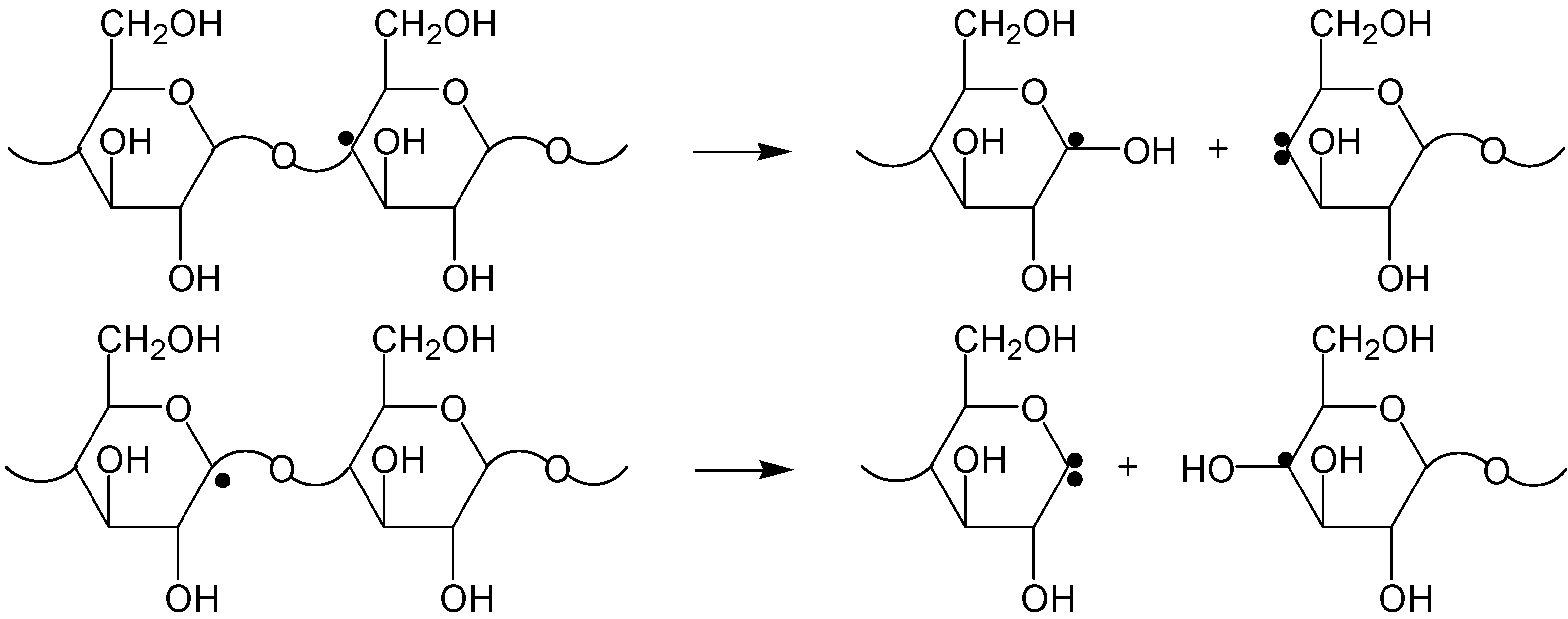
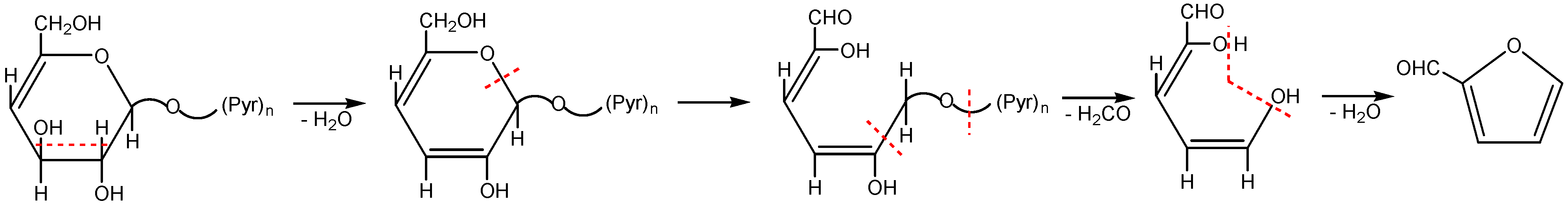
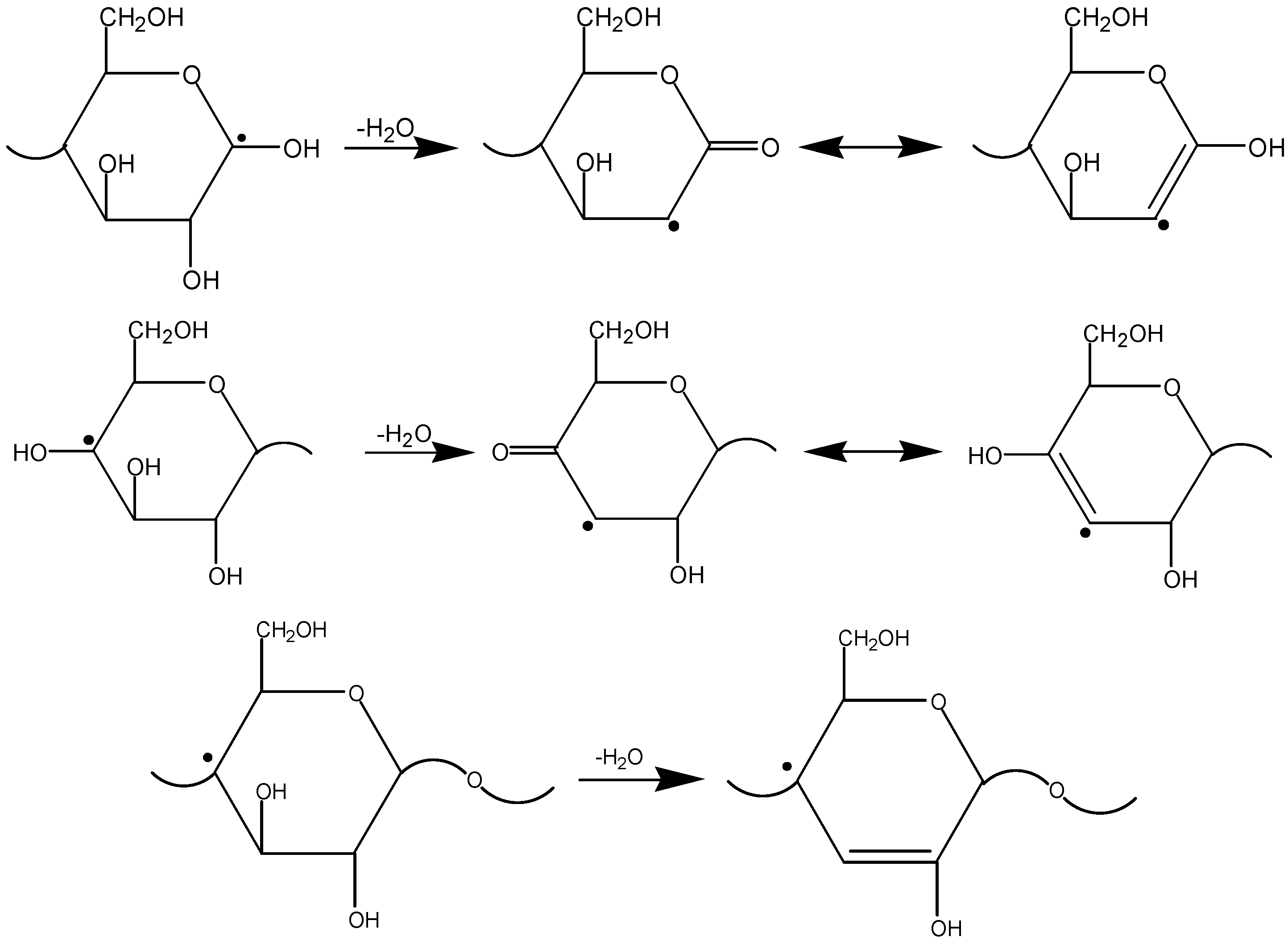
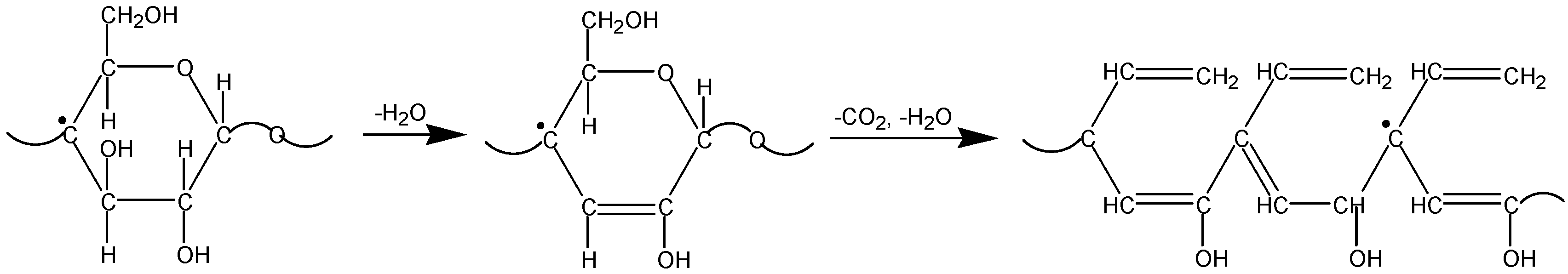
2.2. The Decomposition Mechanism

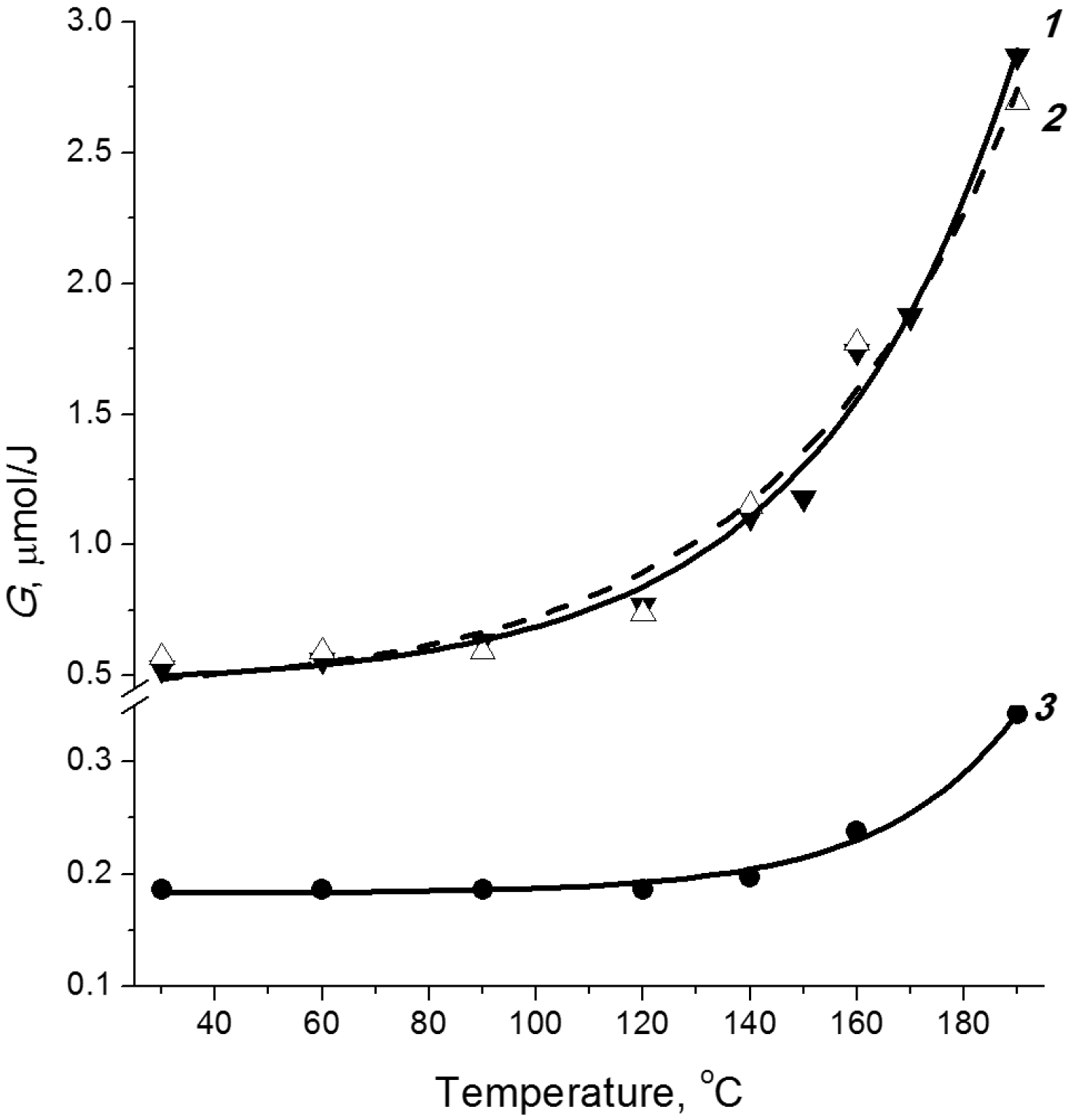
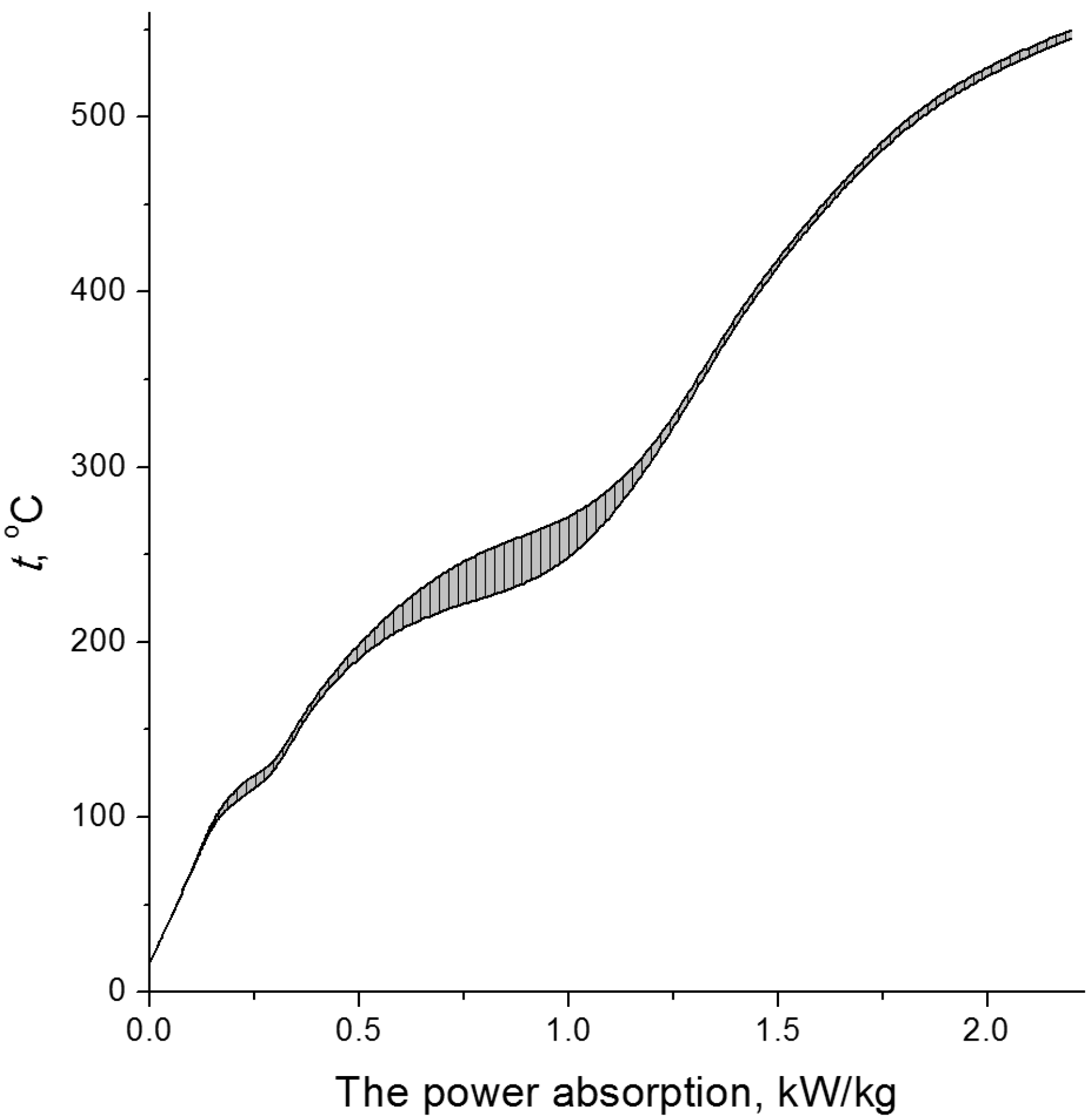
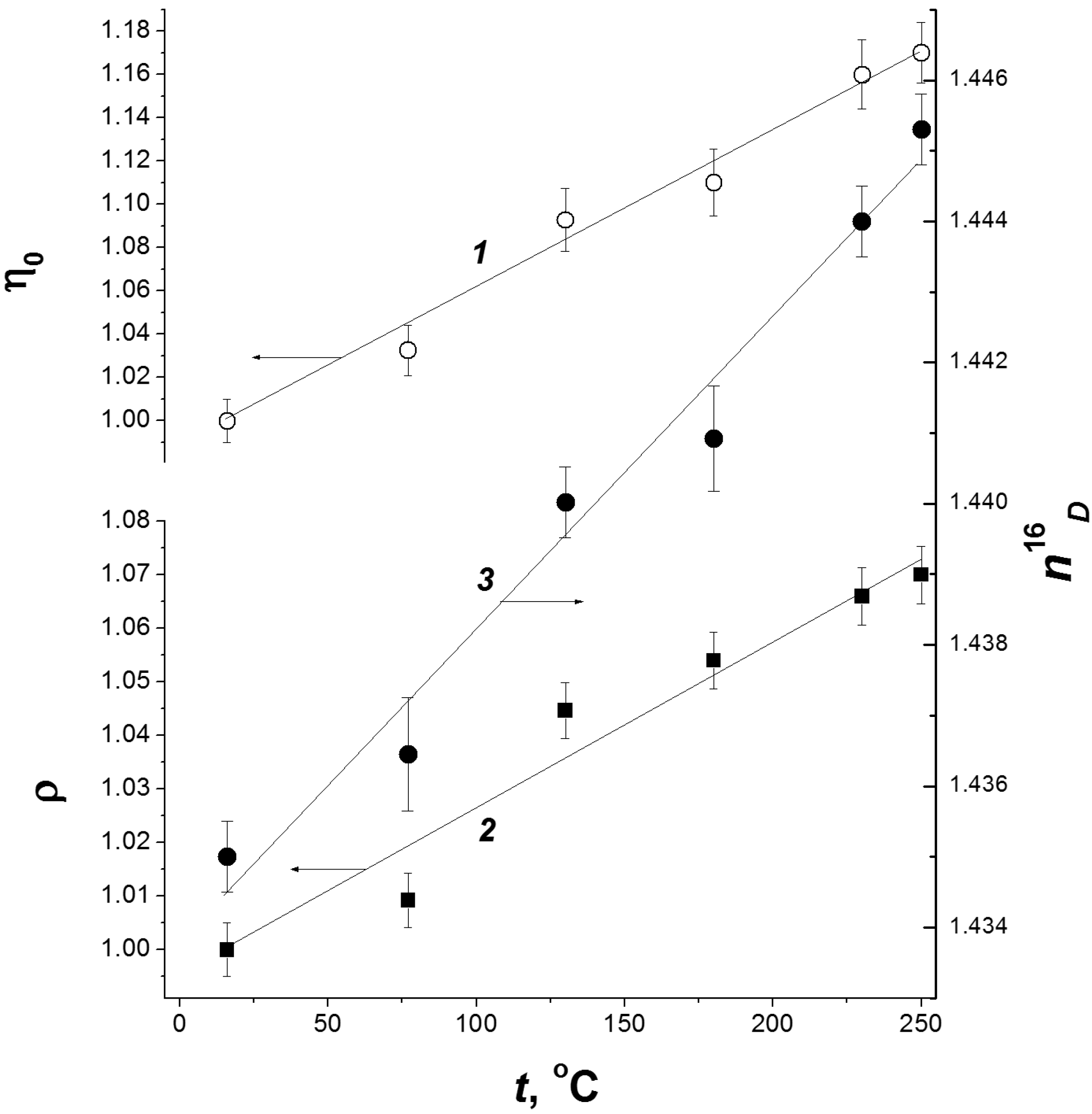
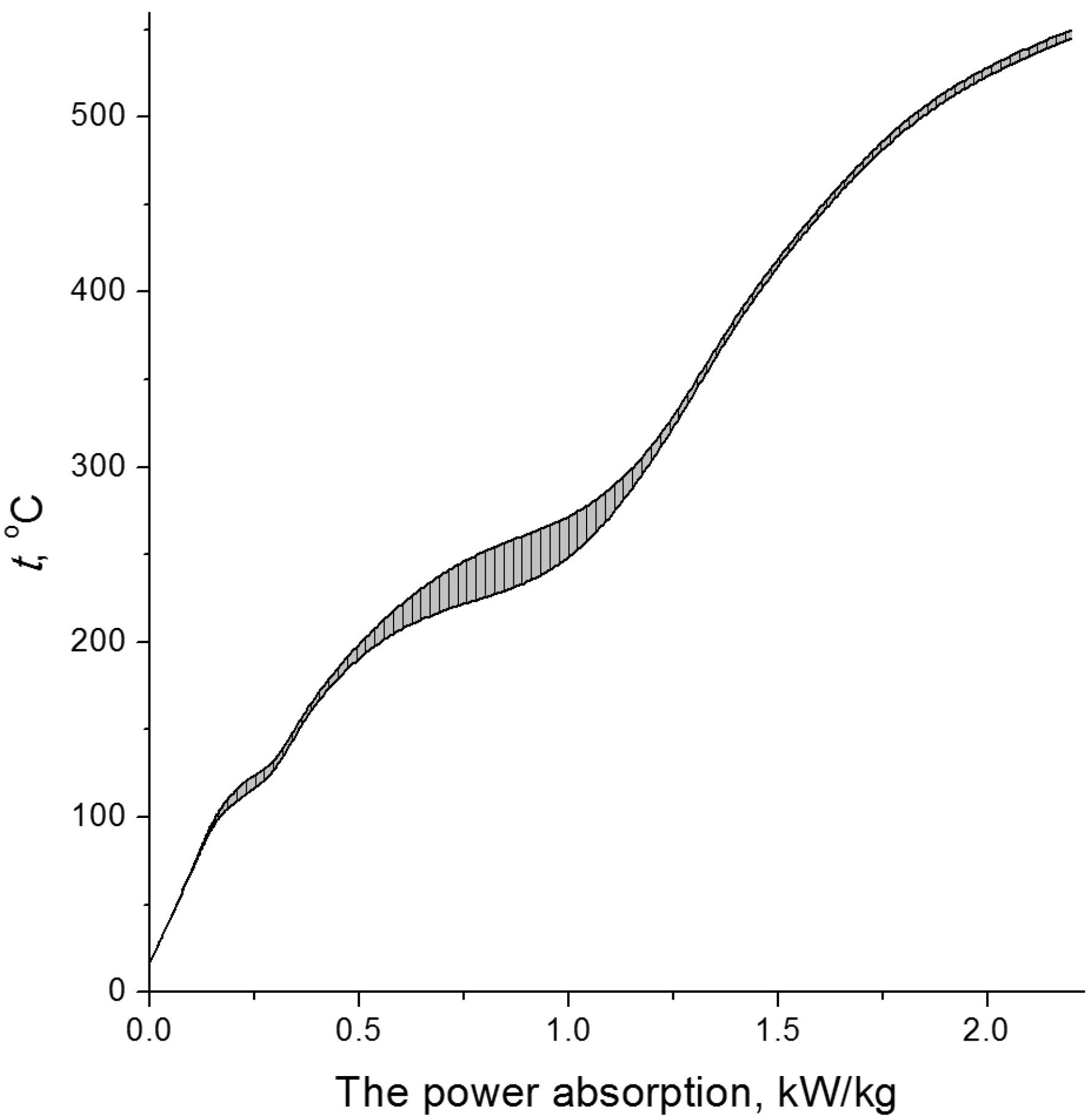
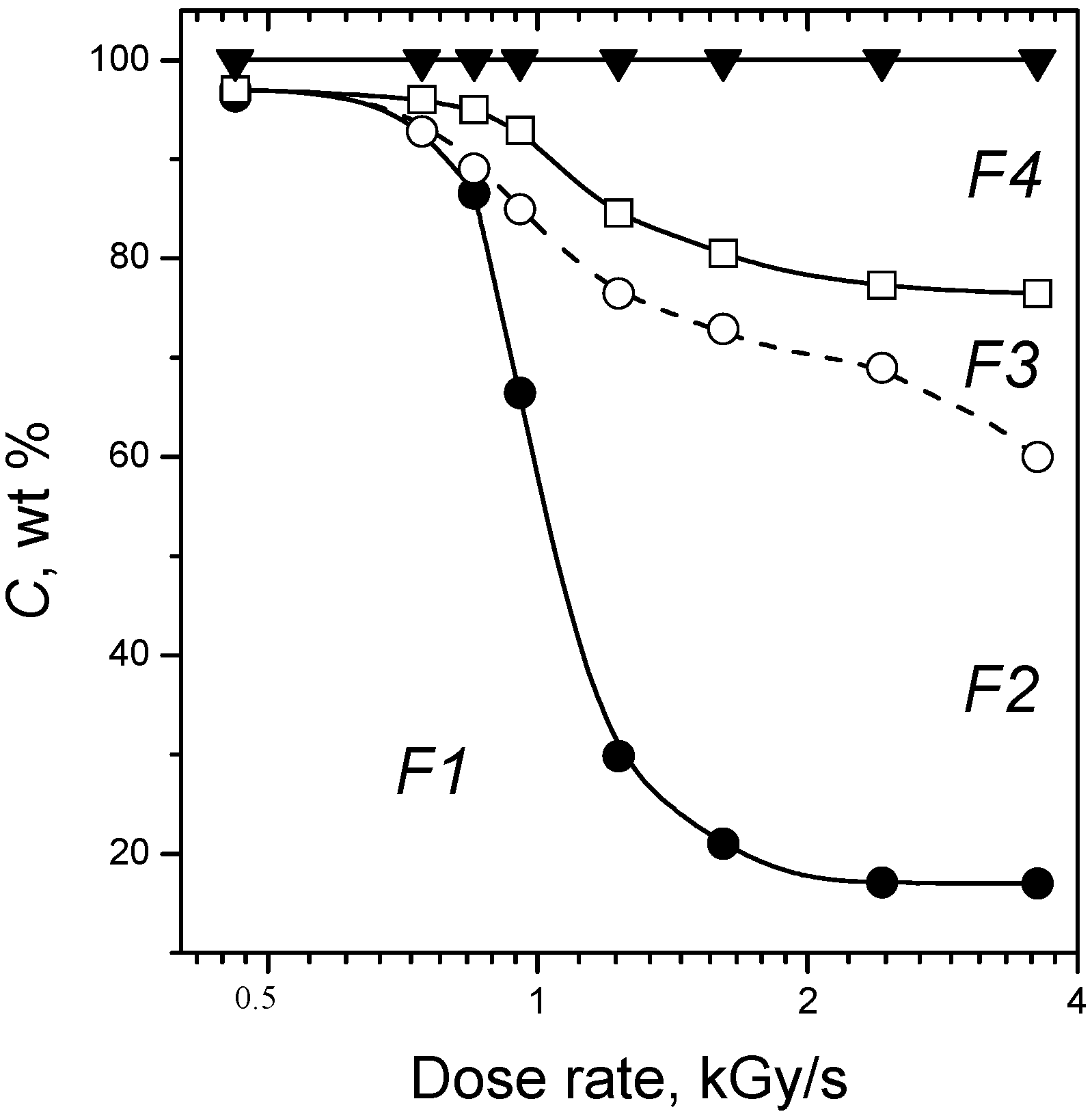
2.3. Temperature Effect
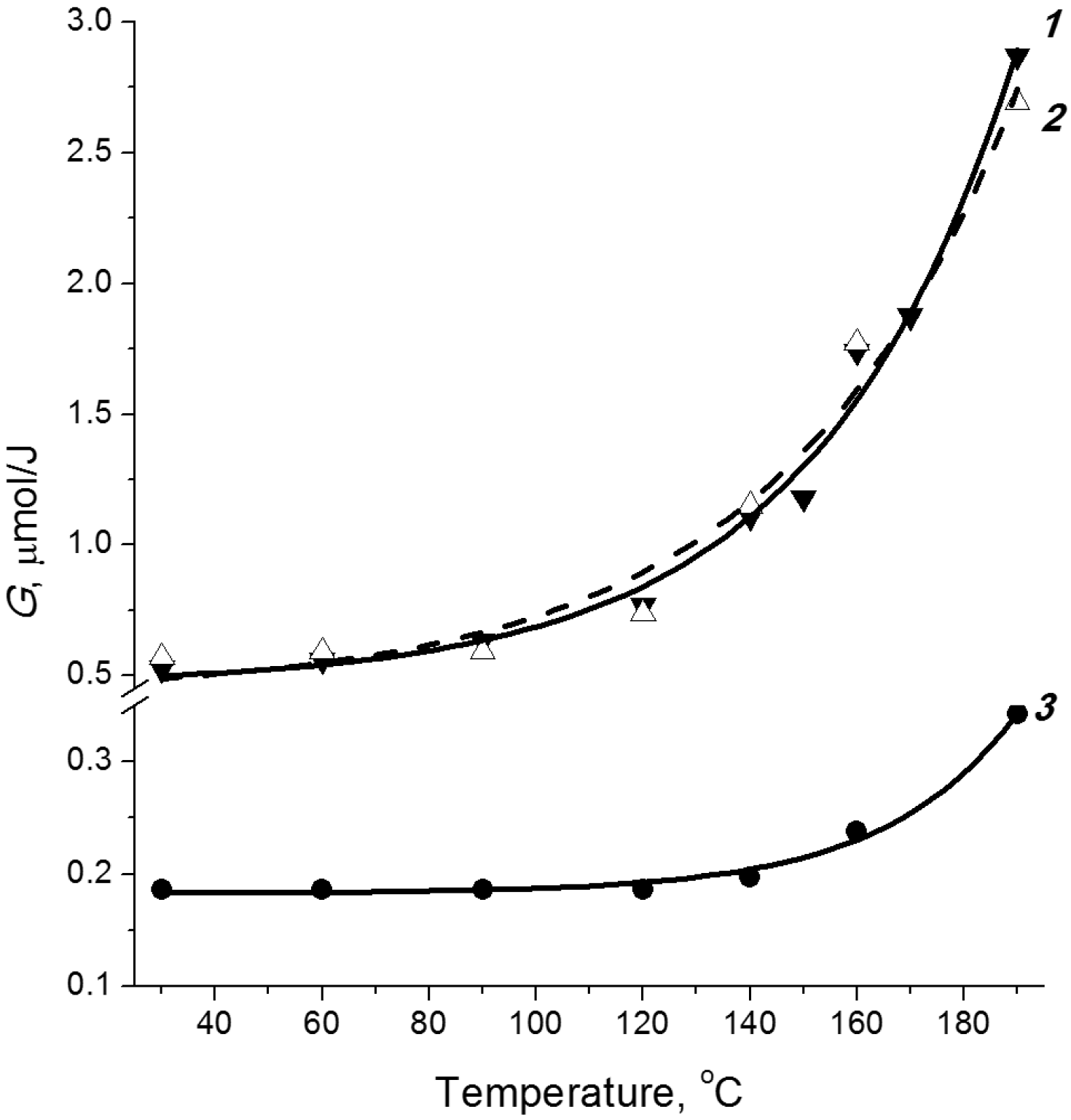
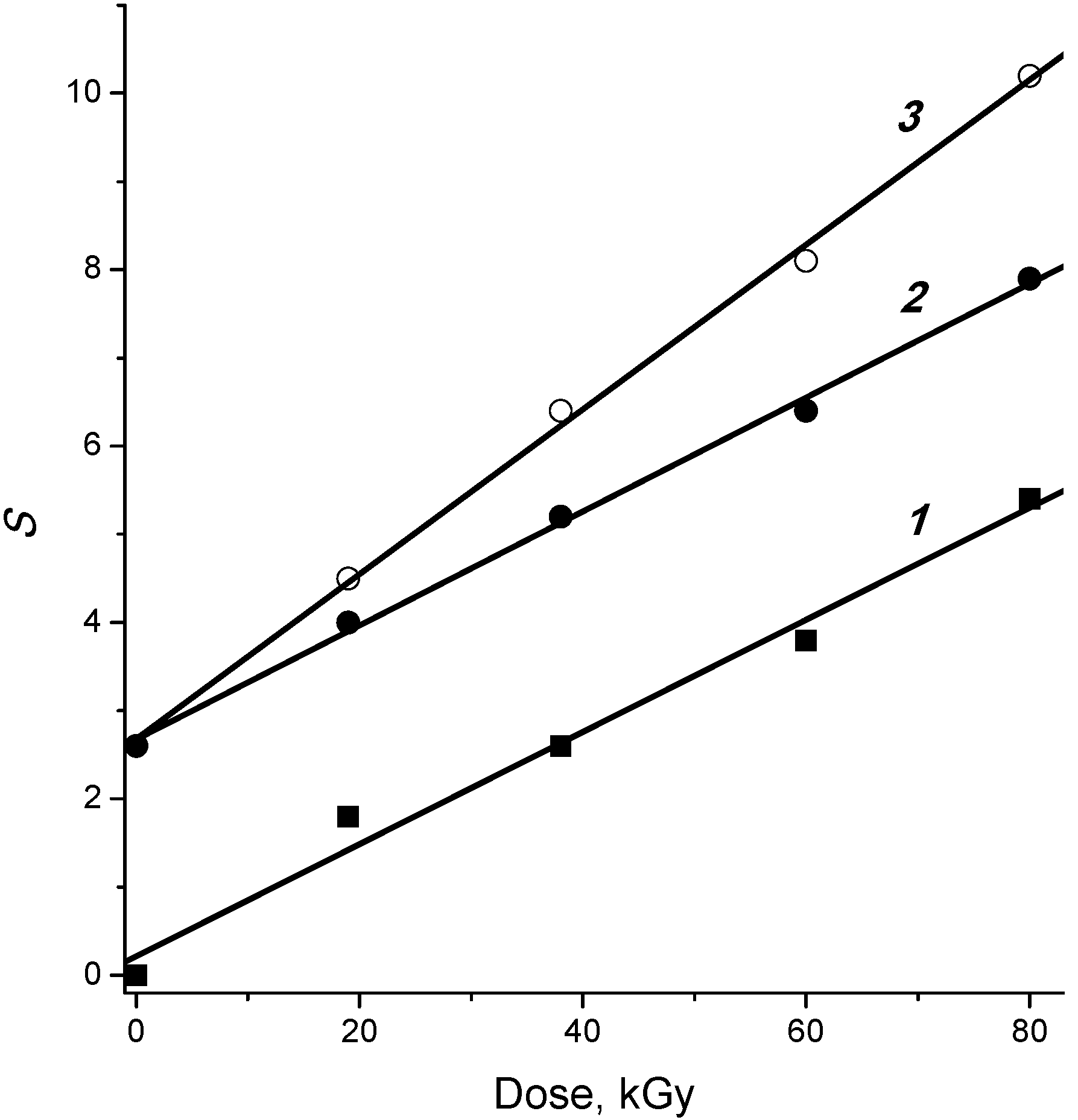
