Five New Iridoids from Roots of Salvia digitaloides

Abstract
:1. Introduction
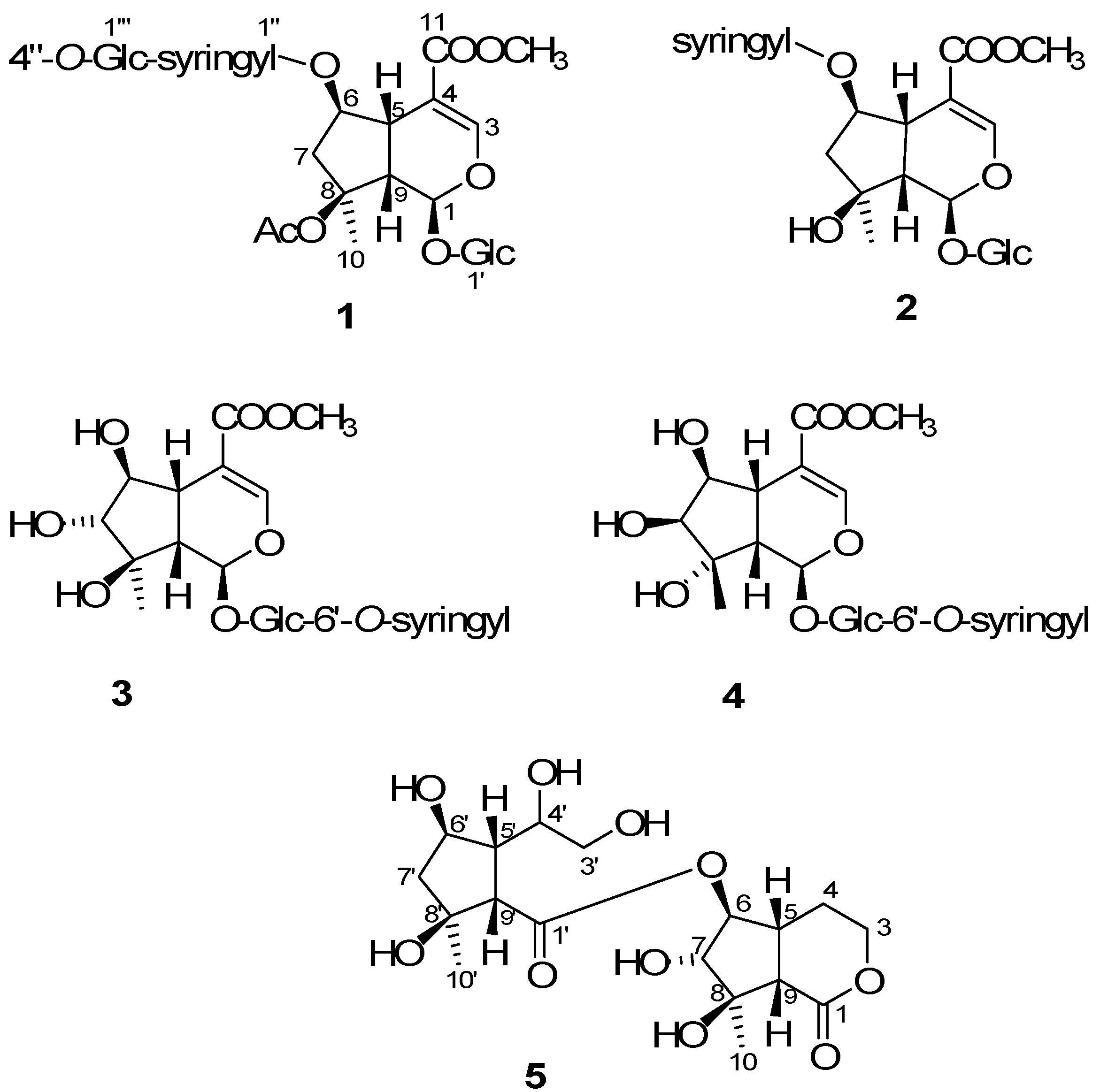
2. Results and Discussion
2.1. Purification and Characterization
2.2. Structural Elucidation of Compounds 1–5
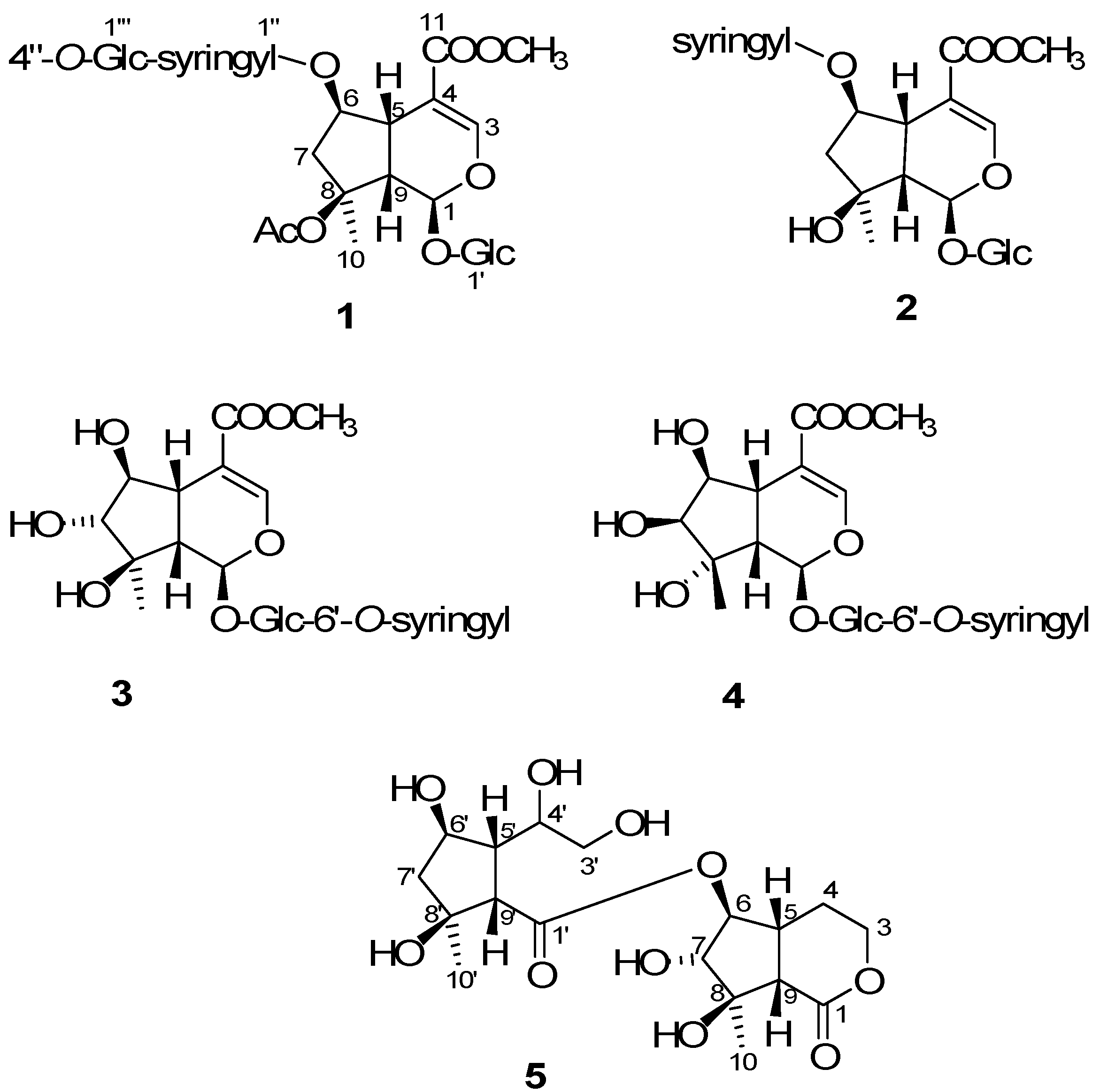
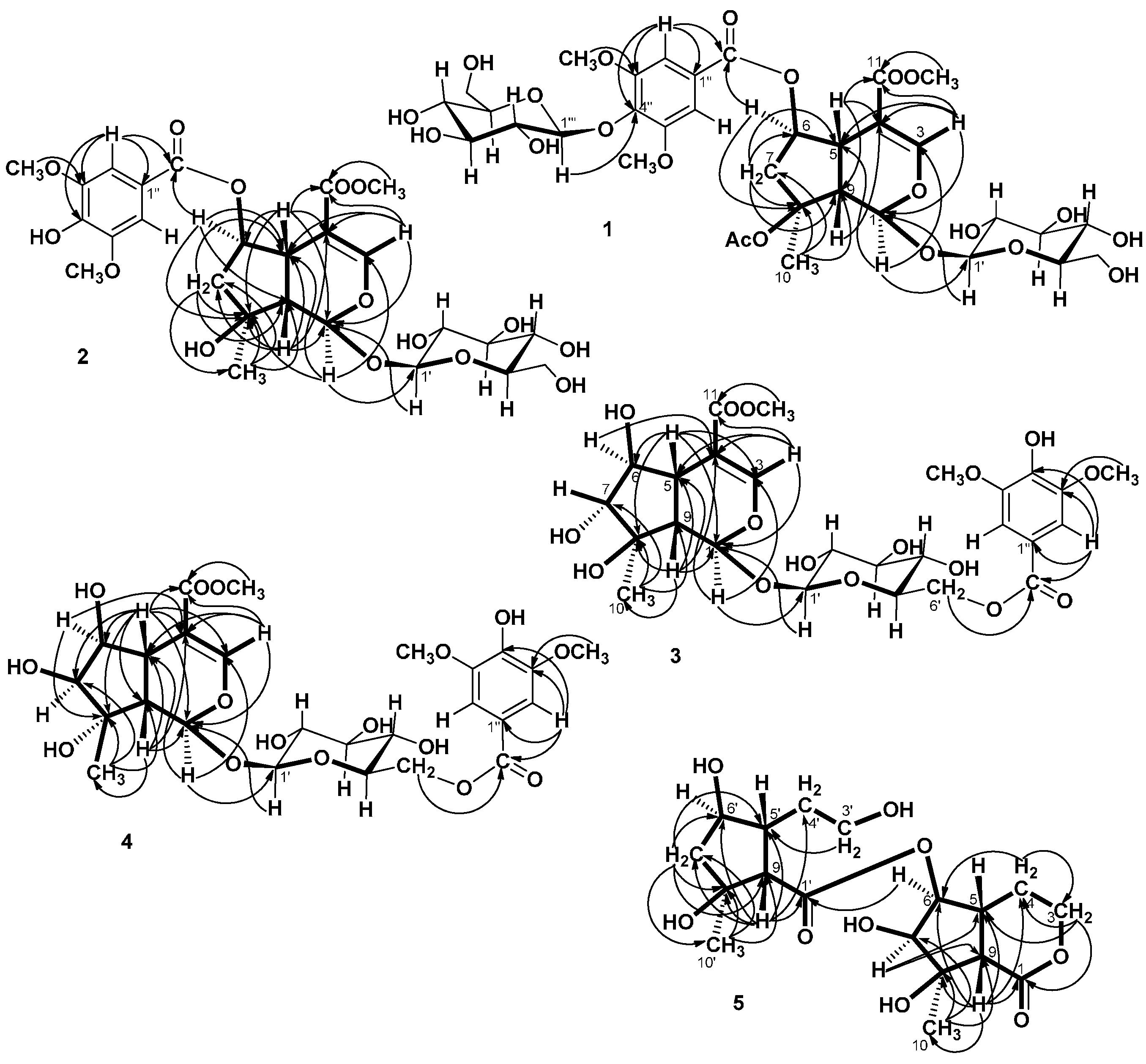
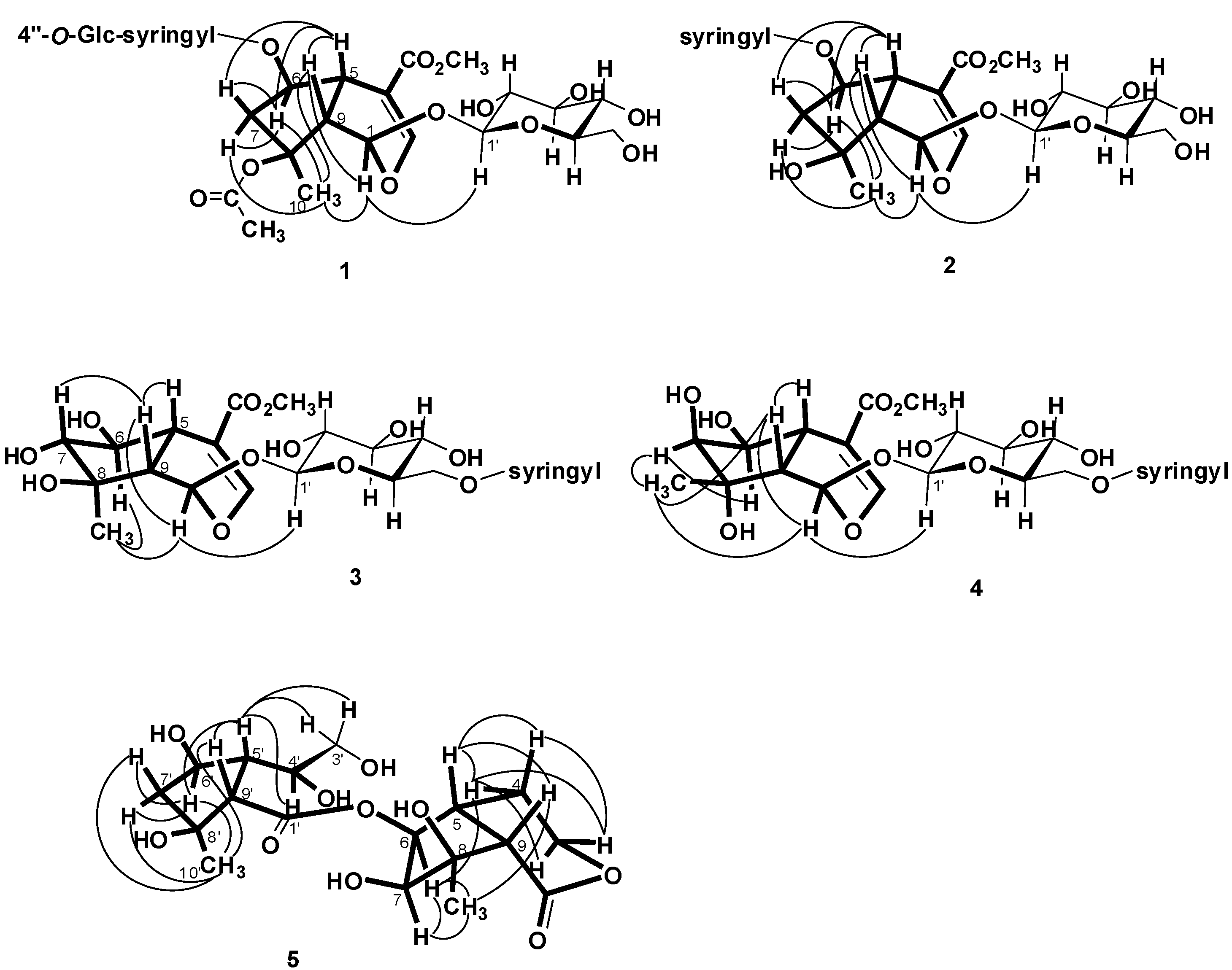
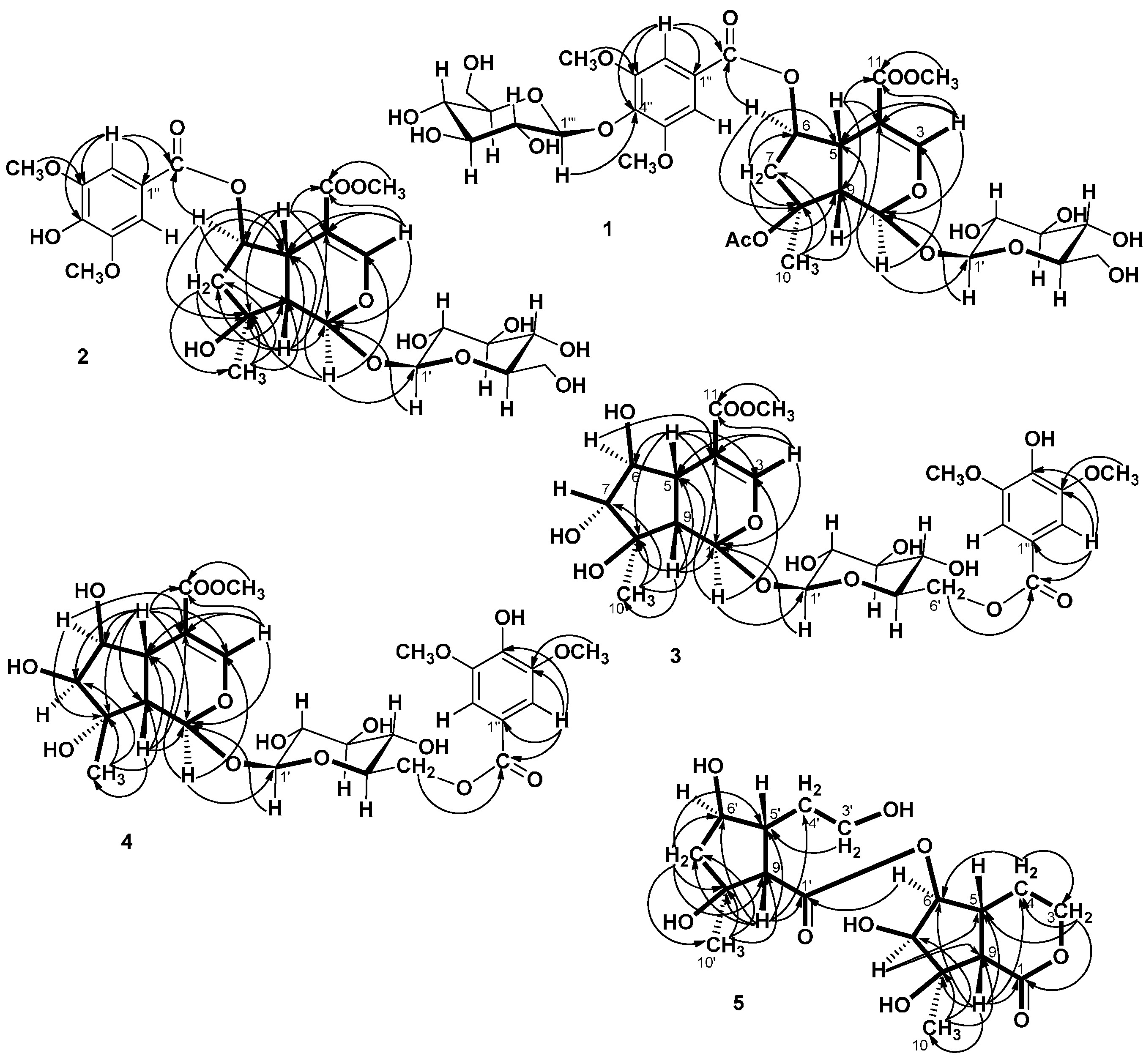
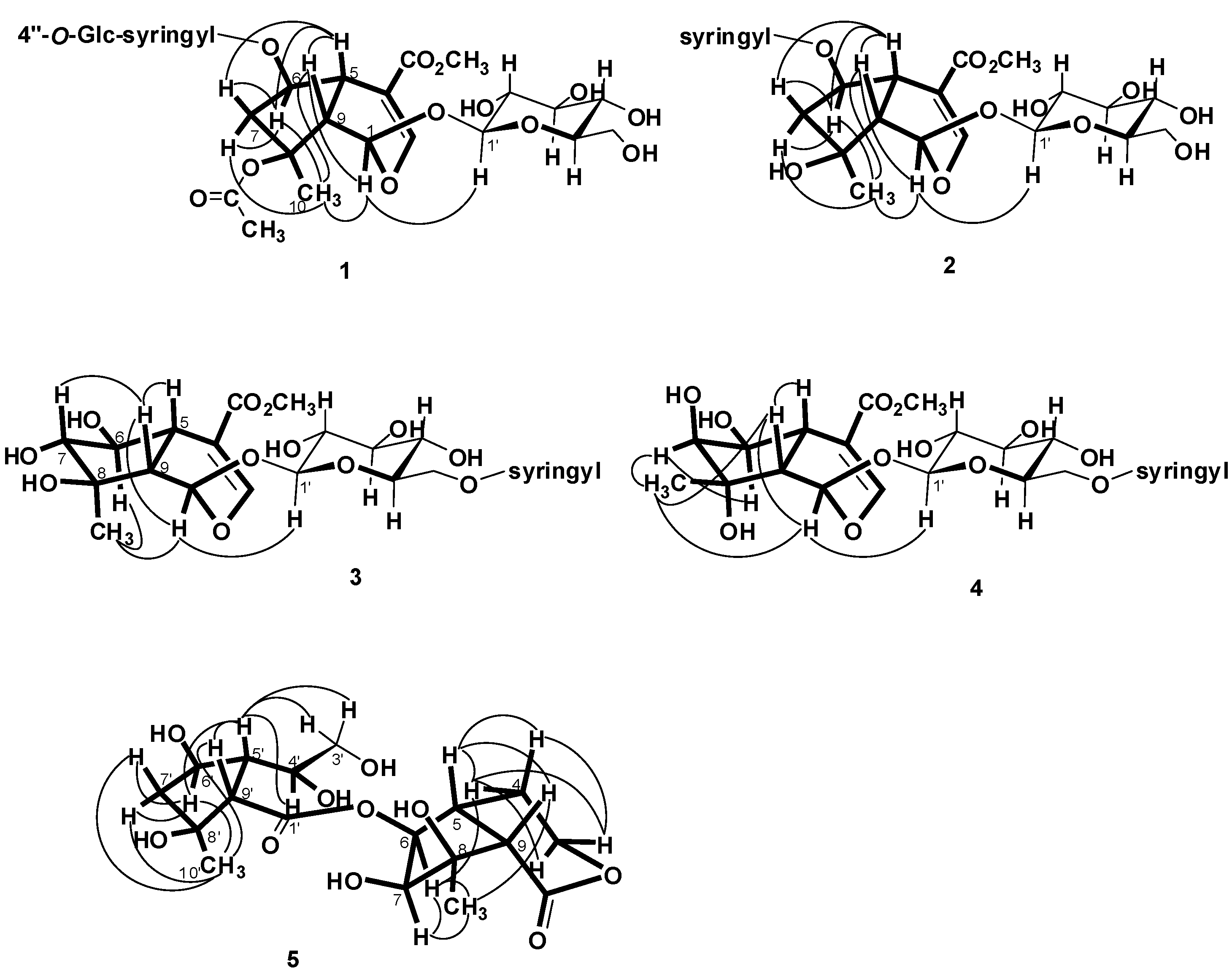
 = −36.0 (c 0.35, MeOH). The HR-ESIMS showed a molecular ion at m/z 813.2424, corresponding to the molecular formula C34H46O21Na. The UV spectrum revealed a very broad absorption band at 263 nm and the IR spectrum exhibited absorption maxima at 3367, 1705 and 1635 cm−1, indicating the presence of hydroxyl, α,β-unsaturated carbonyl and phenyl groups. The 1H- and 13C-NMR spectra showed great similarity with those of iridoid glucoside, 6-O-syringyl-8-O-acetylshanzhiside methyl ester (31). In the NMR spectrum (Table 1), signals for iridoid nucleus presented an enol ether unit [δH 7.57 (H-3); δC 154.5 (C-3), 108.6 (C-4)] with a carbomethoxy group [δH 3.67 (OCH3); δC 168.4 (C-11), 51.9 (OCH3)], an oxygenated methine [δH 5.51 (H-6); δC 79.9 (C-6)], a dioxygenated methine [δH 5.90 (H-1); δC 95.3 (C-1)], two aliphatic methine [δH 3.09 (H-9), 3.48 (H-5); δC 50.4 (C-9), 40.1 (C-5)], a methylene group [δH 2.27, 2.51 (H-7); δC 45.0 (C-9)], and a methyl group [δH 1.63 (H-10); δC 21.8 (C-10)]. Moreover, the iridoid structure was completely verified by the following HMBC correlations (Figure 2): H-1 with C-3/C-5, H-3 with C1/C-4/C-5/C-11, H-5 with C-4/C-9/ C-11, H-6 with C-4/ C-8, H-7 with C-5/C-6/C-8/C-9, and H-10 with C-7/C-8/C-9. The cis-fused cyclopentanodihydropyran ring system in iridoid was confirmed by the strong