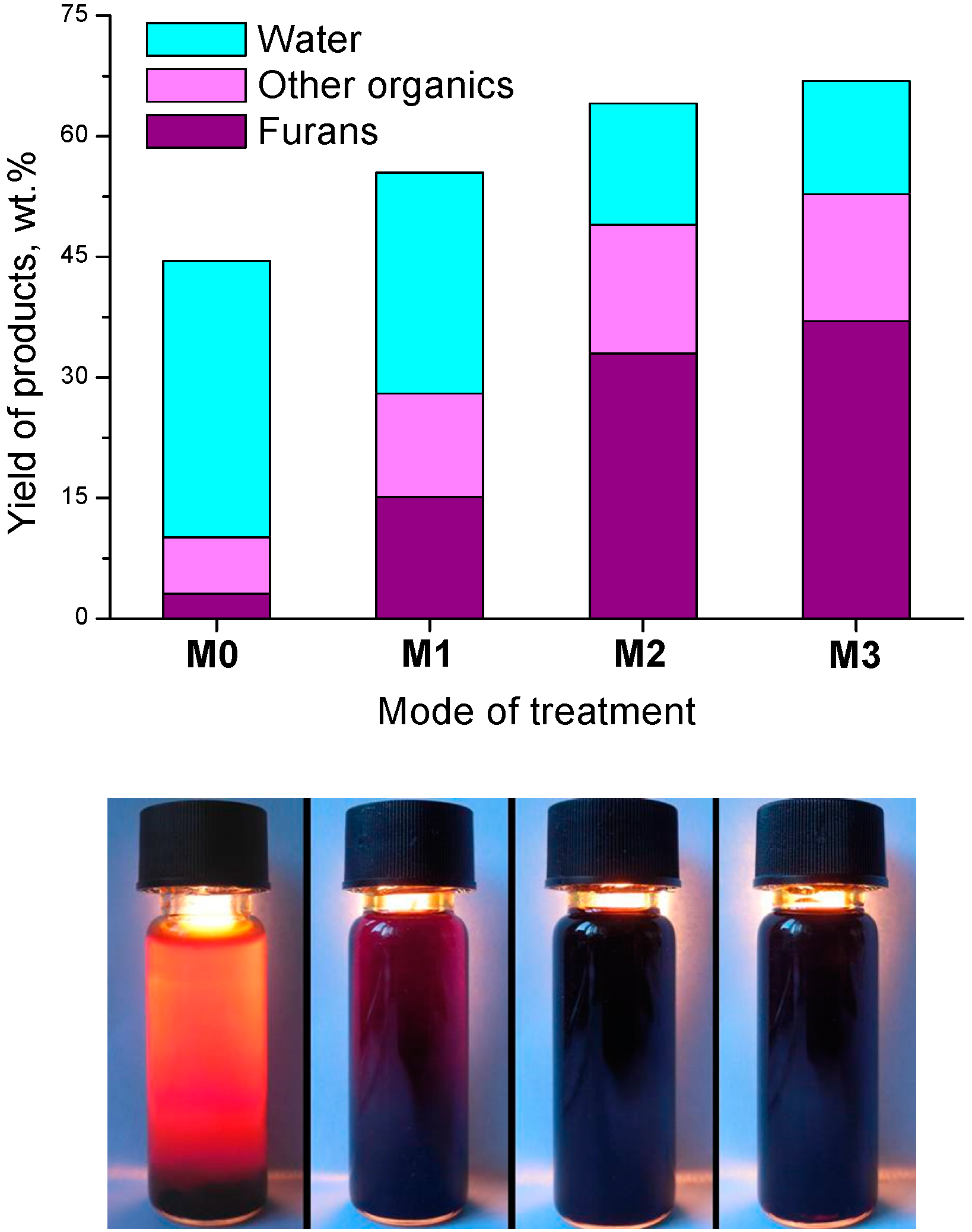
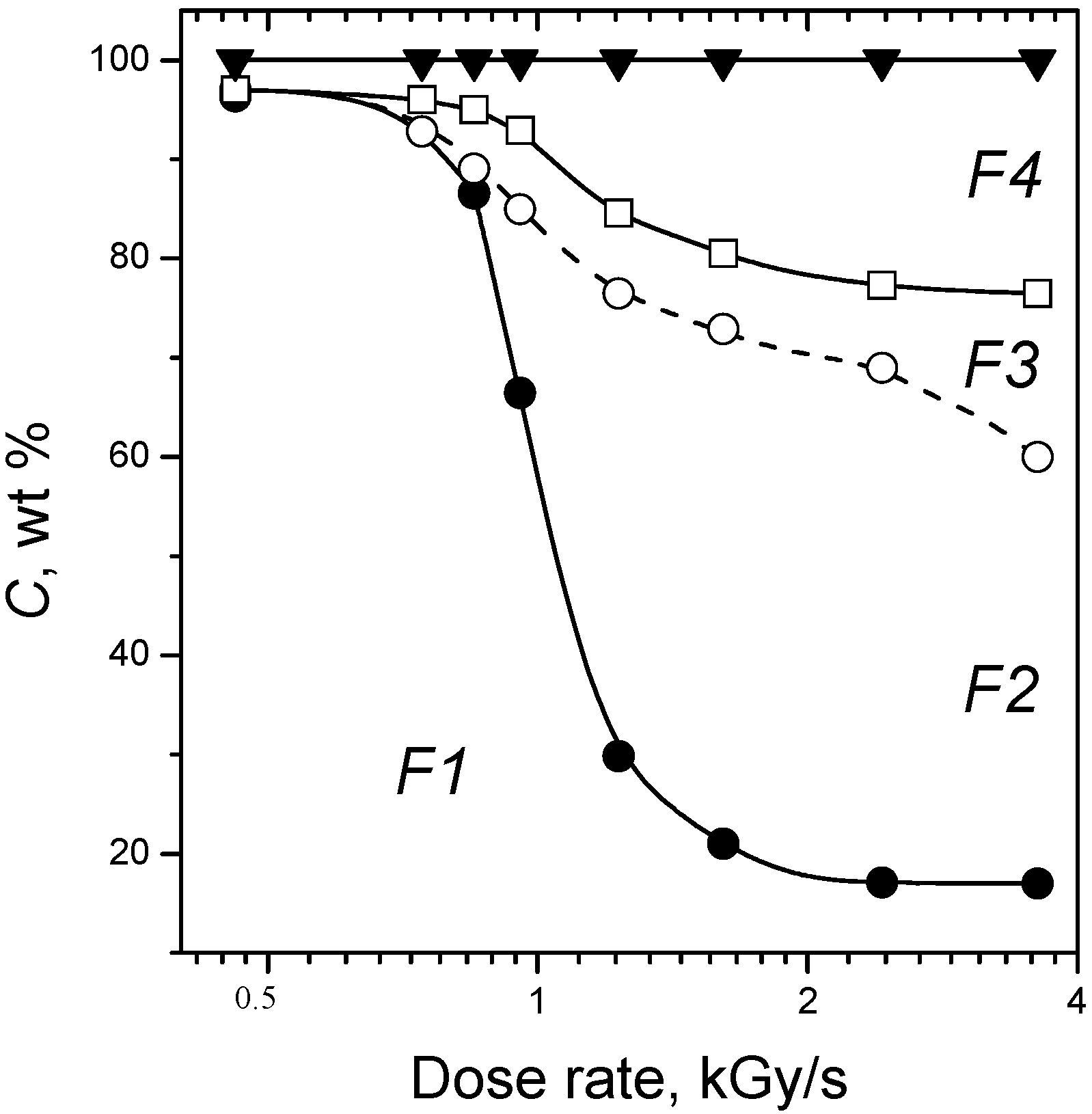
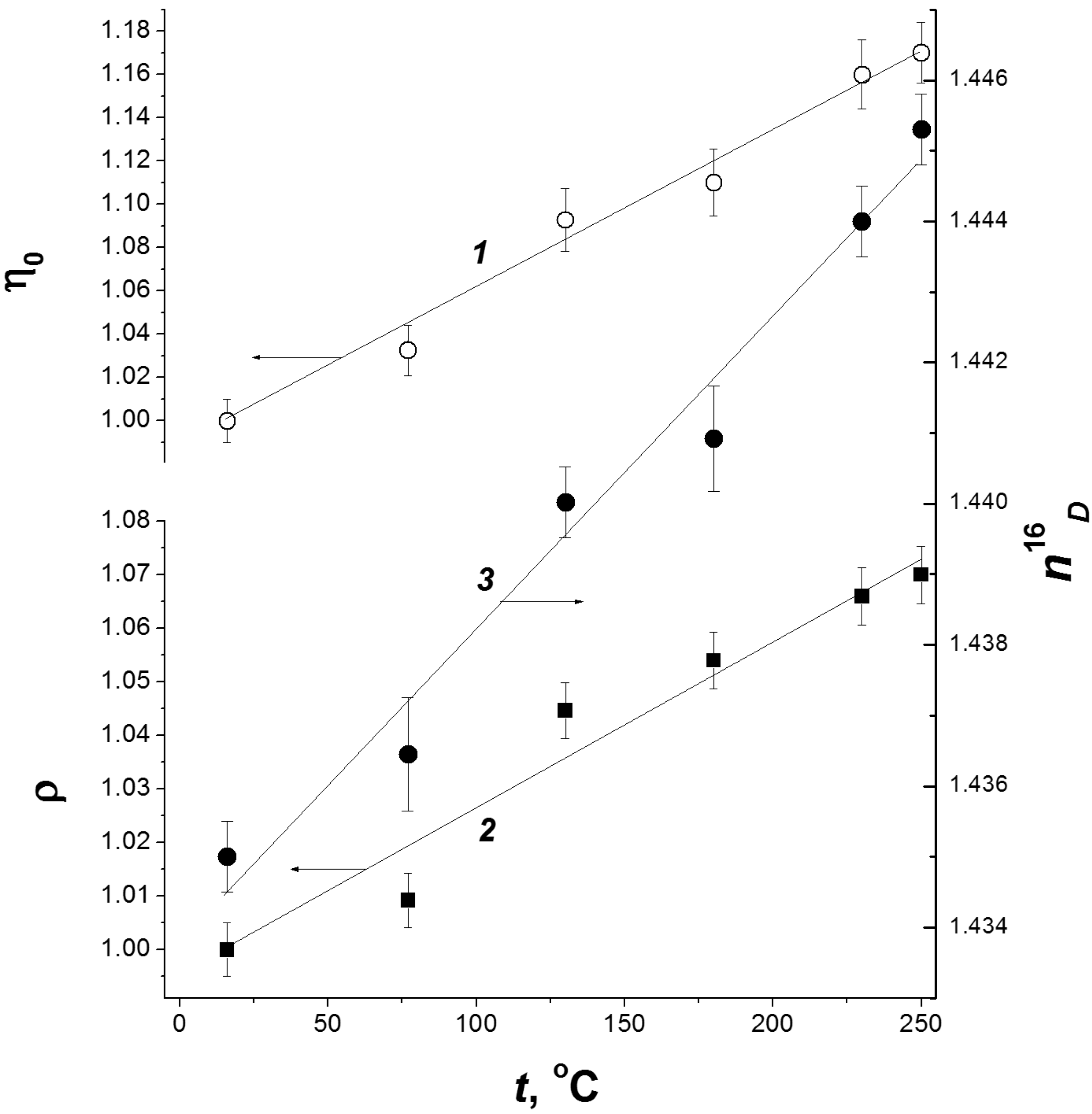
2.4. Post-Radiation Distillation (M1 Mode)
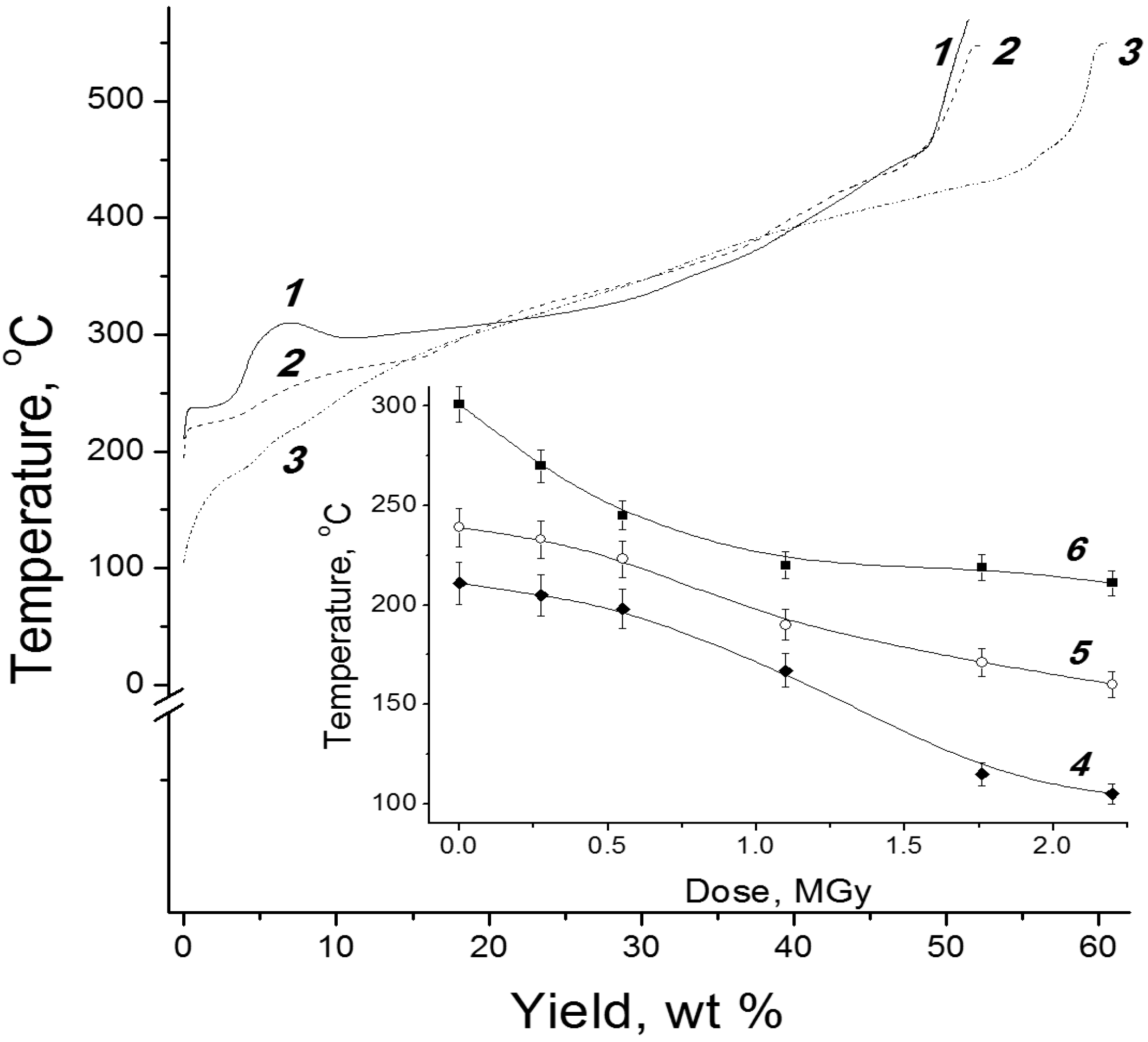
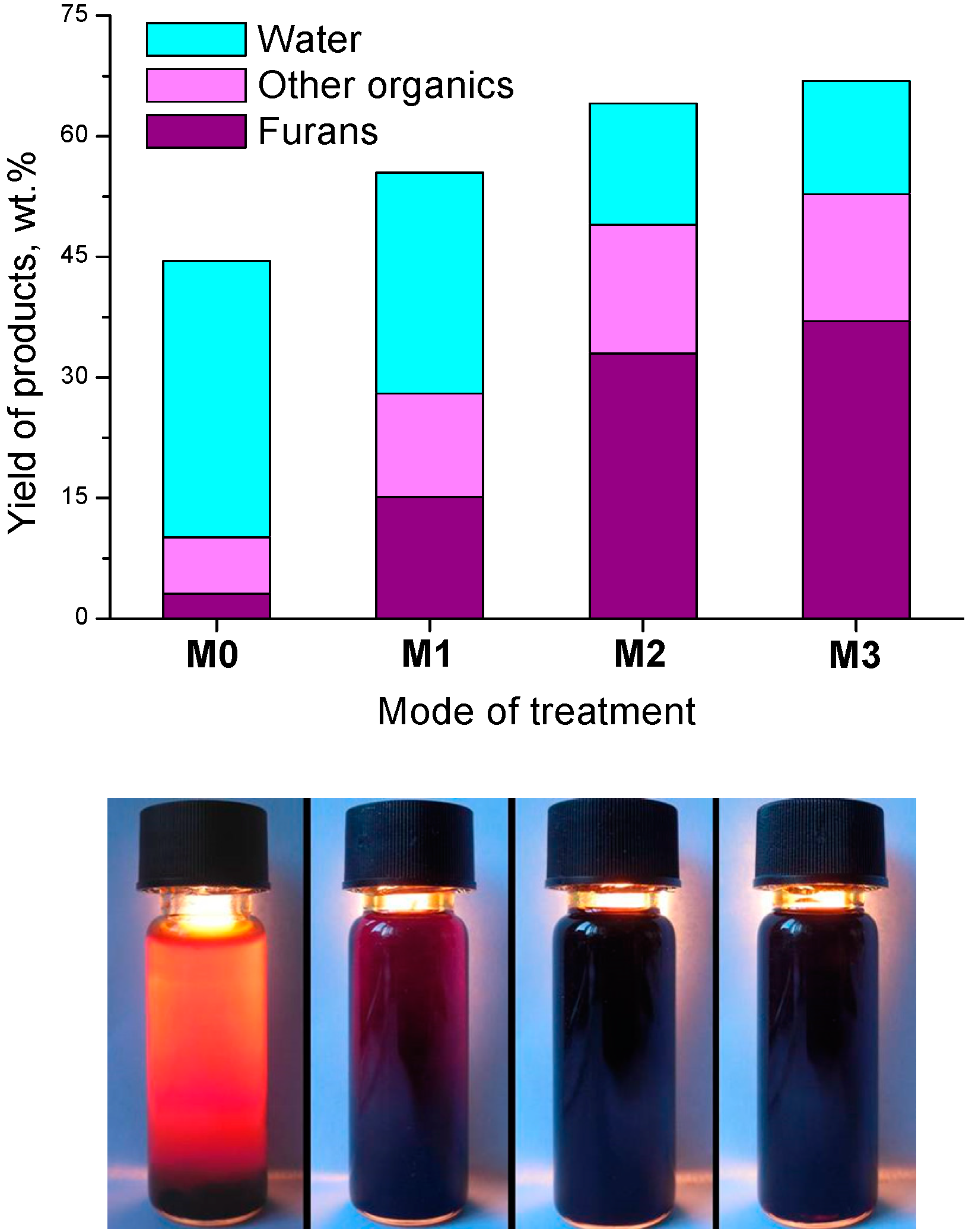
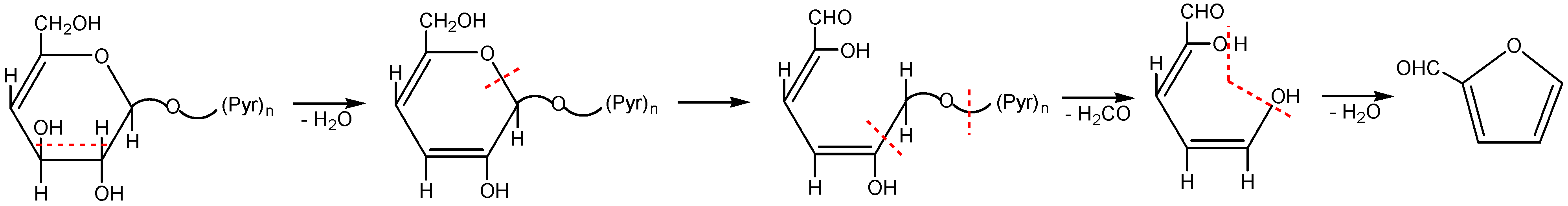
3. Radiation-Thermal Transformations of Cellulose at High Dose Rates
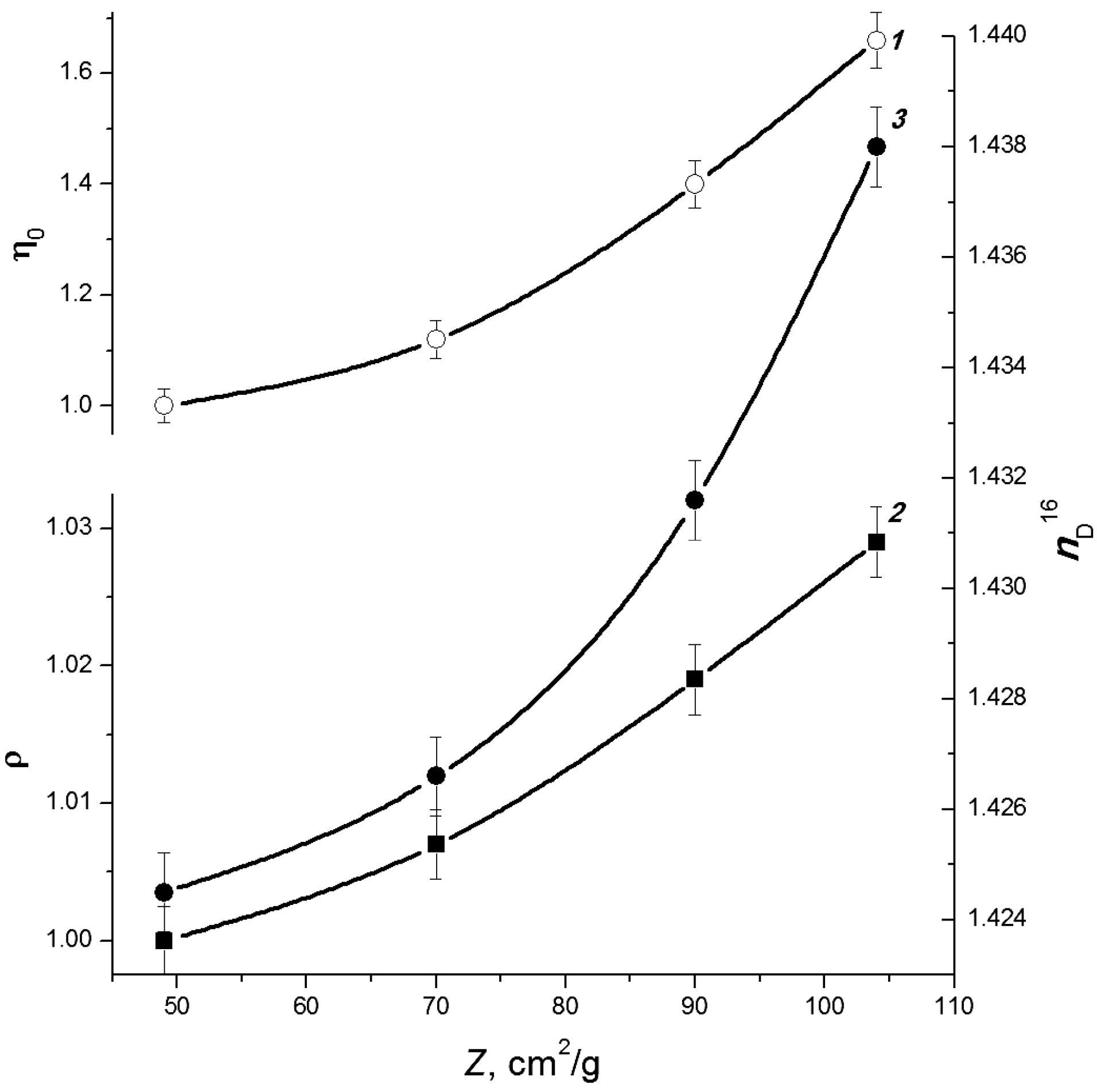
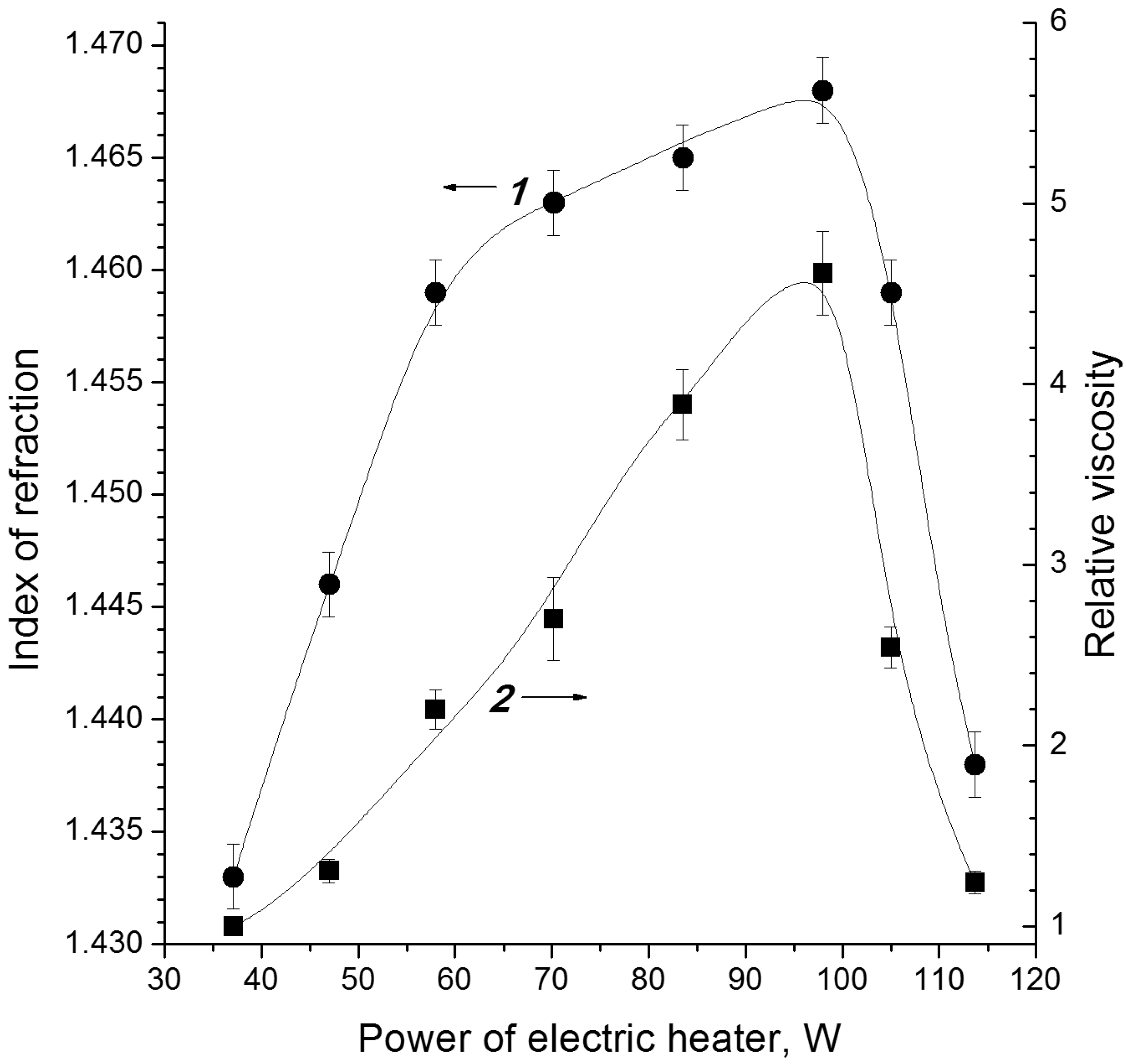
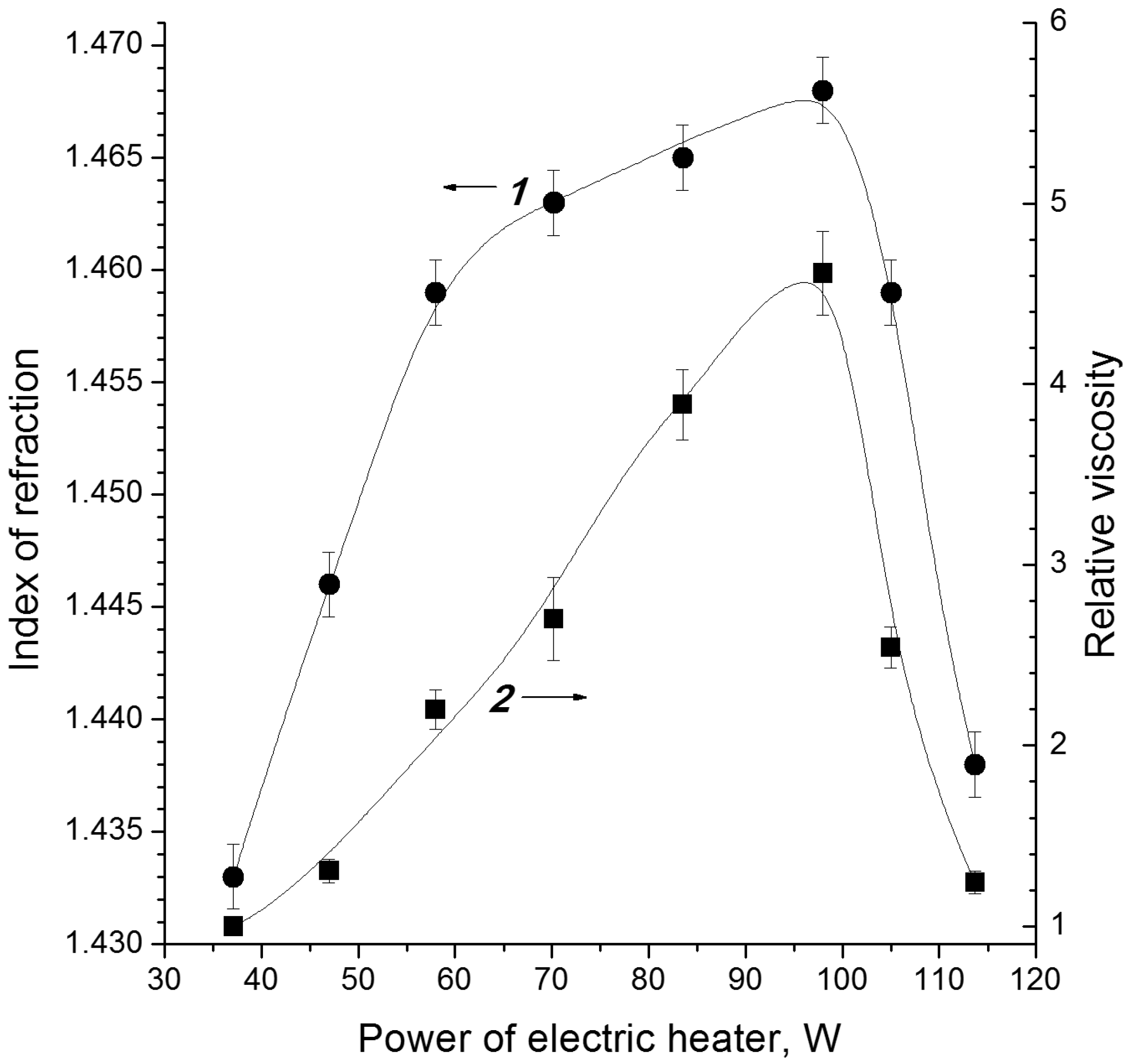
3.1. Electron-Beam Distillation