NOE correlation (Figure 3) between H-5 and H-9. In addition, a,β-glucopyranosyl moiety was characterized by the NMR signals at δH 4.71 (d, J = 8.0 Hz, H-1') and δC 62.5 (C-6'), 71.4 (C-4'), 74.7 (C-2'), 77.9 (C-3'), 78.5 (C-5'), and 100.2 (C-1'). The HMBC correlations (Figure 2) of H-1 with anomeric C-1' and anomeric H-1' with C-1, and the NOE correlation (Figure 3) between iridoid H-1 and glucose H-1' indicated a structure of iridoid 1-glucoside. A set of NMR signals at δH 7.38 (2H, s, H-2" and -6") and 3.93 (6H, s, 3"- and 5"-OCH3) as well as δC 57.3 (3"- and 5"-OCH3), 108.7 (C-2" and -6"), 127.4 (C-1"), 140.6 (C-4"), 154.3 (C-3" and -5"), and 166.8 (C=O) assigned to a syringic acid which made an ester linkage with 6-OH inferred at first from the deshielded H-6 (δ 5.51) and confirmed by the HMBC correlation of H-6 with syringyl C=O. The stereochemistry of H-5, H-9, and 1-O-glucosyl was supposed to adopt universally β-configuration. By careful examination of the NOESY spectrum, the β-face orientations of the 6-O-syringyl and 8-O-acetyl were proved by the presence of weak NOE correlation between the 8-CH3 (H-10) and H-6/H-1/H-7(α). In addition, the unexpected weak NOE correlations between H-9(β) and H-10(α), H-5(β) and H-6(α) were occasionally observed and it was noted that Jensen et al. [40] and Ersoz et al. [41] have claimed that in some conformations they were close in space and should indeed give rise to such NOEs. The remaining 1H- and 13C-NMR signals showed the other set of glucose at δH 5.13 (d, J = 7.8 Hz, H-1'") and δC 63.0 (C-6'"), 71.6 (C-4'"), 75.7 (C-2'"), 77.9 (C-3'"), 78.5 (C-5'"), and 104.3 (C-1'"). Again, the large coupling constant, 7.8 Hz, between H-1'" and H-2'" was in agreement with a β-glucose as the sugar unit.
= −36.0 (c 0.35, MeOH). The HR-ESIMS showed a molecular ion at m/z 813.2424, corresponding to the molecular formula C34H46O21Na. The UV spectrum revealed a very broad absorption band at 263 nm and the IR spectrum exhibited absorption maxima at 3367, 1705 and 1635 cm−1, indicating the presence of hydroxyl, α,β-unsaturated carbonyl and phenyl groups. The 1H- and 13C-NMR spectra showed great similarity with those of iridoid glucoside, 6-O-syringyl-8-O-acetylshanzhiside methyl ester (31). In the NMR spectrum (Table 1), signals for iridoid nucleus presented an enol ether unit [δH 7.57 (H-3); δC 154.5 (C-3), 108.6 (C-4)] with a carbomethoxy group [δH 3.67 (OCH3); δC 168.4 (C-11), 51.9 (OCH3)], an oxygenated methine [δH 5.51 (H-6); δC 79.9 (C-6)], a dioxygenated methine [δH 5.90 (H-1); δC 95.3 (C-1)], two aliphatic methine [δH 3.09 (H-9), 3.48 (H-5); δC 50.4 (C-9), 40.1 (C-5)], a methylene group [δH 2.27, 2.51 (H-7); δC 45.0 (C-9)], and a methyl group [δH 1.63 (H-10); δC 21.8 (C-10)]. Moreover, the iridoid structure was completely verified by the following HMBC correlations (Figure 2): H-1 with C-3/C-5, H-3 with C1/C-4/C-5/C-11, H-5 with C-4/C-9/ C-11, H-6 with C-4/ C-8, H-7 with C-5/C-6/C-8/C-9, and H-10 with C-7/C-8/C-9. The cis-fused cyclopentanodihydropyran ring system in iridoid was confirmed by the strong NOE correlation (Figure 3) between H-5 and H-9. In addition, a,β-glucopyranosyl moiety was characterized by the NMR signals at δH 4.71 (d, J = 8.0 Hz, H-1') and δC 62.5 (C-6'), 71.4 (C-4'), 74.7 (C-2'), 77.9 (C-3'), 78.5 (C-5'), and 100.2 (C-1'). The HMBC correlations (Figure 2) of H-1 with anomeric C-1' and anomeric H-1' with C-1, and the NOE correlation (Figure 3) between iridoid H-1 and glucose H-1' indicated a structure of iridoid 1-glucoside. A set of NMR signals at δH 7.38 (2H, s, H-2" and -6") and 3.93 (6H, s, 3"- and 5"-OCH3) as well as δC 57.3 (3"- and 5"-OCH3), 108.7 (C-2" and -6"), 127.4 (C-1"), 140.6 (C-4"), 154.3 (C-3" and -5"), and 166.8 (C=O) assigned to a syringic acid which made an ester linkage with 6-OH inferred at first from the deshielded H-6 (δ 5.51) and confirmed by the HMBC correlation of H-6 with syringyl C=O. The stereochemistry of H-5, H-9, and 1-O-glucosyl was supposed to adopt universally β-configuration. By careful examination of the NOESY spectrum, the β-face orientations of the 6-O-syringyl and 8-O-acetyl were proved by the presence of weak NOE correlation between the 8-CH3 (H-10) and H-6/H-1/H-7(α). In addition, the unexpected weak NOE correlations between H-9(β) and H-10(α), H-5(β) and H-6(α) were occasionally observed and it was noted that Jensen et al. [40] and Ersoz et al. [41] have claimed that in some conformations they were close in space and should indeed give rise to such NOEs. The remaining 1H- and 13C-NMR signals showed the other set of glucose at δH 5.13 (d, J = 7.8 Hz, H-1'") and δC 63.0 (C-6'"), 71.6 (C-4'"), 75.7 (C-2'"), 77.9 (C-3'"), 78.5 (C-5'"), and 104.3 (C-1'"). Again, the large coupling constant, 7.8 Hz, between H-1'" and H-2'" was in agreement with a β-glucose as the sugar unit.