| Cellulose | G | n18D | ρ, kg/dm3 | η0, mPa·s |
|---|---|---|---|---|
| C1 | 63 | 1.4479 | 1.1639 | 5.96 |
| C2 | 58 | 1.4449 | 1.1560 | 5.67 |
| C3 | 60 | 1.4455 | 1.1594 | 5.79 |
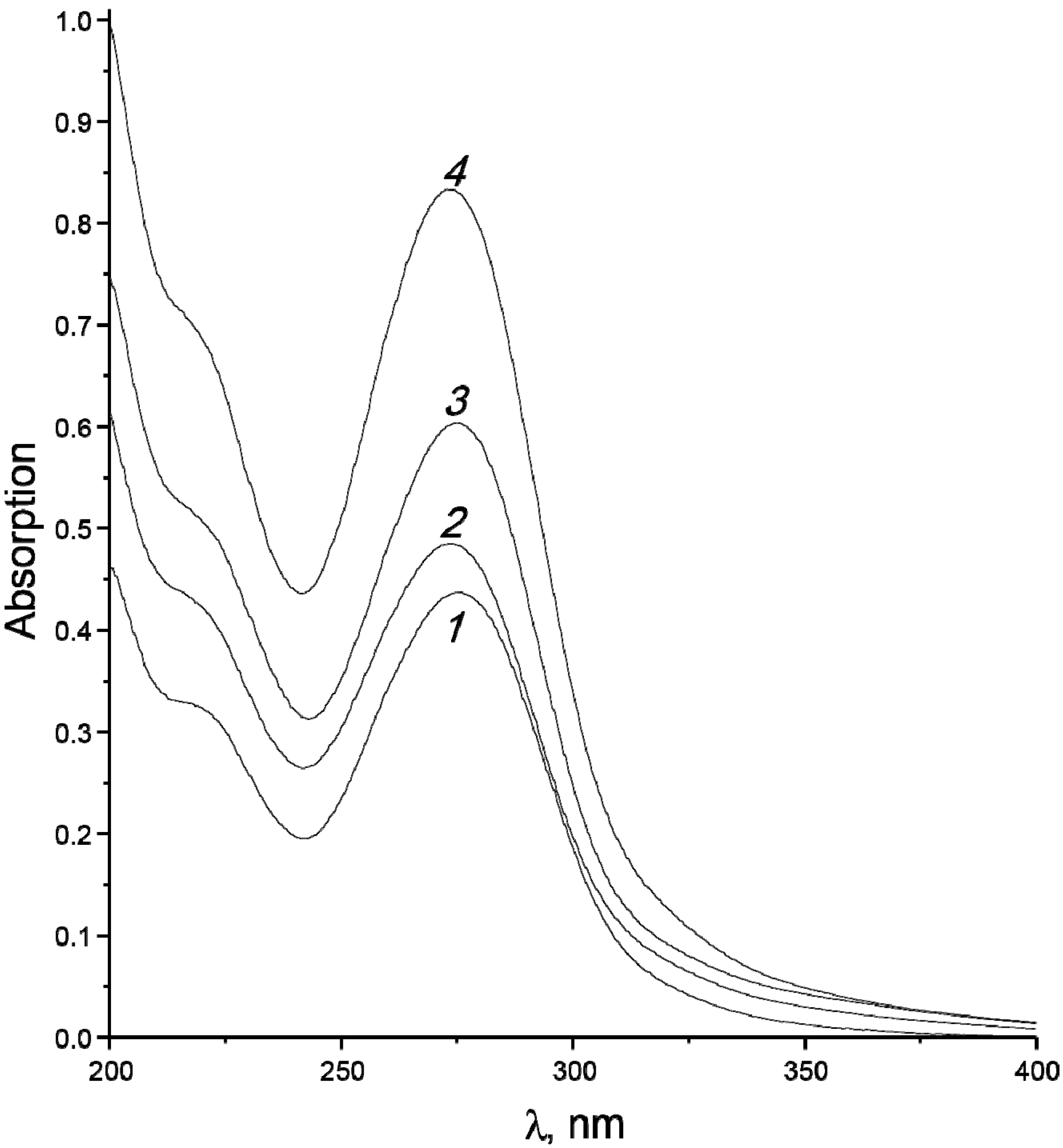
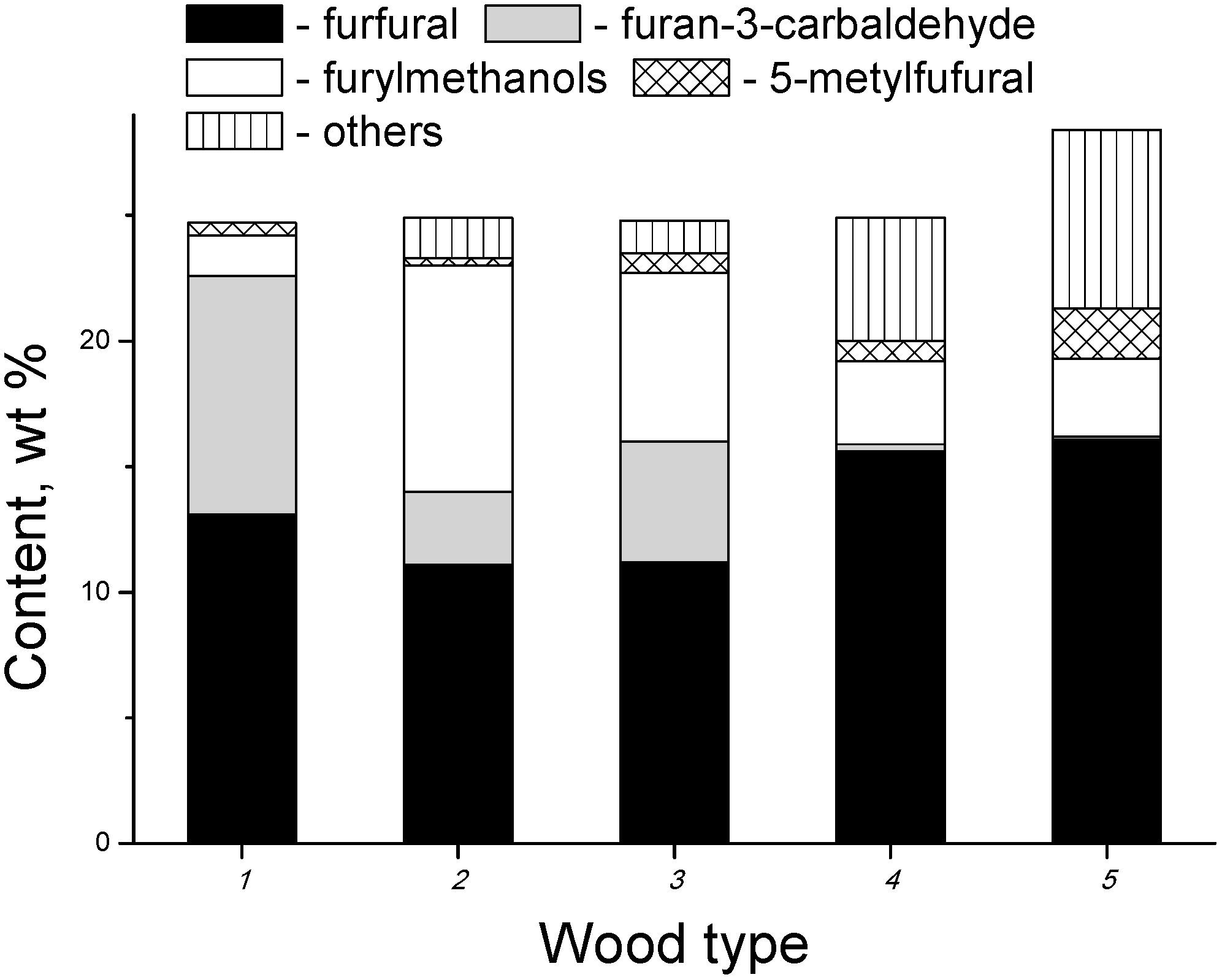
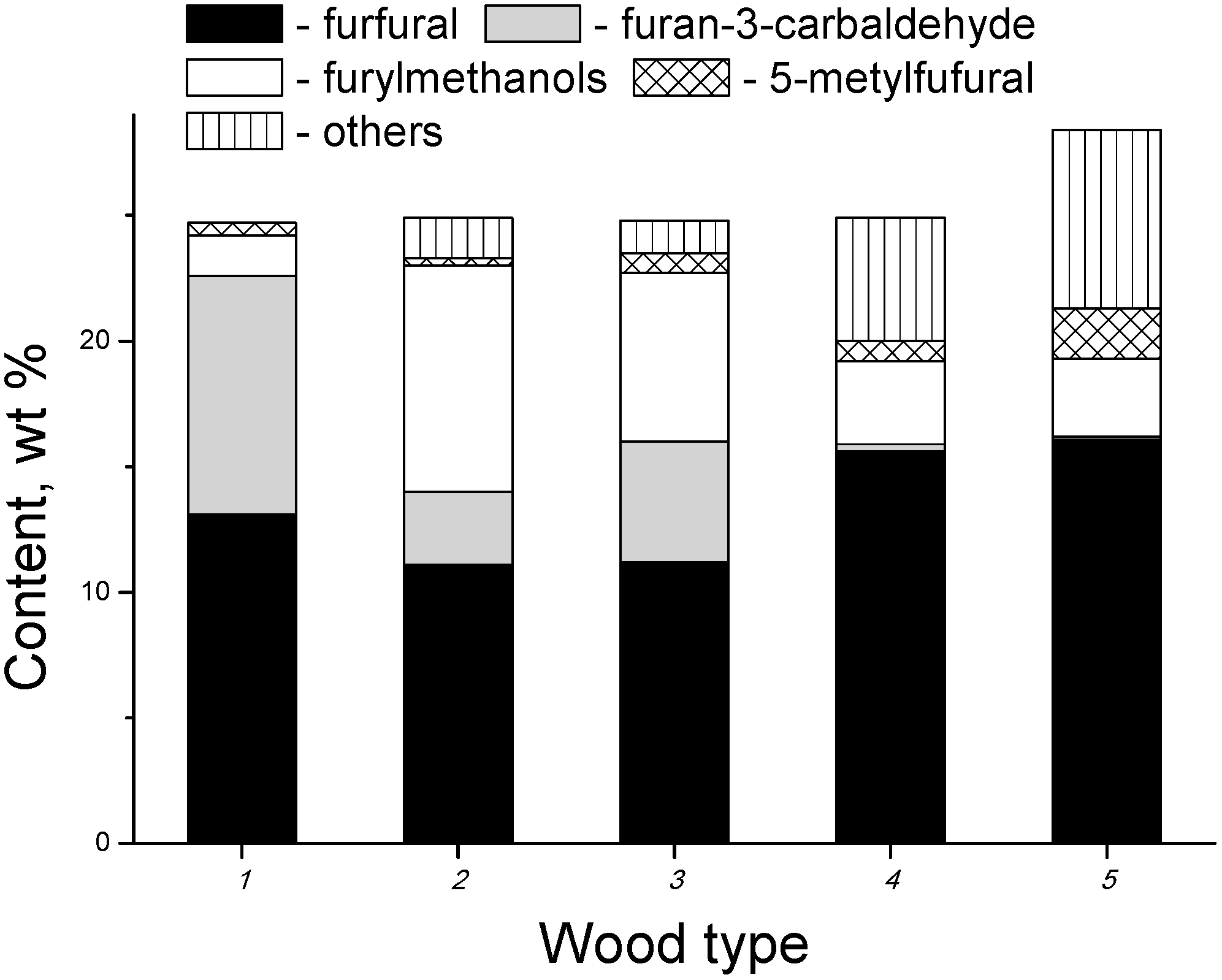
3.2. Products of Electron-Beam Distillation
| Component | Sample | ||
|---|---|---|---|
| C1 | C2 | C3 | |
| Methylformate | 0.90 | 0.80 | 1.30 |
| Acetone | 4.90 | 2.70 | 3.40 |