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |||||||||
| 1 | 5.90 d (3.6) | 95.3 | 5.55 d (4.8) | 93.9 | 5.56 d (1.4) | 92.4 | 5.48 d (2.2) | 95.2 | ||||||||
| 3 | 7.57 d (1.4) | 154.5 | 7.47 d (1.0) | 152.1 | 7.38 d (1.2) | 150.9 | 7.41 d (1.2) | 153.2 | ||||||||
| 4 | 108.6 | 110.7 | 111.1 | 111.9 | ||||||||||||
| 5 | 3.48 ddd (8.8, 2.9, 1.4) | 40.1 | 3.52 ddd (9.0, 3.6, 1.0) | 37.1 | 2.70 ddd (11.2, 4.8, 1.2) | 34.3 | 2.82 ddd (10.9, 5.2, 1.2) | 38.3 | ||||||||
| 6 | 5.51 dd (5.6, 2.9) | 79.9 | 5.52 ddd (7.4, 4.2, 3.6) | 78.4 | 3.48 dd (8.0, 4.8) | 80.5 | 4.06 dd (5.2, 4.4) | 79.5 | ||||||||
| 7 | α: 2.27 dd (15.4, 5.6) β: 2.51 d (15.4) | 45.0 | β: 1.93 dd (14.0, 4.2) α: 2.34 dd (14.0, 7.4) | 46.4 | 3.73 d (8.0) | 84.4 | 3.60 d (4.4) | 81.2 | ||||||||
| 8 | 89.8 | 79.6 | 76.7 | 80.8 | ||||||||||||
| 9 | 3.09 dd (8.8, 3.6) | 50.4 | 2.54 dd (9.0, 4.8) | 50.6 | 2.53 dd (11.2, 1.4) | 45.7 | 2.54 dd (10.9, 2.2) | 47.6 | ||||||||
| 10 | 1.63 s | 21.8 | 1.37 s | 21.9 | 1.03 s | 16.5 | 1.27 s | 23.1 | ||||||||
| 11 | 168.4 | 167.7 | 168.7 | 170.6 | ||||||||||||
| OCH3 | 3.67 s | 51.9 | 3.56 s | 50.4 | 3.72 s | 50.8 | 3.73 s | 52.0 | ||||||||
| 8-OAc | 1.92 s | 22.3, 172.6 | ||||||||||||||
| 1' | 4.71 d (8.0) | 100.2 | 4.71 d (8.0) | 98.9 | 4.66 d (8.0) | 98.3 | 4.69 d (8.0) | 100.2 | ||||||||
| 2' | 3.22 dd (9.2, 8.0) | 74.7 | 3.20 dd (9.0, 8.0) | 73.4 | 3.19 dd (9.2, 8.0) | 73.4 | 3.21 dd (9.2, 8.0) | 74.6 | ||||||||
| 3' | 3.30–3.49 m | 77.9 | 3.37 t (9.0) | 74.6 | 3.41 m | 76.6 | 3.41 m | 77.8 | ||||||||
| 4' | 3.30–3.49 m | 71.4 a | 3.22 t (9.0) | 70.6 | 3.41 m | 70.4 | 3.41 m | 71.8 | ||||||||
| 5' | 3.30–3.49 m | 78.5 | 3.35 m | 76.6 | 3.62 m | 74.6 | 3.68 m | 75.8 | ||||||||
| 6' | 3.66 dd (12.0, 5.6) c 3.79 dd (12.0, 2.3) c | 62.5 b | 3.64 dd (11.8, 6.3) 3.92 dd (11.8, 2.4) | 63.8 | 4.39 dd (12.0, 5.4) 4.66 dd (12.0, 2.0) | 63.8 | 4.44 dd (12.0, 6.1) 4.66 dd (12.0, 2.1) | 65.0 | ||||||||
| 1" | 127.4 | 120.7 | 120.0 | 121.2 | ||||||||||||
| 2", 6" | 7.38 s | 108.7 | 7.39 s | 108.7 | 7.33 s | 107.1 | 7.35 s | 108.3 | ||||||||
| 3", 5" | 154.3 | 147.8 | 147.8 | 148.9 | ||||||||||||
| 4" | 140.6 | 140.6 | 140.6 | 142.1 | ||||||||||||
| 1"-C=O | 166.8 | 166.8 | 166.8 | 167.9 | ||||||||||||
| 3",5"-OCH3 | 3.93 s | 57.3 | 3.89 s | 55.7 | 3.89 s | 55.7 | 3.88 s | 56.9 | ||||||||
| 1'" | 5.13 d (7.8) | 104.3 | ||||||||||||||
| 2'" | 3.52 dd (9.2, 7.8) | 75.7 | ||||||||||||||
| 3'" | 3.30–3.49 m | 77.9 | ||||||||||||||
| 4'" | 3.30–3.49 m | 71.6 a' | ||||||||||||||
| 5'" | 3.30–3.49 m | 78.5 | ||||||||||||||
| 6'" | 3.69 dd (12.2, 5.6) c' 3.94 dd (12.0, 2.2) c' | 63.0 b' | ||||||||||||||
| Position | δH (J in Hz) | δC |
|---|---|---|
| 1 | 174.8 | |
| 3 | β (eq): 4.25 dtd (11.2, 3.6, 1.2) α (ax): 4.50 td (11.2, 2.3) | 67.5 |
| 4 | α (eq): 1.82 dtd (11.2, 3.6, 2.3) β (ax): 2.05 tdd (11.2, 6.5, 3.6) | 26.4 |
| 5 | 2.43 m | 40.8 |
| 6 | 4.28 dd (8.3, 3.8) | 77.8 |
| 7 | 3.56 d (3.8) | 81.2 |
| 8 | 83.0 | |
| 9 | 3.03 d (11.5) | 51.4 |
| 10 | 1.50 s | 22.5 |
| 1' | 178.3 | |
| 3' | 3.64 dd (12.2, 3.8) 3.78 dd (12.2, 2.9) | 64.7 |
| 4' | 4.32 td (3.8, 2.9) | 85.1 |
| 5' | 3.00 ddd (9.6, 3.8, 3.0) | 52.5 |
| 6' | 4.17 dt (5.0, 3.0) | 78.8 |
| 7' | 1.88 dd (13.8, 5.0) 1.96 ddd (13.8, 3.0, 1.2) | 48.4 |
| 8' | 82.1 | |
| 9' | 3.17 dd (9.6, 1.2) | 58.2 |
| 10' | 1.43 s | 24.5 |
3. Experimental Section
3.1. General Information
3.2. Plant Materials
3.3. Extraction and Isolation
3.3.1. Salvialoside A (1)
= −36.0 (c 0.35, MeOH); UV (MeOH), λmax (log ε) 216 (3.95), 263 (3.61) nm; IR(KBr) νmax: 3367, 1705, 1635 cm−1; 1H- and 13C-NMR see Table 1 ; ESIMS m/z (rel. int.): 813 [M+Na]+; HRESIMS m/z: 813.2424 [M+Na]+ (calcd. for C34H46O21Na, 813.2429).3.3.2. Salvialoside B (2)
= −103.4 (c 0.99, MeOH); UV (MeOH), λmax (log ε) 220 (4.41), 277 (4.01 ) nm; IR(KBr) νmax: 3367, 1701 cm−1; 1H- and 13C-NMR see Table 1; ESIMS m/z (rel. int.): 609 [M+Na]+; HRESIMS m/z: 609.1791 [M+Na]+ (calcd. for C26H34O15Na, 609.1795).3.3.3. Salvialoside C (3)
= −80.0 (c 0.24, MeOH); UV (MeOH), λmax (log ε) 220 (4.60), 277 (4.16) nm; IR(KBr) νmax: 3379, 1693 cm−1; 1H- and 13C-NMR see Table 1; ESIMS m/z (rel. int.): 625 [M+Na]+; HRESIMS m/z: 625.1740 [M+Na]+ (calcd. for C26H34O16Na, 625.1742).3.3.4. Salvialoside D (4)
= −14.5 (c 0.42, MeOH); UV (MeOH), λmax (log ε) 220 (4.51), 276 (4.07) nm; IR(KBr) νmax: 3329, 1697 cm−1; 1H- and 13C-NMR see Table 1; ESIMS m/z (rel. int.): 625 [M+Na]+; HRESIMS m/z: 625.1742 [M+Na]+ (calcd. for C26H34O16Na, 625.1744).3.3.5. Salvialoside E (5)
= −0.80 (c 0.48, MeOH); UV (MeOH), λmax (log ε) 217 (3.41) nm; IR(KBr) νmax: 3383,1747, 1708 cm−1; 1H- and 13C-NMR see Table 2; ESIMS m/z (rel. int.): 427 [M+Na]+; HRESIMS m/z: 427.1580 [M+Na]+ (calcd for C18H28O10Na, 427.1582).4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, C.Y.; Li, H.W. On the evolution and distribution in Labeatae. Acta Bot. Yunnan 1982, 4, 97–118. [Google Scholar]
- Ryu, S.Y.; Lee, C.O.; Choi, S.U. In vitro cytotoxicity of tanshinones from Salvia miltiorrhiza. Planta Med. 1997, 63, 339–342. [Google Scholar]
- Ryu, S.Y.; Oak, M.H.; Kim, K.M. Inhibition of mast cell degranulation by tanshinones from the roots of Salvia miltiorrhiza. Planta Med. 1999, 65, 654–655. [Google Scholar]
- Cao, E.H.; Liu, X.Q.; Wang, J.-J.; Xu, N.-F. Effect of natural antioxidant tanshinone II-A on DNA damage by lipid peroxidation in liver cells. Free Radic. Biol. Med. 1996, 20, 801–806. [Google Scholar]
- Honda, G.; Koezuka, Y.; Tabata, M. Isolation of an antidermatophytic substance from the root of Salvia miltiorrhiza. Chem. Pharm. Bull. 1988, 36, 408–411. [Google Scholar]
- Lin, H.C.; Ding, H.Y.; Chang, W.L. Two new fatty diterpenoids from Salvia miltiorrhiza. J. Nat. Prod. 2001, 64, 648–650. [Google Scholar]
- Kunming Institute of Botany, Chinese Academy of Sciences. Flora Yunnannica, Science Press: Beijing, China, 1977; Volume 1, 661.
- Tanaka, T.; Tanaka, O.; Lin, Z.W.; Zhou, J. Sweet and bitter principles of the Chinese plant drug, bai-yun-shen: Revision of the assignment of the source plant and isolation of two new diterpene glycosides. Chem. Pharm. Bull. 1985, 33, 4275–4280. [Google Scholar]
- Tanaka, T.; Tanaka, O.; Lin, Z.W.; Zhou, J.; Ageta, H. Sweet and bitter glycosides of the Chinese plant drug, bai-yun-shen (roots of Salvia digitaloides). Chem. Pharm. Bull. 1983, 31, 780–783. [Google Scholar]
- Zhou, W.; Guo, S. Components of the sclerotia of Polyporus umbellatus. Chem. Nat. Compd. 2009, 45, 124–125. [Google Scholar]
- Wu, T.S.; Hwang, C.C.; Kuo, P.C.; Kuo, T.H.; Damu, A.G.; Su, C.R. New neolignans from Spiraea formosana. Chem. Pharm. Bull. 2004, 52, 1227–1230. [Google Scholar]
- Buchalter, L. Isolation and identification of emodin (1,3,8-trihydroxy-6-methylanthraquinone) from Rumex hymenosepalus, family polygonaceae. J. Pharm. Sci. 1969, 58, 904. [Google Scholar]
- Tan, J.; Bednarek, P.; Liu, J.K.; Schneider, B.; Svatos, A.; Hahlbrock, K. Universally occurring phenylpropanoid and species-specific indolic metabolites in infected and uninfected Arabidopsis thaliana roots and leaves. Phytochemistry 2004, 65, 691–699. [Google Scholar]
- Ioannou, E.; Abdel-Razik, A.F.; Christofidis, D.; Vagias, C.; Roussis, V.; Zervou, M.; Alexi, X.; Alexis, M.N. 5α, 8α-Epidioxysterols from the gorgonian Eunicella cavolini and the ascidian Trididemnum inarmatum: Isolation and evaluation of their antiproliferative activity. Steroids 2009, 74, 73–80. [Google Scholar]
- Pelc, B.; Kodicek, E. [G-3H] ergosterol; location of tritium in rings A and B. J. Chem. Soc. Perkin Trans. 1 1972. [Google Scholar] [CrossRef]
- Songue, J.L.; Kouam; Dongo, E.; Mpondo, T.N.; White, R.L. Chemical constituents from stem bark and roots of Clausena anisata. Molecules 2012, 17, 13673–13686. [Google Scholar]
- Saied, S.; Begum, S. Phytochemical studies of Berberis vulgaris. Chem. Nat. Comp. 2004, 40, 137–140. [Google Scholar]
- Lee, C.K.; Lu, C.K.; Kuo, Y.H.; Chen, J.Z.; Sun, G.Z. New prenylated flavones from the roots of Ficus beecheyana. J. Chin. Chem. Soc. 2004, 51, 437–441. [Google Scholar]
- Wu, S.J.; Chan, H.H.; Hwang, T.L.; Qian, K.; Morris-Natschke, S.; Lee, K.H.; Wu, T.S. Salviatalin A and salvitrijudin A, two diterpenes with novel skeletons from roots of Salvia digitaloides and anti-inflammatory evaluation. Tetrahedron Lett. 2010, 51, 4287–4290. [Google Scholar]
- Mshvildadze, V.; Elais, R.; Faure, R.; Debrauwer, L.; Dekanoside, G.; Kemertelidze, E.; Balansard, G. Triterpenoid saponins from berries of Hedera colchica. Chem. Pharm. Bull. 2001, 49, 752–754. [Google Scholar]
- Dou, H.; Zhou, Y.; Chen, C.; Peng, S.; Liao, X.; Ding, L. Chemical constituents of the aerial parts of Schnabelia tetradonta. J. Nat. Prod. 2001, 65, 1777–1781. [Google Scholar]
- Wu, S.J.; Hwang, C.H.; Chan, Y.Y.; Liao, Y.R.; Hwang, T.L.; Wu, T.S. Two diterpenoids and a cyclopenta[c]pyridine derivative from roots of Salvia digitaloides. Int. J. Mol. Sci. 2014, 15, 11566–11577. [Google Scholar]