| Formic acid | 5.00 | 5.20 | 3.60 |
| Butane-2,3-dione | 1.60 | 3.50 | 1.70 |
| 2-Oxopropanal | 3.40 | 0.60 | 0.80 |
| Acetic acid | 7.30 | 6.00 | 3.10 |
| 1-Hydroxypropan-2-one | 7.30 | 10.70 | 1.50 |
| sec-Butyl acetate | 1.80 | 0.90 | 2.00 |
| Furfural | 40.40 | 42.40 | 48.20 |
| 3-Furancabaldehyde | 2.10 | 1.90 | 2.80 |
| Furylmethanols | 2.00 | 2.80 | 4.70 |
| Methylfuraldehydes | 9.40 | 13.30 | 17.30 |
| Furylacetates | 1.20 | 1.60 | 2.80 |
| 2,2-Dimethyl-3(2H)-furanone | 1.40 | 1.70 | 0.90 |
| Furans, total | 72.50 | 64.40 | 79.00 |
- -
- the content of the fraction of furan derivatives, which represent large fragments of glucopyranose (furancarbaldehydes, furylmethanols, furanones, etc.), increases;
- -
- the content of the fraction of small glucopyranose fragments (1-hydroxyacetone, acetic and formic acids) decreases;
- -
- the content of the fraction of heavy products (furylmethyl acetates), which are formed apparently in the secondary reactions between the major primary products, also decreases.