- Choi, J.N.; Kim, J.; Lee, M.Y.; Park, D.K.; Hong, Y.S.; Lee, C.H. Metabolomics revealed novel isoflavones and optimal cultivation time of Cordyceps militaris fermentation. J. Agric. Food Chem. 2010, 58, 4258–4267. [Google Scholar]
- Sakushima, A.; Coskun, M.; Maoka, T.; Nishibe, S. Dihydrobenzofuran lignans from Boreava orientalis. Phytochemistry 1996, 43, 1349–1354. [Google Scholar]
- Gil, M.; Wang, Y.; Douhal, A. Ultrafast dynamics of lumichrome in solution and in chemical and biological caging media. J. Photochem. Photobiol. A 2012, 234, 146–155. [Google Scholar]
- Ma, S.J.; Mizutani, M.; Hiratake, J.; Hayashi, K.; Yagi, K.; Watanabe, N.; Sakata, K. Substrate specificity of β-primeverosidase, A key enzyme in aroma formation during oolong tea and black tea manufacturing. Biosci. Biotechnol. Biochem. 2001, 65, 2719–2729. [Google Scholar]
- Galve, R.; Camps, F.; Sanchezbaeza, F.; Marco, M.P. Development of an immunochemical technique for the analysis of trichlorophenols using theoretical models. Anal. Chem. 2000, 72, 2237–2246. [Google Scholar]
- Nilsson, M.; Duarte, I.F.; Almeida, C.; Delgadillo, I.; Goodfellow, B.J.; Gil, A.M.; Morris, G.A. High-resolution NMR and diffusion-ordered spectroscopy of port wine. J. Agric. Food Chem. 2004, 52, 3736–3743. [Google Scholar]
- Tan, J.J.; Tan, C.H.; Li, M.; Jiang, S.H.; Zhu, D.Y. Iridoid glycosides from Lamiophlomis rotate. Helv. Chim. Acta 2007, 90, 143–148. [Google Scholar]
- Wang, M.; Kikuzaki, H.; Zhu, N.; Sang, S.; Nakatani, N.; Ho, C.T. Isolation and structural elucidation of two new glycosides from Sage (Salvia officinalis L.). J. Agric. Food Chem. 2000, 48, 235–238. [Google Scholar]
- Ismail, L.D.; El-Azizi, M.M.; Khalifa, T.I.; Stermitz, F.R. Verbascoside derivatives and iridoid glycosides from Penstemon crandallii. Phytochemistry 1995, 39, 1391–1393. [Google Scholar]
- Kijiuma, H.; Ide, T.; Otsuka, H.; Ogimi, C.; Hirata, E.; Takushi, A.; Takeda, Y. Water-soluble phenolic glycoside from leaves of Alangium premnifolium. Phytochemistry 1997, 44, 1551–1557. [Google Scholar]
- Zhao, M.; Bi, L.; Wang, W.; Floch, M.B.; Ju, J.; Peng, S. Synthesis and cytotoxic activities of β-carboline amino acid ester conjugates. Bioorg. Med. Chem. 2006, 14, 6998–7010. [Google Scholar]
- Zhong, C.Z.; Li, C.; Feng, S.I.; Shi, J.G. Iridoid glucosides from Phlomis rotata. Phytochemistry 1991, 30, 4156–4158. [Google Scholar]
- Jia, Z.; Liu, Z.; Wang, C. Phenylpropanoid and iridoid glycosides from Pedicularis lasiophry. Phytochemistry 1992, 31, 263–266. [Google Scholar]
- Bhat, B.; Harrison, D.M.; Lamont, H.M. The biosynthesis of the mould metabolites roquefortine and aszonalenin from l-[2,4,5,6,7–2H5]Tryptophan. Tetrahedron 1993, 49, 10663–10668. [Google Scholar]
- Miles, H.T.; Smyrnitis, P.Z.; Stadtman, E.R. Bacterial degradation products of riboflavin. III. Isolation, structure determination and biological transformations of 1-ribityl-2,3-diketo-1,2,3,4-tetrahydro-6,7-dimtehylquinoxaline. J. Am. Chem. Soc. 1959, 81, 1946–1951. [Google Scholar]
- Capasso, R.; Cristinzio, G.; Evidente, A.; Scognamiglio, F. Isolation, spectroscopy and selective phytotoxic effects of polyphenols from vegetable waste waters. Phytochemistry 1992, 31, 4125–4128. [Google Scholar]
- Morota, T.; Nishimura, H.; Sasaki, H.; Chin, M.; Sugama, K.; Katsuhara, T.; Mitsuhashi, H. Five cyclopentanoid monoterpenes from Rehmannia glutinosa. Phytochemistry 1989, 28, 2385–2391. [Google Scholar]
- Jensen, S.R.; Cü alis, I.; Gotfredsen, C.H.; Søtofte, I. Structural revision of some recently published iridoid glucosides. J. Nat. Prod. 2007, 70, 29–32. [Google Scholar]
- Ersoz, T.; Kaya, D.; Yalcin, F.N.; Kazaz, C.; Palaska, E.; Gotfredsen, C.H.; Jensen, S.R.; Calis, I. Iridoid glucosides from Lamium garganicum subsp. laevigatum. Turk. J. Chem. 2007, 31, 155–162. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.-J.; Chan, Y.-Y. Five New Iridoids from Roots of Salvia digitaloides. Molecules 2014, 19, 15521-15534. https://doi.org/10.3390/molecules191015521
Wu S-J, Chan Y-Y. Five New Iridoids from Roots of Salvia digitaloides. Molecules. 2014; 19(10):15521-15534. https://doi.org/10.3390/molecules191015521
Chicago/Turabian StyleWu, Shwu-Jen, and Yu-Yi Chan. 2014. "Five New Iridoids from Roots of Salvia digitaloides" Molecules 19, no. 10: 15521-15534. https://doi.org/10.3390/molecules191015521
APA StyleWu, S.-J., & Chan, Y.-Y. (2014). Five New Iridoids from Roots of Salvia digitaloides. Molecules, 19(10), 15521-15534. https://doi.org/10.3390/molecules191015521