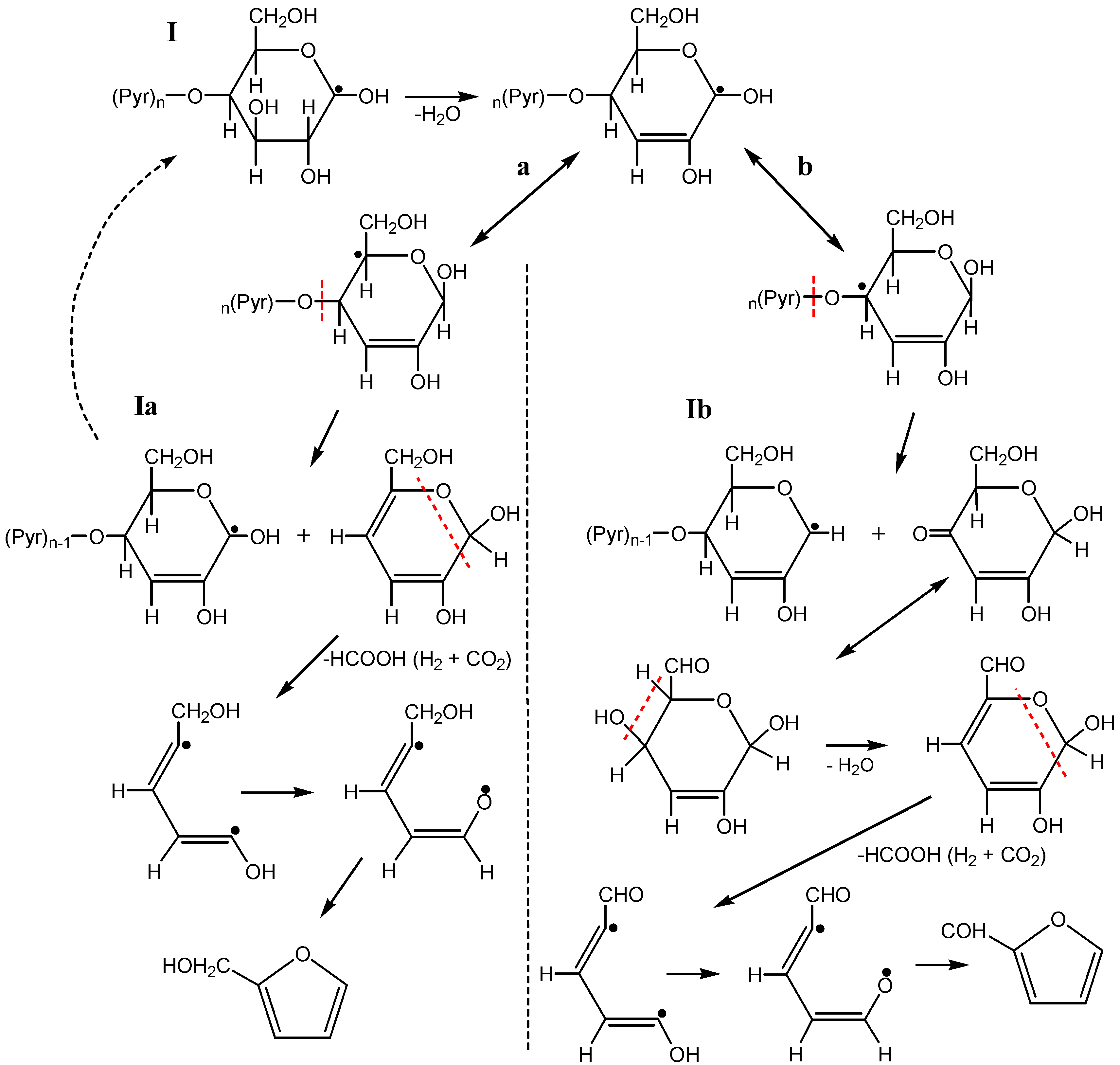
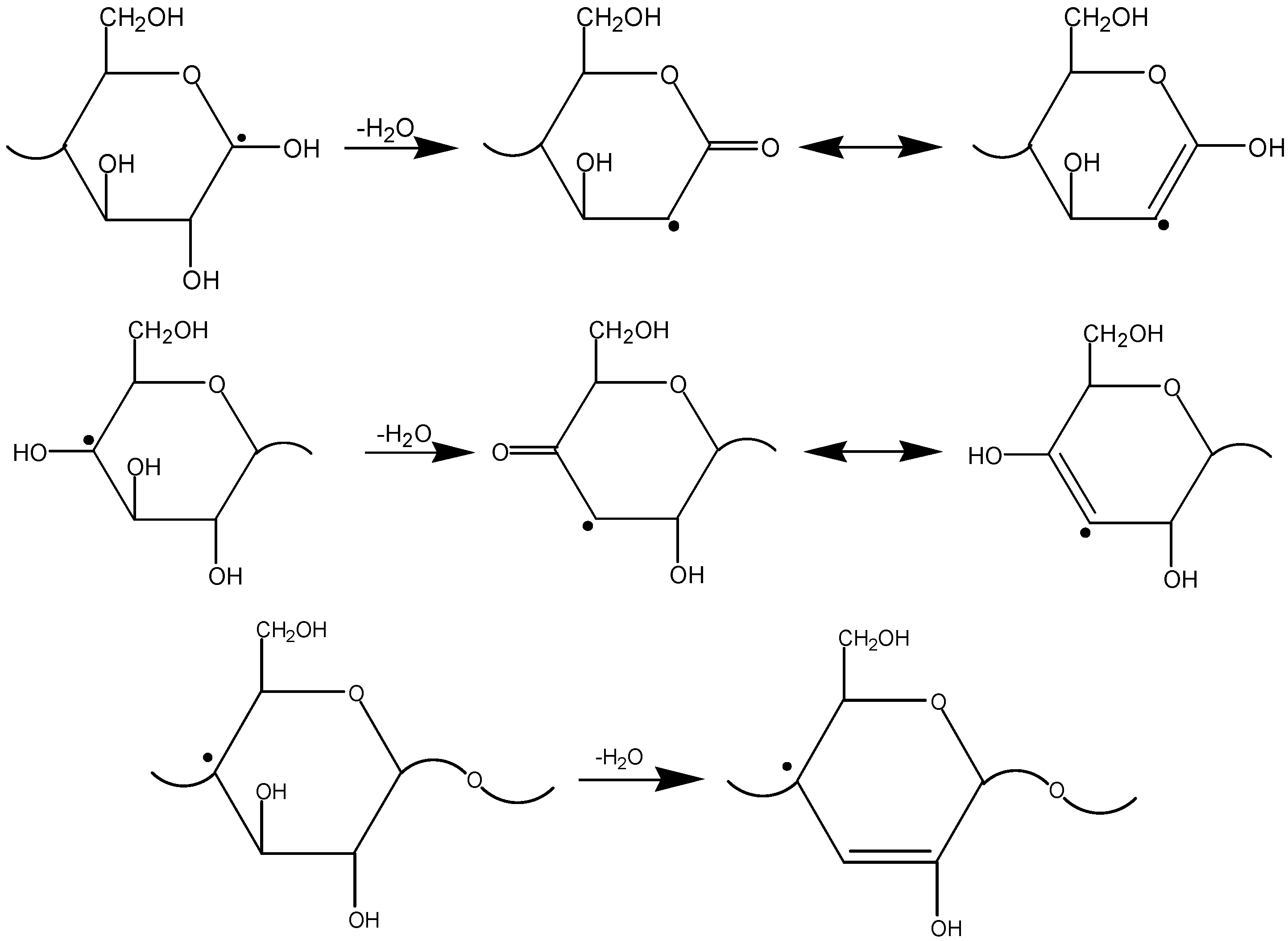
3.3. The Mechanism of Radiation-Thermal Transformations
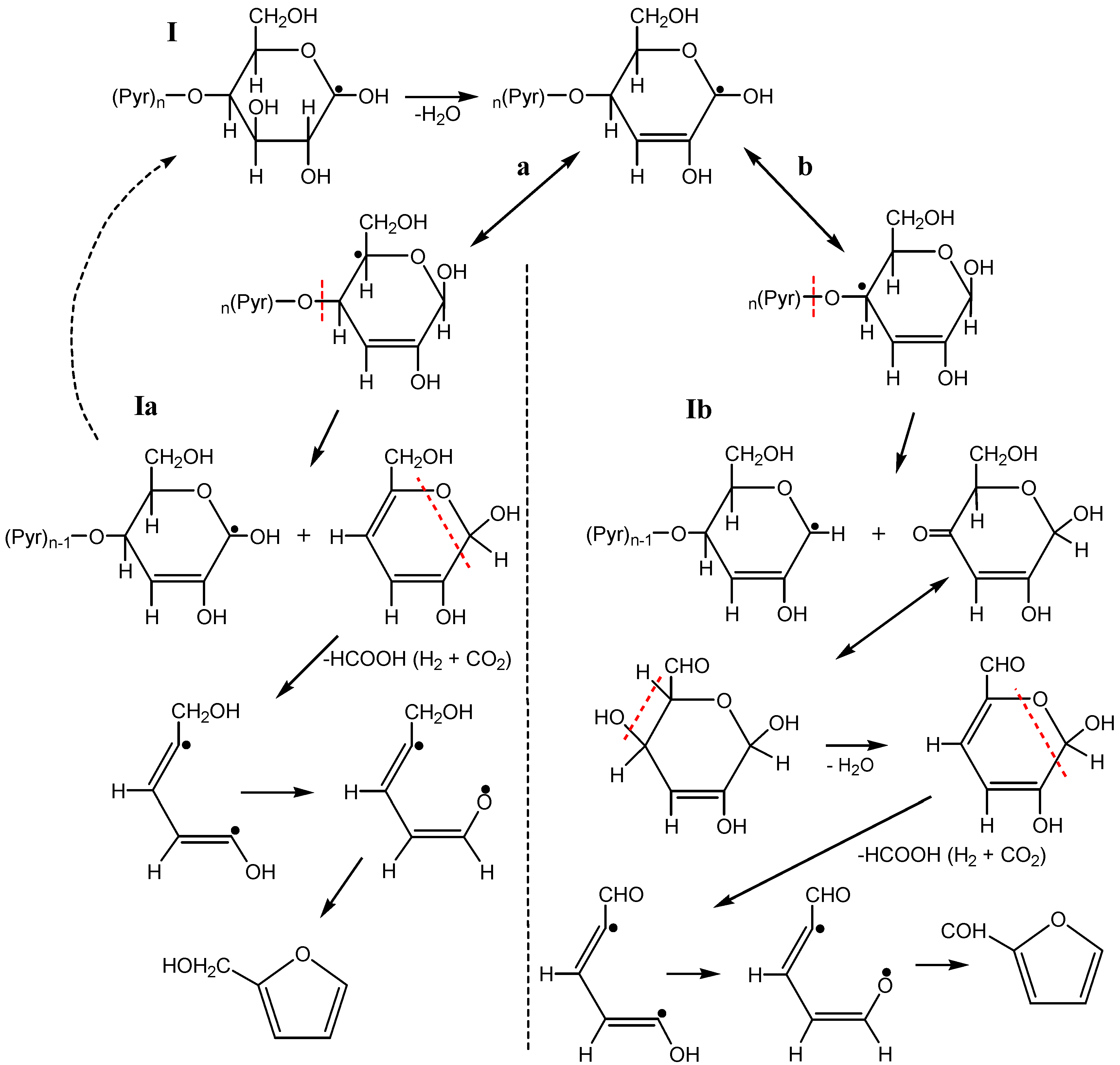
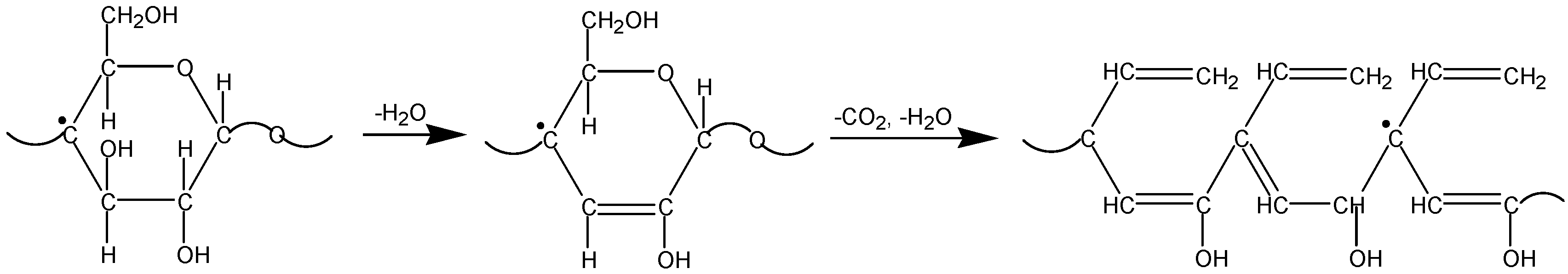
4. Advanced Applications
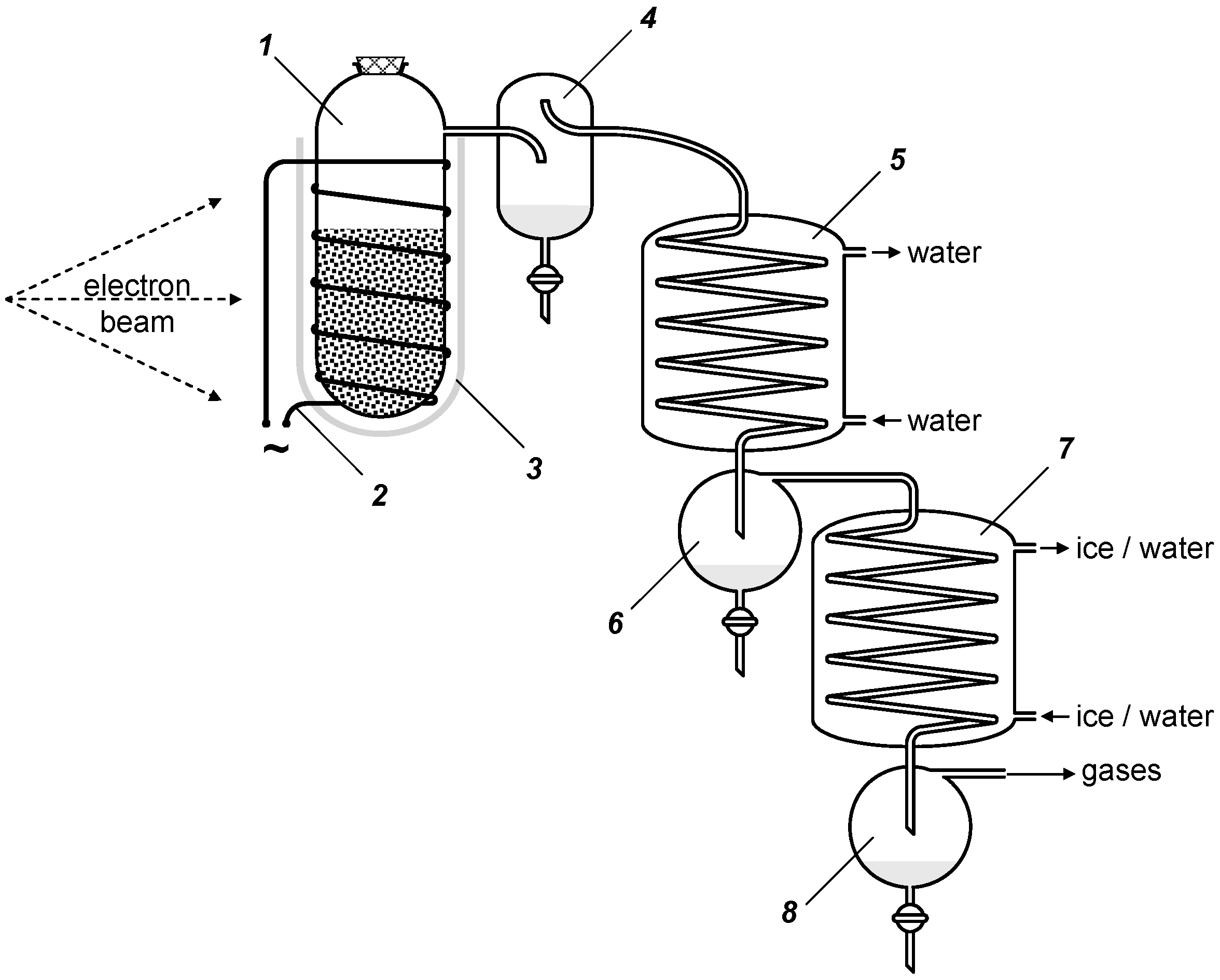
5. Experimental Section
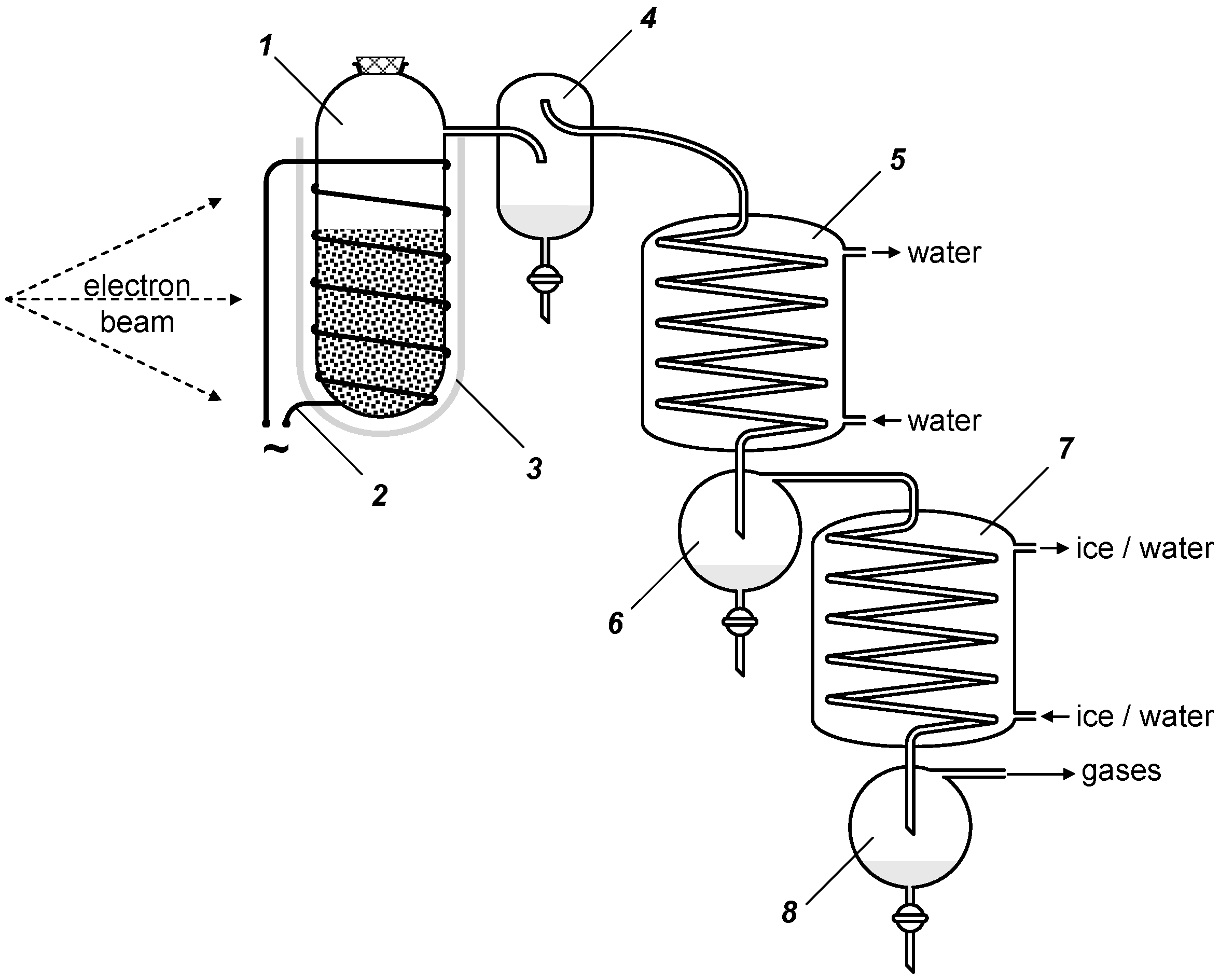
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fengel, D.; Wegener, G. Wood (Chemistry, Ultrastructure, Reactions); Walter de Gruyter: New York, NY, USA, 1984. [Google Scholar]
- Ershov, B.G. Radiation-chemical destruction of cellulose and other polysaccharides. Russ. Chem. Rev. 1998, 67, 353–375. [Google Scholar] [CrossRef]
- Woods, R.J.; Pikaev, A.K. Applied Radiation Chemistry: Radiation Processing; Wiley-Interscience: New York, NY, USA, 1994. [Google Scholar]
- Borsa, J.; Tóth, T.; Takács, E.; Hargittai, P. Radiation modification of swollen and chemically modified cellulose. Radiat. Phys. Chem. 2003, 67, 509–512. [Google Scholar] [CrossRef]
- Driscoll, M.; Stipanovic, A.; Winter, W.; Cheng, K.; Manning, M.; Spiese, M.J.; Galloway, R.A.; Cleland, M.R. Electron beam irradiation of cellulose. Radiat. Phys. Chem. 2009, 78, 539–542. [Google Scholar] [CrossRef]
- Földvary, C.M.; Takács, E.; Wojnárovits, L. Effect of high-energy radiation and alkali treatment on the properties of cellulose. Radiat. Phys. Chem. 2003, 67, 505–508. [Google Scholar] [CrossRef]
- Sun, J.; Xu, L.; Ge, M.; Zhai, M. Radiation degradation microcrystalline cellulose in solid status. J. Appl. Polym. Sci. 2013, 127, 1630–1636. [Google Scholar] [CrossRef]
- Takács, E.; Wojnárovits, L.; Borsa, J.; Földvary, C.; Hargittai, P.; Zöld, O. Effect of gamma-irradiation on cotton-cellulose. Radiat. Phys. Chem. 1999, 55, 663–666. [Google Scholar] [CrossRef]
- Bikales, N.; Segal, L. (Eds.) Cellulose and Cellulose Derivatives; Wiley: New York, NY, USA, 1974; Volume 2.
- Ershov, B.G.; Klimentov, A.S. The Radiation Chemistry of Cellulose. Russ. Chem. Rev. 1984, 53, 1195–1207. [Google Scholar] [CrossRef]
- Kabanov, V.Y.; Fel’dman, V.I.; Ershov, B.G.; Polikarpov, A.I.; Kiryukhin, D.P.; Apel, P.Y. Erratum to: “Radiation Chemistry of Polymers”. High Energy Chem. 2009, 43, 425. [Google Scholar] [CrossRef]
- Ponomarev, A.V.; Makarov, I.E.; Tananaev, I.G.; Myasoedov, B.F. Method of Biomass Processing. Russian Patent 2338769, 2008. [Google Scholar]
- Ponomarev, A.V. Electron-beam decomposition of phytogenous substances: Solid-to-liquid conversion. Radiat. Phys. Chem. 2009, 78, 345–350. [Google Scholar] [CrossRef]
- Komarov, V.B.; Gordeev, A.V.; Samuilova, S.D.; Fadin, A.V.; Ershov, B.G. Radiation-thermal destruction of cellulose by electron-beam irradiation. High Energy Chem. 1999, 33, 156–160. [Google Scholar]
- Imamura, R.; Uena, T.; Murukami, K. Depolymerization of Cellulose by Electron Beam Irradiation. Bull. Inst. Res. Kyoto Univ. 1972, 50, 51–63. [Google Scholar]
- Sultanov, K.; Azizov, U.A.; Usmanov, K.U. γ-Radiolysis of cellulose: Mechanism of the formation of volatile products. Doklady Chem. 1989, 309, 981–983. [Google Scholar]
- Gaylord, N.G.; Maiti, S.; Dixit, S.S. Cellulose graft copolymers. IV. Role of aldehydes in uncatalyzed polymerization of methyl methacrylate in the presence of cellulose-water. J. Polym. Sci. Polym. Lett. 1972, 10, 855–861. [Google Scholar]
- Kusama, Y.; Kageyama, E.; Shimada, M.; Nakamura, Y. Molecular weights and molecular weight distributions of irradiated cellulose fibers by gel permeation chromatography. J. Appl. Polym. Sci. 1976, 20, 1679–1688. [Google Scholar] [CrossRef]
- Kuzina, S.I.; Mikhailov, A.I. The Oxidation and Thermal Transformations of Macroradicals in Gamma Irradiated Cellulose. Russ. J. Phys. Chem. 2006, 80, 1666–1670. [Google Scholar] [CrossRef]
- Ershov, B.G.; Samuilova, S.D.; Petropavlovskii, G.A.; Vasil’eva, G.G. Radiation-thermal decomposition of cellulose. Dokl. Akad. Nauk SSSR 1984, 274, 102–106. [Google Scholar]
- Ershov, B.G.; Isakova, O.V.; Matyushkina, E.P.; Samuilova, S.D. Mechanism of radiation-chemical transformations of cellulose. High Energy Chem. 1986, 20, 142–147. [Google Scholar]
- Blouin, F.A.; Arthur, J.C. The Effects of Gamma Radiation on Cotton: Part I: Some of the Properties of Purified Cotton Irradiated in Oxygen and Nitrogen Atmospheres. Text. Res. J. 1958, 28, 198–204. [Google Scholar] [CrossRef]
- Arthur, J.C.; Blouin, F.A.; Demint, J.R. The effects of gamma radiation on cotton cellulose. Am. Dyest. Rep. 1960, 49, 21–26. [Google Scholar]
- Bludovsky, R.; Duchacek, V. Some aspects of the mechanism of cellulose radiolysis. Radiochem. Radioanal. Lett. 1979, 38, 21–29. [Google Scholar]
- Duchacek, V.; Bludovsky, R. Gamma irradiation of cellulose and some problems of its utilization. Radiochem. Radioanal. Lett. 1979, 38, 31–38. [Google Scholar]
- Bludovsky, R.; Prochazka, M.; Kopoldova, J. The influence of oxygen on the radiolytical products of cellulose. J. Radioanal. Nucl. Chem. 1984, 87, 69–79. [Google Scholar] [CrossRef]
- Komarov, V.B.; Samuilova, S.D.; Kirsanova, L.S.; Morozov, V.A.; Kuleshova, T.M.; Smirnov, A.G.; Ershov, B.G. Preparation of nitrate esters from irradiated cellulose. Russ. J. Appl. Chem. 1993, 66, 323–328. [Google Scholar]
- Horio, M.; Imamura, R.; Mizukami, H. Effect of Gamma Irradiation upon Cellulose. Bull. Inst. Chem. Res. Kyoto Univ. 1963, 41, 17–38. [Google Scholar]
- Dilli, S.; Garnett, J.L. Effect of ionizing radiation on crystalline carbohydrates. Chem. Ind. 1963, 10, 409–410. [Google Scholar]
- Florin, R.; Wall, L.; Brown, D. Free radicals in gamma-irradiated polystyrenes. Trans. Faraday Soc. 1960, 56, 1304–1310. [Google Scholar] [CrossRef]
- Baugh, P.J.; Hinojosa, O.; Arthur, J.C. ESR study of post-irradiation reactions of cellulose and acrylonitrile. Appl. Polym. Sci. 1967, 11, 1139–1153. [Google Scholar] [CrossRef]
- Arthur, J.C.; Hinojosa, O.; Tripp, V.W. Effect of crystalline structure on the trapped radical spectra of irradiated cellulose. J. Appl. Polym. Sci. 1969, 13, 1497–1507. [Google Scholar] [CrossRef]
- Arthur, J.C.; Mares, T.; Hinojosa, O. ESR Spectra of Gamma-Irradiated Cotton Cellulose I and II. Text. Res. J. 1966, 36, 630–635. [Google Scholar] [CrossRef]
- Dilly, S.; Ernst, J.T.; Garnet, J. Radiation-induced reactions with cellulose. IV. Electron paramagnetic resonance studies of radical formation. Aust. J. Chem. 1967, 20, 911–927. [Google Scholar] [CrossRef]
- Hinojosa, O.; Arthur, J.C., Jr. Propagating radical in copolymerization of methacrylic acid with γ-irradiated cellulose. J. Polym. Sci. Part B Polym. Lett. 1972, 10, 161–165. [Google Scholar] [CrossRef]
- Shimada, M.; Nakamura, Y.; Kusama, T.; Matsuda, O.; Kageyama, E. Electron spin resonance studies of γ-irradiated cellulose. II. Free radicals in accessible regions to water in cellulose I and cellulose II. J. Appl. Polym. Sci. 1974, 18, 3387–3396. [Google Scholar] [CrossRef]
- Kholodkova, E.M.; Ponomarev, A.V. Phase distribution of the products of radiation and postradiation distillation of cellulose. High Energy Chem. 2011, 45, 386–389. [Google Scholar] [CrossRef]
- Golova, O.P. Chemical Effects of Heat on Cellulose. Russ. Chem. Rev. 1975, 44, 687–704. [Google Scholar] [CrossRef]
- Ohno, M.; Kakuta, A.; Ozaki, O.; Kimura, S.; Miyauchi, T. Measurement of Neutron Dose with Boron-Doped Cellulose Nitrate Film. J. Nucl. Sci. Technol. 1977, 14, 673–679. [Google Scholar] [CrossRef]
- Kholodkova, E.M.; Ponomarev, A.V.; Metreveli, P.K.; Metreveli, A.K. Effect of absorbed dose on the postradiation dry distillation of cellulose and lignin. High Energy Chem. 2013, 47, 286–290. [Google Scholar] [CrossRef]
- Frolova, S.V.; Kuvshinova, L.A.; Bugaeva, A.Y.; Kuchin, A.V. Thermal analysis of powder cellulose, obtained by destruction of sulphate cellulose titanium tetrachloride. Chem. Plant Raw Mater. 2011, 2, 43–46. [Google Scholar]
- Dejneko, I.P. Chemical Transformations of Cellulose under Pyrolysis. Izv. Vyssh. Uchebn. Zaved. Lesn. Zh. 2004, 4, 96–112. [Google Scholar]
- Bogolitsyn, K.G.; Lunin, V.V.; Kosyakov, D.S. Physical Chemistry of Lignin; Akademkniga: Moscow, Russia, 2010. [Google Scholar]
- Strizhakov, D.A.; Solntsev, A.P.; Agabekov, V.E.; Sel’kin, V.P.; Pleskachevsky, Y.M. Effect of g-irradiation on composition of pine sawdust pyrolysis liquid products. Proc. Natl. Acad. Sci. Belarus Series Chem. Sci. 2011, 3, 64–69. [Google Scholar]
- Kholodkova, E.M.; Bludenko, A.V.; Chulkov, V.N.; Ponomarev, A.V. Cellulose destruction by electron-beam-induced heating. Russ. Chem. Bull. 2010, 59, 1827–1833. [Google Scholar] [CrossRef]
- Ponomarev, A.V.; Kholodkova, E.M.; Metreveli, A.K.; Metreveli, P.K.; Erasov, V.S.; Bludenko, A.V.; Chulkov, V.N. Phase distribution of products of radiation and post–radiation distillation of biopolymers: Cellulose, lignin and chitin. Radiat. Phys. Chem. 2011, 80, 1186–1194. [Google Scholar] [CrossRef]
- Kholodkova, E.M.; Bludenko, A.V.; Chulkov, V.N.; Ponomarev, A.V.; Tananaev, I.G. Effect of temperature and dispersity on electron-beam distillation of cellulose. In Actual Problems of High Energy Chemistry, Proceedings of the Fourth All-Russian Conference, Moscow, Russia, 2–3 November 2009.
- Ponomarev, A.V.; Bludenko, A.V.; Chulkov, V.N.; Tananaev, I.G.; Myasoedov, B.F.; Tsivadze, A.Y. Electron beam conversion of cellulose into liquid organic products. High Energy Chem. 2009, 43, 350–353. [Google Scholar] [CrossRef]
- Jones, S.B.; Valkenburg, C.; Walton, C.W.; Elliott, D.C.; Holladay, J.E.; Stevens, D.J.; Kinchin, C.; Czernik, S. Producrion of Gasoline and Diesel from Biomass via Fast Pyrolysis, Hydrogenating and Hydrocracking; Pacific Northwest National Laboratory Report, PNNL-18284; Pacific Northwest National Laboratory: Richland, WA, USA, 2009. [Google Scholar]
- Ponomarev, A.V.; Holodkova, E.M.; Ershov, B.G. Electron-beam synthesis of fuel in the gas phase. Radiat. Phys. Chem. 2012, 81, 1440–1444. [Google Scholar] [CrossRef]
- Shcherbakov, A.A. Furfural; Gos. Izd. Tekhn. Lit. USSR: Kiev, Ukraine, 1962. [Google Scholar]
- Ponomarev, A.V.; Ershov, B.G. Fundamental aspects of radiation-thermal transformations of cellulose and plant biomass. Russ. Chem. Rev. 2012, 81, 918–935. [Google Scholar] [CrossRef]
- Livingston, R.; Zeldes, H. Paramagnetic Resonance Study of Liquids during Photolysis. III. Aqueous Solutions of Alcohols with Hydrogen Peroxide. J. Am. Chem. Soc. 1966, 88, 4333–4336. [Google Scholar] [CrossRef]
- Sharpatyi, V.A. Radiation Chemistry of Biopolymers; Energoatomizdat: Moscow, Russia, 1981. [Google Scholar]
- Kuzina, S.I.; Mikhailov, A.I. Formation of Free Radicals during the Photolysis and Radiolysis of Cellulose. Russ. J. Phys. Chem. A 2005, 79, 973–980. [Google Scholar]
- Ficher, K.; Goldberg, W.; Wilke, M. Strahlenvorbehandlung von Zellstoff für die Regeneratfaserherstellung. Lenzing. Ber. 1985, 59, 32–39. [Google Scholar]
- Campbell, F.J. Radiation damage in organic materials. Radiat. Phys. Chem. 1977, 18, 109–123. [Google Scholar] [CrossRef]
- Bludenko, A.V.; Ponomarev, A.V.; Chulkov, V.N. Electron-beam degradation of wood: 1. Dry distillation products. High Energy Chem. 2009, 43, 83–86. [Google Scholar] [CrossRef]
- Parikka, M. Global biomass fuel resources. Biomass Bioenergy 2004, 27, 613–620. [Google Scholar] [CrossRef]
- Schell, C.; Riley, C.; Petersen, G.R. Pathways for development of a biorenewables industry. Bioresour. Technol. 2008, 99, 5160–5164. [Google Scholar] [CrossRef]
- Klemm, D.; Heublein, B.; Fink, H.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef]
- Szuromi, P.; Jasny, B.; Clery, D.; Austin, J.; Hanson, B. Energy for the long haul. Science 2007, 315, 781. [Google Scholar] [CrossRef]
- Kramer, G.J.; Haigh, M. No quick switch to low-carbon energy. Nature 2009, 462, 568–569. [Google Scholar] [CrossRef]
- Hoogwijk, M.; Faaij, A.; Eickhout, B.; de Vries, B.; Turkenburg, W. Potential of biomass energy out to 2100, for four IPCC SRES land-use scenarios. Biomass Bioenergy 2005, 29, 225–257. [Google Scholar] [CrossRef]
- Huber, G.W.; Chheda, J.N.; Barrett, C.J.; Dumesic, J.A. Production of liquid alkanes by aqueous-phase processing of biomassderived carbohydrates. Science 2005, 308, 1446–1450. [Google Scholar] [CrossRef]
- Fischer, G.; Schrattenholzer, L. Global bioenergy potentials through 2050. Biomass Bioenergy 2001, 20, 151–159. [Google Scholar] [CrossRef]
- Lynd, L.R.; Larson, E.; Greene, N.; Laser, M.; Sheehan, J.; Dale, B.E.; McLaughlin, S.; Wang, M. The role of biomass in America’s energy future: framing the analysis. Biofuels Bioprod. Biorefin. 2009, 3, 113–123. [Google Scholar] [CrossRef]
- Magnusson, L.; Islam, R.; Cicek, N.; Sparling, R.; Levin, D.B. Direct hydrogen production from cellulosic waste materials with a single-step dark fermentation process. Int. J. Hydrog. Energy 2008, 33, 5398–5403. [Google Scholar] [CrossRef]
- Talebnia, F.; Karakashev, D.; Angelidaki, I. Production of bioethanol from wheat straw: An overview on pretreatment, hydrolysis and fermentation. Bioresour. Technol. 2009, 101, 4744–4753. [Google Scholar] [CrossRef]
- Yamamoto, H.; Fujino, J.; Yamaji, K. Evaluation of bioenergy potential with a multi-regional global-land-use-and-energy model. Biomass Bioenergy 2001, 21, 185–203. [Google Scholar] [CrossRef]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–777. [Google Scholar] [CrossRef]
- Lynd, L.R.; Zyl, W.; McBride, J.; Laser, M. Consolidated bioprocessing of cellulosic biomass: An update. Curr. Opin. Biotechnol. 2005, 16, 577–583. [Google Scholar] [CrossRef]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef]
- Lin, C.Y.; Hung, W.C. Enhancement of fermentative hydrogen/ethanol production from cellulose using mixed anaerobic cultures. Int. J. Hydrog. Energy 2008, 33, 3660–3667. [Google Scholar] [CrossRef]
- Beguin, P.; Aubert, J.P. The biological degradation of cellulose. FEMS Microbiol. Rev. 1994, 13, 25–58. [Google Scholar] [CrossRef]
- David, K.; Ragauskas, A.J. Switchgrass as an energy crop for biofuel production: A review of its ligno-cellulosic chemical properties. Energy Environ. Sci. 2010, 3, 1182–1190. [Google Scholar] [CrossRef]
- Lynd, L.R.; Cushman, J.H.; Nichols, R.J.; Wyman, C.E. Fuel ethanol from cellulosic biomass. Science 1991, 251, 1318–1323. [Google Scholar] [CrossRef]
- Wyman, C.E. What is (and is not) vital to advancing cellulosic ethanol. Trends Biotechnol. 2007, 25, 153–157. [Google Scholar] [CrossRef]
- Balat, M. Production of bioethanol from lignocellulosic materials via the biochemical pathway: A review. Energy Convers. Manag. 2011, 52, 858–75. [Google Scholar] [CrossRef]
- Alan, E.W.; Basso, L.C.; Alves, D.M.G.; Amorim, H.V. Fuel ethanol after 25 years. Trends Biotechnol. 1999, 17, 482–487. [Google Scholar] [CrossRef]
- Sanchez, O.J.; Cardona, C.A. Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresour. Technol. 2008, 99, 5270–5295. [Google Scholar] [CrossRef]
- Hamelinck, C.N.; Hooijdonk, G.; Faaij, A.P.C. Ethanol from lignocellulosic biomass: Techno-economic performance in short-, middle- and long-term. Biomass Bioenergy 2005, 28, 384–410. [Google Scholar] [CrossRef]
- Carroll, A.; Somerville, C. Cellulosic biofuels. Annu. Rev. Plant Biol. 2009, 60, 165–182. [Google Scholar] [CrossRef]
- Hahn-Hägerdal, B.; Galbe, M.; Gorwa-Grauslund, M.F.; Liden, G.; Zacchi, G. Bio-ethanol: The fuel of tomorrow from the residues of today. Trends Biotechnol. 2006, 24, 549–556. [Google Scholar] [CrossRef]
- Kim, S.; Dale, B.E. Global potential bioethanol production from wasted crops and crop residues. Biomass Bioenergy 2004, 26, 361–375. [Google Scholar] [CrossRef]
- Lin, Y.; Tanaka, S. Ethanol fermentation from biomass resources: Current state and prospects. Appl. Microbiol. Biotechnol. 2006, 69, 627–642. [Google Scholar] [CrossRef]
- Ponomarev, A.V.; Tsivadze, A.Y. Wood radiolysis: Some aspects of liquid fuel manufacturing. Doklady Phys. Chem. 2009, 424, 1–4. [Google Scholar]
- Ponomarev, A.V.; Ershov, B.G. The deep decomposition of wood: light products of electron-beam fragmentation. In Wood Types, Properties and Uses; Botannini, L.F., Ed.; Nova Science Publishers: New York, NY, USA, 2010. [Google Scholar]
- Chulkov, V.N.; Bludenko, A.V.; Ponomarev, A.V.; Ershov, B.G. Electron beam distillation of straw. High Energy Chem. 2009, 43, 423–424. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Paul, S.F. Alternative Fuel. U.S. Patent 1997. [Google Scholar]
- Ponomarev, A.V.; Ershov, B.G. Radiation chemistry for biomass processing. In Actual Problems of High Energy Chemistry, Proceedings of the Fourth All-Russian Conference, Moscow, Russia, 2–3 November 2009.
- Ponomarev, A.V. Electron–beam radiolysis of gaseous alkanes under circulation conditions: Gas–to–liquid transformation. Radiat. Phys. Chem. 2009, 78, 48–56. [Google Scholar] [CrossRef]
- Ponomarev, A.V. Gas–to–liquid transformation of alkanes by electron–beam irradiation. Mendeleev Commun. 2006, 16, 256–258. [Google Scholar] [CrossRef]
- Kholodkova, E.M.; Tananaev, I.G.; Ponomarev, A.V. Electron-beam synthesis of carbon sorbents. In Proceedings of the Fifth Anniversary Conference “Ural Nuclear Industrial Complex: Problems and Perspectives”, Ozersk, Russia, 21–23 April 2009. Abstracts of Reports.
- Sazonov, A.B.; Tkhun, A.D.; Magomedbekov, E.P.; Ponomarev, A.V.; Tananaev, I.G.; Myasoedov, B.F. Carbon sorbents for fixation of hyzone-containing oil waste. Ross. Khim. Zh. 2010, 54, 94. [Google Scholar]
- Sample Availability: Samples of the condensates and charcoals are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarev, A.V.; Ershov, B.G. Radiation-Induced High-Temperature Conversion of Cellulose. Molecules 2014, 19, 16877-16908. https://doi.org/10.3390/molecules191016877
Ponomarev AV, Ershov BG. Radiation-Induced High-Temperature Conversion of Cellulose. Molecules. 2014; 19(10):16877-16908. https://doi.org/10.3390/molecules191016877
Chicago/Turabian StylePonomarev, Alexander V., and Boris G. Ershov. 2014. "Radiation-Induced High-Temperature Conversion of Cellulose" Molecules 19, no. 10: 16877-16908. https://doi.org/10.3390/molecules191016877
APA StylePonomarev, A. V., & Ershov, B. G. (2014). Radiation-Induced High-Temperature Conversion of Cellulose. Molecules, 19(10), 16877-16908. https://doi.org/10.3390/molecules191016877